
cfDNA Sequencing: Technological Approaches and Bioinformatic Issues


Abstract
1. Introduction
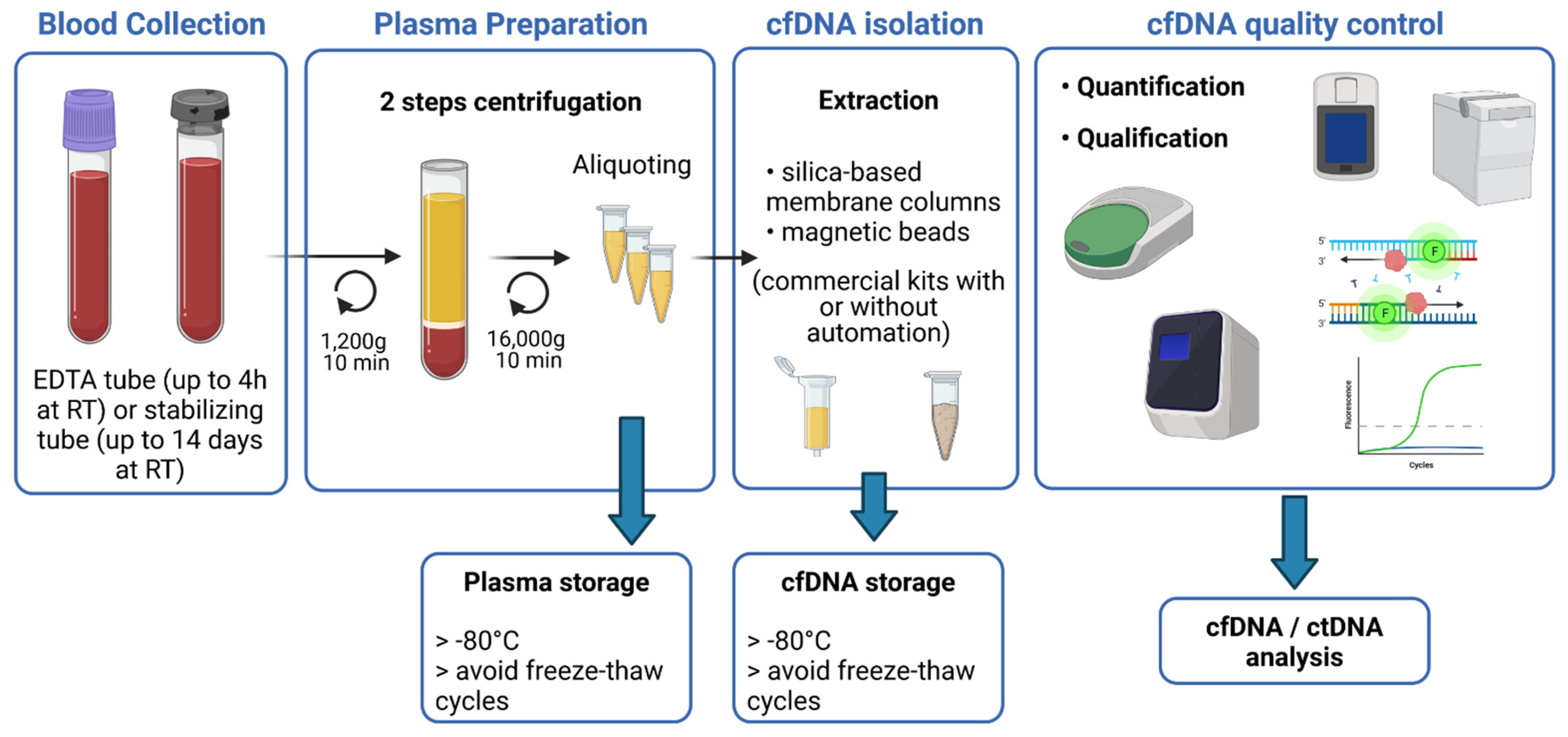
2. Pre-Analytical Requirements
3. Detection of ctDNA by Sequencing Technologies
3.1. PCR-Based Methods
3.1.1. Quantitative PCR
3.1.2. Digital PCR
3.1.3. PCR Coupled with Mass Spectrometry
3.2. Targeted NGS-Based Methods
- Tagged-amplicon deep sequencing (Tam-Seq)
- Safe-Sequencing System (Safe-SeqS)
- Duplex sequencing
- Targeted error correction sequencing (TEC-Seq)
- Single primer extension (SPE) with unique molecular barcode
- Cancer Personalized Profiling by Deep Sequencing (CAPP-Seq)
- Immunoglobulin high-throughput sequencing (Ig-HTS)
3.3. Untargeted NGS-Based Methods
4. Bioinformatical Methods
4.1. Adapter Contamination
4.2. Library Biases and Molecular Barcoding
4.3. Bioinformatics Processing
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mandel, P.; Metais, P. Nuclear acids in human blood plasma. C. R. Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar]
- Koffler, D.; Agnello, V.; Winchester, R.; Kunkel, H.G. The Occurrence of Single-Stranded DNA in the Serum of Patients with Systemic Lupus Erythematosus and Other Diseases. J. Clin. Investig. 1973, 52, 198–204. [Google Scholar] [CrossRef]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar] [PubMed]
- Stroun, M.; Anker, P.; Maurice, P.; Lyautey, J.; Lederrey, C.; Beljanski, M. Neoplastic Characteristics of the DNA Found in the Plasma of Cancer Patients. Oncology 1989, 46, 318–322. [Google Scholar] [CrossRef]
- Sidransky, D.; Von Eschenbach, A.; Tsai, Y.C.; Jones, P.; Summerhayes, I.; Marshall, F.; Paul, M.; Green, P.; Hamilton, S.R.; Frost, P.; et al. Identification of p53 gene mutations in bladder cancers and urine samples. Science 1991, 252, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Vasioukhin, V.; Anker, P.; Maurice, P.; Lyautey, J.; Lederrey, C.; Stroun, M. Point mutations of the N-ras gene in the blood plasma DNA of patients with myelodysplastic syndrome or acute myelogenous leukaemia. Br. J. Haematol. 1994, 86, 774–779. [Google Scholar] [CrossRef]
- Anker, P.; Lefort, F.; Vasioukhin, V.; Lyautey, J.; Lederrey, C.; Chen, X.Q.; Stroun, M.; Mulcahy, H.E.; Farthing, M.J. K-ras mutations are found in DNA extracted from the plasma of patients with colorectal cancer. Gastroenterology 1997, 112, 1114–1120. [Google Scholar] [CrossRef]
- Austrup, F.; Uciechowski, P.; Eder, C.; Böckmann, B.; Suchy, B.; Driesel, G.; Jäckel, S.; Kusiak, I.; Grill, H.J.; Giesing, M. Prognostic value of genomic alterations in minimal residual cancer cells purified from the blood of breast cancer patients. Br. J. Cancer 2000, 83, 1664–1673. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bobillo, S.; Crespo, M.; Escudero, L.; Mayor, R.; Raheja, P.; Carpio, C.; Rubio-Perez, C.; Tazón-Vega, B.; Palacio, C.; Carabia, J.; et al. Cell free circulating tumor DNA in cerebrospinal fluid detects and monitors central nervous system involvement of B-cell lymphomas. Haematologica 2020, 106, 513–521. [Google Scholar] [CrossRef]
- Spina, V.; Bruscaggin, A.; Cuccaro, A.; Martini, M.; Di Trani, M.; Forestieri, G.; Manzoni, M.; Condoluci, A.; Arribas, A.; Terzi-Di-Bergamo, L.; et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood 2018, 131, 2413–2425. [Google Scholar] [CrossRef]
- Camus, V.; Viennot, M.; LeQuesne, J.; Viailly, P.-J.; Bohers, E.; Bessi, L.; Marcq, B.; Etancelin, P.; Dubois, S.; Picquenot, J.-M.; et al. Targeted genotyping of circulating tumor DNA for classical Hodgkin lymphoma monitoring: A prospective study. Haematologica 2020, 106, 154–162. [Google Scholar] [CrossRef]
- Satyal, U.; Srivastava, A.; Abbosh, P.H. Urine Biopsy—Liquid Gold for Molecular Detection and Surveillance of Bladder Cancer. Front. Oncol. 2019, 9, 1266. [Google Scholar] [CrossRef]
- Esposito, A.; Criscitiello, C.; Trapani, D.; Curigliano, G. The Emerging Role of “Liquid Biopsies,” Circulating Tumor Cells, and Circulating Cell-Free Tumor DNA in Lung Cancer Diagnosis and Identification of Resistance Mutations. Curr. Oncol. Rep. 2017, 19, 1. [Google Scholar] [CrossRef]
- Elazezy, M.; Joosse, S.A. Techniques of Using Circulating Tumor DNA as a Liquid Biopsy Component in Cancer Management. Comput. Struct. Biotechnol. J. 2018, 16, 370–378. [Google Scholar] [CrossRef]
- Zill, O.A.; Banks, K.; Fairclough, S.R.; Mortimer, S.A.; Vowles, J.V.; Mokhtari, R.; Gandara, D.R.; Mack, P.C.; Odegaard, J.I.; Nagy, R.J.; et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin. Cancer Res. 2018, 24, 3528–3538. [Google Scholar] [CrossRef]
- Stanta, G.; Bonin, S. Overview on Clinical Relevance of Intra-Tumor Heterogeneity. Front. Med. 2018, 5, 85. [Google Scholar] [CrossRef]
- Scherer, F.; Kurtz, D.M.; Newman, A.M.; Craig, M.A.; Stehr, H.; Zhou, L.; Glover, C.; Kohrt, H.; Levy, R.; Diehn, M.; et al. Noninvasive Detection of Ibrutinib Resistance in Non-Hodgkin Lymphoma Using Cell-Free DNA. Blood 2016, 128, 1752. [Google Scholar] [CrossRef]
- Bohers, E.; Viailly, P.-J.; Becker, S.; Marchand, V.; Ruminy, P.; Maingonnat, C.; Bertrand, P.; Etancelin, P.; Picquenot, J.-M.; Camus, V.; et al. Non-invasive monitoring of diffuse large B-cell lymphoma by cell-free DNA high-throughput targeted sequencing: Analysis of a prospective cohort. Blood Cancer J. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Thompson, J.R.; Menon, S.P. Liquid Biopsies and Cancer Immunotherapy. Cancer J. 2018, 24, 78–83. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Scherer, F.; Jin, M.C.; Soo, J.; Craig, A.F.; Esfahani, M.S.; Chabon, J.J.; Stehr, H.; Liu, C.L.; Tibshirani, R.; et al. Circulating Tumor DNA Measurements As Early Outcome Predictors in Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2018, 36, 2845–2853. [Google Scholar] [CrossRef]
- Van der Pol, Y.; Mouliere, F. Toward the Early Detection of Cancer by Decoding the Epigenetic and Environmental Fingerprints of Cell-Free DNA. Cancer Cell 2019, 36, 350–368. [Google Scholar] [CrossRef]
- Cheng, F.; Su, L.; Qian, C. Circulating tumor DNA: A promising biomarker in the liquid biopsy of cancer. Oncotarget 2016, 7, 48832–48841. [Google Scholar] [CrossRef]
- Meddeb, R.; Pisareva, E.; Thierry, A.R. Guidelines for the Preanalytical Conditions for Analyzing Circulating Cell-Free DNA. Clin. Chem. 2019, 65, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Diaz, I.M.; Nocon, A.; Mehnert, D.H.; Fredebohm, J.; Diehl, F.; Holtrup, F. Performance of Streck cfDNA Blood Collection Tubes for Liquid Biopsy Testing. PLoS ONE 2016, 11, e0166354. [Google Scholar] [CrossRef]
- Alidousty, C.; Brandes, D.; Heydt, C.; Wagener, S.; Wittersheim, M.; Schäfer, S.C.; Holz, B.; Merkelbach-Bruse, S.; Büttner, R.; Fassunke, J.; et al. Comparison of Blood Collection Tubes from Three Different Manufacturers for the Collection of Cell-Free DNA for Liquid Biopsy Mutation Testing. J. Mol. Diagn. 2017, 19, 801–804. [Google Scholar] [CrossRef]
- Gahlawat, A.W.; Lenhardt, J.; Witte, T.; Keitel, D.; Kaufhold, A.; Maass, K.K.; Pajtler, K.W.; Sohn, C.; Schott, S. Evaluation of Storage Tubes for Combined Analysis of Circulating Nucleic Acids in Liquid Biopsies. Int. J. Mol. Sci. 2019, 20, 704. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Chen, P.; Li, S.; Luo, J.; Xia, H. Performance comparison of blood collection tubes as liquid biopsy storage system for minimizing cfDNA contamination from genomic DNA. J. Clin. Lab. Anal. 2019, 33, e22670. [Google Scholar] [CrossRef] [PubMed]
- El Messaoudi, S.; Rolet, F.; Mouliere, F.; Thierry, A.R. Circulating cell free DNA: Preanalytical considerations. Clin. Chim. Acta 2013, 424, 222–230. [Google Scholar] [CrossRef]
- Sorber, L.; Zwaenepoel, K.; Jacobs, J.; De Winne, K.; Goethals, S.; Reclusa, P.; Van Casteren, K.; Augustus, E.; Lardon, F.; Roeyen, G.; et al. Circulating Cell-Free DNA and RNA Analysis as Liquid Biopsy: Optimal Centrifugation Protocol. Cancers 2019, 11, 458. [Google Scholar] [CrossRef]
- Diefenbach, R.J.; Lee, J.; Kefford, R.; Rizos, H. Evaluation of commercial kits for purification of circulating free DNA. Cancer Genet. 2018, 228–229, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Van Dessel, L.F.; Vitale, S.R.; Helmijr, J.C.A.; Wilting, S.M.; Vlugt-Daane, M.; Hoop, E.O.-D.; Sleijfer, S.; Martens, J.W.M.; Jansen, M.P.H.M.; Lolkema, M.P.; et al. High-throughput isolation of circulating tumor DNA: A comparison of automated platforms. Mol. Oncol. 2018, 13, 392–402. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Henikoff, S.; Church, G.M. Simultaneous Discovery of Cell-Free DNA and the Nucleosome Ladder. Genetics 2018, 209, 27–29. [Google Scholar] [CrossRef]
- Lapin, M.; Oltedal, S.; Tjensvoll, K.; Buhl, T.; Smaaland, R.; Garresori, H.; Javle, M.; Glenjen, N.I.; Abelseth, B.K.; Gilje, B.; et al. Fragment size and level of cell-free DNA provide prognostic information in patients with advanced pancreatic cancer. J. Transl. Med. 2018, 16, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, S.; Lemmens, L.; Koessler, T.; Blouin, J.-L.; Nouspikel, T. Circulating tumoral DNA: Preanalytical validation and quality control in a diagnostic laboratory. Anal. Biochem. 2018, 542, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Devonshire, A.S.; Whale, A.S.; Gutteridge, A.; Jones, G.; Cowen, S.; Foy, C.A.; Huggett, J.F. Towards standardisation of cell-free DNA measurement in plasma: Controls for extraction efficiency, fragment size bias and quantification. Anal. Bioanal. Chem. 2014, 406, 6499–6512. [Google Scholar] [CrossRef]
- Alcaide, M.; Cheung, M.; Hillman, J.; Rassekh, S.R.; Deyell, R.; Batist, G.; Karsan, A.; Wyatt, A.W.; Johnson, N.; Scott, D.W.; et al. Evaluating the quantity, quality and size distribution of cell-free DNA by multiplex droplet digital PCR. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hohaus, S.; Giachelia, M.; Massini, G.; Mansueto, G.; Vannata, B.; Bozzoli, V.; Criscuolo, M.; D’Alò, F.; Martini, M.; Larocca, L.M.; et al. Cell-free circulating DNA in Hodgkin’s and non-Hodgkin’s lymphomas. Ann. Oncol. 2009, 20, 1408–1413. [Google Scholar] [CrossRef]
- Aung, K.L.; Donald, E.; Ellison, G.; Bujac, S.; Fletcher, L.; Cantarini, M.; Brady, G.; Orr, M.; Clack, G.; Ranson, M.; et al. Analytical Validation of BRAF Mutation Testing from Circulating Free DNA Using the Amplification Refractory Mutation Testing System. J. Mol. Diagn. 2014, 16, 343–349. [Google Scholar] [CrossRef]
- Siggillino, A.; Ulivi, P.; Pasini, L.; Reda, M.S.; Chiadini, E.; Tofanetti, F.R.; Baglivo, S.; Metro, G.; Crinó, L.; Delmonte, A.; et al. Detection of EGFR Mutations in Plasma Cell-Free Tumor DNA of TKI-Treated Advanced-NSCLC Patients by Three Methodologies: Scorpion-ARMS, PNAClamp, and Digital PCR. Diagnostics 2020, 10, 1062. [Google Scholar] [CrossRef]
- Zhang, X.; Chang, N.; Yang, G.; Zhang, Y.; Ye, M.; Cao, J.; Xiong, J.; Han, Z.; Wu, S.; Shang, L.; et al. A comparison of ARMS-Plus and droplet digital PCR for detecting EGFR activating mutations in plasma. Oncotarget 2017, 8, 112014–112023. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Fukuhara, T.; Tsukita, Y.; Morita, M.; Suzuki, A.; Tanaka, N.; Terasaki, H.; Nukiwa, T.; Maemondo, M. EGFR Mutation Analysis of Circulating Tumor DNA Using an Improved PNA-LNA PCR Clamp Method. Can. Respir. J. 2016, 2016, 1–7. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, Z.; Huang, C.; Ding, C.; Li, C.; Chen, J.; Zhao, J.; Miao, L. Ultrasensitive and quantitative detection ofEGFRmutations in plasma samples from patients with non-small-cell lung cancer using a dual PNA clamping-mediated LNA-PNA PCR clamp. Analyst 2019, 144, 1718–1724. [Google Scholar] [CrossRef]
- Milbury, C.A.; Li, J.; Liu, P.; Makrigiorgos, G.M. COLD-PCR: Improving the sensitivity of molecular diagnostics assays. Expert Rev. Mol. Diagn. 2011, 11, 159–169. [Google Scholar] [CrossRef]
- Galbiati, S.; Damin, F.; Burgio, V.; Brisci, A.; Soriani, N.; Belcastro, B.; Di Resta, C.; Gianni, L.; Chiari, M.; Ronzoni, M.; et al. Evaluation of three advanced methodologies, COLD-PCR, microarray and ddPCR, for identifying the mutational status by liquid biopsies in metastatic colorectal cancer patients. Clin. Chim. Acta 2019, 489, 136–143. [Google Scholar] [CrossRef]
- Wilson, W.H.; Young, R.M.; Schmitz, R.; Yang, Y.; Pittaluga, S.; Wright, G.; Lih, C.-J.; Williams, P.M.; Shaffer, A.L.; Gerecitano, J.; et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat. Med. 2015, 21, 922–926. [Google Scholar] [CrossRef]
- Hiemcke-Jiwa, L.S.; Minnema, M.C.; Loon, J.H.R.-V.; Jiwa, N.M.; De Boer, M.; Leguit, R.J.; De Weger, R.A.; Huibers, M.M. The use of droplet digital PCR in liquid biopsies: A highly sensitive technique for MYD88 p.(L265P) detection in cerebrospinal fluid. Hematol. Oncol. 2018, 36, 429–435. [Google Scholar] [CrossRef]
- Chen, K.; Ma, Y.; Ding, T.; Zhang, X.; Chen, B.; Guan, M. Effectiveness of digital PCR for MYD88L265P detection in vitreous fluid for primary central nervous system lymphoma diagnosis. Exp. Ther. Med. 2020, 20, 301–308. [Google Scholar] [CrossRef]
- Dressman, D.; Yan, H.; Traverso, G.; Kinzler, K.W.; Vogelstein, B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc. Natl. Acad. Sci. USA 2003, 100, 8817–8822. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Holdhoff, M.; Schmidt, K.; Donehower, R.; Diaz, L.A. Analysis of Circulating Tumor DNA to Confirm Somatic KRAS Mutations. J. Natl. Cancer Inst. 2009, 101, 1284–1285. [Google Scholar] [CrossRef]
- O’Leary, B.; Hrebien, S.; Beaney, M.; Fribbens, C.; Garcia-Murillas, I.; Jiang, J.; Li, Y.; Bartlett, C.H.; Andre, F.; Loibl, S.; et al. Comparison of BEAMing and Droplet Digital PCR for Circulating Tumor DNA Analysis. Clin. Chem. 2019, 65, 1405–1413. [Google Scholar] [CrossRef]
- Garcia, J.; Gauthier, A.; Lescuyer, G.; Barthelemy, D.; Geiguer, F.; Balandier, J.; Edelstein, D.L.; Jones, F.S.; Holtrup, F.; Duruisseau, M.; et al. Routine Molecular Screening of Patients with Advanced Non-SmallCell Lung Cancer in Circulating Cell-Free DNA at Diagnosis and During Progression Using OncoBEAMTM EGFR V2 and NGS Technologies. Mol. Diagn. Ther. 2021, 25, 239–250. [Google Scholar] [CrossRef]
- Butler, T.; Spellman, P.T.; Gray, J. Circulating-tumor DNA as an early detection and diagnostic tool. Curr. Opin. Genet. Dev. 2017, 42, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Milbury, C.A.; Zhong, Q.; Lin, J.; Williams, M.; Olson, J.; Link, D.R.; Hutchison, B. Determining lower limits of detection of digital PCR assays for cancer-related gene mutations. Biomol. Detect. Quantif. 2014, 1, 8–22. [Google Scholar] [CrossRef]
- Shoda, K.; Ichikawa, D.; Fujita, Y.; Masuda, K.; Hiramoto, H.; Hamada, J.; Arita, T.; Konishi, H.; Komatsu, S.; Shiozaki, A.; et al. Monitoring the HER2 copy number status in circulating tumor DNA by droplet digital PCR in patients with gastric cancer. Gastric Cancer 2016, 20, 126–135. [Google Scholar] [CrossRef]
- Lee, K.S.; Nam, S.K.; Seo, S.H.; Park, K.U.; Oh, H.-K.; Kim, D.-W.; Kang, S.-B.; Kim, W.H.; Lee, H.S. Digital polymerase chain reaction for detecting c-MYC copy number gain in tissue and cell-free plasma samples of colorectal cancer patients. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Delfau-Larue, M.-H.; Van Der Gucht, A.; Dupuis, J.; Jais, J.-P.; Nel, I.; Beldi-Ferchiou, A.; Hamdane, S.; Benmaad, I.; Laboure, G.; Verret, B.; et al. Total metabolic tumor volume, circulating tumor cells, cell-free DNA: Distinct prognostic value in follicular lymphoma. Blood Adv. 2018, 2, 807–816. [Google Scholar] [CrossRef]
- Pott, C.; Brüggemann, M.; Ritgen, M.; Van Der Velden, V.H.J.; Van Dongen, J.J.M.; Kneba, M. MRD Detection in B-Cell Non-Hodgkin Lymphomas Using Ig Gene Rearrangements and Chromosomal Translocations as Targets for Real-Time Quantitative PCR. Meth. Mol. Biol. 2019, 199–228. [Google Scholar] [CrossRef]
- Pyrak, E.; Krajczewski, J.; Kowalik, A.; Kudelski, A.; Jaworska, A. Surface Enhanced Raman Spectroscopy for DNA Biosensors—How Far Are We? Molecular 2019, 24, 4423. [Google Scholar] [CrossRef] [PubMed]
- Wee, E.J.; Wang, Y.; Tsao, S.C.-H.; Trau, M. Simple, Sensitive and Accurate Multiplex Detection of Clinically Important Melanoma DNA Mutations in Circulating Tumour DNA with SERS Nanotags. Theranostics 2016, 6, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Lyu, N.; Rajendran, V.K.; Diefenbach, R.; Charles, K.; Clarke, S.J.; Engel, A.; Rizos, H.; Molloy, M.P.; Wang, Y. Sydney 1000 Colorectal Cancer Study Investigators* Multiplex detection of ctDNA mutations in plasma of colorectal cancer patients by PCR/SERS assay. Nanotheranostics 2020, 4, 224–232. [Google Scholar] [CrossRef]
- Gray, E.S.; Witkowski, T.; Pereira, M.; Calapre, L.; Herron, K.; Irwin, D.; Chapman, B.; Khattak, M.A.; Raleigh, J.; Hatzimihalis, A.; et al. Genomic Analysis of Circulating Tumor DNA Using a Melanoma-Specific UltraSEEK Oncogene Panel. J. Mol. Diagn. 2019, 21, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Lamy, P.-J.; Van Der Leest, P.; Lozano, N.; Becht, C.; Duboeuf, F.; Groen, H.J.M.; Hilgers, W.; Pourel, N.; Rifaela, N.; Schuuring, E.; et al. Mass Spectrometry as a Highly Sensitive Method for Specific Circulating Tumor DNA Analysis in NSCLC: A Comparison Study. Cancers 2020, 12, 3002. [Google Scholar] [CrossRef] [PubMed]
- Mamanova, L.; Coffey, A.J.; Scott, C.E.; Kozarewa, I.; Turner, E.; Kumar, A.; Howard, E.; Shendure, J.; Turner, D.J. Target-enrichment strategies for next-generation sequencing. Nat. Methods 2010, 7, 111–118. [Google Scholar] [CrossRef]
- Deveson, I.W.; Gong, B.; Lai, K.; LoCoco, J.S.; Richmond, T.A.; Schageman, J.; Zhang, Z.; Novoradovskaya, N.; Willey, J.C.; Jones, W.; et al. Evaluating the analytical validity of circulating tumor DNA sequencing assays for precision oncology. Nat. Biotechnol. 2021, 1–14. [Google Scholar] [CrossRef]
- Glenn, T.C. Field guide to next-generation DNA sequencers. Mol. Ecol. Resour. 2011, 11, 759–769. [Google Scholar] [CrossRef]
- Loman, N.J.; Misra, R.V.; Dallman, T.J.; Constantinidou, C.; Gharbia, S.E.; Wain, J.; Pallen, M.J. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 2012, 30, 434–439. [Google Scholar] [CrossRef]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017, 9, eaan2415. [Google Scholar] [CrossRef]
- Xu, C.; Ranjbar, M.R.N.; Wu, Z.; Dicarlo, J.; Wang, Y. Detecting very low allele fraction variants using targeted DNA sequencing and a novel molecular barcode-aware variant caller. BMC Genom. 2017, 18, 5. [Google Scholar] [CrossRef]
- Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat. Rev. Genet. 2018, 19, 269–285. [Google Scholar] [CrossRef]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.Y.; Kaper, F.; Dawson, S.-J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive Identification and Monitoring of Cancer Mutations by Targeted Deep Sequencing of Plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef]
- Gale, D.; Lawson, A.R.J.; Howarth, K.; Madi, M.; Durham, B.; Smalley, S.; Calaway, J.; Blais, S.; Jones, G.; Clark, J.; et al. Development of a highly sensitive liquid biopsy platform to detect clinically-relevant cancer mutations at low allele fractions in cell-free DNA. PLoS ONE 2018, 13, e0194630. [Google Scholar] [CrossRef] [PubMed]
- Fostira, F.; Oikonomopoulou, P.; Kladi, A.; Edelstein, D.; Stieler, K.; Heim, D.; Gkotzamanidou, M.; Anastasiou, M.; Kotsantis, I.; Kavourakis, G.; et al. Blood-based testing of mutations in patients with head and neck squamous cell carcinoma (HNSCC) using highly sensitive SafeSEQ technology. Ann. Oncol. 2019, 30, v469. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Lo, S.N.; Wang, Y.; Li, L.; Christie, M.; Lee, M.; Wong, R.; Kosmider, S.; Skinner, I.; et al. Prognostic significance of postsurgery circulating tumor DNA in nonmetastatic colorectal cancer: Individual patient pooled analysis of three cohort studies. Int. J. Cancer 2021, 148, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.W.; Kennedy, S.R.; Salk, J.J.; Fox, E.; Hiatt, J.B.; Loeb, L.A. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 14508–14513. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.R.; Schmitt, M.W.; Fox, E.J.; Kohrn, B.; Salk, J.J.; Ahn, E.H.; Prindle, M.J.; Kuong, K.J.; Shen, J.-C.; Risques, R.-A.; et al. Detecting ultralow-frequency mutations by Duplex Sequencing. Nat. Protoc. 2014, 9, 2586–2606. [Google Scholar] [CrossRef]
- Costello, M.; Pugh, T.J.; Fennell, T.J.; Stewart, C.; Lichtenstein, L.; Meldrim, J.C.; Fostel, J.L.; Friedrich, D.C.; Perrin, D.; Dionne, D.; et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 2013, 41, e67. [Google Scholar] [CrossRef]
- Alcaide, M.; Yu, S.; Davidson, J.; Albuquerque, M.; Bushell, K.; Fornika, D.; Arthur, S.; Grande, B.M.; McNamara, S.; du Tertre, M.C.; et al. Targeted error-suppressed quantification of circulating tumor DNA using semi-degenerate barcoded adapters and biotinylated baits. Sci. Rep. 2017, 7, 10574. [Google Scholar] [CrossRef]
- Ren, Y.; Zhang, Y.; Wang, D.; Liu, F.; Fu, Y.; Xiang, S.; Su, L.; Li, J.; Dai, H.; Huang, B. SinoDuplex: An Improved Duplex Sequencing Approach to Detect Low-frequency Variants in Plasma cfDNA Samples. Genom. Proteom. Bioinform. 2020, 18, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Mallampati, S.; Zalles, S.; Duose, D.Y.; Hu, P.C.; Medeiros, L.J.; Wistuba, I.I.; Kopetz, S.; Luthra, R. Development and Application of Duplex Sequencing Strategy for Cell-Free DNA–Based Longitudinal Monitoring of Stage IV Colorectal Cancer. J. Mol. Diagn. 2019, 21, 994–1009. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Xu, C.; Kim, D.; Lewis, M.; Dicarlo, J.; Wang, Y. Targeted Single Primer Enrichment Sequencing with Single End Duplex-UMI. Sci. Rep. 2019, 9, 4810. [Google Scholar] [CrossRef] [PubMed]
- Sim, W.C.; Loh, C.H.; Toh, G.L.-X.; Lim, C.W.; Chopra, A.; Chang, A.Y.C.; Goh, L.L. Non-invasive detection of actionable mutations in advanced non-small-cell lung cancer using targeted sequencing of circulating tumor DNA. Lung Cancer 2018, 124, 154–159. [Google Scholar] [CrossRef]
- Zou, D.; Day, R.; Cocadiz, J.A.; Parackal, S.; Mitchell, W.; Black, M.A.; Lawrence, B.; Fitzgerald, S.; Print, C.; Jackson, C.; et al. Circulating tumor DNA is a sensitive marker for routine monitoring of treatment response in advanced colorectal cancer. Carcinogenesis 2020, 41, 1507–1517. [Google Scholar] [CrossRef]
- Viailly, P.-J.; Sater, V.; Viennot, M.; Bohers, E.; Vergne, N.; Berard, C.; Dauchel, H.; Lecroq, T.; Celebi, A.; Ruminy, P.; et al. Improving high-resolution copy number variation analysis from next generation sequencing using unique molecular identifiers. BMC Bioinform. 2021, 22, 1–15. [Google Scholar] [CrossRef]
- Newman, A.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Bratman, S.V.; Newman, A.; Alizadeh, A.A.; Diehn, M. Potential clinical utility of ultrasensitive circulating tumor DNA detection with CAPP-Seq. Expert Rev. Mol. Diagn. 2015, 15, 715–719. [Google Scholar] [CrossRef]
- Klass, D.; Newman, A.; Lovejoy, A.; Zhou, L.; Stehr, H.; Xu, T.; He, J.; Komaki, R.; Liao, Z.; Maru, D.; et al. Analysis of Circulating Tumor DNA in Esophageal Carcinoma Patients Treated with Chemoradiation Therapy. Int. J. Radiat. Oncol. 2015, 93, S104–S105. [Google Scholar] [CrossRef]
- Scherer, F.; Kurtz, D.M.; Newman, A.; Stehr, H.; Craig, A.F.M.; Esfahani, M.S.; Lovejoy, A.F.; Chabon, J.J.; Klass, D.M.; Liu, C.L.; et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci. Transl. Med. 2016, 8, 364ra155. [Google Scholar] [CrossRef]
- Dudley, J.C.; Schroers-Martin, J.; Lazzareschi, D.V.; Shi, W.Y.; Chen, S.B.; Esfahani, M.S.; Trivedi, D.; Chabon, J.J.; Chaudhuri, A.A.; Stehr, H.; et al. Detection and Surveillance of Bladder Cancer Using Urine Tumor DNA. Cancer Discov. 2019, 9, 500–509. [Google Scholar] [CrossRef]
- Newman, A.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef]
- Roschewski, M.; Dunleavy, K.; Pittaluga, S.; Moorhead, M.; Pepin, F.; Kong, K.; Shovlin, M.; Jaffe, E.S.; Staudt, L.M.; Lai, C.; et al. Circulating tumour DNA and CT monitoring in patients with untreated diffuse large B-cell lymphoma: A correlative biomarker study. Lancet Oncol. 2015, 16, 541–549. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Green, M.R.; Bratman, S.V.; Scherer, F.; Liu, C.L.; Kunder, C.A.; Takahashi, K.; Glover, C.; Keane, C.; Kihira, S.; et al. Noninvasive monitoring of diffuse large B-cell lymphoma by immunoglobulin high-throughput sequencing. Blood 2015, 125, 3679–3687. [Google Scholar] [CrossRef]
- Sarkozy, C.; Huet, S.; Carlton, V.E.; Fabiani, B.; Delmer, A.; Jardin, F.; Delfau-Larue, M.-H.; Hacini, M.; Ribrag, V.; Guidez, S.; et al. The prognostic value of clonal heterogeneity and quantitative assessment of plasma circulating clonal IG-VDJ sequences at diagnosis in patients with follicular lymphoma. Oncotarget 2017, 8, 8765–8774. [Google Scholar] [CrossRef]
- Hossain, N.M.; Dahiya, S.; Le, R.; Abramian, A.M.; Kong, K.A.; Muffly, L.S.; Miklos, D.B. Circulating tumor DNA assessment in patients with diffuse large B-cell lymphoma following CAR T-cell therapy. Leuk. Lymphoma 2019, 60, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Chabner, B.A. Application of Cell-free DNA Analysis to Cancer Treatment. N. Engl. J. Med. 2018, 379, 1754–1765. [Google Scholar] [CrossRef] [PubMed]
- Bos, M.K.; Angus, L.; Nasserinejad, K.; Jager, A.; Jansen, M.P.; Martens, J.W.; Sleijfer, S. Whole exome sequencing of cell-free DNA—A systematic review and Bayesian individual patient data meta-analysis. Cancer Treat. Rev. 2020, 83, 101951. [Google Scholar] [CrossRef]
- Choi, M.; Scholl, U.I.; Ji, W.; Liu, T.; Tikhonova, I.R.; Zumbo, P.; Nayir, A.; Bakkaloğlu, A.; Özen, S.; Sanjad, S.; et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. USA 2009, 106, 19096–19101. [Google Scholar] [CrossRef]
- Murtaza, M.; Dawson, S.-J.; Tsui, D.W.Y.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.-F.; Kingsbury, Z.; Wong, A.S.C.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nat. Cell Biol. 2013, 497, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Heidary, M.; Auer, M.; Ulz, P.; Heitzer, E.; Petru, E.; Gasch, C.; Riethdorf, S.; Mauermann, O.; Lafer, I.; Pristauz, G.; et al. The dynamic range of circulating tumor DNA in metastatic breast cancer. Breast Cancer Res. 2014, 16, 1–10. [Google Scholar] [CrossRef]
- Murtaza, M.; Dawson, S.; Pogrebniak, K.; Rueda, O.M.; Provenzano, E.; Grant, J.; Chin, S.-F.; Tsui, D.W.Y.; Marass, F.; Gale, D.; et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat. Commun. 2015, 6, 8760. [Google Scholar] [CrossRef]
- Lebofsky, R.; Decraene, C.; Bernard, V.; Kamal, M.; Blin, A.; Leroy, Q.; Frio, T.R.; Pierron, G.; Callens, C.; Bieche, I.; et al. Circulating tumor DNA as a non-invasive substitute to metastasis biopsy for tumor genotyping and personalized medicine in a prospective trial across all tumor types. Mol. Oncol. 2015, 9, 783–790. [Google Scholar] [CrossRef]
- Chicard, M.; Colmet-Daage, L.; Clement, N.; Danzon, A.; Bohec, M.; Bernard, V.; Baulande, S.; Bellini, A.; Eve, L.; Pierron, G.; et al. Whole-Exome Sequencing of Cell-Free DNA Reveals Temporo-spatial Heterogeneity and Identifies Treatment-Resistant Clones in Neuroblastoma. Clin. Cancer Res. 2018, 24, 939–949. [Google Scholar] [CrossRef]
- Heitzer, E.; Ulz, P.; Belic, J.; Gutschi, S.; Quehenberger, F.; Fischereder, K.; Benezeder, T.; Auer, M.; Pischler, C.; Mannweiler, S.; et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med. 2013, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Raman, L.; Van Der Linden, M.; De Vriendt, C.; Broeck, B.V.D.; Muylle, K.; Deeren, D.; Dedeurwaerdere, F.; Verbeke, S.; Dendooven, A.; De Grove, K.; et al. Shallow-depth sequencing of cell-free DNA for Hodgkin and diffuse large B-cell lymphoma (differential) diagnosis: A standardized approach with underappreciated potential. Haematologica 2020. [Google Scholar] [CrossRef]
- Kinde, I.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. FAST-SeqS: A Simple and Efficient Method for the Detection of Aneuploidy by Massively Parallel Sequencing. PLoS ONE 2012, 7, e41162. [Google Scholar] [CrossRef] [PubMed]
- Douville, C.; Springer, S.; Kinde, I.; Cohen, J.D.; Hruban, R.H.; Lennon, A.M.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Detection of aneuploidy in patients with cancer through amplification of long interspersed nucleotide elements (LINEs). Proc. Natl. Acad. Sci. USA 2018, 115, 1871–1876. [Google Scholar] [CrossRef] [PubMed]
- Leary, R.J.; Kinde, I.; Diehl, F.; Schmidt, K.; Clouser, C.; Duncan, C.; Antipova, A.; Lee, C.; McKernan, K.; De La Vega, F.M.; et al. Development of Personalized Tumor Biomarkers Using Massively Parallel Sequencing. Sci. Transl. Med. 2010, 2, 20ra14. [Google Scholar] [CrossRef]
- Leary, R.J.; Sausen, M.; Kinde, I.; Papadopoulos, N.; Carpten, J.D.; Craig, D.; O’Shaughnessy, J.; Kinzler, K.W.; Parmigiani, G.; Vogelstein, B.; et al. Detection of Chromosomal Alterations in the Circulation of Cancer Patients with Whole-Genome Sequencing. Sci. Transl. Med. 2012, 4, 162ra154. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-W.; Song, Y.; Kim, H.-S.; Sim, H.W.; Poojan, S.; Eom, B.W.; Kook, M.-C.; Joo, J.; Hong, K.-M.; Kim, Y.-H. Monitoring circulating tumor DNA by analyzing personalized cancer-specific rearrangements to detect recurrence in gastric cancer. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018, 10, eaat4921. [Google Scholar] [CrossRef]
- Cristiano, S.; Leal, A.; Phallen, J.; Fiksel, J.; Adleff, V.; Bruhm, D.C.; Jensen, S.Ø.; Medina, J.E.; Hruban, C.; White, J.R.; et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nat. Cell Biol. 2019, 570, 385–389. [Google Scholar] [CrossRef]
- Mehrmohamadi, M.; Esfahani, M.S.; Soo, J.; Scherer, F.; Schroers-Martin, J.G.; Chen, B.; Kurtz, D.M.; Hamilton, E.; Liu, C.L.; Diehn, M.; et al. Distinct Chromatin Accessibility Profiles of Lymphoma Subtypes Revealed By Targeted Cell Free DNA Profiling. Blood 2018, 132, 672. [Google Scholar] [CrossRef]
- Ørntoft, M.-B.W.; Jensen, S.Ø.; Hansen, T.B.; Bramsen, J.B.; Andersen, C.L. Comparative analysis of 12 different kits for bisulfite conversion of circulating cell-free DNA. Epigenetics 2017, 12, 626–636. [Google Scholar] [CrossRef]
- Cirillo, M.; Craig, A.F.; Borchmann, S.; Kurtz, D.M. Liquid biopsy in lymphoma: Molecular methods and clinical applications. Cancer Treat. Rev. 2020, 91, 102106. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.Y.; Singhania, R.; Fehringer, G.; Chakravarthy, A.; Roehrl, M.H.A.; Chadwick, D.; Zuzarte, P.C.; Borgida, A.; Wang, T.T.; Li, T.; et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nat. Cell Biol. 2018, 563, 579–583. [Google Scholar] [CrossRef]
- Chen, X.; Gole, J.; Gore, A.; He, Q.; Lu, M.; Min, J.; Yuan, Z.; Yang, X.; Jiang, Y.; Zhang, T.; et al. Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.S.; Hansen, J.W.; Kristensen, S.S.; Tholstrup, D.; Harsløf, L.B.S.; Pedersen, O.B.; Brown, P.D.N.; Grønbæk, K. Aberrant methylation of cell-free circulating DNA in plasma predicts poor outcome in diffuse large B cell lymphoma. Clin. Epigenet. 2016, 8, 1–11. [Google Scholar] [CrossRef]
- Wedge, E.; Hansen, J.W.; Garde, C.; Asmar, F.; Tholstrup, D.; Kristensen, S.S.; Munch-Petersen, H.D.; Ralfkiaer, E.; Brown, P.; Grønbaek, K.; et al. Global hypomethylation is an independent prognostic factor in diffuse large B cell lymphoma. Am. J. Hematol. 2017, 92, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra, 1st ed.; O’Reilly Media: Cambridge, MA, USA, 2020. [Google Scholar]
- Kechin, A.; Boyarskikh, U.; Kel, A.; Filipenko, M. cutPrimers: A New Tool for Accurate Cutting of Primers from Reads of Targeted Next Generation Sequencing. J. Comput. Biol. 2017, 24, 1138–1143. [Google Scholar] [CrossRef]
- Schmieder, R.; Lim, Y.W.; Rohwer, F.; Edwards, R. TagCleaner: Identification and removal of tag sequences from genomic and metagenomic datasets. BMC Bioinform. 2010, 11, 341. [Google Scholar] [CrossRef]
- Babraham Bioinformatics—Trim Galore! Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 11 May 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.; Heger, A.; Sudbery, I. UMI-tools: Modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017, 27, 491–499. [Google Scholar] [CrossRef]
- Sater, V.; Viailly, P.-J.; Lecroq, T.; Prieur-Gaston, É.; Bohers, É.; Viennot, M.; Ruminy, P.; Dauchel, H.; Vera, P.; Jardin, F. UMI-VarCal: A new UMI-based variant caller that efficiently improves low-frequency variant detection in paired-end sequencing NGS libraries. Bioinform. Oxf. Engl. 2020, 36, 2718–2724. [Google Scholar] [CrossRef]
- Boeva, V.; Popova, T.; Lienard, M.; Toffoli, S.; Kamal, M.; Le Tourneau, C.; Gentien, D.; Servant, N.; Gestraud, P.; Frio, T.R.; et al. Multi-factor data normalization enables the detection of copy number aberrations in amplicon sequencing data. Bioinform. Oxf. Engl. 2014, 30, 3443–3450. [Google Scholar] [CrossRef] [PubMed]
- Sater, V.; Viailly, P.-J.; Lecroq, T.; Ruminy, P.; Bérard, C.; Prieur-Gaston, É.; Jardin, F. UMI-Gen: A UMI-based read simulator for variant calling evaluation in paired-end sequencing NGS libraries. Comput. Struct. Biotechnol. J. 2020, 18, 2270–2280. [Google Scholar] [CrossRef]
- Volckmar, A.-L.; Sültmann, H.; Riediger, A.; Fioretos, T.; Schirmacher, P.; Endris, V.; Stenzinger, A.; Dietz, S. A field guide for cancer diagnostics using cell-free DNA: From principles to practice and clinical applications. Genes Chromosom. Cancer 2018, 57, 123–139. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Analysis Type | Technique | Sensitivity (LoD) | Targets | Applications | Advantages | Limitations | |
|---|---|---|---|---|---|---|---|
| PCR based methods | qPCR | ARMS-PCR | 0.01–0.1% | Hotspot mutation | Cancer detection and monitoring, targetable alterations, some assays approved for clinical use | High specificity and sensitivity, cost effective, rapid, ease of use | No multiplexing, limited to detection of known mutations |
| PNA-LNA Clamp PCR | |||||||
| COLD PCR | |||||||
| digital PCR | ddPCR | 0.01–0.1% | Hotspot mutations, gene fusions, CNV | Cancer detection and monitoring, targetable alterations, some assays approved for clinical use | Up to 5 targets, high sensitivity and specificity, absolute quantification, single molecule analysis, cost effective, rapid, ease of use | Limited multiplexing (number of fluorescent colors), limited to detection of known mutations | |
| BEAMing | |||||||
| PCR coupled to spectrometry | SERS | 0.1–1% | Known mutations | Cancer detection and monitoring, targetable alterations, for research use | Multiplexing capacity | Limited to detection of known mutations | |
| PCR based methods | UltraSEEK | ||||||
| NGS based methods | targeted | Tam-Seq | 2% | Known and unknown mutations, indels, CNV, chromosomal rearrangements (capture) | Cancer detection and monitoring, classification, targetable alterations, for research use | High specificity | Amplicon methods by multiplex PCR (depend on fragment size), no error correction |
| eTam-Seq | 0.02% | Error correction | Amplicon methods by multiplex PCR | ||||
| Safe-SeqS | 0.01–0.05% | Error correction by SSCS | Amplicon methods by multiplex PCR | ||||
| Duplex sequencing | 0.0001–0.1% | Error correction by DSCS | Amplicon methods by multiplex PCR | ||||
| TEC-Seq | 0.05–0.1% | Error correction by SSCS, Hybrid capture method (not dependent on fragment size) | Less comprehensive than WGS or WES | ||||
| single primer extension (SPE) | 0.5–1% | Amplicon methods by SPE (not dependent on fragment size), error correction by SSCS | Less comprehensive than WGS or WES | ||||
| SPE-duplex UMI | 0.1–0.2% | Error correction by DSCS | Less comprehensive than WGS or WES | ||||
| CAPP-Seq | 0.02% | Hybrid capture method (not dependent on fragment size) | Need large input, allelic bias (capture), stereotypical errors (hybridization step), less comprehensive than WGS or WES | ||||
| iDES eCAPP-Seq | 0.00025–0.004% | Error correction by DSCS and correction of stereotypical errors | Less comprehensive than WGS or WES | ||||
| Ig-HTS | 0.001% | VDJ rearrangements | Non-invasive monitoring, approved for clinical use | Very high sensitivity | Tissue biopsy needed | ||
| Untargeted | WES | 5% | Coding regions, intron-exon junctions, promoters, untranslated regions, non-coding DNA of miRNA genes | Cancer detection, monitoring of resistant clones in metastasis, for research use | Mutation discovery and signatures, detection of CNV, fusion genes, rearrangements, predicted neoantigens and Tumor Mutational Burden | Low sensitivity (increasing depth lead to high cost), need bioinformatic expertise | |
| WGS | 5–10% | Structural variants (fragmentation pattern, genome-wide CNV, methylation profile) | Cancer localization and origin, early detection (early and late stage), for research use | Shallow sequencing, genome wide profiling, identification of cancer signatures | Expensive, variable sensitivity (low) and specificity, need bioinformatic expertise, lots of data generated | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bohers, E.; Viailly, P.-J.; Jardin, F. cfDNA Sequencing: Technological Approaches and Bioinformatic Issues. Pharmaceuticals 2021, 14, 596. https://doi.org/10.3390/ph14060596
Bohers E, Viailly P-J, Jardin F. cfDNA Sequencing: Technological Approaches and Bioinformatic Issues. Pharmaceuticals. 2021; 14(6):596. https://doi.org/10.3390/ph14060596
Chicago/Turabian StyleBohers, Elodie, Pierre-Julien Viailly, and Fabrice Jardin. 2021. "cfDNA Sequencing: Technological Approaches and Bioinformatic Issues" Pharmaceuticals 14, no. 6: 596. https://doi.org/10.3390/ph14060596
APA StyleBohers, E., Viailly, P.-J., & Jardin, F. (2021). cfDNA Sequencing: Technological Approaches and Bioinformatic Issues. Pharmaceuticals, 14(6), 596. https://doi.org/10.3390/ph14060596