
Characteristics of the New Insulin-Resistant Zebrafish Model

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Short-Term Insulin Treatment
2.2. Long-Term Insulin Treatment
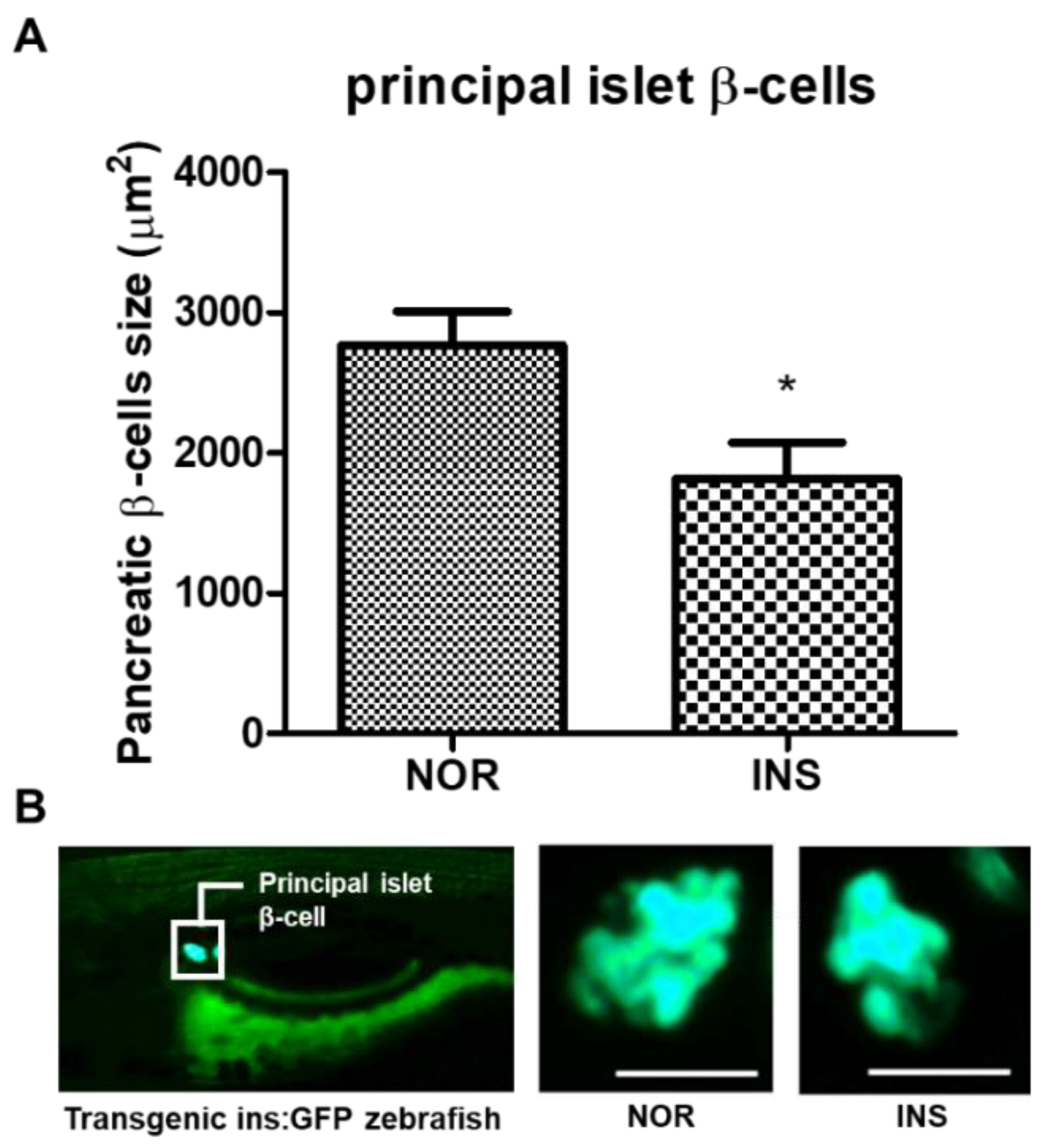
2.3. Change of the Islet β-Cells with Excess Insulin Treatment
2.4. Glucose Tolerance
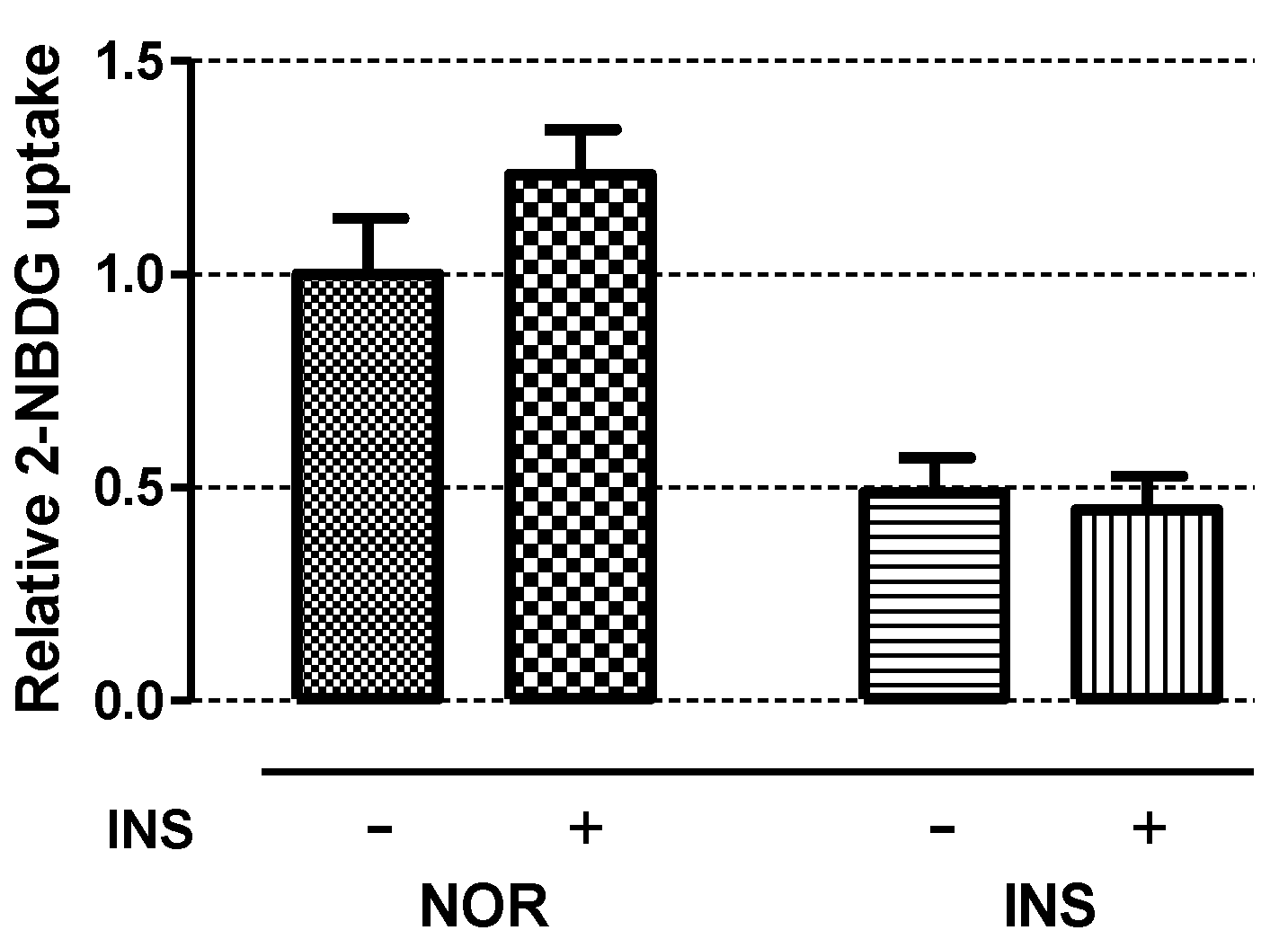
2.5. Insulin Sensitivity
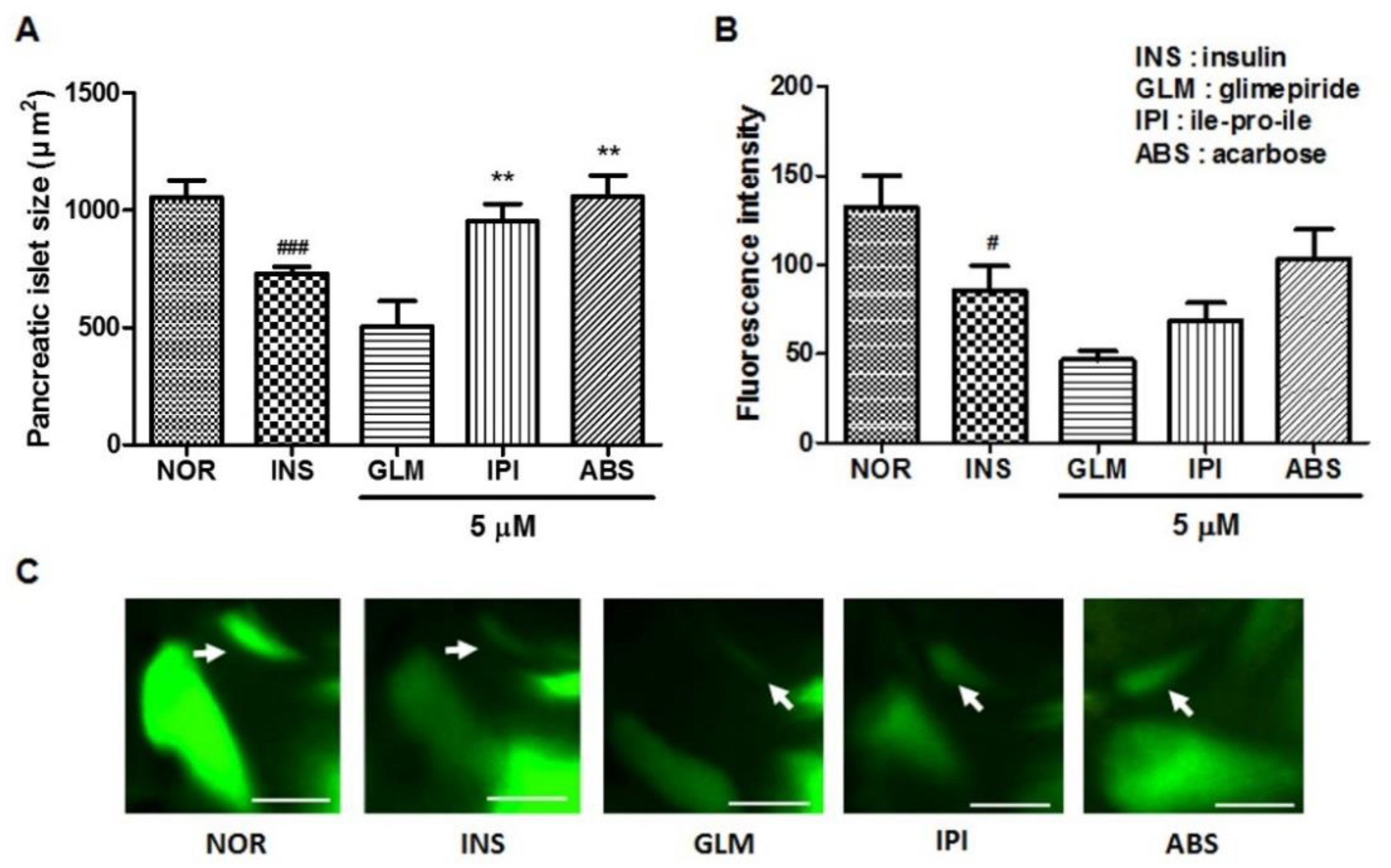
2.6. Efficacy of Glimepiride (GLM), ile-pro-ile (IPI) and Acarbose (ABS) on Insulin-Treated Zebrafish Larvae
2.7. Efficacy of Pioglitazone (PIO) on Insulin-Treated Zebrafish Larvae
2.8. Free Glucose Level in Insulin-Treated Zebrafish Larvae
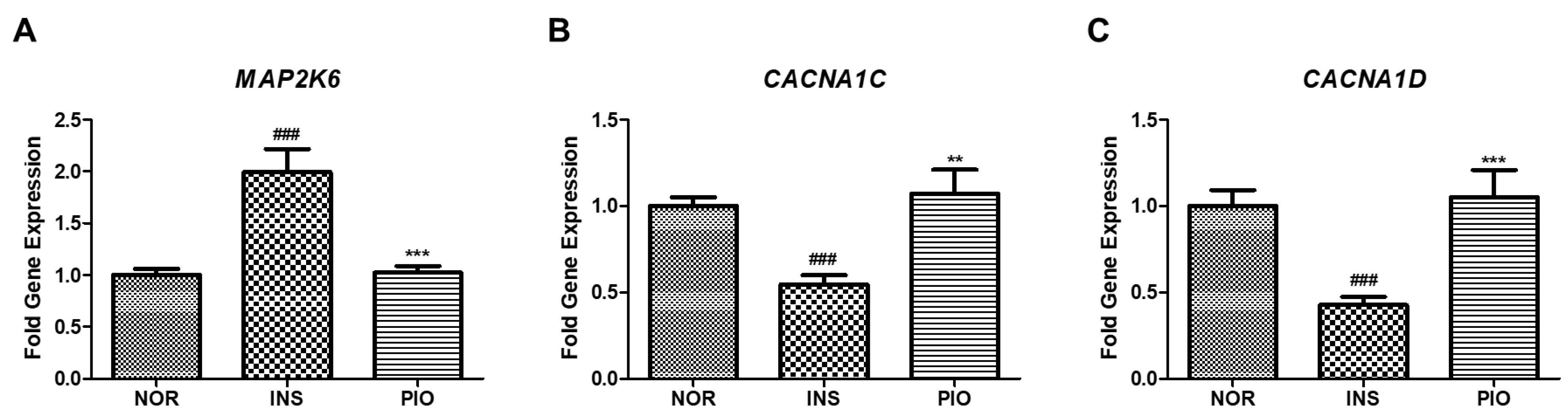
2.9. Differential Gene Expression Induced by Insulin-Treated Zebrafish
2.10. Effect of PIO in Altered Gene Expression on Insulin-Treated Zebrafish
3. Discussion
4. Materials and Methods
4.1. Reagents and Equipment
4.2. Zebrafish Maintenance and Embryo Collection
4.3. Short-Term Insulin Treatment
4.4. Long-Term Insulin Treatment
4.5. Change of Islet β-Cells by Excess Insulin Treatment
4.6. Glucose Tolerance Test
4.7. Insulin Sensitivity
4.8. Treatment of Glimepiride (GLM), ile-pro-ile (IPI), Acarbose (ABS), Pioglitazone (PIO) on Excess Insulin-Treated Zebrafish Larvae
4.9. Free Glucose Assay
4.10. mRNA Sequencing and Pathway Analysis
4.11. Real Time-qPCR
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kahn, B.B.; Flier, J.S. Obesity and insulin resistance. J. Clin. Investig. 2000, 106, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Mei, S.; Gu, H.; Guo, H.; Zha, L.; Cai, J.; Li, X.; Liu, Z.; Cao, W. Exposure to excess insulin (glargine) induces type 2 diabetes mellitus in mice fed on a chow diet. J. Endocrinol. 2014, 221, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Olefsky, J.M. Effect of experimental hyperinsulinemia on insulin binding and glucose transport in isolated rat adipocytes. Am. J. Physiol. 1978, 235, E53–E62. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Desai, K.S.; Steiner, G. Receptor and postreceptor insulin resistance induced by in vivo hyperinsulinemia. Can. J. Physiol. Pharmacol. 1983, 61, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Nolan, C.J. Islet β cell failure in type 2 diabetes. J. Clin. Investig. 2006, 116, 1802–1812. [Google Scholar] [CrossRef]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Flamment, M.; Hajduch, E.; Ferré, P.; Foufelle, F. New insights into ER stress-induced insulin resistance. Trends Endocrinol. Metab. 2012, 23, 381–390. [Google Scholar] [CrossRef]
- Samuel, V.T.; Petersen, K.F.; Shulman, G.I. Lipid-induced insulin resistance: Unravelling the mechanism. Lancet 2010, 375, 2267–2277. [Google Scholar] [CrossRef]
- Ozcan, L.; Tabas, I. Calcium signalling and ER stress in insulin resistance and atherosclerosis. J. Intern. Med. 2016, 280, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Nam, Y.H.; Hong, B.N.; Rodriguez, I.; Ji, M.G.; Kim, K.; Kim, U.J.; Kang, T.H. Synergistic Potentials of Coffee on Injured Pancreatic Islets and Insulin Action via KATP Channel Blocking in Zebrafish. J. Agric. Food Chem. 2015, 63, 5612–5621. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M. ATP-sensitive potassium channelopathies: Focus on insulin secretion. J. Clin. Investig. 2005, 115, 2047–2058. [Google Scholar] [CrossRef] [PubMed]
- Huopio, H.; Shyng, S.L.; Otonkoski, T.; Nichols, C.G. K(ATP) channels and insulin secretion disorders. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E207–E216. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Khan, A.H.; Pessin, J.E. Insulin regulation of glucose uptake: A complex interplay of intracellular signalling pathways. Diabetologia 2002, 45, 1475–1483. [Google Scholar]
- Seth, A.; Stemple, D.L.; Barroso, I. The emerging use of zebrafish to model metabolic disease. Dis. Model Mech. 2013, 6, 1080–1088. [Google Scholar] [CrossRef] [PubMed]
- Olsen, A.S.; Sarras Jr, M.P.; Intine, R.V. Limb regeneration is impaired in an adult zebrafish model of diabetes mellitus. Wound Repair. Regen. 2010, 18, 532–542. [Google Scholar] [CrossRef]
- Pisharath, H.; Rhee, J.M.; Swanson, M.A.; Leach, S.D.; Parsons, M.J. Targeted ablation of beta cells in the embryonic zebrafish pancreas using E. coli nitroreductase. Mech. Dev. 2007, 124, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, R.A.; Dobler, S.; Schmitner, N.; Walsen, T.; Freudenblum, J.; Meyer, D. Diabetic pdx1-mutant zebrafish show conserved responses to nutrient overload and anti-glycemic treatment. Sci. Rep. 2015, 5, 14241. [Google Scholar] [CrossRef] [PubMed]
- Dalgin, G.; Prince, V.E. Differential levels of Neurod establish zebrafish endocrine pancreas cell fates. Dev. Biol. 2015, 402, 81–97. [Google Scholar] [CrossRef]
- Cassar, S.; Adatto, I.; Freeman, J.L.; Gamse, J.T.; Iturria, I.; Lawrence, C.; Muriana, A.; Peterson, R.T.; Cruchten, S.V.; Zon, L.I. Use of Zebrafish in Drug Discovery Toxicology. Chem. Res. Toxicol. 2020, 33, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Eames, S.C.; Philipson, L.H.; Prince, V.E.; Kinkel, M.D. Blood sugar measurement in zebrafish reveals dynamics of glucose homeostasis. Zebrafish 2010, 7, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Jurczyk, A.; Roy, N.; Bajwa, R.; Gut, P.; Lipson, K.; Yang, C.; Covassin, L.; Racki, W.J.; Rossini, A.A.; Phillips, N.; et al. Dynamic glucoregulation and mammalian-like responses to metabolic and developmental disruption in zebrafish. Gen. Comp. Endocrinol. 2011, 170, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Field, H.A.; Dong, P.D.; Beis, D.; Stainier, D.Y. Formation of the digestive system in zebrafish. II. Pancreas morphogenesis. Dev. Biol. 2003, 261, 197–208. [Google Scholar] [CrossRef]
- Matsuda, H.; Mullapudi, S.T.; Zhang, Y.; Hesselson, D.; Stainier, D.Y.R. Thyroid Hormone Coordinates Pancreatic Islet Maturation During the Zebrafish Larval-to-Juvenile Transition to Maintain Glucose Homeostasis. Diabetes 2017, 66, 2623–2635. [Google Scholar] [CrossRef]
- Zang, L.; Shimada, Y.; Nishimura, N. Development of a Novel Zebrafish Model for Type 2 Diabetes Mellitus. Sci. Rep. 2017, 7, 1461. [Google Scholar] [CrossRef] [PubMed]
- Marín-Juez, R.; Jong-Raadsen, S.; Yang, S.; Spaink, H.P. Hyperinsulinemia induces insulin resistance and immune suppression via Ptpn6/Shp1 in zebrafish. J. Endocrinol. 2014, 222, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Zang, L.; Maddison, L.A.; Chen, W. Zebrafish as a model for obesity and diabetes. Front. Cell Dev. Biol. 2018, 6, 91. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Um, J.I.; Jo, A.; Lee, J.; Jung, D.W.; Williams, D.R.; Park, S.B. Impact of molecular charge on GLUT-specific cellular uptake of glucose bioprobes and in vivo application of the glucose bioprobe, GB2-Cy3. Chem. Commun. 2014, 50, 9251–9254. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Saito, M.; Matsuoka, H.; Inagaki, N. A real-time method of imaging glucose uptake in single, living mammalian cells. Nat. Protoc. 2007, 2, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Manaph, N.P.A.; Sivanathan, K.N.; Nitschke, J.; Zhou, X.F.; Coates, P.T.; Drogemuller, C.J. An overview on small molecule-induced differentiation of mesenchymal stem cells into beta cells for diabetic therapy. Stem. Cell Res. Ther. 2019, 10, 293. [Google Scholar]
- Hernandez, R.E.; Galitan, L.; Cameron, J.; Goodwin, N.; Ramakrishnan, L. Delay of Initial Feeding of Zebrafish Larvae Until 8 Days Postfertilization Has No Impact on Survival or Growth Through the Juvenile Stage. Zebrafish 2018, 15, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Kasuga, M. Insulin resistance and pancreatic β cell failure. J. Clin. Investig. 2006, 116, 1756–1760. [Google Scholar] [CrossRef] [PubMed]
- Beith, J.L.; Alejandro, E.U.; Johnson, J.D. Insulin stimulates primary beta-cell proliferation via Raf-1 kinase. Endocrinology 2008, 149, 2251–2260. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M. Role of insulin resistance in human disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Kemnitz, J.W.; Elson, D.F.; Roecker, E.B.; Baum, S.T.; Bergman, R.N.; Meglasson, M.D. Pioglitazone increase insulin sensitivity, reduces blood glucose, insulin, and lipid levels, and lowers blood pressure in obese, insulin-resistant rhesus monkeys. Diabetes 1994, 43, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Matsuda, M.; DeFronzo, R.A. Dose-response effect of pioglitazone on insulin sensitivity and insulin secretion in type 2 diabetes. Diabetes Care 2002, 25, 517–523. [Google Scholar] [CrossRef]
- Umezawa, H.; Aoyagi, T.; Ogawa, K.; Naganawa, H.; Hamada, M.; Takeuchi, T. Diprotins A and B, inhibitors of dipeptidyl aminopeptidase IV, produced by bacteria. J. Antibiot. 1984, 37, 422–425. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A.; Gallwitz, B. Mechanism of action of inhibitors of dipeptidyl-peptidase-4 (DPP-4). Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 479–486. [Google Scholar] [CrossRef]
- Bischoff, H. Pharmacology of alpha-glucosidase inhibition. Eur. J. Clin. Investig. 1994, 24, 3–10. [Google Scholar]
- Shafrir, E. Contribution of animal models to the research of the causes of diabetes. World J. Diabetes 2010, 1, 137–140. [Google Scholar] [CrossRef]
- Ikeda, H.; Taketomi, S.; Sugiyama, Y.; Shimura, Y.; Sohda, T.; Meguro, K.; Fujita, T. Effects of pioglitazone on glucose and lipid metabolism in normal and insulin resistant animals. Arzneimittelforschung 1990, 40, 156–162. [Google Scholar]
- Brown, A.E.; Palsgaard, J.; Borup, R.; Avery, P.; Gunn, D.A.; Meyts, P.D.; Yeaman, S.J.; Walker, M. p38 MAPK activation upregulates proinflammatory pathways in skeletal muscle cells from insulin-resistant type 2 diabetic patients. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E63–E70. [Google Scholar] [CrossRef]
- Grayson, B.L.; Wang, L.; Aune, T.M. Peripheral blood gene expression profiles in metabolic syndrome, coronary artery disease and type 2 diabetes. Genes Immun. 2011, 12, 341–351. [Google Scholar] [CrossRef]
- Kumphune, S.; Chattipakorn, S.; Chattipakorn, N. Roles of p38-MAPK in insulin resistant heart: Evidence from bench to future bedside application. Curr. Pharm. Des. 2013, 19, 5742–5754. [Google Scholar] [CrossRef]
- Liu, Z.; Cao, W. p38 mitogen-activated protein kinase: A critical node linking insulin resistance and cardiovascular diseases in type 2 diabetes mellitus. Endocr. Metab. Immune Disord. Drug Targets 2009, 9, 38–46. [Google Scholar] [CrossRef]
- Reinbothe, T.M.; Alkayyali, S.; Ahlqvist, E.; Tuomi, T.; Isomaa, B.; Lyssenko, V.; Renström, E. The human L-type calcium channel Cav1. 3 regulates insulin release and polymorphisms in CACNA1D associate with type 2 diabetes. Diabetologia 2013, 56, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Hernandez, A.; Verkhratsky, A. Calcium signalling in diabetes. Cell Calcium 2014, 56, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Kinkel, M.D.; Prince, V.E. On the diabetic menu: Zebrafish as a model for pancreas development and function. Bioessays 2009, 31, 139–152. [Google Scholar] [CrossRef]
- Li, M.; Dean, E.D.; Zhao, L.; Nicholson, W.E.; Powers, A.C.; Chen, W. Glucagon receptor inactivation leads to α-cell hyperplasia in zebrafish. J. Endocrinol. 2015, 227, 93–103. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Sequence | Reverse Sequence |
|---|---|---|
| β-Actin | CGA GCA GGA GAT GGG AAC C | CAA CGG AAA CGC TCA TTG C |
| map2k6 | CCA CAG CAA TCT GTC AGT C | CTC TGG GTT GAT TCT CTC AG |
| cacna1c | TAC TGC TGC TCT TCC TCT TC | ACA TCA CAG AGT TCC AGT CC |
| cacna1d | CAG GGA GAG AAG GAG TAC AA | TCA GAC TGA CCA TAG TGC TG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, Y.H.; Rodriguez, I.; Shin, S.W.; Shim, J.H.; Kim, N.W.; Kim, M.C.; Jeong, S.Y.; Nuankaew, W.; Hong, B.N.; Kim, H.; et al. Characteristics of the New Insulin-Resistant Zebrafish Model. Pharmaceuticals 2021, 14, 642. https://doi.org/10.3390/ph14070642
Nam YH, Rodriguez I, Shin SW, Shim JH, Kim NW, Kim MC, Jeong SY, Nuankaew W, Hong BN, Kim H, et al. Characteristics of the New Insulin-Resistant Zebrafish Model. Pharmaceuticals. 2021; 14(7):642. https://doi.org/10.3390/ph14070642
Chicago/Turabian StyleNam, Youn Hee, Isabel Rodriguez, Sung Woo Shin, Ji Heon Shim, Na Woo Kim, Min Cheol Kim, Seo Yule Jeong, Wanlapa Nuankaew, Bin Na Hong, Hyunggun Kim, and et al. 2021. "Characteristics of the New Insulin-Resistant Zebrafish Model" Pharmaceuticals 14, no. 7: 642. https://doi.org/10.3390/ph14070642
APA StyleNam, Y. H., Rodriguez, I., Shin, S. W., Shim, J. H., Kim, N. W., Kim, M. C., Jeong, S. Y., Nuankaew, W., Hong, B. N., Kim, H., & Kang, T. H. (2021). Characteristics of the New Insulin-Resistant Zebrafish Model. Pharmaceuticals, 14(7), 642. https://doi.org/10.3390/ph14070642