
Hypomethylating Chemotherapeutic Agents as Therapy for Myelodysplastic Syndromes and Prevention of Acute Myeloid Leukemia

 ,
,  ,
,
Abstract
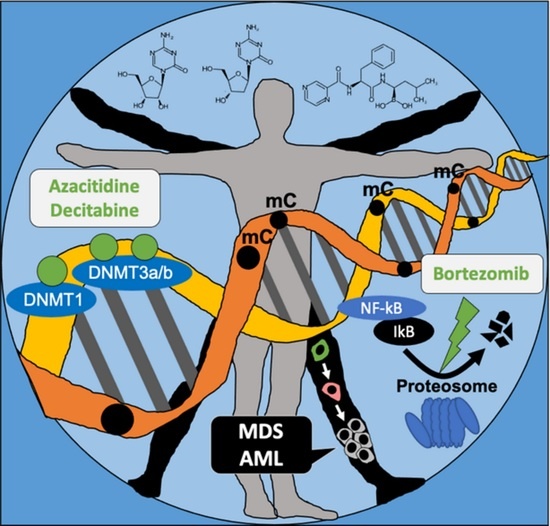
1. Introduction
2. Main Body of Review
2.1. Decitabine (5-Aza-2′-Deoxycytidine)
2.1.1. Mechanism of Action
2.1.2. Decitabine Hematological Malignancies
2.1.3. Side Effects and Complications
2.2. Azacitidine
2.2.1. Mechanism of Action
2.2.2. Azacitidine in Hematological Malignancies
2.2.3. Side Effects and Complications
2.3. Bortezomib (Velcade)
2.3.1. Mechanism of Action
2.3.2. Bortezomib and Hematological Malignancies
2.3.3. Side Effects and Complications
3. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Bond, D.R.; Lee, H.J.; Enjeti, A.K. Unravelling the Epigenome of Myelodysplastic Syndrome: Diagnosis, Prognosis, and Response to Therapy. Cancers 2020, 12, 3128. [Google Scholar] [CrossRef]
- Cogle, C.R. Incidence and Burden of the Myelodysplastic Syndromes. Curr. Hematol. Malig. Rep. 2015, 10, 272–281. [Google Scholar] [CrossRef]
- Germing, U.; Kobbe, G.; Haas, R.; Gattermann, N. Myelodysplastic syndromes: Diagnosis, prognosis, and treatment. Dtsch. Arztebl. Int. 2013, 110, 783–790. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Tefferi, A. Refractory anemia with ring sideroblasts (RARS) and RARS with thrombocytosis (RARS-T): 2017 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2017, 92, 297–310. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Saultz, J.N.; Garzon, R. Acute Myeloid Leukemia: A Concise Review. J. Clin. Med. 2016, 5, 33. [Google Scholar] [CrossRef]
- Hassan, H.T.; Rees, J.K. Auer bodies in acute myeloid leukaemia patients. Pathol. Res. Pract. 1990, 186, 293–295. [Google Scholar] [CrossRef]
- Ogawa, S. Genetics of MDS. Blood 2019, 133, 1049–1059. [Google Scholar] [CrossRef]
- Byrd, J.C.; Mrózek, K.; Dodge, R.K.; Carroll, A.J.; Edwards, C.G.; Arthur, D.C.; Pettenati, M.J.; Patil, S.R.; Rao, K.W.; Watson, M.S.; et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: Results from Cancer and Leukemia Group B (CALGB 8461). Blood 2002, 100, 4325–4336. [Google Scholar] [CrossRef]
- Duy, C.; Teater, M.; Garrett-Bakelman, F.E.; Lee, T.C.; Meydan, C.; Glass, J.L.; Li, M.; Hellmuth, J.C.; Mohammad, H.P.; Smitheman, K.N.; et al. Rational Targeting of Cooperating Layers of the Epigenome Yields Enhanced Therapeutic Efficacy against AML. Cancer Discov. 2019, 9, 872–889. [Google Scholar] [CrossRef]
- Pliml, J.; Šorm, F. Synthesis of a 2-deoxy-D-ribofuranosyl-5-azacytosine. Collect. Czech. Chem. Commun. 1964, 29, 2576–2578. [Google Scholar] [CrossRef]
- Veselý, J.; Cihák, A.; Sorm, F. Characteristics of mouse leukemic cells resistant to 5-azacytidine and 5-aza-2′-deoxycytidine. Cancer Res. 1968, 28, 1995–2000. [Google Scholar]
- Momparier, R.L.; Gonzales, F.A. Effect of intravenous infusion of 5-aza-2′-deoxycytidine on survival time of mice with L1210 leukemia. Cancer Res. 1978, 38, 2673–2678. [Google Scholar] [PubMed]
- Wilson, V.L.; Jones, P.A.; Momparler, R.L. Inhibition of DNA methylation in L1210 leukemic cells by 5-aza-2′-deoxycytidine as a possible mechanism of chemotherapeutic action. Cancer Res. 1983, 43, 3493–3496. [Google Scholar] [PubMed]
- de Vos, D.; van Overveld, W. Decitabine: A historical review of the development of an epigenetic drug. Ann. Hematol. 2005, 84 (Suppl. S1), 3–8. [Google Scholar] [CrossRef] [PubMed]
- Schwartsmann, G.; Schunemann, H.; Gorini, C.N.; Filho, A.F.; Garbino, C.; Sabini, G.; Muse, I.; DiLeone, L.; Mans, D.R. A phase I trial of cisplatin plus decitabine, a new DNA-hypomethylating agent, in patients with advanced solid tumors and a follow-up early phase II evaluation in patients with inoperable non-small cell lung cancer. Investig. New Drugs 2000, 18, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Thibault, A.; Figg, W.D.; Bergan, R.C.; Lush, R.M.; Myers, C.E.; Tompkins, A.; Reed, E.; Samid, D. A phase II study of 5-aza-2′deoxycytidine (decitabine) in hormone independent metastatic (D2) prostate cancer. Tumori 1998, 84, 87–89. [Google Scholar] [CrossRef]
- Hagemann, S.; Heil, O.; Lyko, F.; Brueckner, B. Azacytidine and decitabine induce gene-specific and non-random DNA demethylation in human cancer cell lines. PLoS ONE 2011, 6, e17388. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Lugthart, S.; Li, Y.; Erpelinck-Verschueren, C.; Deng, X.; Christos, P.J.; Schifano, E.; Booth, J.; van Putten, W.; Skrabanek, L.; et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 2010, 17, 13–27. [Google Scholar] [CrossRef]
- Kantarjian, H.; Oki, Y.; Garcia-Manero, G.; Huang, X.; O’Brien, S.; Cortes, J.; Faderl, S.; Bueso-Ramos, C.; Ravandi, F.; Estrov, Z.; et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood 2007, 109, 52–57. [Google Scholar] [CrossRef]
- Jabbour, E.; Issa, J.P.; Garcia-Manero, G.; Kantarjian, H. Evolution of decitabine development: Accomplishments, ongoing investigations, and future strategies. Cancer 2008, 112, 2341–2351. [Google Scholar] [CrossRef]
- Gnyszka, A.; Jastrzebski, Z.; Flis, S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res. 2013, 33, 2989–2996. [Google Scholar]
- Pískala, A.; Šorm, F. Nucleic acids components and their analogues. Synthesis of 1-glycosyl derivatives of 5 azauracil and 5-azacytosine. Collect. Czech. Chem. Commun. 1964, 29, 2060–2076. [Google Scholar] [CrossRef]
- Krečmerová, M.; Otmar, M. 5-azacytosine compounds in medicinal chemistry: Current stage and future perspectives. Future Med. Chem. 2012, 4, 991–1005. [Google Scholar] [CrossRef] [PubMed]
- Hanka, L.J.; Evans, J.S.; Mason, D.J.; Dietz, A. Microbiological production of 5-azacytidine. I. Production and biological activity. Antimicrob. Agents Chemother. 1966, 6, 619–624. [Google Scholar]
- Kaminskas, E.; Farrell, A.; Abraham, S.; Baird, A.; Hsieh, L.S.; Lee, S.L.; Leighton, J.K.; Patel, H.; Rahman, A.; Sridhara, R.; et al. Approval summary: Azacitidine for treatment of myelodysplastic syndrome subtypes. Clin. Cancer Res. 2005, 11, 3604–3608. [Google Scholar] [CrossRef] [PubMed]
- Almasri, J.; Alkhateeb, H.B.; Firwana, B.; Sonbol, M.B.; Damlaj, M.; Wang, Z.; Murad, M.H.; Al-Kali, A. A systematic review and network meta-analysis comparing azacitidine and decitabine for the treatment of myelodysplastic syndrome. Syst. Rev. 2018, 7, 144. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Stoltz, M.L.; Ward, M.R.; Kantarjian, H.; Sharma, S. A pilot pharmacokinetic study of oral azacitidine. Leukemia 2008, 22, 1680–1684. [Google Scholar] [CrossRef]
- Roboz, G.J.; Montesinos, P.; Selleslag, D.; Wei, A.; Jang, J.H.; Falantes, J.; Voso, M.T.; Sayar, H.; Porkka, K.; Marlton, P.; et al. Design of the randomized, Phase III, QUAZAR AML Maintenance trial of CC-486 (oral azacitidine) maintenance therapy in acute myeloid leukemia. Future Oncol. 2016, 12, 293–302. [Google Scholar] [CrossRef]
- Wei, A.H.; Döhner, H.; Pocock, C.; Montesinos, P.; Afanasyev, B.; Dombret, H.; Ravandi, F.; Sayar, H.; Jang, J.H.; Porkka, K.; et al. The QUAZAR AML-001 Maintenance Trial: Results of a Phase III International, Randomized, Double-Blind, Placebo-Controlled Study of CC-486 (Oral Formulation of Azacitidine) in Patients with Acute Myeloid Leukemia (AML) in First Remission. Blood 2019, 134, LBA-3. [Google Scholar] [CrossRef]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Adams, J. Development of the proteasome inhibitor PS-341. Oncologist 2002, 7, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Ri, M. Mechanism of action of bortezomib in multiple myeloma thera. Int. J. Myeloma 2016, 6, 1–6. [Google Scholar]
- Adams, J. The proteasome: A suitable antineoplastic target. Nat. Rev. Cancer 2004, 4, 349–360. [Google Scholar] [CrossRef]
- King, R.W.; Deshaies, R.J.; Peters, J.M.; Kirschner, M.W. How proteolysis drives the cell cycle. Science 1996, 274, 1652–1659. [Google Scholar] [CrossRef]
- Dulić, V.; Kaufmann, W.K.; Wilson, S.J.; Tlsty, T.D.; Lees, E.; Harper, J.W.; Elledge, S.J.; Reed, S.I. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell 1994, 76, 1013–1023. [Google Scholar] [CrossRef]
- Adams, J.; Palombella, V.J.; Sausville, E.A.; Johnson, J.; Destree, A.; Lazarus, D.D.; Maas, J.; Pien, C.S.; Prakash, S.; Elliott, P.J. Proteasome inhibitors: A novel class of potent and effective antitumor agents. Cancer Res. 1999, 59, 2615–2622. [Google Scholar]
- Daher, M.; Hidalgo Lopez, J.E.; Randhawa, J.K.; Jabbar, K.J.; Wei, Y.; Pemmaraju, N.; Borthakur, G.; Kadia, T.; Konopleva, M.; Kantarjian, H.M.; et al. An exploratory clinical trial of bortezomib in patients with lower risk myelodysplastic syndromes. Am. J. Hematol. 2017, 92, 674–682. [Google Scholar] [CrossRef]
- Terpos, E.; Verrou, E.; Banti, A.; Kaloutsi, V.; Lazaridou, A.; Zervas, K. Bortezomib is an effective agent for MDS/MPD syndrome with 5q− anomaly and thrombocytosis. Leuk. Res. 2007, 31, 559–562. [Google Scholar] [CrossRef]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.W.; Irwin, D.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef]
- Narayanan, S.; Cai, C.Y.; Assaraf, Y.G.; Guo, H.Q.; Cui, Q.; Wei, L.; Huang, J.J.; Ashby, C.R., Jr.; Chen, Z.S. Targeting the ubiquitin-proteasome pathway to overcome anti-cancer drug resistance. Drug Resist. Updat. 2020, 48, 100663. [Google Scholar] [CrossRef]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef]
- Momparler, R.L. Pharmacology of 5-Aza-2′-deoxycytidine (decitabine). Semin. Hematol. 2005, 42, S9–S16. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Taylor, S.M. Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980, 20, 85–93. [Google Scholar] [CrossRef]
- Griffiths, E.A.; Choy, G.; Redkar, S.; Taverna, P.; Azab, M.; Karpf, A.R. SGI-110: DNA Methyltransferase Inhibitor Oncolytic. Drugs Future 2013, 38, 535–543. [Google Scholar] [PubMed]
- Santi, D.V.; Norment, A.; Garrett, C.E. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc. Natl. Acad. Sci. USA 1984, 81, 6993–6997. [Google Scholar] [CrossRef]
- Christman, J.K. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, C.S. Clinical development of decitabine as a prototype for an epigenetic drug program. Semin. Oncol. 2005, 32, 465–472. [Google Scholar] [CrossRef]
- Toyota, M.; Kopecky, K.J.; Toyota, M.O.; Jair, K.W.; Willman, C.L.; Issa, J.P. Methylation profiling in acute myeloid leukemia. Blood 2001, 97, 2823–2829. [Google Scholar] [CrossRef]
- Shimamoto, T.; Ohyashiki, J.H.; Ohyashiki, K. Methylation of p15(INK4b) and E-cadherin genes is independently correlated with poor prognosis in acute myeloid leukemia. Leuk. Res. 2005, 29, 653–659. [Google Scholar] [CrossRef]
- Buelow, D.R.; Anderson, J.T.; Pounds, S.B.; Shi, L.; Lamba, J.K.; Hu, S.; Gibson, A.A.; Goodwin, E.A.; Sparreboom, A.; Baker, S.D. DNA Methylation-Based Epigenetic Repression of SLC22A4 Promotes Resistance to Cytarabine in Acute Myeloid Leukemia. Clin. Transl. Sci. 2021, 14, 137–142. [Google Scholar] [CrossRef]
- Zhang, Z.; He, Q.; Tao, Y.; Guo, J.; Xu, F.; Wu, L.-Y.; Zhao, Y.-S.; Wu, D.; Zhou, L.-Y.; Su, J.-Y.; et al. Decitabine treatment sensitizes tumor cells to T-cell-mediated cytotoxicity in patients with myelodysplastic syndromes. Am. J. Transl. Res. 2017, 9, 454–465. [Google Scholar] [PubMed]
- Krishnadas, D.K.; Bao, L.; Bai, F.; Chencheri, S.C.; Lucas, K. Decitabine facilitates immune recognition of sarcoma cells by upregulating CT antigens, MHC molecules, and ICAM-1. Tumour Biol. 2014, 35, 5753–5762. [Google Scholar] [CrossRef]
- Gupta, N.; Miller, A.; Gandhi, S.; Ford, L.A.; Vigil, C.E.; Griffiths, E.A.; Thompson, J.E.; Wetzler, M.; Wang, E.S. Comparison of epigenetic versus standard induction chemotherapy for newly diagnosed acute myeloid leukemia patients ≥60 years old. Am. J. Hematol. 2015, 90, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Tamm, I.; Sattler, N.; Wagner, M.; Lübbert, M.; leCoutre, P.; Dörken, B.; Schmelz, K. Decitabine: Where Is the Target? Blood 2005, 106, 495. [Google Scholar] [CrossRef]
- Joeckel, T.E.; Lübbert, M. Clinical results with the DNA hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in patients with myelodysplastic syndromes: An update. Semin. Hematol. 2012, 49, 330–341. [Google Scholar] [CrossRef]
- Blum, W.; Schwind, S.; Tarighat, S.S.; Geyer, S.; Eisfeld, A.K.; Whitman, S.; Walker, A.; Klisovic, R.; Byrd, J.C.; Santhanam, R.; et al. Clinical and pharmacodynamic activity of bortezomib and decitabine in acute myeloid leukemia. Blood 2012, 119, 6025–6031. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef]
- Jabbour, E.; Short, N.J.; Montalban-Bravo, G.; Huang, X.; Bueso-Ramos, C.; Qiao, W.; Yang, H.; Zhao, C.; Kadia, T.; Borthakur, G.; et al. Randomized phase 2 study of low-dose decitabine vs. low-dose azacitidine in lower-risk MDS and MDS/MPN. Blood 2017, 130, 1514–1522. [Google Scholar] [CrossRef]
- Wu, D.; Du, X.; Jin, J.; Xiao, Z.; Shen, Z.; Shao, Z.; Li, X.; Huang, X.; Liu, T.; Yu, L.; et al. Decitabine for Treatment of Myelodysplastic Syndromes in Chinese Patients: An Open-Label, Phase-3b Study. Adv. Ther. 2015, 32, 1140–1159. [Google Scholar] [CrossRef]
- Becker, H.; Suciu, S.; Rüter, B.H.; Platzbecker, U.; Giagounidis, A.; Selleslag, D.; Labar, B.; Germing, U.; Salih, H.R.; Muus, P.; et al. Decitabine versus best supportive care in older patients with refractory anemia with excess blasts in transformation (RAEBt)—Results of a subgroup analysis of the randomized phase III study 06011 of the EORTC Leukemia Cooperative Group and German MDS Study Group (GMDSSG). Ann. Hematol. 2015, 94, 2003–2013. [Google Scholar] [CrossRef]
- Kantarjian, H.; Issa, J.P.; Rosenfeld, C.S.; Bennett, J.M.; Albitar, M.; DiPersio, J.; Klimek, V.; Slack, J.; de Castro, C.; Ravandi, F.; et al. Decitabine improves patient outcomes in myelodysplastic syndromes: Results of a phase III randomized study. Cancer 2006, 106, 1794–1803. [Google Scholar] [CrossRef]
- Lübbert, M.; Suciu, S.; Hagemeijer, A.; Rüter, B.; Platzbecker, U.; Giagounidis, A.; Selleslag, D.; Labar, B.; Germing, U.; Salih, H.R.; et al. Decitabine improves progression-free survival in older high-risk MDS patients with multiple autosomal monosomies: Results of a subgroup analysis of the randomized phase III study 06011 of the EORTC Leukemia Cooperative Group and German MDS Study Group. Ann. Hematol. 2016, 95, 191–199. [Google Scholar] [CrossRef]
- Lübbert, M.; Ihorst, G.; Sander, P.N.; Bogatyreva, L.; Becker, H.; Wijermans, P.W.; Suciu, S.; Bissé, E.; Claus, R. Elevated fetal haemoglobin is a predictor of better outcome in MDS/AML patients receiving 5-aza-2′-deoxycytidine (Decitabine). Br. J. Haematol. 2017, 176, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Griffiths, E.A.; Steensma, D.P.; Roboz, G.J.; Wells, R.; McCloskey, J.; Odenike, O.; DeZern, A.E.; Yee, K.; Busque, L.; et al. Oral cedazuridine/decitabine for MDS and CMML: A phase 2 pharmacokinetic/pharmacodynamic randomized crossover study. Blood 2020, 136, 674–683. [Google Scholar] [CrossRef]
- Momparler, R.L.; Côté, S.; Momparler, L.F.; Idaghdour, Y. Inhibition of DNA and Histone Methylation by 5-Aza-2′-Deoxycytidine (Decitabine) and 3-Deazaneplanocin-A on Antineoplastic Action and Gene Expression in Myeloid Leukemic Cells. Front. Oncol. 2017, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P.; Kantarjian, H.M. Targeting DNA methylation. Clin. Cancer Res. 2009, 15, 3938–3946. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Jelinek, J.; Si, J.; Shu, J.; Issa, J.P. Mechanisms of resistance to 5-aza-2′-deoxycytidine in human cancer cell lines. Blood 2009, 113, 659–667. [Google Scholar] [CrossRef]
- Diesch, J.; Zwick, A.; Garz, A.K.; Palau, A.; Buschbeck, M.; Götze, K.S. A clinical-molecular update on azanucleoside-based therapy for the treatment of hematologic cancers. Clin. Epigenet. 2016, 8, 71. [Google Scholar] [CrossRef]
- Bejar, R.; Lord, A.; Stevenson, K.; Bar-Natan, M.; Pérez-Ladaga, A.; Zaneveld, J.; Wang, H.; Caughey, B.; Stojanov, P.; Getz, G.; et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood 2014, 124, 2705–2712. [Google Scholar] [CrossRef]
- Traina, F.; Visconte, V.; Elson, P.; Tabarroki, A.; Jankowska, A.M.; Hasrouni, E.; Sugimoto, Y.; Szpurka, H.; Makishima, H.; O’Keefe, C.L.; et al. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia 2014, 28, 78–87. [Google Scholar] [CrossRef]
- Strebhardt, K. Multifaceted polo-like kinases: Drug targets and antitargets for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 643–660. [Google Scholar] [CrossRef]
- Degenhardt, Y.; Lampkin, T. Targeting Polo-like kinase in cancer therapy. Clin. Cancer Res. 2010, 16, 384–389. [Google Scholar] [CrossRef]
- Talati, C.; Griffiths, E.A.; Wetzler, M.; Wang, E.S. Polo-like kinase inhibitors in hematologic malignancies. Crit. Rev. Oncol. Hematol. 2016, 98, 200–210. [Google Scholar] [CrossRef]
- Hollenbach, P.W.; Nguyen, A.N.; Brady, H.; Williams, M.; Ning, Y.; Richard, N.; Krushel, L.; Aukerman, S.L.; Heise, C.; MacBeth, K.J. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS ONE 2010, 5, e9001. [Google Scholar] [CrossRef]
- Sullivan, M.; Hahn, K.; Kolesar, J.M. Azacitidine: A novel agent for myelodysplastic syndromes. Am. J. Health Syst. Pharm. 2005, 62, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Israili, Z.H.; Vogler, W.R.; Mingioli, E.S.; Pirkle, J.L.; Smithwick, R.W.; Goldstein, J.H. The disposition and pharmacokinetics in humans of 5-azacytidine administered intravenously as a bolus or by continuous infusion. Cancer Res. 1976, 36, 1453–1461. [Google Scholar]
- Yang, X.; Lay, F.; Han, H.; Jones, P.A. Targeting DNA methylation for epigenetic therapy. Trends Pharmacol. Sci. 2010, 31, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M. Azacitidine: A review of its use in the management of myelodysplastic syndromes/acute myeloid leukaemia. Drugs 2012, 72, 1111–1136. [Google Scholar] [CrossRef]
- Chen, L.; MacMillan, A.M.; Chang, W.; Ezaz-Nikpay, K.; Lane, W.S.; Verdine, G.L. Direct identification of the active-site nucleophile in a DNA (cytosine-5)-methyltransferase. Biochemistry 1991, 30, 11018–11025. [Google Scholar] [CrossRef]
- Cechova, H.; Lassuthova, P.; Novakova, L.; Belickova, M.; Stemberkova, R.; Jencik, J.; Stankova, M.; Hrabakova, P.; Pegova, K.; Zizkova, H.; et al. Monitoring of methylation changes in 9p21 region in patients with myelodysplastic syndromes and acute myeloid leukemia. Neoplasma 2012, 59, 168–174. [Google Scholar] [CrossRef]
- Quesnel, B.; Guillerm, G.; Vereecque, R.; Wattel, E.; Preudhomme, C.; Bauters, F.; Vanrumbeke, M.; Fenaux, P. Methylation of the p15(INK4b) gene in myelodysplastic syndromes is frequent and acquired during disease progression. Blood 1998, 91, 2985–2990. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Kinoshita, T.; Nagai, H.; Nakahara, Y.; Saito, H.; Hotta, T.; Murate, T. Hypermethylation of the p15INK4B gene in myelodysplastic syndromes. Blood 1997, 90, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Ohyashiki, K.; Nishimaki, J.; Shoji, N.; Miyazawa, K.; Kimura, Y.; Ohyashiki, J.H. Re-evaluation of refractory anemia with excess blasts in transformation. Leuk. Res. 2001, 25, 933–939. [Google Scholar] [CrossRef]
- Tran, H.T.T.; Kim, H.N.; Lee, I.-K.; Kim, Y.-K.; Ahn, J.-S.; Yang, D.-H.; Lee, J.-J.; Kim, H.-J. DNA methylation changes following 5-azacitidine treatment in patients with myelodysplastic syndrome. J. Korean Med. Sci. 2011, 26, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Raj, K.; John, A.; Ho, A.; Chronis, C.; Khan, S.; Samuel, J.; Pomplun, S.; Thomas, N.S.B.; Mufti, G.J. CDKN2B methylation status and isolated chromosome 7 abnormalities predict responses to treatment with 5-azacytidine. Leukemia 2007, 21, 1937–1944. [Google Scholar] [CrossRef][Green Version]
- Kimura, S.; Kuramoto, K.; Homan, J.; Naruoka, H.; Ego, T.; Nogawa, M.; Sugahara, S.; Naito, H. Antiproliferative and antitumor effects of azacitidine against the human myelodysplastic syndrome cell line SKM-1. Anticancer Res. 2012, 32, 795–798. [Google Scholar]
- Follo, M.Y.; Finelli, C.; Mongiorgi, S.; Clissa, C.; Bosi, C.; Testoni, N.; Chiarini, F.; Ramazzotti, G.; Baccarani, M.; Martelli, A.M.; et al. Reduction of phosphoinositide-phospholipase C beta1 methylation predicts the responsiveness to azacitidine in high-risk MDS. Proc. Natl. Acad. Sci. USA 2009, 106, 16811–16816. [Google Scholar] [CrossRef]
- Follo, M.Y.; Finelli, C.; Bosi, C.; Martinelli, G.; Mongiorgi, S.; Baccarani, M.; Manzoli, L.; Blalock, W.L.; Martelli, A.M.; Cocco, L. PI-PLCβ1 and activated Akt levels are linked to azacitidine responsiveness in high-risk myelodysplastic syndromes. Leukemia 2008, 22, 198–200. [Google Scholar] [CrossRef]
- Khan, C.; Pathe, N.; Fazal, S.; Lister, J.; Rossetti, J.M. Azacitidine in the management of patients with myelodysplastic syndromes. Ther. Adv. Hematol. 2012, 3, 355–373. [Google Scholar] [CrossRef]
- Silverman, L.R.; McKenzie, D.R.; Peterson, B.L.; Holland, J.F.; Backstrom, J.T.; Beach, C.L.; Larson, R.A. Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: Studies 8421, 8921, and 9221 by the Cancer and Leukemia Group B. J. Clin. Oncol. 2006, 24, 3895–3903. [Google Scholar] [CrossRef]
- Silverman, L.R.; Demakos, E.P.; Peterson, B.L.; Kornblith, A.B.; Holland, J.C.; Odchimar-Reissig, R.; Stone, R.M.; Nelson, D.; Powell, B.L.; DeCastro, C.M.; et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: A study of the cancer and leukemia group B. J. Clin. Oncol. 2002, 20, 2429–2440. [Google Scholar] [CrossRef]
- Silverman, L.R.; Holland, J.F.; Weinberg, R.S.; Alter, B.P.; Davis, R.B.; Ellison, R.R.; Demakos, E.P.; Cornell, C.J., Jr.; Carey, R.W.; Schiffer, C.; et al. Effects of treatment with 5-azacytidine on the in vivo and in vitro hematopoiesis in patients with myelodysplastic syndromes. Leukemia 1993, 7 (Suppl. S1), 21–29. [Google Scholar] [PubMed]
- DeSimone, J.; Heller, P.; Schimenti, J.C.; Duncan, C.H. Fetal hemoglobin production in adult baboons by 5-azacytidine or by phenylhydrazine-induced hemolysis is associated with hypomethylation of globin gene DNA. Prog. Clin. Biol. Res. 1983, 134, 489–500. [Google Scholar] [PubMed]
- Campos, L.; Rouault, J.P.; Sabido, O.; Oriol, P.; Roubi, N.; Vasselon, C.; Archimbaud, E.; Magaud, J.P.; Guyotat, D. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 1993, 81, 3091–3096. [Google Scholar] [CrossRef]
- Karakas, T.; Miething, C.C.; Maurer, U.; Weidmann, E.; Ackermann, H.; Hoelzer, D.; Bergmann, L. The coexpression of the apoptosis-related genes bcl-2 and wt1 in predicting survival in adult acute myeloid leukemia. Leukemia 2002, 16, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.V.; Shukla, S.N.; Vora, H.H. Overexpression of Bcl2 protein predicts chemoresistance in acute myeloid leukemia: Its correlation with FLT3. Neoplasma 2013, 60, 666–675. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. International phase 3 study of azacitidine vs. conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef]
- Grövdal, M.; Karimi, M.; Khan, R.; Aggerholm, A.; Antunovic, P.; Astermark, J.; Bernell, P.; Engström, L.M.; Kjeldsen, L.; Linder, O.; et al. Maintenance treatment with azacytidine for patients with high-risk myelodysplastic syndromes (MDS) or acute myeloid leukaemia following MDS in complete remission after induction chemotherapy. Br. J. Haematol. 2010, 150, 293–302. [Google Scholar] [CrossRef]
- Krecmerová, M.; Masojídková, M.; Holý, A. Acyclic nucleoside phosphonates with 5-azacytosine base moiety substituted in C-6 position. Bioorg. Med. Chem. 2010, 18, 387–395. [Google Scholar] [CrossRef]
- Helbig, G.; Chromik, K.; Woźniczka, K.; Kopińska, A.J.; Boral, K.; Dworaczek, M.; Koclęga, A.; Armatys, A.; Panz-Klapuch, M.; Markiewicz, M. Real Life Data on Efficacy and Safety of Azacitidine Therapy for Myelodysplastic Syndrome, Chronic Myelomonocytic Leukemia and Acute Myeloid Leukemia. Pathol. Oncol. Res. 2019, 25, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, F.; Hodgson, J.G.; Eden, A.; Jackson-Grusby, L.; Dausman, J.; Gray, J.W.; Leonhardt, H.; Jaenisch, R. Induction of tumors in mice by genomic hypomethylation. Science 2003, 300, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Howard, G.; Eiges, R.; Gaudet, F.; Jaenisch, R.; Eden, A. Activation and transposition of endogenous retroviral elements in hypomethylation induced tumors in mice. Oncogene 2008, 27, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Rius, M.; Stresemann, C.; Keller, D.; Brom, M.; Schirrmacher, E.; Keppler, D.; Lyko, F. Human concentrative nucleoside transporter 1-mediated uptake of 5-azacytidine enhances DNA demethylation. Mol. Cancer Ther. 2009, 8, 225–231. [Google Scholar] [CrossRef]
- Sripayap, P.; Nagai, T.; Uesawa, M.; Kobayashi, H.; Tsukahara, T.; Ohmine, K.; Muroi, K.; Ozawa, K. Mechanisms of resistance to azacitidine in human leukemia cell lines. Exp. Hematol. 2014, 42, 294–306.e2. [Google Scholar] [CrossRef]
- Cluzeau, T.; Robert, G.; Mounier, N.; Karsenti, J.M.; Dufies, M.; Puissant, A.; Jacquel, A.; Renneville, A.; Preudhomme, C.; Cassuto, J.-P.; et al. BCL2L10 is a predictive factor for resistance to azacitidine in MDS and AML patients. Oncotarget 2012, 3, 490–501. [Google Scholar] [CrossRef]
- Cheng, J.X.; Chen, L.; Li, Y.; Cloe, A.; Yue, M.; Wei, J.; Watanabe, K.A.; Shammo, J.M.; Anastasi, J.; Shen, Q.J.; et al. RNA cytosine methylation and methyltransferases mediate chromatin organization and 5-azacytidine response and resistance in leukaemia. Nat. Commun. 2018, 9, 1163. [Google Scholar] [CrossRef] [PubMed]
- Gruber, E.; Franich, R.L.; Shortt, J.; Johnstone, R.W.; Kats, L.M. Distinct and overlapping mechanisms of resistance to azacytidine and guadecitabine in acute myeloid leukemia. Leukemia 2020, 34, 3388–3392. [Google Scholar] [CrossRef]
- Unnikrishnan, A.; Papaemmanuil, E.; Beck, D.; Deshpande, N.P.; Verma, A.; Kumari, A.; Woll, P.S.; Richards, L.A.; Knezevic, K.; Chandrakanthan, V.; et al. Integrative Genomics Identifies the Molecular Basis of Resistance to Azacitidine Therapy in Myelodysplastic Syndromes. Cell Rep. 2017, 20, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Daher-Reyes, G.S.; Merchan, B.M.; Yee, K.W.L. Guadecitabine (SGI-110): An investigational drug for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Expert Opin. Investig. Drugs 2019, 28, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Sébert, M.; Renneville, A.; Bally, C.; Peterlin, P.; Beyne-Rauzy, O.; Legros, L.; Gourin, M.-P.; Sanhes, L.; Wattel, E.; Gyan, E.; et al. A phase II study of guadecitabine in higher-risk myelodysplastic syndrome and low blast count acute myeloid leukemia after azacitidine failure. Haematologica 2019, 104, 1565–1571. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.J.; Roboz, G.; Rizzieri, D.; Jabbour, E.; Stock, W.; O’Connell, C.; Yee, K.; Tibes, R.; Griffiths, E.A.; Walsh, K.; et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: A multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 2015, 16, 1099–1110. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Roboz, G.J.; Kropf, P.L.; Yee, K.W.L.; O’Connell, C.L.; Tibes, R.; Walsh, K.J.; Podoltsev, N.A.; Griffiths, E.A.; Jabbour, E.; et al. Guadecitabine (SGI-110) in treatment-naive patients with acute myeloid leukaemia: Phase 2 results from a multicentre, randomised, phase 1/2 trial. Lancet Oncol. 2017, 18, 1317–1326. [Google Scholar] [CrossRef]
- Groll, M.; Berkers, C.R.; Ploegh, H.L.; Ovaa, H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure 2006, 14, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef]
- Hideshima, T.; Chauhan, D.; Hayashi, T.; Akiyama, M.; Mitsiades, N.; Mitsiades, C.; Podar, K.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Proteasome inhibitor PS-341 abrogates IL-6 triggered signaling cascades via caspase-dependent downregulation of gp130 in multiple myeloma. Oncogene 2003, 22, 8386–8393. [Google Scholar] [CrossRef]
- Noborio-Hatano, K.; Kikuchi, J.; Takatoku, M.; Shimizu, R.; Wada, T.; Ueda, M.; Nobuyoshi, M.; Oh, I.; Sato, K.; Suzuki, T.; et al. Bortezomib overcomes cell-adhesion-mediated drug resistance through downregulation of VLA-4 expression in multiple myeloma. Oncogene 2009, 28, 231–242. [Google Scholar] [CrossRef]
- Adams, J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell 2004, 5, 417–421. [Google Scholar] [CrossRef]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007, 12, 115–130. [Google Scholar] [CrossRef]
- Li, C.C.; Dai, R.M.; Longo, D.L. Inactivation of NF-kappa B inhibitor I kappa B alpha: Ubiquitin-dependent proteolysis and its degradation product. Biochem. Biophys. Res. Commun. 1995, 215, 292–301. [Google Scholar] [CrossRef]
- Li, Z.W.; Chen, H.; Campbell, R.A.; Bonavida, B.; Berenson, J.R. NF-kappaB in the pathogenesis and treatment of multiple myeloma. Curr. Opin. Hematol. 2008, 15, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D.; Edwards, N.L.; Duckett, C.S.; Agranoff, A.B.; Schmid, R.M.; Nabel, G.J. A cooperative interaction between NF-kappa B and Sp1 is required for HIV-1 enhancer activation. EMBO J. 1993, 12, 3551–3558. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, Z.; Xie, Z.; Pang, J.; Yu, J.; Lehmann, E.; Huynh, L.; Vukosavljevic, T.; Takeki, M.; Klisovic, R.B.; et al. Bortezomib induces DNA hypomethylation and silenced gene transcription by interfering with Sp1/NF-κB–dependent DNA methyltransferase activity in acute myeloid leukemia. Blood 2008, 111, 2364–2373. [Google Scholar] [CrossRef]
- Mitsiades, N.; Mitsiades, C.S.; Richardson, P.G.; Poulaki, V.; Tai, Y.T.; Chauhan, D.; Fanourakis, G.; Gu, X.; Bailey, C.; Joseph, M.; et al. The proteasome inhibitor PS-341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents: Therapeutic applications. Blood 2003, 101, 2377–2380. [Google Scholar] [CrossRef]
- Velegzhaninov, I.O.; Ievlev, V.A.; Pylina, Y.I.; Shadrin, D.M.; Vakhrusheva, O.M. Programming of Cell Resistance to Genotoxic and Oxidative Stress. Biomedicines 2018, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Cerruti, F.; Jocollè, G.; Salio, C.; Oliva, L.; Paglietti, L.; Alessandria, B.; Mioletti, S.; Donati, G.; Numico, G.; Cenci, S.; et al. Proteasome stress sensitizes malignant pleural mesothelioma cells to bortezomib-induced apoptosis. Sci. Rep. 2017, 7, 17626. [Google Scholar] [CrossRef]
- Hazlehurst, L.A.; Valkov, N.; Wisner, L.; Storey, J.A.; Boulware, D.; Sullivan, D.M.; Dalton, W.S. Reduction in drug-induced DNA double-strand breaks associated with beta1 integrin-mediated adhesion correlates with drug resistance in U937 cells. Blood 2001, 98, 1897–1903. [Google Scholar] [CrossRef]
- Rosenquist, T.A.; Zharkov, D.O.; Grollman, A.P. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc. Natl. Acad. Sci. USA 1997, 94, 7429–7434. [Google Scholar] [CrossRef] [PubMed]
- Łuczkowska, K.; Rogińska, D.; Ulańczyk, Z.; Paczkowska, E.; Schmidt, C.A.; Machaliński, B. Molecular Mechanisms of Bortezomib Action: Novel Evidence for the miRNA-mRNA Interaction Involvement. Int. J. Mol. Sci. 2020, 21, 350. [Google Scholar] [CrossRef]
- Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: An expanding universe. Nat. Rev. Genet. 2009, 10, 94–108. [Google Scholar] [CrossRef]
- Balamurugan, K.; Sterneck, E. The many faces of C/EBPδ and their relevance for inflammation and cancer. Int. J. Biol. Sci. 2013, 9, 917–933. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.Y.; Ko, C.Y.; Wang, S.M.; Lin, P.I.; Wang, H.Y.; Lin, W.C.; Wu, D.Y.; Wang, L.H.; Wang, J.M. Bortezomib-induced miRNAs direct epigenetic silencing of locus genes and trigger apoptosis in leukemia. Cell Death Dis. 2017, 8, e3167. [Google Scholar] [CrossRef]
- Agrawal, S.; Hofmann, W.K.; Tidow, N.; Ehrich, M.; van den Boom, D.; Koschmieder, S.; Berdel, W.E.; Serve, H.; Müller-Tidow, C. The C/EBPdelta tumor suppressor is silenced by hypermethylation in acute myeloid leukemia. Blood 2007, 109, 3895–3905. [Google Scholar] [CrossRef] [PubMed]
- Li, C.F.; Tsai, H.H.; Ko, C.Y.; Pan, Y.C.; Yen, C.J.; Lai, H.Y.; Yuh, C.H.; Wu, W.C.; Wang, J.M. HMDB and 5-AzadC Combination Reverses Tumor Suppressor CCAAT/Enhancer-Binding Protein Delta to Strengthen the Death of Liver Cancer Cells. Mol. Cancer Ther. 2015, 14, 2623–2633. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Li, C.F.; Chu, Y.Y.; Wang, Y.H.; Hour, T.C.; Yen, C.J.; Chang, W.C.; Wang, J.M. Inhibition of the EGFR/STAT3/CEBPD Axis Reverses Cisplatin Cross-resistance with Paclitaxel in the Urothelial Carcinoma of the Urinary Bladder. Clin. Cancer Res. 2017, 23, 503–513. [Google Scholar] [CrossRef]
- Ko, J.H.; Lee, J.H.; Jung, S.H.; Lee, S.G.; Chinnathambi, A.; Alharbi, S.A.; Yang, W.M.; Um, J.Y.; Sethi, G.; Ahn, K.S. 2,5-Dihydroxyacetophenone Induces Apoptosis of Multiple Myeloma Cells by Regulating the MAPK Activation Pathway. Molecules 2017, 22, 1157. [Google Scholar] [CrossRef] [PubMed]
- Boutros, T.; Chevet, E.; Metrakos, P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: Roles in cell growth, death, and cancer. Pharmacol. Rev. 2008, 60, 261–310. [Google Scholar] [CrossRef]
- Obata, T.; Brown, G.E.; Yaffe, M.B. MAP kinase pathways activated by stress: The p38 MAPK pathway. Crit. Care Med. 2000, 28, N67–N77. [Google Scholar] [CrossRef]
- Shah, S.A.; Potter, M.W.; McDade, T.P.; Ricciardi, R.; Perugini, R.A.; Elliott, P.J.; Adams, J.; Callery, M.P. 26S proteasome inhibition induces apoptosis and limits growth of human pancreatic cancer. J. Cell Biochem. 2001, 82, 110–122. [Google Scholar] [CrossRef]
- Frankel, A.; Man, S.; Elliott, P.; Adams, J.; Kerbel, R.S. Lack of multicellular drug resistance observed in human ovarian and prostate carcinoma treated with the proteasome inhibitor PS-341. Clin. Cancer Res. 2000, 6, 3719–3728. [Google Scholar]
- Hideshima, T.; Richardson, P.; Chauhan, D.; Palombella, V.J.; Elliott, P.J.; Adams, J.; Anderson, K.C. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001, 61, 3071–3076. [Google Scholar]
- Fang, J.; Rhyasen, G.; Bolanos, L.; Rasch, C.; Varney, M.; Wunderlich, M.; Goyama, S.; Jansen, G.; Cloos, J.; Rigolino, C.; et al. Cytotoxic effects of bortezomib in myelodysplastic syndrome/acute myeloid leukemia depend on autophagy-mediated lysosomal degradation of TRAF6 and repression of PSMA1. Blood 2012, 120, 858–867. [Google Scholar] [CrossRef]
- Schnerch, D.; Schüler, J.; Follo, M.; Felthaus, J.; Wider, D.; Klingner, K.; Greil, C.; Duyster, J.; Engelhardt, M.; Wäsch, R. Proteasome inhibition enhances the efficacy of volasertib-induced mitotic arrest in AML in vitro and prolongs survival in vivo. Oncotarget 2017, 8, 21153–21166. [Google Scholar] [CrossRef] [PubMed]
- Jagannath, S.; Durie, B.G.; Wolf, J.; Camacho, E.; Irwin, D.; Lutzky, J.; McKinley, M.; Gabayan, E.; Mazumder, A.; Schenkein, D.; et al. Bortezomib therapy alone and in combination with dexamethasone for previously untreated symptomatic multiple myeloma. Br. J. Haematol. 2005, 129, 776–783. [Google Scholar] [CrossRef] [PubMed]
- San Miguel, J.F.; Schlag, R.; Khuageva, N.K.; Dimopoulos, M.A.; Shpilberg, O.; Kropff, M.; Spicka, I.; Petrucci, M.T.; Palumbo, A.; Samoilova, O.S.; et al. Persistent overall survival benefit and no increased risk of second malignancies with bortezomib-melphalan-prednisone versus melphalan-prednisone in patients with previously untreated multiple myeloma. J. Clin. Oncol. 2013, 31, 448–455. [Google Scholar] [CrossRef]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Aplenc, R.; Meshinchi, S.; Sung, L.; Alonzo, T.; Choi, J.; Fisher, B.; Gerbing, R.; Hirsch, B.; Horton, T.; Kahwash, S.; et al. Bortezomib with standard chemotherapy for children with acute myeloid leukemia does not improve treatment outcomes: A report from the Children’s Oncology Group. Haematologica 2020, 105, 1879–1886. [Google Scholar] [CrossRef]
- San-Miguel, J.F.; Richardson, P.G.; Günther, A.; Sezer, O.; Siegel, D.; Bladé, J.; LeBlanc, R.; Sutherland, H.; Sopala, M.; Mishra, K.K.; et al. Phase Ib study of panobinostat and bortezomib in relapsed or relapsed and refractory multiple myeloma. J. Clin. Oncol. 2013, 31, 3696–3703. [Google Scholar] [CrossRef]
- Pancheri, E.; Guglielmi, V.; Wilczynski, G.M.; Malatesta, M.; Tonin, P.; Tomelleri, G.; Nowis, D.; Vattemi, G. Non-Hematologic Toxicity of Bortezomib in Multiple Myeloma: The Neuromuscular and Cardiovascular Adverse Effects. Cancers 2020, 12, 2540. [Google Scholar] [CrossRef]
- Argyriou, C.; D’Agostino, M.D.; Braverman, N. Peroxisome biogenesis disorders. Transl. Sci. Rare Dis. 2016, 1, 111–144. [Google Scholar] [CrossRef] [PubMed]
- Cavaletti, G.; Gilardini, A.; Canta, A.; Rigamonti, L.; Rodriguez-Menendez, V.; Ceresa, C.; Marmiroli, P.; Bossi, M.; Oggioni, N.; D’Incalci, M.; et al. Bortezomib-induced peripheral neurotoxicity: A neurophysiological and pathological study in the rat. Exp. Neurol. 2007, 204, 317–325. [Google Scholar] [CrossRef]
- Enrico, O.; Gabriele, B.; Nadia, C.; Sara, G.; Daniele, V.; Giulia, C.; Antonio, S.; Mario, P. Unexpected cardiotoxicity in haematological bortezomib treated patients. Br. J. Haematol. 2007, 138, 396–397. [Google Scholar] [CrossRef]
- Jerkins, J.H.; Suciu, A.; Mazimba, S.; Calvo, A. Bortezomib-induced Severe Congestive Heart Failure. Cardiol. Res. 2010, 1, 20–23. [Google Scholar] [CrossRef][Green Version]
- Franke, N.E.; Niewerth, D.; Assaraf, Y.G.; van Meerloo, J.; Vojtekova, K.; van Zantwijk, C.H.; Zweegman, S.; Chan, E.T.; Kirk, C.J.; Geerke, D.P.; et al. Impaired bortezomib binding to mutant β5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia 2012, 26, 757–768. [Google Scholar] [CrossRef]
- de Wilt, L.H.; Jansen, G.; Assaraf, Y.G.; van Meerloo, J.; Cloos, J.; Schimmer, A.D.; Chan, E.T.; Kirk, C.J.; Peters, G.J.; Kruyt, F.A. Proteasome-based mechanisms of intrinsic and acquired bortezomib resistance in non-small cell lung cancer. Biochem. Pharmacol. 2012, 83, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; et al. Molecular basis of bortezomib resistance: Proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499. [Google Scholar] [CrossRef] [PubMed]
- Allmeroth, K.; Horn, M.; Kroef, V.; Miethe, S.; Müller, R.-U.; Denzel, M.S. Bortezomib resistance mutations in PSMB5 determine response to second-generation proteasome inhibitors in multiple myeloma. Leukemia 2021, 35, 887–892. [Google Scholar] [CrossRef]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Lawrie, C.H.; Green, T.M. Oncogenic Roles and Inhibitors of DNMT1, DNMT3A, and DNMT3B in Acute Myeloid Leukaemia. Biomark. Insights 2019, 14, 1177271919846454. [Google Scholar] [CrossRef]
- Huang, S.; Stillson, N.J.; Sandoval, J.E.; Yung, C.; Reich, N.O. A novel class of selective non-nucleoside inhibitors of human DNA methyltransferase 3A. Bioorg. Med. Chem. Lett. 2021, 40, 127908. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Itzykson, R.; Renneville, A.; de Renzis, B.; Dreyfus, F.; Laribi, K.; Bouabdallah, K.; Vey, N.; Toma, A.; Recher, C.; et al. Molecular predictors of response to decitabine in advanced chronic myelomonocytic leukemia: A phase 2 trial. Blood 2011, 118, 3824–3831. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Patel, K.P.; Garcia-Manero, G.; Luthra, R.; Pierce, S.; Borthakur, G.; Jabbour, E.; Kadia, T.; Pemmaraju, N.; Konopleva, M.; et al. Lack of association of IDH1, IDH2 and DNMT3A mutations with outcome in older patients with acute myeloid leukemia treated with hypomethylating agents. Leuk. Lymphoma 2014, 55, 1925–1929. [Google Scholar] [CrossRef] [PubMed]
- Metzeler, K.H.; Walker, A.; Geyer, S.; Garzon, R.; Klisovic, R.B.; Bloomfield, C.D.; Blum, W.; Marcucci, G. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia 2012, 26, 1106–1107. [Google Scholar] [CrossRef]
- Voso, M.T.; Santini, V.; Fabiani, E.; Fianchi, L.; Criscuolo, M.; Falconi, G.; Guidi, F.; Hohaus, S.; Leone, G. Why methylation is not a marker predictive of response to hypomethylating agents. Haematologica 2014, 99, 613–619. [Google Scholar] [CrossRef]
- Pan, F.; Peng, S.; Fleurence, R.; Linnehan, J.E.; Knopf, K.; Kim, E. Economic analysis of decitabine versus best supportive care in the treatment of intermediate- and high-risk myelodysplastic syndromes from a US payer perspective. Clin. Ther. 2010, 32, 2444–2456. [Google Scholar] [CrossRef]
- Damaraju, V.L.; Mowles, D.; Yao, S.; Ng, A.; Young, J.D.; Cass, C.E.; Tong, Z. Role of human nucleoside transporters in the uptake and cytotoxicity of azacitidine and decitabine. Nucleosides Nucleotides Nucleic Acids 2012, 31, 236–255. [Google Scholar] [CrossRef] [PubMed]
- Koczor, C.A.; Torres, R.A.; Lewis, W. The role of transporters in the toxicity of nucleoside and nucleotide analogs. Expert Opin. Drug Metab. Toxicol. 2012, 8, 665–676. [Google Scholar] [CrossRef]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Selene, I.I.; Jose, J.A.; Sardar, M.; Shah, Z.; Shafqat, M.; Faridi, W.; Malik, M.N.; Yasir, M.; Khalil, M.J.; Aslam, S.; et al. Histone Deacetylase Inhibitors in Myelodysplastic Syndrome. Blood 2018, 132, 5528. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Kaźmierczak, M.; Fong, C.Y.; Montesinos, P.; Venditti, A.; Mappa, S.; Spezia, R.; Ades, L. A Phase 3 Randomized Study (PRIMULA) of the Epigenetic Combination of Pracinostat, a Pan-Histone Deacetylase (HDAC) Inhibitor, with Azacitidine (AZA) in Patients with Newly Diagnosed Acute Myeloid Leukemia (AML) Unfit for Standard Intensive Chemotherapy (IC). Blood 2019, 134, 2652. [Google Scholar] [CrossRef]
- Zhuang, L.; Ma, Y.; Wang, Q.; Zhang, J.; Zhu, C.; Zhang, L.; Xu, X. Atg3 Overexpression Enhances Bortezomib-Induced Cell Death in SKM-1 Cell. PLoS ONE 2016, 11, e0158761. [Google Scholar] [CrossRef]
- Ettou, S.; Humbrecht, C.; Benet, B.; Billot, K.; d’Allard, D.; Mariot, V.; Goodhardt, M.; Kosmider, O.; Mayeux, P.; Solary, E.; et al. Epigenetic control of NF-κB-dependent FAS gene transcription during progression of myelodysplastic syndromes. Mol. Cancer Res. 2013, 11, 724–735. [Google Scholar] [CrossRef]
- Ettou, S.; Audureau, E.; Humbrecht, C.; Benet, B.; Jammes, H.; Clozel, T.; Bardet, V.; Lacombe, C.; Dreyfus, F.; Mayeux, P.; et al. Fas expression at diagnosis as a biomarker of azacitidine activity in high-risk MDS and secondary AML. Leukemia 2012, 26, 2297–2299. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xie, H.; Gu, Y.; Wang, W.; Wang, X.; Ye, X.; Xin, C.; Lu, M.; Reddy, B.A.; Shu, P. Silencing of SENP2 in Multiple Myeloma Induces Bortezomib Resistance by Activating NF-κB Through the Modulation of IκBα Sumoylation. Sci. Rep. 2020, 10, 766. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, J.J.; Pelluru, D.; Lefkimmiatis, K.; Fulciniti, M.; Prabhala, R.H.; Greipp, P.R.; Barlogie, B.; Tai, Y.T.; Anderson, K.C.; Shaughnessy, J.D., Jr.; et al. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood 2010, 115, 2827–2834. [Google Scholar] [CrossRef] [PubMed]
- De Smedt, E.; Lui, H.; Maes, K.; De Veirman, K.; Menu, E.; Vanderkerken, K.; De Bruyne, E. The Epigenome in Multiple Myeloma: Impact on Tumor Cell Plasticity and Drug Response. Front. Oncol. 2018, 8, 566. [Google Scholar] [CrossRef]
- Hideshima, T.; Bradner, J.E.; Wong, J.; Chauhan, D.; Richardson, P.; Schreiber, S.L.; Anderson, K.C. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl. Acad. Sci. USA 2005, 102, 8567–8572. [Google Scholar] [CrossRef]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef]
- Mishima, Y.; Santo, L.; Eda, H.; Cirstea, D.; Nemani, N.; Yee, A.J.; O’Donnell, E.; Selig, M.K.; Quayle, S.N.; Arastu-Kapur, S.; et al. Ricolinostat (ACY-1215) induced inhibition of aggresome formation accelerates carfilzomib-induced multiple myeloma cell death. Br. J. Haematol. 2015, 169, 423–434. [Google Scholar] [CrossRef]
- Deng, C.; Lipstein, M.R.; Scotto, L.; Jirau Serrano, X.O.; Mangone, M.A.; Li, S.; Vendome, J.; Hao, Y.; Xu, X.; Deng, S.X.; et al. Silencing c-Myc translation as a therapeutic strategy through targeting PI3Kδ and CK1ε in hematological malignancies. Blood 2017, 129, 88–99. [Google Scholar] [CrossRef]
- Guo, M.; Sun, D.; Fan, Z.; Yuan, Y.; Shao, M.; Hou, J.; Zhu, Y.; Wei, R.; Zhu, Y.; Qian, J.; et al. Targeting MK2 Is a Novel Approach to Interfere in Multiple Myeloma. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Zaal, E.A.; Wu, W.; Jansen, G.; Zweegman, S.; Cloos, J.; Berkers, C.R. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab. 2017, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Antao, A.M.; Tyagi, A.; Kim, K.-S.; Ramakrishna, S. Advances in Deubiquitinating Enzyme Inhibition and Applications in Cancer Therapeutics. Cancers 2020, 12, 1579. [Google Scholar] [CrossRef] [PubMed]





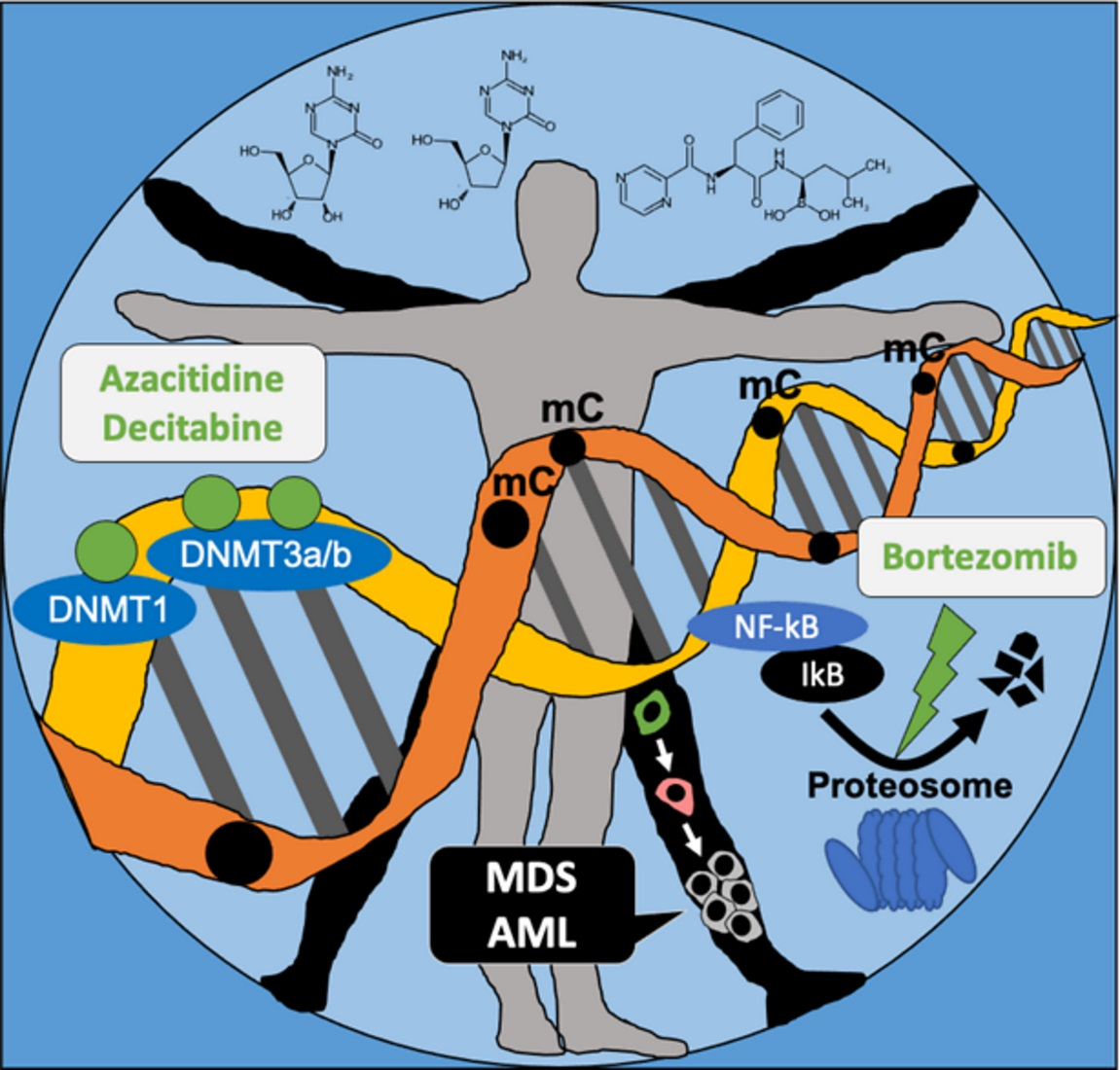{kind=link}
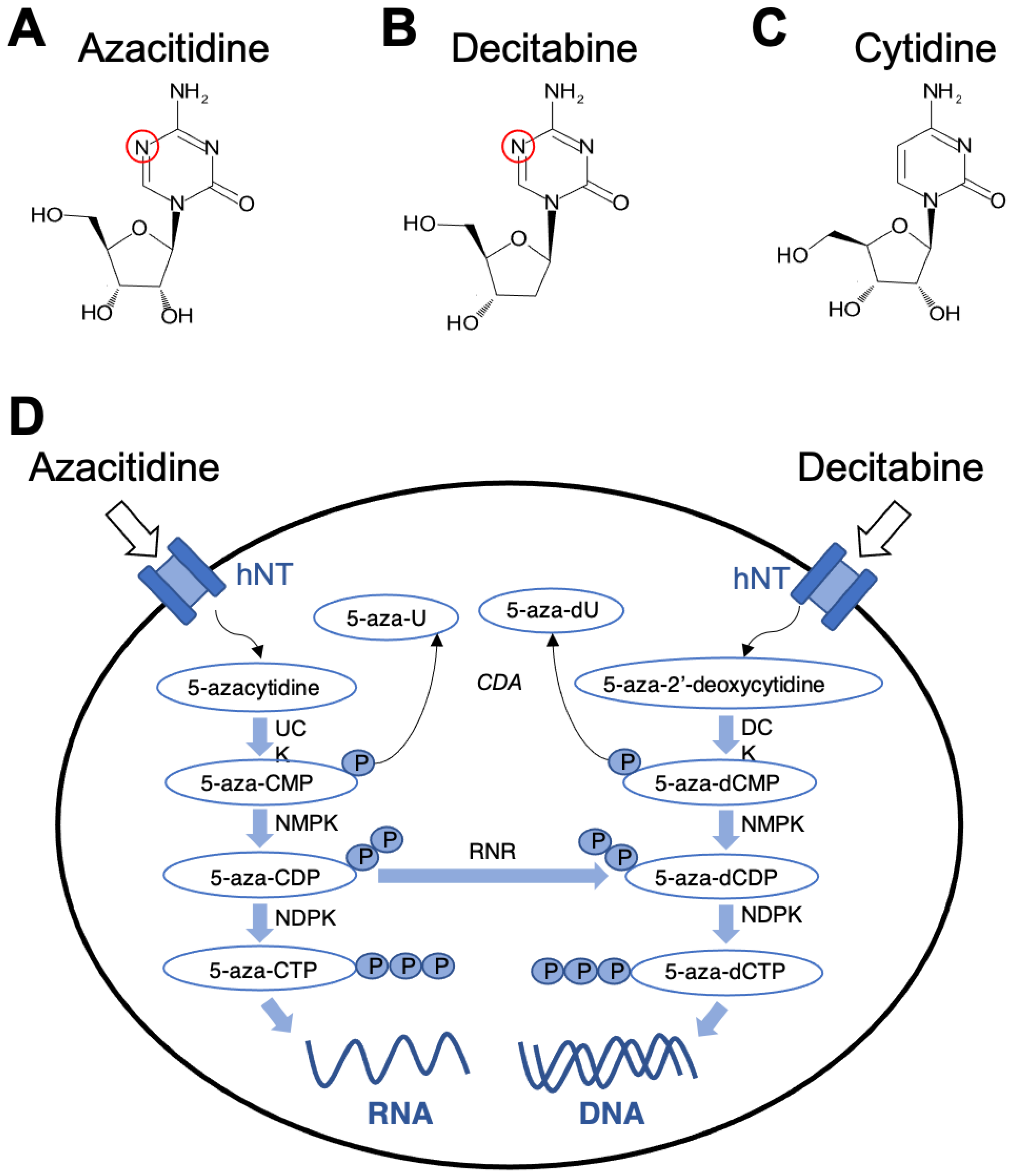{kind=link}
{kind=link}
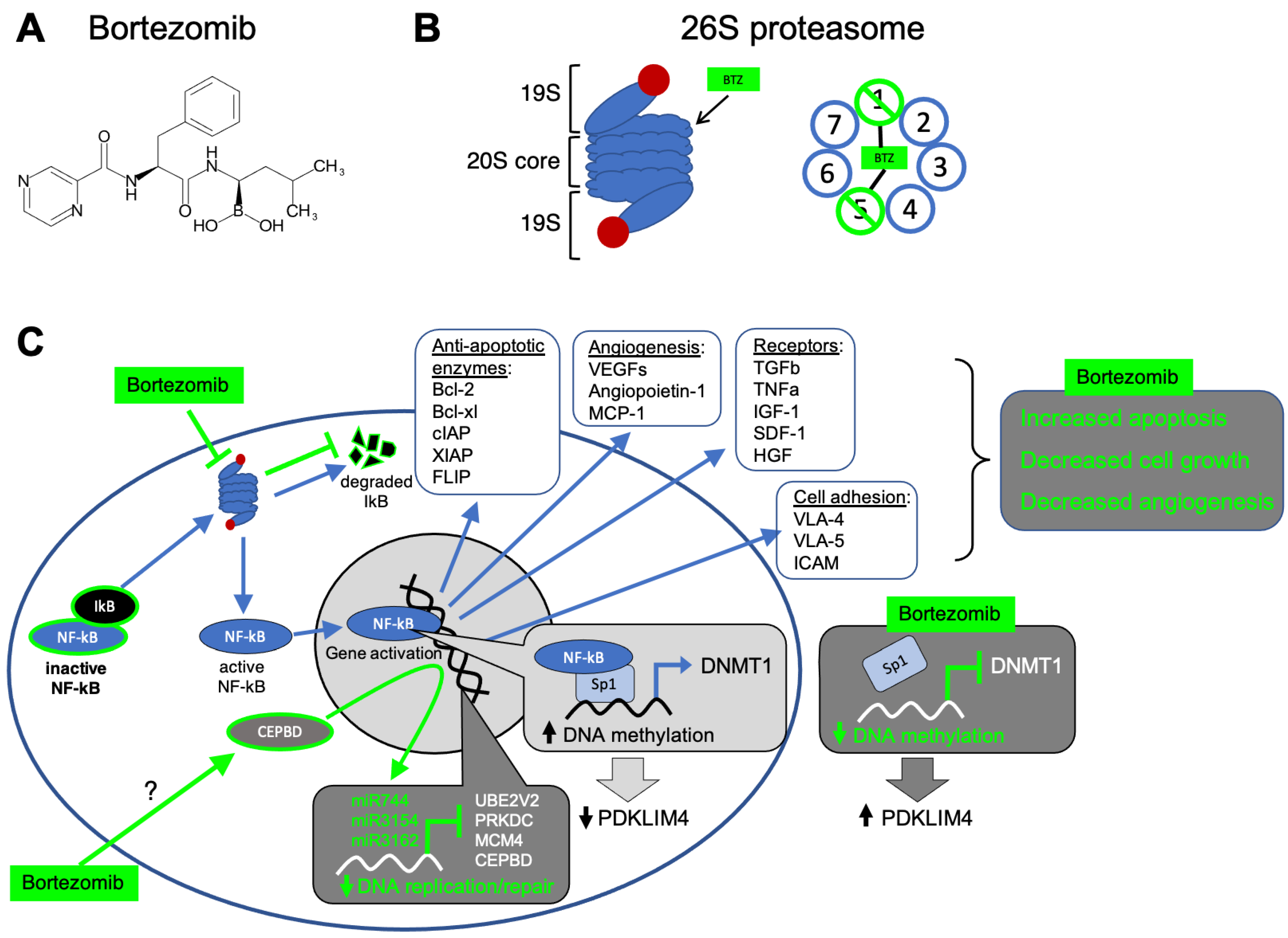{kind=link}
{kind=link}
| Study Number | Status | Drug | Study Title | Phase | Masking | Indication/ Condition | No./Type of Participants | Start/Completion Dates |
|---|---|---|---|---|---|---|---|---|
| NCT01809392 | Unknown | Decitabine | Decitabine Augments for Post Allogeneic Stem Cell Transplantation in Patients With Acute Myeloid Leukemia and Myelodysplastic Syndrome | 2, 3 | Open label, non-randomized | Acute Myeloid Leukemia, Myelodysplastic Syndromes | 15 | Jan 2013–Dec 2015 |
| NCT03377725 | Unknown | Decitabine, Arsenic Trioxide | Decitabine and Arsenic Trioxide for Myelodysplastic Syndrome (MDS) | 3 | Single, randomized | Myelodysplastic Syndromes, P53 mutation | 300 | March 2018–Nov 2020 |
| NCT02744742 | Unknown | Decitabine, Busulfan (BU), Cyclophosphamide (CY), Granulocyte Colony-Stimulating Factor(G-CSF) | G-CSF+Decitabine+BUCY vs. BUCY Conditioning Regimen for RAEB-1, REAB-2 and AML Secondary to MDS Undergoing Allo-HSCT | 2, 3 | Open label, randomized | Myelodysplastic Syndrome, Allogeneic Hematopoietic Stem Cell Transplantation, Conditioning | 122 | March 2019–March 2020 |
| NCT02214407 | Active, not recruiting | Decitabine, Hydroxyurea | Randomized Phase III Study of Decitabine +/− Hydroxyurea (HY) Versus HY in Advanced Proliferative CMML (GFM-DAC-CMML) | 3 | Open label, randomized | Myelodysplastic Syndrome | 168 | Oct 2014–Oct 2021 |
| NCT02272478 | Recruiting | Arm A: Mylotarg plus DA Versus CPX-351, Arm B: Vosaroxin and Decitabine, Arm D: Small molecule or Not, Arm C: DA V FLAG-Ida V DAC, Arm E: CPX-351 (200 V 300), Arm F: DA V IDAC | Trial to Test the Effects of Adding 1 of 2 New Treatment Agents to Commonly Used Chemotherapy Combinations (AML18) | 2, 3 | Open label, randomized | Acute Myeloid Leukemia, Myelodysplastic Syndrome | 1600 | Feb 2021–Feb 2022 |
| NCT03306264 | Recruiting | ASTX727, Dacogen (decitabine) | Study of ASTX727 vs. IV Decitabine in MDS, CMML, and AML | 3 | Open label, randomized | Myelodysplastic Syndromes, Chronic Myelomonocytic Leukemia, Acute Myeloid Leukemia | 200 | Feb 2018–May 2022 |
| NCT02172872 | Active, not recruiting | Standard combination chemotherapy, decitabine | “InDACtion” vs. “3 + 7” Induction in AML | 3 | Open label, randomized | Acute Myeloid Leukemia | 606 | Nov 2014–Dec 2022 |
| NCT04713956 | Recruiting | Granulocyte Colony-Stimulating Factor(G-CSF), Decitabine (DAC), Busulfan (BU), Cyclophosphamide (CY), Fludarabine (FLU) | G-CSF+DAC+BUCY vs. G-CSF+DAC+BF Conditioning Regimen for RAEB-1,REAB-2 and AML Secondary to MDS Undergoing Allo-HSCT | 2, 3 | Open label, randomized | Myelodysplastic Syndrome, Allogeneic Hematopoietic Stem Cell Transplantation, Conditioning | 242 | Jan 2021–July 2024 |
| NCT02085408 | Active, not recruiting | Clofarabine, daunorubicin hydrochloride, cytarabine, decitabine | Clofarabine or Daunorubicin Hydrochloride and Cytarabine Followed By Decitabine or Observation in Treating Older Patients With Newly Diagnosed Acute Myeloid Leukemia | 3 | Open label, randomized | Acute Myeloid Leukemia | 727 | Dec 2010–Oct 2024 |
| NCT04173533 | Recruiting | Oral Azacitidine, Matched placebo | Randomized Study of Oral Azacitidine vs. Placebo Maintenance in AML or MDS Patients After Allo-SCT (AMADEUS) | III | Double, Randomized | AML, Myelodysplasia | 324 | Jun 2019–Jun 2024 |
| NCT03092674 | Active, not recruiting | Azacitidine, Cytarabine, Decitabine, Midostaurin | Azacitidine With or Without Nivolumab or Midostaurin, or Decitabine and Cytarabine Alone in Treating Older Patients With Newly Diagnosed Acute Myeloid Leukemia or High-Risk Myelodysplastic Syndrome | II, III | Open Label, Randomized | AML, Myelodysplastic Syndrome | 1670 | Dec 2017–Aug 2023 |
| NCT03268954 | Recruiting | Azacitidine, Pevonedistat | Pevonedistat Plus Azacitidine Versus Single-Agent Azacitidine as First-Line Treatment for Participants With Higher-Risk Myelodysplastic Syndromes (HR MDS), Chronic Myelomonocytic Leukemia (CMML), or Low-Blast Acute Myelogenous Leukemia (AML) (PANTHER) | III | Open Label, Randomized | Myelodysplastic Syndrome (Leukemia, Myelonocytic, Chronic Leukemia, Myeloid, Acute) | 502 | Nov 2017–Feb 2025 |
| NCT00071799 | Completed | Azacitidine | A Survival Study in Patients With High Risk Myelodysplastic Syndromes Comparing Azacitidine Versus Conventional Care | III | Open Label, Randomized | Myelodysplastic Syndromes | 358 | Nov 2003–July 2007 |
| NCT04256317 | Recruiting | ASTX030 (Cedazuridine + Azacitidine) | A Study of ASTX030 (Cedazuridine in Combination With Azacitidine) in MDS, CMML, or AML | II, III | Open Label, Randomized | MDS, CMML, AML | 245 | May 2020–Apr 2023 |
| NCT03978364 | Recruiting | Azacitidine combined HHT, Azacitidine regimen | A Study of Azacitidine for Patients With Int/High -Risk MDS and AML-MRC | III | Open Label, Randomized | Myelodysplastic Syndrome, AML | 100 | Jun 2019–Dec 2022 |
| NCT00887068 | Completed | Azacitidine | Controlled Study of Post-transplant Azacitidine for Prevention of Acute Myelogenous Leukemia and Myelodysplastic Syndrome Relapse (VZ-AML-PI-0129) | III | Open Label, Randomized | AML, MDS | 187 | Apr 2009–Aug 2018 |
| NCT03173248 | Recruiting | AG-120 (ivosidenib) with Azacitidine, Placebo with Azacitidine | Study of AG-120 (Ivosidenib) vs. Placebo in Combination With Azacitidine in Patients With Previously Untreated Acute Myeloid Leukemia With an IDH1 Mutation (AGILE) | III | Triple. Randomized | AML (Newly Diagnosed, Untreated AML, AML arising from MDS, Leukemia, Myeloid, Acute | 200 | Jun 2017–Jun 2022 |
| NCT04842604 | New, not yet recruiting | Glasdegib, Azacitidine | Continuation Study of B1371019(NCT03416179) and B1371012(NCT02367456) Evaluating Azacitidine With Or Without Glasdegib In Patients With Previously Untreated AML, MDS or CMML | III | Open Label, Non-Randomized | AML, MDS, CMML | 37 | Apr 2021–Dec 2022 |
| NCT04401748 | Recruiting | Venetoclax, Azacitidine, Placebo | Study Of Venetoclax Tablet With Intravenous or Subcutaneous Azacitidine to Assess Change in Disease Activity In Adult Participants With Newly Diagnosed Higher-Risk Myelodysplastic Syndrome (Verona) | III | Quadruple, Randomized | MDS | 500 | Sep 2020–Feb 2025 |
| NCT01566695 | Active, not recruiting | Venetoclax, Azacitidine, Placebo | The Efficacy and Safety of Oral Azacitidine Plus Best Supportive Care Versus Placebo and Best Supportive Care in Subjects With Red Blood Cell (RBC) Transfusion-Dependent Anemia and Thrombocytopenia Due to International Prognostic Scoring System (IPSS) Low Risk Myelodysplastic Syndrome (MDS) | III | Quadruple, Randomized | Myelodysplastic Syndrome | 216 | Apr 2013–Dec 2021 |
| NCT04313881 | Recruiting | Magrolimab, Azacitidine, Placebo | Magrolimab + Azacitidine Versus Azacitidine + Placebo in Untreated Participants With Myelodysplastic Syndrome (MDS) (ENHANCE) | III | Double, Randomized | Myelodysplastic Syndrome | 520 | Sep 2020–Aug 2025 |
| NCT03416179 | Active, not recruiting | Glasdegib, Daunorubicin + Cytarabine, Azacitidine, Placebo | A Study Evaluating Intensive Chemotherapy With or Without Glasdegib or Azacitidine With or Without Glasdegib In Patients With Previously Untreated Acute Myeloid Leukemia (BRIGHT AML1019) | III | Quadruple, Randomized | Untreated AML, Leukemia, Myeloid, Acute | 731 | Apr 2018–Dec 2022 |
| NCT01109004 | Completed | Lenalidomide, bortezomib, dexamethasone | Stem Cell Transplant With Lenalidomide Maintenance in Patients With Multiple Myeloma (BMT CTN 0702) | 3 | Open label, randomized | Multiple Myeloma | 758 | May 2010–March 2018 |
| NCT02811978 | Completed | Bortezomib (Velcade), Dexamethasone | Study of Subcutaneous and Intravenous Velcade in Combination With Dexamethasone in Chinese Subjects With Relapsed and Refractory Multiple Myeloma | 3 | Open label, randomized | Multiple Myeloma | 81 | Sept 2016–Nov 2018 |
| NCT01146834 | Completed | Bortezomib (Velcade), cyclophosphamide, G-CSF, Plerixafor | Trial of Three Stem Cell Mobilization Regimens for Multiple Myeloma | 3 | Open label, randomized | Multiple Myeloma | 47 | March 2011–Feb 2019 |
| NCT02112916 | Active, not recruiting | Bortezomib | Combination Chemotherapy With or Without Bortezomib in Treating Younger Patients With Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia or Stage II-IV T-Cell Lymphoblastic Lymphoma | 3 | Open label, randomized | T-Cell Acute Lymphoblastic Leukemia, Stage II-IV T-Cell Lymphoblastic Lymphoma | 844 | Sept 2014–March 2020 |
| NCT02195479 | Active, not recruiting | Velcade, Melphalan, Prednisone, Daratumumab IV and SC, Dexamethasone | A Study of Combination of Daratumumab and Velcade (Bortezomib) Melphalan-Prednisone (DVMP) Compared to Velcade Melphalan-Prednisone (VMP) in Participants With Previously Untreated Multiple Myeloma | 3 | Open label, randomized | Multiple myeloma | 706 | Dec 2014–Oct 2021 |
| NCT02136134 | Active, not recruiting | Bortezomib, Daratumumab | Addition of Daratumumab to Combination of Bortezomib and Dexamethasone in Participants With Relapsed or Refractory Multiple Myeloma | 3 | Open label, randomized | Multiple Myeloma | 499 | Jan 2014–Sept 2021 |
| NCT01208662 | Active, not recruiting | Lenalidomide, Bortezomib, Dexamethasone | Randomized Trial of Lenalidomide, Bortezomib, Dexamethasone vs. High-Dose Treatment With SCT in MM Patients up to Age 65 (DFCI 10-106) | 3 | Open label, randomized | Multiple Myeloma | 660 | Sept 2010–Sept 2023 |
| NCT03110562 | Active, not recruiting | Bortezomib, Selinexor, Dexamethasone | Bortezomib, Selinexor, and Dexamethasone in Patients with Multiple Myeloma (BOSTON) | 3 | Open label, randomized | Multiple Myeloma | 402 | May 2017–Sept 2023 |
| NCT01371981 | Active, not recruiting | Bortezomib, Sorafenib Tosylate | Bortezomib and Sorafenib Tosylate in Treating Patients With Newly Diagnosed Acute Myeloid Leukemia | 3 | Open label, randomized | Acute Myeloid Leukemia | 1645 | June 2011–Sept 2027 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorrentino, V.G.; Thota, S.; Gonzalez, E.A.; Rameshwar, P.; Chang, V.T.; Etchegaray, J.-P. Hypomethylating Chemotherapeutic Agents as Therapy for Myelodysplastic Syndromes and Prevention of Acute Myeloid Leukemia. Pharmaceuticals 2021, 14, 641. https://doi.org/10.3390/ph14070641
Sorrentino VG, Thota S, Gonzalez EA, Rameshwar P, Chang VT, Etchegaray J-P. Hypomethylating Chemotherapeutic Agents as Therapy for Myelodysplastic Syndromes and Prevention of Acute Myeloid Leukemia. Pharmaceuticals. 2021; 14(7):641. https://doi.org/10.3390/ph14070641
Chicago/Turabian StyleSorrentino, Vincent G., Srijan Thota, Edward A. Gonzalez, Pranela Rameshwar, Victor T. Chang, and Jean-Pierre Etchegaray. 2021. "Hypomethylating Chemotherapeutic Agents as Therapy for Myelodysplastic Syndromes and Prevention of Acute Myeloid Leukemia" Pharmaceuticals 14, no. 7: 641. https://doi.org/10.3390/ph14070641
APA StyleSorrentino, V. G., Thota, S., Gonzalez, E. A., Rameshwar, P., Chang, V. T., & Etchegaray, J.-P. (2021). Hypomethylating Chemotherapeutic Agents as Therapy for Myelodysplastic Syndromes and Prevention of Acute Myeloid Leukemia. Pharmaceuticals, 14(7), 641. https://doi.org/10.3390/ph14070641