
Potentiating Therapeutic Effects of Epidermal Growth Factor Receptor Inhibition in Triple-Negative Breast Cancer

 , , ,
, , ,
Abstract

1. Introduction
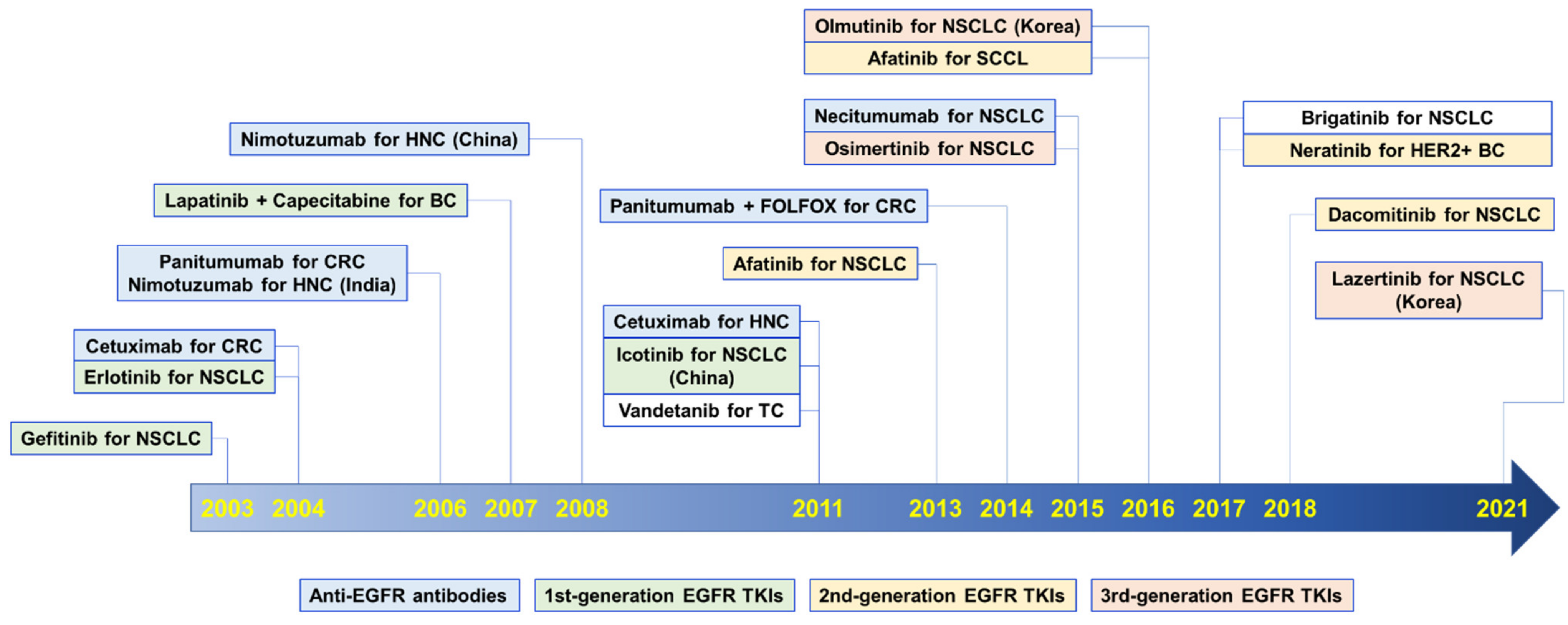
2. Anti-EGFR Therapeutics
2.1. Anti-EGFR Antibody Therapeutics

{kind=link}
{kind=link}
{kind=link}
| First Appr | Anti-EGFR Antibodies | KD1 in nM | Developer | Indications Approved by the US FDA 2 | |
|---|---|---|---|---|---|
| 2004 | Cetuximab (Erbitux®, ICM-C225) | 0.201 [40]; 1.8 [46] | ImClone Systems | 2004 | Advanced CRC 3 |
| 2011 | Late-stage HNC 4 | ||||
| 2006 | Panitumumab (Vectibix®, ABX-EGF) | 0.05 [47] | Abgenix | 2006 | Metastatic CRC |
| 2014 | First-line use of panitumumab and FOLFOX for wild-type KRAS metastatic CRC | ||||
| 2017 | Wild-type KRAS metastatic CRC | ||||
| 2006 | Nimotuzumab (TheraCIM, h-R3, BIOMAb EGFR) | 21 [46] | CIMYM BioSciences | 2006 | HNC by the Central Drugs Standard Control Organization (CDSCO), India |
| 2008 | HNC by the China Food and Drug Administration (CFDA) 5 | ||||
| 2015 | Necitumumab (Portrazza®) | 6.1 [48] | Eli Lilly | 2015 | Advanced squamous NSCLC |
2.2. Small-Molecule TKIs
2.2.1. First-Generation EGFR TKIs
2.2.2. Second-Generation EGFR TKIs
2.2.3. Third-Generation EGFR TKIs
2.2.4. Fourth-Generation EGFR TKIs
2.2.5. Multitargeting TKIs
| First Appr | Gen 1 | EGFRi | Known Targets (IC50 or EC50 nM) | Developer | Indications Approved by the US FDA 2 | |
|---|---|---|---|---|---|---|
| 2003 | 1st | Gefitinib (Iressa®, ZD-1839) | EGFR (37) [92] | AstraZeneca | 2003 | Advanced NSCLC 3 |
| 2015 | First-line treatment of metastatic NSCLC | |||||
| 2004 | 1st | Erlotinib (Tarceva®, OSI-774, CP358774, NSC 718781) | EGFR (2) [58] | OSI Pharmaceuticals | 2004 | NSCLC |
| 2010 | Advanced NSCLC | |||||
| 2013 | First-line therapy of EGFR-mutant NSCLC [50] | |||||
| 2007 | 1st | Lapatinib (Tykerb®, GSK572016,GW2016) | HER2 (9.2), EGFR (10.8), HER4 (367) [61] | GlaxoSmithKline | 2007 | Advanced or metastatic breast cancer in combination with Xeloda (capecitabine) |
| 2010 | First-line combination treatment of metastatic breast cancer | |||||
| 2011 | - | Vandetanib (Caprelsa®, Zactima®, ZD6474, ZD6) | VEGFR2 (40), VEGFR3 (110), RET (130), EGFR (500) [88,89] | AstraZeneca | 2011 | Advanced medullary thyroid cancer |
| 2011 | 1st | Icotinib (Conmana, BPI-2009H) | EGFR (5) [93] | Beta Pharma | 2011 | Solely approved for NSCLC by CFDA [50] |
| 2014 | First line treatment for advanced NSCLC patients with EGFR mutation by CFDA [65] | |||||
| 2013 | 2nd | Afatinib (Gilotriff®, BIBW2992) | EGFRL858R (0.4), EGFR (0.5), HER4 (1), EGFRL858R/T790M (10), HER2 (14) [94,95] | Boehringer Ingelheim | 2013 | late stage NSCLC |
| 2016 | squamous cell carcinoma of the lung | |||||
| 2018 | EGFR mutation-positive NSCLC | |||||
| 2015 | 3rd | Osimertinib (Tagrisso®, AZD9291) | EGFRex19del/T790M (3), EGFRL858R (9), EGFRex19del (12), EGFRL858R/T790M (13) [96] | AstraZeneca | 2015 | EGFRT790M mutation-positive NSCLC |
| 2017 | Full approval | |||||
| 2018 | First-line treatment for EGFR-mutated NSCLC | |||||
| 2020 | Adjuvant treatment of patients with early-stage, EGFR-mutated NSCLC | |||||
| 2016 | 3rd | Olmutinib (OlitaTM, HM61713, BI 1482694) | EGFRL858R/T790M (18) [78]; BTK (13.9), EGFR (17.6) [97] | Hanmi Pharmaceutical | 2016 | Approved for locally advanced or metastatic EGFRT790M-mutated NSCLC by the Korea MFDS 4 [78] |
| 2017 | - | Brigatinib (Alunbrig®, AP26113) | ALK (0.37), ROS1 (1.9), FLT3 (2.1) IGF1R (24.9), EGFRC797S/ex9del (39.9), EGFRex19del (43.7), EGFRC797S/T790M/ex19del (67.2) [86,87] | ARIAD Pharmaceuticals | 2017 | Accelerated approval for patients with ALK+ NSCLC |
| 2020 | First-line treatment option for patients with ALK+ metastatic NSCLC | |||||
| 2017 | 2nd | Neratinib (Nerlynx®, HKI-272) | HER2 (59), EGFR (92) [70] | Puma Biotechnology | 2017 | Extended adjuvant treatment of early stage HER2+ breast cancer |
| 2020 | HER2+ metastatic breast cancer | |||||
| 2018 | 2nd | Dacomitinib (Vizimpro®, PF00299804) | EGFR (6.0), HER2 (45.7), HER4 (73.7) [74] | Pfizer | 2018 | First-line treatment of EGFR-mutated metastatic NSCLC |
| 2021 | 3rd | Lazertinib (Leclaza®, YH25448,GNS-1480) | EGFRex19del/T790M (1.7), EGFRL858R/T790M (2), EGFRex19del (5.3), EGFRL858R (20.6), EGFR (76) [79] | Genosco/Yuhan | 2021 | Approved for EGFRT790M-mutated NSCLC by the Korea MFDS |
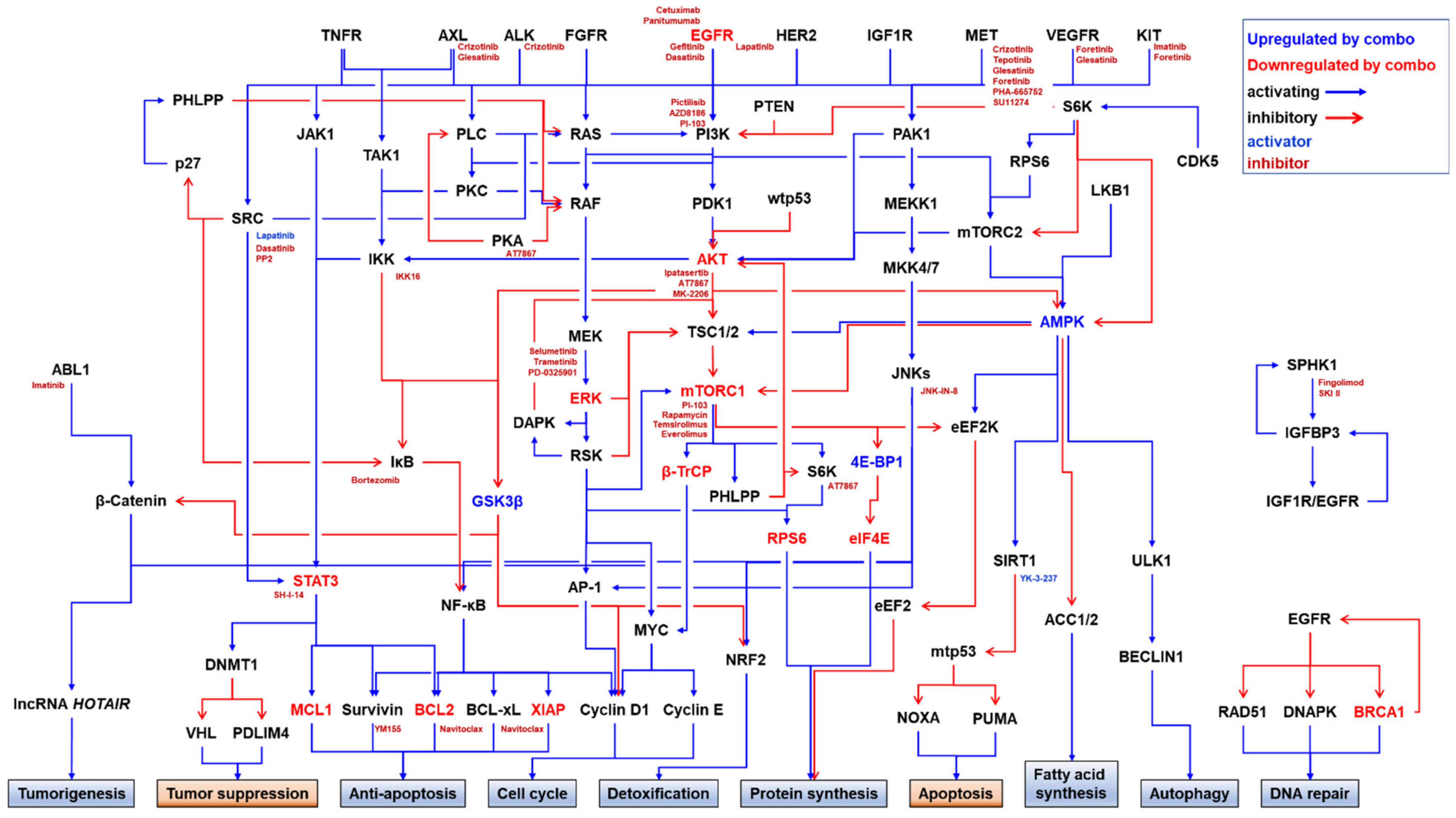
3. Resistance to Anti-EGFR Therapeutics
3.1. EGFR Mutations or Amplification
3.2. Activation of Bypass Signaling Pathways
3.3. Activation of Downstream Pathways
3.3.1. Activation of the RAS/RAF/MEK/ERK Pathway
3.3.2. Activation of the PI3K/AKT/mTORC1 Pathway
3.3.3. Activation of the NF-κB Pathway
3.3.4. Activation of the c-Jun N-Terminal Kinase (JNK) Pathway
3.3.5. Activation of the Notch Pathway
3.4. Others Resistance Mechanisms
3.4.1. Expression of Mutant p53 (mtp53)
3.4.2. Overexpression of Anti-apoptotic Proteins
3.4.3. Contributions of Phosphatases
3.4.4. Overexpression of the Heat Shock Protein 90 (HSP90)
3.4.5. Overexpression of the Estrogen Receptor Alpha (ERα) Variant
3.4.6. Overexpression of Adenosine Diphosphate Ribosylation Factor (ARF)
3.4.7. Dysregulation of Reactive Oxygen Species (ROS)
3.4.8. Expression of the Cluster of Differentiation 44 (CD44)
3.4.9. Overexpression of Annexin A2 (ANAX2)
3.4.10. Induction of Autophagy
3.4.11. Contribution of Secretomes
4. Combination Strategy for Overcoming EGFRi Resistance in TNBC
4.1. PKIs
4.1.1. Combination with Other EGFRis
4.1.2. Combination with MET Inhibitors (METis)
4.1.3. Combination with PI3K/AKT/mTORC1 Inhibitors
4.1.4. Combination with RAS/RAF/MEK/ERK Inhibitors
4.1.5. Combination with IKK/NF-κB Inhibitors
4.1.6. Combination with JNK Inhibitors
4.1.7. Combination with CDK Inhibitors
4.1.8. Combination with SFK Inhibitors
4.1.9. Combination with ABL1 Inhibitors
4.1.10. Combination with Focal Adhesion Kinase (FAK) Inhibitors
4.1.11. Combination with Rho-Associated, Coiled-Coil-Containing Protein Kinase (ROCK) Inhibitors
4.1.12. Triple Combination of PKIs
4.2. Combination with Other Targeted Therapeutics
4.2.1. Combination with PARP Inhibitors (PARPis)
4.2.2. Combination with Inhibitors of Anti-Apoptotic Proteins
4.2.3. Combination with Sphingosine Kinase (SPHK) Inhibitors
4.2.4. Combination with ARF Exchange Activity Inhibitors (ARFis)
4.2.5. Combination with Protein–Protein Interaction Inhibitors (PPIis)
4.3. Combination with Conventional Chemotherapeutics
4.3.1. Combination with Microtubule Stabilizers
4.3.2. Combination with DNA-Damaging Agents
4.3.3. Combination with Nucleoside Analogues and Nucleobases
4.3.4. Combination with Multiple Chemotherapeutics
4.4. Other Combination Strategies
4.4.1. Combination of Anti-EGFR Antibodies
4.4.2. Combination in a Single Molecule: Bispecific Antibody
4.4.3. Combination with Autophagy Inhibitors
4.4.4. Combination with Antioxidants
4.4.5. Combination with Natural Products
4.4.6. Combination with Gene Therapy
4.5. Immuno-Oncological Approaches
4.5.1. ADCC
4.5.2. Chimeric Antigen Receptor (CAR)-Engineered Cell Therapies
4.5.3. Immune Checkpoint Inhibitors (ICIs)
4.6. Use of EGFR as a Docking Protein for Targeted Drug Delivery
4.6.1. Direct Drug Conjugates
4.6.2. EGFR-Targeting Nanocarriers
5. Clinical Studies
5.1. Anti-EGFR Antibodies in Combination Therapy in Clinical Studies of TNBC
| NCT Number (Publication Year) | EGFRi | Comb Drug | Phase | Clinical Outcomes | Ref | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Treatment | Enrolled | ||||||||||
| Status | |||||||||||
| Sponsor | |||||||||||
| NCT00232505(2012) | Cetuximab | Carboplatin | Phase 2 | Combo (n = 71) | Cetuximab (n = 31) | [635] | |||||
| QW 1, IV 2, first: 400 mg/m2; subsequently, 250 mg/m2 | AUC 3 of 2 IVs on days 1, 8, and 15 of each 28-day cycle | 102 | |||||||||
| Competed | CR 4 | 1 (1%) | 0 (0%) | ||||||||
| Bristol–Myers Squibb | PR 5 | 11 (16%) | 2 (6%) | ||||||||
| SD 6 | 15 (21%) | 3 (10%) | |||||||||
| PD 7 | 38 (54%) | 26 (84%) | |||||||||
| NE 8 | 6 (8%) | 0 (0%) | |||||||||
| TTP 9 | 2.1 months | 1.4 months | |||||||||
| Median OS 10 | 10.4 months | 7.5 months | |||||||||
| NCT00463788 (2013) | Cetuximab | Cisplatin | Phase 2 | Combo (n = 115) | Cisplatin (n = 58) | [636] | |||||
| QW, IV, first: 400 mg/m2; subsequently, 250 mg/m2 | Q3W 11, IV, 75 mg/m2 on day 1, 6 cycles | 173 | |||||||||
| Completed | CR | 2 (2%) | 1 (2%) | ||||||||
| Merck KgaA | PR | 21 (18%) | 5 (9%) | ||||||||
| SD | 48 (42%) | 18 (31%) | |||||||||
| PD | 34 (30%) | 31(53%) | |||||||||
| NE | 10 (9%) | 3 (5%)) | |||||||||
| Median PFS 12 | 3.7 months | 1.5 months | |||||||||
| Median OS | 12.9 months | 9.4 months | |||||||||
| NCT00600249 (2016) | Cetuximab | Docetaxel | Phase 2 | Combo (n = 25) | [637] | ||||||
| QW, 18 IVs, first: 400 mg/m2; subsequently: 250 mg/m2 | Q3W, (100 mg/m2) on day 1, 6 cycles | ||||||||||
| 25 | |||||||||||
| Completed | pCR | 6 (24%) | |||||||||
| Merch Serono and Sanofi-Aventis | cCR 13 | 22% | |||||||||
| NCT00633464 (2015) | Cetuximab | Ixabepilone | Phase 2 | Combo (n = 39) | Ixabepilone (n = 40) | [638] | |||||
| QW, IV, first: 400 mg/m2; subsequently, 250 mg/m2 | Q3W, IV, 400 mg/m2 | 79 | |||||||||
| Completed | CR | 0 | 3 (7.5%) | ||||||||
| Bristol–Myers Squibb | PR | 14 (35.9%) | 9 (22.5%) | ||||||||
| SD | 12 (30.8%) | 17 (42.5%) | |||||||||
| PD | 10 (25.6%) | 9 (22.5%) | |||||||||
| NE | 1 (2.6%) | 2 (5.0%) | |||||||||
| ORR | 14 (35.95) | 12 (30.0%) | |||||||||
| Median PFS | 4.1 months | 4.1 months | |||||||||
| NCT00275041 (2016) | Cetuximab | Irinotecan | Phase 2 | Combo (n = 19) | [639] | ||||||
| QW, IV, first: 400 mg/m2; subsequently 250 mg/m2 | IV, 80 mg/m2 on days 1 and 8 of a 21-day cycle. | ||||||||||
| 19 | |||||||||||
| Completed | CR | 1 | |||||||||
| NCI and Alliance for Clinical Trials in Oncology | PR | 1 | |||||||||
| ORR | 11% | ||||||||||
| RR | TNBC, 18% vs. non-TNBC, 0% | ||||||||||
| Median OS | 9.4 months | ||||||||||
| Median TTP | 1.4 months | ||||||||||
| NCT00933517 (2014) | Panitumumab | FEC 14 and docetaxel | Phase 2 | Combo (n = 47) | [640] | ||||||
| 62 | |||||||||||
| Q3W, IV, 9 mg/kg, 8 cycles | Q3W, IV, FEC: 500/100/500 mg/m2, 4 cycles followed by Q3W, IV docetaxel: 100 mg/m2, 4 cycles | Completed | CR | 28 (59.6%) | |||||||
| Centre Jean Perrin | PR | 3 (6.4%) | |||||||||
| SD | 3 (6.4%) | ||||||||||
| Progression | 2 (4.3) | ||||||||||
| NE | 11 (23.4) | ||||||||||
| NCT01009983 (2015) | Panitumumab | Paclitaxel Carboplatin | Phase 2 | Combo (n = 14) | [641] | ||||||
| 6 mg/kg on days 1 and 15 | 80 mg/m2 paclitaxel and carboplatin AUC of 2 on days 1, 8, 15 | 14 | CR | 2 (14.3%) | |||||||
| Terminated | PR | 4 (28.6%) | |||||||||
| Wake Forest Univ Health Sci | SD | 3 (21.4%) | |||||||||
| PD | 4 (28.6%) | ||||||||||
| NE | 1 (7.1%) | ||||||||||
| 28-day cycle | |||||||||||
| NCT01036087 (2018) | Panitumumab | Nab-paclitaxel + carboplatin | Phase 2 | TNBC n = 19 | HR(+)/HER2(−) n = 21 | [645] | |||||
| 1 dose of panitumumab (2.5 mg/kg), then QW, panitumumab (2.5 mg/kg) + nab-paclitaxel (100 mg/m2) + carboplatin, 4 cycle followed by Q3W, FU (500 mg/m2) + epirubicin (100 mg/m2) + cyclophosphamide (500 mg/m2), 4 cycles | 40 | pCR | 8 (42.1%) | 3 (14.2%) | |||||||
| Completed | |||||||||||
| Celgene Corp and Amgen | |||||||||||
| NCT00894504 (2016) | Panitumumab | Gemcitabine + Carboplatin | Phase 2 | Combo (n = 71) | [646] | ||||||
| Q2W 15, IV, 6 mg/kg, 3 cycles | Q2W, IV, Gemcitabine, 1500 mg/m2 + Carboplatin, AUC = 2.5 IV, 3 cycles | 71 | Median PFS | 4.4 months | |||||||
| Completed | ORR | 42% | |||||||||
| SCRI 16, Amgen and Eli Lilly | |||||||||||
| NCT00239343 (2011) | Gefitinib | Epirubicin + Cyclophosphamide | Phase 2 | Combo (n = 71) | Epirubicin + Cyclophosphamide(n = 73) | [647] | |||||
| 144 | |||||||||||
| 250 mg, daily, 12 weeks | Q3W, epirubicin 90 mg/m2 + cyclophosphamide 600 mg/m2, 4 cycles | Completed | |||||||||
| AstraZeneca | pCR | 12 (17%) | 9 (12%) | ||||||||
| CR | 7 (10%) | 7 (10%) | |||||||||
| PR | 41 (58%) | 38 (52%) | |||||||||
| SD | 17 (24%) | 26 (43%) | |||||||||
| PD | 5 (7%) | 2 (2.7%) | |||||||||
| NE | 1 (1.4%) | 0 (0%) | |||||||||
| NCT02158507 (2021) | Lapatinib | Veliparib | NA | Combo (n = 17) | [648] | ||||||
| 1250 mg, daily, 28 days, starting at cycle 1 day 1 | 200 mg, every 12 h for 28 days, starting at cycle 1 day 2 | 23 | PR | 4 (23.5%) | |||||||
| Completed | SD | 2 (11.8%) | |||||||||
| GSK and AbbVie | PD | 11 (64.7%) | |||||||||
| >2 cycles | |||||||||||
5.2. EGFR TKIs in Combination Therapy in Clinical Studies of TNBC
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Robinson, D.R.; Wu, Y.-M.; Lin, S.-F. The Protein Tyrosine Kinase Family of the Human Genome. Oncogene 2000, 19, 5548–5557. [Google Scholar] [CrossRef] [PubMed]
- Wieduwilt, M.J.; Moasser, M.M. The Epidermal Growth Factor Receptor Family: Biology Driving Targeted Therapeutics. Cell Mol. Life Sci. 2008, 65, 1566–1584. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Dunn, E.F.; Harari, P.M. Understanding Resistance to EGFR Inhibitors—Impact on Future Treatment Strategies. Nat. Rev. Clin. Oncol. 2010, 7, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Eccles, S.A. The Epidermal Growth Factor Receptor/Erb-B/HER Family in Normal and Malignant Breast Biology. Int. J. Dev. Biol. 2011, 55, 685–696. [Google Scholar] [CrossRef]
- Yarden, Y.; Pines, G. The ERBB Network: At Last, Cancer Therapy Meets Systems Biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Scaltriti, M.; Baselga, J. The Epidermal Growth Factor Receptor Pathway: A Model for Targeted Therapy. Clin. Cancer Res. 2006, 12, 5268–5272. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB Signalling Network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Zulkifli, A.A.; Tan, F.H.; Putoczki, T.L.; Stylli, S.S.; Luwor, R.B. STAT3 Signaling Mediates Tumour Resistance to EGFR Targeted Therapeutics. Mol. Cell Endocrinol. 2017, 451, 15–23. [Google Scholar] [CrossRef]
- Normanno, N.; Bianco, C.; Luca, A.D.; Maiello, M.R.; Salomon, D.S. Target-Based Agents against ErbB Receptors and Their Ligands: A Novel Approach to Cancer Treatment. Endocr. Relat. Cancer 2003, 10, 1–21. [Google Scholar] [CrossRef]
- Bartholomeusz, C.; Yamasaki, F.; Saso, H.; Kurisu, K.; Horto-Bagyi, G.N.; Ueno, N.T. Gemcitabine Overcomes Erlotinib Resistance in EGFR-Overexpressing Cancer Cells through Downregulation of Akt. J. Cancer 2011, 435. [Google Scholar] [CrossRef]
- Knebel, F.H.; Bettoni, F.; Shimada, A.K.; Cruz, M.; Alessi, J.V.; Negrão, M.V.; Reis, L.F.L.; Katz, A.; Camargo, A.A. Sequential Liquid Biopsies Reveal Dynamic Alterations of EGFR Driver Mutations and Indicate EGFR Amplification as a New Mechanism of Resistance to Osimertinib in NSCLC. Lung Cancer 2017, 108, 238–241. [Google Scholar] [CrossRef]
- Xu, M.; Xie, Y.; Ni, S.; Liu, H. The Latest Therapeutic Strategies after Resistance to First Generation Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors (EGFR TKIs) in Patients with Non-Small Cell Lung Cancer (NSCLC). Ann. Transl. Med. 2015, 3, 96. [Google Scholar] [CrossRef]
- Patel, H.; Pawara, R.; Ansari, A.; Surana, S. Recent Updates on Third Generation EGFR Inhibitors and Emergence of Fourth Generation EGFR Inhibitors to Combat C797S Resistance. Eur. J. Med. Chem. 2017, 142, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.W.; You, K.; Bae, E.J.; Kwak, S.-J.; Seong, Y.-S.; Bae, I. Dual Inhibition of EGFR and MET Induces Synthetic Lethality in Triple-Negative Breast Cancer Cells through Downregulation of Ribosomal Protein S6. Int. J. Oncol. 2015, 47, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.K.; Cryns, V.L.; Symmans, W.F.; Sneige, N. Triple Negative Breast Carcinoma and the Basal Phenotype: From Expression Profiling to Clinical Practice. Adv. Anat. Pathol. 2007, 14, 419–430. [Google Scholar] [CrossRef]
- Huynh, M.; Pambid, M.R.; Jayanthan, A.; Dorr, A.; Los, G.; Dunn, S.E. The Dawn of Targeted Therapies for Triple Negative Breast Cancer (TNBC): A Snapshot of Investigational Drugs in Phase I and II Trials. Expert Opin. Inv. Drug 2020, 29, 1199–1208. [Google Scholar] [CrossRef]
- Brenton, J.D.; Carey, L.A.; Ahmed, A.A.; Caldas, C. Molecular Classification and Molecular Forecasting of Breast Cancer: Ready for Clinical Application? J. Clin. Oncol. 2005, 23, 7350–7360. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of Human Triple-Negative Breast Cancer Subtypes and Preclinical Models for Selection of Targeted Therapies. J. Clin. Invest. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Pietenpol, J.A. Identification and Use of Biomarkers in Treatment Strategies for Triple-negative Breast Cancer Subtypes. J. Pathol. 2014, 232, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Nakai, K.; Hung, M.-C.; Yamaguchi, H. A Perspective on Anti-EGFR Therapies Targeting Triple-Negative Breast Cancer. Am. J. Cancer Res. 2016, 6, 1609–1623. [Google Scholar]
- You, K.S.; Yi, Y.W.; Cho, J.; Seong, Y.-S. Dual Inhibition of AKT and MEK Pathways Potentiates the Anti-Cancer Effect of Gefitinib in Triple-Negative Breast Cancer Cells. Cancers 2021, 13, 1205. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Truica, C.I. Challenges in the Development of Anti-Epidermal Growth Factor Receptor Therapies in Breast Cancer. Semin. Oncol. 2004, 31, 3–8. [Google Scholar] [CrossRef]
- Liu, D.; He, J.; Yuan, Z.; Wang, S.; Peng, R.; Shi, Y.; Teng, X.; Qin, T. EGFR Expression Correlates with Decreased Disease-Free Survival in Triple-Negative Breast Cancer: A Retrospective Analysis Based on a Tissue Microarray. Med. Oncol. 2012, 29, 401–405. [Google Scholar] [CrossRef]
- Nogi, H.; Kobayashi, T.; Suzuki, M.; Tabei, I.; Kawase, K.; Toriumi, Y.; Uchida, H.; Fukushima, K. EGFR as Paradoxical Predictor of Chemosensitivity and Outcome among Triple-Negative Breast Cancer. Oncol. Rep. 2009, 21, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Pietenpol, J.A.; Tan, A.R. Triple-Negative Breast Cancer: Molecular Subtypes and New Targets for Therapy. Am. Soc. Clin. Oncol. Educ. Book 2015, e31–e39. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Ezenwajiaku, N.; Ma, C.X.; Ademuyiwa, F.O. Updates on Molecular Classification of Triple Negative Breast Cancer. Curr. Breast Cancer Rep. 2018, 10, 289–295. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S. Triple-Negative Breast Cancer Molecular Subtyping and Treatment Progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.; Shah, A.N.; Santa-Maria, C.A.; Cruz, M.R.; Mahalingam, D.; Carneiro, B.A.; Chae, Y.K.; Cristofanilli, M.; Gradishar, W.J.; Giles, F.J. Targeting Epidermal Growth Factor Receptor in Triple Negative Breast Cancer: New Discoveries and Practical Insights for Drug Development. Cancer Treat. Rev. 2017, 53, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.R.; Yang, X.; Hewitt, S.M.; Berman, A.; Lepper, E.R.; Sparreboom, A.; Parr, A.L.; Figg, W.D.; Chow, C.; Steinberg, S.M.; et al. Evaluation of Biologic End Points and Pharmacokinetics in Patients with Metastatic Breast Cancer After Treatment with Erlotinib, an Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor. J. Clin. Oncol. 2004, 22, 3080–3090. [Google Scholar] [CrossRef]
- Baselga, J.; Albanell, J.; Ruiz, A.; Lluch, A.; Gascón, P.; Guillém, V.; González, S.; Sauleda, S.; Marimón, I.; Tabernero, J.M.; et al. Phase II and Tumor Pharmacodynamic Study of Gefitinib in Patients with Advanced Breast Cancer. J. Clin. Oncol. 2005, 23, 5323–5333. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; D’Andrea, G.; Norton, L.; Yao, T.J.; Caravelli, J.; Rosen, P.P.; Hudis, C.; Seidman, A.D. A Phase I Study of Cetuximab/Paclitaxel in Patients with Advanced-Stage Breast Cancer. Clin. Breast Cancer 2006, 7, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Dickler, M.N.; Cobleigh, M.A.; Miller, K.D.; Klein, P.M.; Winer, E.P. Efficacy and Safety of Erlotinib in Patients with Locally Advanced or Metastatic Breast Cancer. Breast Cancer Res. Treat. 2009, 115, 115–121. [Google Scholar] [CrossRef]
- Normanno, N.; Tejpar, S.; Morgillo, F.; Luca, A.D.; Cutsem, E.V.; Ciardiello, F. Implications for KRAS Status and EGFR-Targeted Therapies in Metastatic CRC. Nat. Rev. Clin. Oncol. 2009, 6, 519–527. [Google Scholar] [CrossRef]
- Cai, W.-Q.; Zeng, L.-S.; Wang, L.-F.; Wang, Y.-Y.; Cheng, J.-T.; Zhang, Y.; Han, Z.-W.; Zhou, Y.; Huang, S.-L.; Wang, X.-W.; et al. The Latest Battles Between EGFR Monoclonal Antibodies and Resistant Tumor Cells. Front. Oncol. 2020, 10, 1249. [Google Scholar] [CrossRef]
- Yazdi, M.H.; Faramarzi, M.A.; Nikfar, S.; Abdollahi, M. A Comprehensive Review of Clinical Trials on EGFR Inhibitors Such as Cetuximab and Panitumumab as Monotherapy and in Combination for Treatment of Metastatic Colorectal Cancer. Avicenna J. Med. Biotechnol. 2015, 7, 134–144. [Google Scholar]
- Fala, L.; Writer, M. Portrazza (Necitumumab), an IgG1 Monoclonal Antibody, FDA Approved for Advanced Squamous Non–Small-Cell Lung Cancer. Am. Health Drug Benefits 2016, 9, 119–122. [Google Scholar]
- Goldstein, N.I.; Prewett, M.; Zuklys, K.; Rockwell, P.; Mendelsohn, J. Biological Efficacy of a Chimeric Antibody to the Epidermal Growth Factor Receptor in a Human Tumor Xenograft Model. Clin. Cancer Res. 1995, 1, 1311–1318. [Google Scholar]
- Cho, J. Mechanistic Insights into Differential Requirement of Receptor Dimerization for Oncogenic Activation of Mutant EGFR and Its Clinical Perspective. BMB Rep. 2020, 53, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Mazorra, Z.; Chao, L.; Lavastida, A.; Sanchez, B.; Ramos, M.; Iznaga, N.; Crombet, T. Nimotuzumab: Beyond the EGFR Signaling Cascade Inhibition. Semin. Oncol. 2018, 45, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Giusti, R.M.; Shastri, K.; Pilaro, A.M.; Fuchs, C.; Cordoba-Rodriguez, R.; Koti, K.; Rothmann, M.; Men, A.Y.; Zhao, H.; Hughes, M.; et al. U.S. Food and Drug Administration Approval: Panitumumab for Epidermal Growth Factor Receptor–Expressing Metastatic Colorectal Carcinoma with Progression Following Fluoropyrimidine-, Oxaliplatin-, and Irinotecan-Containing Chemotherapy Regimens. Clin. Cancer Res. 2008, 14, 1296–1302. [Google Scholar] [CrossRef]
- Yang, X.-D.; Jia, X.-C.; Corvalan, J.R.F.; Wang, P.; Davis, C.G. Development of ABX-EGF, a Fully Human Anti-EGF Receptor Monoclonal Antibody, for Cancer Therapy. Crit Rev. Oncol. Hemat. 2001, 38, 17–23. [Google Scholar] [CrossRef]
- Ramakrishnan, M.S.; Eswaraiah, A.; Crombet, T.; Piedra, P.; Saurez, G.; Iyer, H.; Arvind, A.S. Nimotuzumab, a Promising Therapeutic Monoclonal for Treatment of Tumors of Epithelial Origin. Mabs 2014, 1, 41–48. [Google Scholar] [CrossRef]
- Talavera, A.; Friemann, R.; Gómez-Puerta, S.; Martinez-Fleites, C.; Garrido, G.; Rabasa, A.; López-Requena, A.; Pupo, A.; Johansen, R.F.; Sánchez, O.; et al. Nimotuzumab, an Antitumor Antibody That Targets the Epidermal Growth Factor Receptor, Blocks Ligand Binding While Permitting the Active Receptor Conformation. Cancer Res. 2009, 69, 5851–5859. [Google Scholar] [CrossRef]
- Yang, X.D.; Jia, X.C.; Corvalan, J.R.; Wang, P.; Davis, C.G.; Jakobovits, A. Eradication of Established Tumors by a Fully Human Monoclonal Antibody to the Epidermal Growth Factor Receptor without Concomitant Chemotherapy. Cancer Res. 1999, 59, 1236–1243. [Google Scholar] [PubMed]
- Bagchi, A.; Haidar, J.N.; Eastman, S.W.; Vieth, M.; Topper, M.; Iacolina, M.D.; Walker, J.M.; Forest, A.; Shen, Y.; Novosiadly, R.D.; et al. Molecular Basis for Necitumumab Inhibition of EGFR Variants Associated with Acquired Cetuximab Resistance. Mol. Cancer 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2021 Update. Pharm. Res. 2021, 105463. [Google Scholar] [CrossRef] [PubMed]
- Karachaliou, N.; Fernandez-Bruno, M.; Bracht, J.W.P.; Rosell, R. EGFR First- and Second-Generation TKIs—There Is Still Place for Them in EGFR -Mutant NSCLC Patients. Transl. Cancer Res. 2018, 1, S23–S47. [Google Scholar] [CrossRef]
- Reardon, D.A.; Wen, P.Y.; Mellinghoff, I.K. Targeted Molecular Therapies against Epidermal Growth Factor Receptor: Past Experiences and Challenges. Neuro Oncol. 2014, 16, viii7–viii13. [Google Scholar] [CrossRef]
- Peters, S.; Zimmermann, S.; Adjei, A.A. Oral Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors for the Treatment of Non-Small Cell Lung Cancer: Comparative Pharmacokinetics and Drug–Drug Interactions. Cancer Treat. Rev. 2014, 40, 917–926. [Google Scholar] [CrossRef]
- Kazandjian, D.; Blumenthal, G.M.; Yuan, W.; He, K.; Keegan, P.; Pazdur, R. FDA Approval of Gefitinib for the Treatment of Patients with Metastatic EGFR Mutation–Positive Non–Small Cell Lung Cancer. Clin. Cancer Res. 2016, 22, 1307–1312. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus Cisplatin plus Docetaxel in Patients with Non-Small-Cell Lung Cancer Harbouring Mutations of the Epidermal Growth Factor Receptor (WJTOG3405): An Open Label, Randomised Phase 3 Trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Fukuoka, M.; Wu, Y.-L.; Thongprasert, S.; Sunpaweravong, P.; Leong, S.-S.; Sriuranpong, V.; Chao, T.-Y.; Nakagawa, K.; Chu, D.-T.; Saijo, N.; et al. Biomarker Analyses and Final Overall Survival Results from a Phase III, Randomized, Open-Label, First-Line Study of Gefitinib Versus Carboplatin/Paclitaxel in Clinically Selected Patients with Advanced Non–Small-Cell Lung Cancer in Asia (IPASS). J. Clin. Oncol. 2011, 29, 2866–2874. [Google Scholar] [CrossRef]
- Inoue, A.; Kobayashi, K.; Maemondo, M.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Updated Overall Survival Results from a Randomized Phase III Trial Comparing Gefitinib with Carboplatin–Paclitaxel for Chemo-Naïve Non-Small Cell Lung Cancer with Sensitive EGFR Gene Mutations (NEJ002). Ann. Oncol. 2013, 24, 54–59. [Google Scholar] [CrossRef]
- Douillard, J.-Y.; Ostoros, G.; Cobo, M.; Ciuleanu, T.; McCormack, R.; Webster, A.; Milenkova, T. First-Line Gefitinib in Caucasian EGFR Mutation-Positive NSCLC Patients: A Phase-IV, Open-Label, Single-Arm Study. Br. J. Cancer 2014, 110, 55–62. [Google Scholar] [CrossRef]
- Moyer, J.D.; Barbacci, E.G.; Iwata, K.K.; Arnold, L.; Boman, B.; Cunningham, A.; DiOrio, C.; Doty, J.; Morin, M.J.; Moyer, M.P.; et al. Induction of Apoptosis and Cell Cycle Arrest by CP-358,774, an Inhibitor of Epidermal Growth Factor Receptor Tyrosine Kinase. Cancer Res. 1997, 57, 4838–4848. [Google Scholar]
- Johnson, J.R.; Cohen, M.; Sridhara, R.; Chen, Y.-F.; Williams, G.M.; Duan, J.; Gobburu, J.; Booth, B.; Benson, K.; Leighton, J.; et al. Approval Summary for Erlotinib for Treatment of Patients with Locally Advanced or Metastatic Non–Small Cell Lung Cancer after Failure of at Least One Prior Chemotherapy Regimen. Clin. Cancer Res. 2005, 11, 6414–6421. [Google Scholar] [CrossRef]
- Shepherd, F.A.; Pereira, J.R.; Ciuleanu, T.; Tan, E.H.; Hirsh, V.; Thongprasert, S.; Campos, D.; Maoleekoonpiroj, S.; Smylie, M.; Martins, R.; et al. Erlotinib in Previously Treated Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2005, 353, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Rusnak, D.W.; Lackey, K.; Affleck, K.; Wood, E.R.; Alligood, K.J.; Rhodes, N.; Keith, B.R.; Murray, D.M.; Knight, W.B.; Mullin, R.J.; et al. The Effects of the Novel, Reversible Epidermal Growth Factor Receptor/ErbB-2 Tyrosine Kinase Inhibitor, GW2016, on the Growth of Human Normal and Tumor-Derived Cell Lines in Vitro and in Vivo. Mol. Cancer 2001, 1, 85–94. [Google Scholar]
- Higa, G.M.; Abraham, J. Lapatinib in the Treatment of Breast Cancer. Expert Rev. Anticancer Ther. 2014, 7, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A Unique Structure for Epidermal Growth Factor Receptor Bound to GW572016 (Lapatinib) Relationships among Protein Conformation, Inhibitor Off-Rate, and Receptor Activity in Tumor Cells. Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, L.; Liu, X.; Zhou, C.; Zhang, L.; Zhang, S.; Wang, D.; Li, Q.; Qin, S.; Hu, C.; et al. Icotinib versus Gefitinib in Previously Treated Advanced Non-Small-Cell Lung Cancer (ICOGEN): A Randomised, Double-Blind Phase 3 Non-Inferiority Trial. Lancet Oncol. 2013, 14, 953–961. [Google Scholar] [CrossRef]
- Shi, Y.; Sun, Y.; Ding, C.; Wang, Z.; Wang, C.; Wang, Z.; Bai, C.; Bai, C.; Feng, J.; Liu, X.; et al. China Experts Consensus on Icotinib for Non-Small Cell Lung Cancer Treatment (2015 Version). J. Thorac. Dis 2015, 7, E468–E472. [Google Scholar] [CrossRef]
- Liao, B.-C.; Lin, C.-C.; Yang, J.C.-H. Second and Third-Generation Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Advanced Nonsmall Cell Lung Cancer. Curr. Opin. Oncol. 2015, 27, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Target Binding Properties and Cellular Activity of Afatinib (BIBW 2992), an Irreversible ErbB Family Blocker. J. Pharm. Exp. 2012, 343, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Yang, J.C.-H.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.-M.; Boyer, M.; et al. Phase III Study of Afatinib or Cisplatin Plus Pemetrexed in Patients with Metastatic Lung Adenocarcinoma with EGFR Mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Zhou, C.; Hu, C.-P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus Cisplatin plus Gemcitabine for First-Line Treatment of Asian Patients with Advanced Non-Small-Cell Lung Cancer Harbouring EGFR Mutations (LUX-Lung 6): An Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2014, 15, 213–222. [Google Scholar] [CrossRef]
- Rabindran, S.K.; Discafani, C.M.; Rosfjord, E.C.; Baxter, M.; Floyd, M.B.; Golas, J.; Hallett, W.A.; Johnson, B.D.; Nilakantan, R.; Overbeek, E.; et al. Antitumor Activity of HKI-272, an Orally Active, Irreversible Inhibitor of the HER-2 Tyrosine Kinase. Cancer Res. 2004, 64, 3958–3965. [Google Scholar] [CrossRef]
- Dhillon, S. Neratinib in Early-Stage Breast Cancer: A Profile of Its Use in the EU. Clin. Drug Invest. 2019, 39, 221–229. [Google Scholar] [CrossRef]
- Saura, C.; Oliveira, M.; Feng, Y.-H.; Dai, M.-S.; Chen, S.-W.; Hurvitz, S.A.; Kim, S.-B.; Moy, B.; Delaloge, S.; Gradishar, W.; et al. Neratinib Plus Capecitabine Versus Lapatinib Plus Capecitabine in HER2-Positive Metastatic Breast Cancer Previously Treated with ≥ 2 HER2-Directed Regimens: Phase III NALA Trial. J. Clin. Oncol. 2020, 38, 3138–3149. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, A.J.; Hook, K.E.; Althaus, I.W.; Ellis, P.A.; Trachet, E.; Delaney, A.M.; Harvey, P.J.; Ellis, T.A.; Amato, D.M.; Nelson, J.M.; et al. Antitumor Activity and Pharmacokinetic Properties of PF-00299804, a Second-Generation Irreversible Pan-ErbB Receptor Tyrosine Kinase Inhibitor. Mol. Cancer 2008, 7, 1880–1889. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Gale, C.-M.; Lifshits, E.; Gonzales, A.J.; Shimamura, T.; Zhao, F.; Vincent, P.W.; Naumov, G.N.; Bradner, J.E.; et al. PF00299804, an Irreversible Pan-ERBB Inhibitor, Is Effective in Lung Cancer Models with EGFR and ERBB2 Mutations That Are Resistant to Gefitinib. Cancer Res. 2007, 67, 11924–11932. [Google Scholar] [CrossRef] [PubMed]
- Lavacchi, D.; Mazzoni, F.; Giaccone, G. Clinical Evaluation of Dacomitinib for the Treatment of Metastatic Non-Small Cell Lung Cancer (NSCLC): Current Perspectives. Drug Des. Dev. 2019, 13, 3187–3198. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Ke, L.; Zhang, Z.; Yu, J.; Meng, X. Development of EGFR TKIs and Options to Manage Resistance of Third-Generation EGFR TKI Osimertinib: Conventional Ways and Immune Checkpoint Inhibitors. Front. Oncol. 2020, 10, 602762. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance Mechanisms to Osimertinib in EGFR-Mutated Non-Small Cell Lung Cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef]
- Kim, E.S. Olmutinib: First Global Approval. Drugs 2016, 76, 1153–1157. [Google Scholar] [CrossRef]
- Yun, J.; Hong, M.H.; Kim, S.-Y.; Park, C.W.; Kim, S.-Y.; Yun, M.R.; Kang, H.N.; Pyo, K.H.; Lee, S.S.; Koh, J.S.; et al. YH25448, an Irreversible EGFR-TKI with Potent Intracranial Activity in EGFR Mutant Non-Small-Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 2575–2587. [Google Scholar] [CrossRef]
- Ahn, M.-J.; Han, J.-Y.; Lee, K.H.; Kim, S.-W.; Kim, D.-W.; Lee, Y.-G.; Cho, E.K.; Kim, J.-H.; Lee, G.-W.; Lee, J.-S.; et al. Lazertinib in Patients with EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer: Results from the Dose Escalation and Dose Expansion Parts of a First-in-Human, Open-Label, Multicentre, Phase 1–2 Study. Lancet Oncol. 2019, 20, 1681–1690. [Google Scholar] [CrossRef]
- Park, S.; Ku, B.M.; Jung, H.A.; Sun, J.-M.; Ahn, J.S.; Lee, S.-H.; Park, K.; Ahn, M.-J. EGFR C797S as a Resistance Mechanism of Lazertinib in Non-Small Cell Lung Cancer with EGFR T790M Mutation. Cancer Res. Treat. 2020, 52, 1288–1290. [Google Scholar] [CrossRef]
- Piper-Vallillo, A.J.; Sequist, L.V.; Piotrowska, Z. Emerging Treatment Paradigms for EGFR-Mutant Lung Cancers Progressing on Osimertinib: A Review. J. Clin. Oncol. 2020, 38, 2926–2936. [Google Scholar] [CrossRef]
- Jia, Y.; Yun, C.-H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) Resistance with Mutant-Selective Allosteric Inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef]
- Wang, S.; Song, Y.; Liu, D. EAI045: The Fourth-Generation EGFR Inhibitor Overcoming T790M and C797S Resistance. Cancer Lett. 2017, 385, 51–54. [Google Scholar] [CrossRef]
- To, C.; Jang, J.; Chen, T.; Park, E.; Mushajiang, M.; Clercq, D.J.H.D.; Xu, M.; Wang, S.; Cameron, M.D.; Heppner, D.E.; et al. Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor. Cancer Discov. 2019, 9, 926–943. [Google Scholar] [CrossRef]
- Huang, W.-S.; Liu, S.; Zou, D.; Thomas, M.; Wang, Y.; Zhou, T.; Romero, J.; Kohlmann, A.; Li, F.; Qi, J.; et al. Discovery of Brigatinib (AP26113), a Phosphine Oxide-Containing, Potent, Orally Active Inhibitor of Anaplastic Lymphoma Kinase. J. Med. Chem. 2016, 59, 4948–4964. [Google Scholar] [CrossRef]
- Uchibori, K.; Inase, N.; Araki, M.; Kamada, M.; Sato, S.; Okuno, Y.; Fujita, N.; Katayama, R. Brigatinib Combined with Anti-EGFR Antibody Overcomes Osimertinib Resistance in EGFR-Mutated Non-Small-Cell Lung Cancer. Nat. Commun. 2017, 8, 14768. [Google Scholar] [CrossRef]
- Wedge, S.R.; Ogilvie, D.J.; Dukes, M.; Kendrew, J.; Chester, R.; Jackson, J.A.; Boffey, S.J.; Valentine, P.J.; Curwen, J.O.; Musgrove, H.L.; et al. ZD6474 Inhibits Vascular Endothelial Growth Factor Signaling, Angiogenesis, and Tumor Growth Following Oral Administration. Cancer Res. 2002, 62, 4645–4655. [Google Scholar] [PubMed]
- Morabito, A.; Piccirillo, M.C.; Falasconi, F.; Feo, G.D.; Giudice, A.D.; Bryce, J.; Maio, M.D.; Maio, E.D.; Normanno, N.; Perrone, F. Vandetanib (ZD6474), a Dual Inhibitor of Vascular Endothelial Growth Factor Receptor (VEGFR) and Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinases: Current Status and Future Directions. Oncologist 2009, 14, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Cabanillas, M.E.; Ryder, M.; Jimenez, C. Targeted Therapy for Advanced Thyroid Cancer: Kinase Inhibitors and Beyond. Endocr. Rev. 2019, 40, 1573–1604. [Google Scholar] [CrossRef] [PubMed]
- Hatem, R.; Labiod, D.; Château-Joubert, S.; de Plater, L.; Botty, R.E.; Vacher, S.; Bonin, F.; Servely, J.; Dieras, V.; Bièche, I.; et al. Vandetanib as a Potential New Treatment for Estrogen Receptor-negative Breast Cancers. Int. J. Cancer 2016, 138, 2510–2521. [Google Scholar] [CrossRef]
- Pedersen, M.W.; Pedersen, N.; Ottesen, L.H.; Poulsen, H.S. Differential Response to Gefitinib of Cells Expressing Normal EGFR and the Mutant EGFRvIII. Br. J. Cancer 2005, 93, 915–923. [Google Scholar] [CrossRef]
- Tan, F.; Shen, X.; Wang, D.; Xie, G.; Zhang, X.; Ding, L.; Hu, Y.; He, W.; Wang, Y.; Wang, Y. Icotinib (BPI-2009H), a Novel EGFR Tyrosine Kinase Inhibitor, Displays Potent Efficacy in Preclinical Studies. Lung Cancer 2012, 76, 177–182. [Google Scholar] [CrossRef]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.R.; Padera, R.F.; Shapiro, G.I.; Baum, A.; Himmelsbach, F.; et al. BIBW2992, an Irreversible EGFR/HER2 Inhibitor Highly Effective in Preclinical Lung Cancer Models. Oncogene 2008, 27, 4702–4711. [Google Scholar] [CrossRef]
- Ferrarotto, R.; Gold, K.A. Afatinib in the Treatment of Head and Neck Squamous Cell Carcinoma. Expert Opin. Inv. Drug 2013, 23, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Yasuda, H.; Tani, T.; Hamamoto, J.; Oashi, A.; Ishioka, K.; Arai, D.; Nukaga, S.; Miyawaki, M.; Kawada, I.; et al. In Vitro Modeling to Determine Mutation Specificity of EGFR Tyrosine Kinase Inhibitors against Clinically Relevant EGFR Mutants in Non-Small-Cell Lung Cancer. Oncotarget 2015, 6, 38789–38803. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, L.; Yu, J.; Li, H.; He, S.; Tang, W.; Zuo, J.; Lu, W. Discovery of New BTK Inhibitors with B Cell Suppression Activity Bearing a 4,6-Substituted Thieno [3,2- d ]Pyrimidine Scaffold. Rsc Adv. 2017, 7, 26060–26069. [Google Scholar] [CrossRef]
- Aldea, M.; Andre, F.; Marabelle, A.; Dogan, S.; Barlesi, F.; Soria, J.-C. Overcoming Resistance to Tumor-Targeted and Immune-Targeted Therapies. Cancer Discov. 2021, 11, 874–899. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, R.P.; He, J.; van der Noord, V.E.; Redel, J.; Foekens, J.A.; Martens, J.W.M.; Smid, M.; Zhang, Y.; van de Water, B. A Kinase Inhibitor Screen Identifies a Dual Cdc7/CDK9 Inhibitor to Sensitise Triple-Negative Breast Cancer to EGFR-Targeted Therapy. Breast Cancer Res. 2019, 21, 77. [Google Scholar] [CrossRef]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef]
- Chong, C.R.; Jänne, P.A. The Quest to Overcome Resistance to EGFR-Targeted Therapies in Cancer. Nat. Med. 2013, 19, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.K.; Kholodenko, B.N. Feedback Regulation in Cell Signalling: Lessons for Cancer Therapeutics. Semin Cell Dev. Biol. 2016, 50, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Cremers, C.G.; Nguyen, L.K. Network Rewiring, Adaptive Resistance and Combating Strategies in Breast Cancer. Cancer Drug Resist. 2019. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib Is Associated with a Second Mutation in the EGFR Kinase Domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M Mutation in EGFR Kinase Causes Drug Resistance by Increasing the Affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S Mutation Mediates Resistance to AZD9291 in Non–Small Cell Lung Cancer Harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients with EGFR T790M–Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527. [Google Scholar] [CrossRef]
- Song, H.-N.; Jung, K.S.; Yoo, K.H.; Cho, J.; Lee, J.Y.; Lim, S.H.; Kim, H.S.; Sun, J.-M.; Lee, S.-H.; Ahn, J.S.; et al. Acquired C797S Mutation upon Treatment with a T790M-Specific Third-Generation EGFR Inhibitor (HM61713) in Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2016, 11, e45–e47. [Google Scholar] [CrossRef]
- Chabon, J.J.; Simmons, A.D.; Lovejoy, A.F.; Esfahani, M.S.; Newman, A.M.; Haringsma, H.J.; Kurtz, D.M.; Stehr, H.; Scherer, F.; Karlovich, C.A.; et al. Circulating Tumour DNA Profiling Reveals Heterogeneity of EGFR Inhibitor Resistance Mechanisms in Lung Cancer Patients. Nat. Commun 2016, 7, 11815. [Google Scholar] [CrossRef]
- Tan, D.S.-W.; Kim, D.-W.; Leighl, N.B.; Riely, G.J.; Yang, J.C.-H.; Wolf, J.; Seto, T.; Felip, E.; Aix, S.P.; Jonnaert, M.; et al. Genomic Profiling of Resistant Tumor Samples Following Progression on EGF816, a Third Generation, Mutant-Selective EGFR Tyrosine Kinase Inhibitor (TKI), in Advanced Non-Small Cell Lung Cancer (NSCLC). J. Clin. Oncol. 2017, 35, 11506. [Google Scholar] [CrossRef]
- Kim, A.; Jang, M.H.; Lee, S.J.; Bae, Y.K. Mutations of the Epidermal Growth Factor Receptor Gene in Triple-Negative Breast Cancer. J. Breast Cancer 2017, 20, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.H.-F.; Tan, W.-J.; Thike, A.-A.; Cheok, P.-Y.; Tse, G.M.-K.; Wong, N.-S.; Yip, G.W.-C.; Bay, B.-H.; Tan, P.-H. Mutations in the Epidermal Growth Factor Receptor (EGFR) Gene in Triple Negative Breast Cancer: Possible Implications for Targeted Therapy. Breast Cancer Res. 2011, 13, R35. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.T.; Vyse, S.; Huang, P.H. Rare Epidermal Growth Factor Receptor (EGFR) Mutations in Non-Small Cell Lung Cancer. Semin. Cancer Biol. 2019, 61, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, L.; Qi, Y.; Stemke-Hale, K.; Wang, B.; Young, E.J.; Booser, D.J.; Holmes, F.A.; O’Shaughnessy, J.; Hellerstedt, B.; Pippen, J.; et al. Mutation Profiling Identifies Numerous Rare Drug Targets and Distinct Mutation Patterns in Different Clinical Subtypes of Breast Cancers. Breast Cancer Res. Treat. 2012, 134, 333–343. [Google Scholar] [CrossRef]
- Tilch, E.; Seidens, T.; Cocciardi, S.; Reid, L.E.; Byrne, D.; Simpson, P.T.; Vargas, A.C.; Cummings, M.C.; Fox, S.B.; Lakhani, S.R.; et al. Mutations in EGFR, BRAF and RAS Are Rare in Triple-Negative and Basal-like Breast Cancers from Caucasian Women. Breast Cancer Res. Treat. 2014, 143, 385–392. [Google Scholar] [CrossRef]
- Reis-Filho, J.; Pinheiro, C.; Lambros, M.; Milanezi, F.; Carvalho, S.; Savage, K.; Simpson, P.; Jones, C.; Swift, S.; Mackay, A.; et al. EGFR Amplification and Lack of Activating Mutations in Metaplastic Breast Carcinomas. J. Pathol. 2006, 209, 445–453. [Google Scholar] [CrossRef]
- Jacot, W.; Lopez-Crapez, E.; Thezenas, S.; Senal, R.; Fina, F.; Bibeau, F.; Romieu, G.; Lamy, P.-J. Lack of EGFR-Activating Mutations in European Patients with Triple-Negative Breast Cancer Could Emphasise Geographic and Ethnic Variations in Breast Cancer Mutation Profiles. Breast Cancer Res. 2011, 13, R133. [Google Scholar] [CrossRef]
- Secq, V.; Villeret, J.; Fina, F.; Carmassi, M.; Carcopino, X.; Garcia, S.; Metellus, I.; Boubli, L.; Iovanna, J.; Charpin, C. Triple Negative Breast Carcinoma EGFR Amplification Is Not Associated with EGFR, Kras or ALK Mutations. Br. J. Cancer 2014, 110, 1045–1052. [Google Scholar] [CrossRef]
- Grob, T.J.; Heilenkötter, U.; Geist, S.; Paluchowski, P.; Wilke, C.; Jaenicke, F.; Quaas, A.; Wilczak, W.; Choschzick, M.; Sauter, G.; et al. Rare Oncogenic Mutations of Predictive Markers for Targeted Therapy in Triple-Negative Breast Cancer. Breast Cancer Res. Treat. 2012, 134, 561–567. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, J.; Lee, H.-D.; Jeong, J.; Lee, W.; Lee, K.-A. Spectrum of EGFR Gene Copy Number Changes and KRAS Gene Mutation Status in Korean Triple Negative Breast Cancer Patients. PLoS ONE 2013, 8, e79014. [Google Scholar] [CrossRef]
- Park, H.S.; Jang, M.H.; Kim, E.J.; Kim, H.J.; Lee, H.J.; Kim, Y.J.; Kim, J.H.; Kang, E.; Kim, S.-W.; Kim, I.A.; et al. High EGFR Gene Copy Number Predicts Poor Outcome in Triple-Negative Breast Cancer. Mod. Pathol. 2014, 27, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- Lv, N.; Xie, X.; Ge, Q.; Lin, S.; Wang, X.; Kong, Y.; Shi, H.; Xie, X.; Wei, W. Epidermal Growth Factor Receptor in Breast Carcinoma: Association between Gene Copy Number and Mutations. Diagn. Pathol. 2011, 6, 118. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Botta, F.; Zanellato, E.; Molinari, F.; Crippa, S.; Mazzucchelli, L.; Frattini, M. Molecular Characterization of EGFR and EGFR-Downstream Pathways in Triple Negative Breast Carcinomas with Basal like Features. Histol. Histopathol. 2012, 27, 785–792. [Google Scholar] [PubMed]
- Toyama, T.; Yamashita, H.; Kondo, N.; Okuda, K.; Takahashi, S.; Sasaki, H.; Sugiura, H.; Iwase, H.; Fujii, Y. Frequently Increased Epidermal Growth Factor Receptor (EGFR) Copy Numbers and Decreased BRCA1 MRNA Expression in Japanese Triple-Negative Breast Cancers. BMC Cancer 2008, 8, 309. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Ishikawa, Y.; Furuya, M.; Sano, T.; Ohno, Y.; Horiguchi, J.; Oyama, T. Protein Expression, Gene Amplification, and Mutational Analysis of EGFR in Triple-Negative Breast Cancer. Breast Cancer 2014, 21, 66–74. [Google Scholar] [CrossRef]
- Lo, H.-W.; Hsu, S.-C.; Ali-Seyed, M.; Gunduz, M.; Xia, W.; Wei, Y.; Bartholomeusz, G.; Shih, J.-Y.; Hung, M.-C. Nuclear Interaction of EGFR and STAT3 in the Activation of the INOS/NO Pathway. Cancer Cell 2005, 7, 575–589. [Google Scholar] [CrossRef]
- Lo, H.-W.; Hung, M.-C. Nuclear EGFR Signalling Network in Cancers: Linking EGFR Pathway to Cell Cycle Progression, Nitric Oxide Pathway and Patient Survival. Br. J. Cancer 2006, 94, 184–188. [Google Scholar] [CrossRef]
- Huang, W.-C.; Chen, Y.-J.; Li, L.-Y.; Wei, Y.-L.; Hsu, S.-C.; Tsai, S.-L.; Chiu, P.-C.; Huang, W.-P.; Wang, Y.-N.; Chen, C.-H.; et al. Nuclear Translocation of Epidermal Growth Factor Receptor by Akt-Dependent Phosphorylation Enhances Breast Cancer-Resistant Protein Expression in Gefitinib-Resistant Cells. J. Biol. Chem. 2011, 286, 20558–20568. [Google Scholar] [CrossRef] [PubMed]
- Brand, T.M.; Iida, M.; Dunn, E.F.; Luthar, N.; Kostopoulos, K.T.; Corrigan, K.L.; Wleklinski, M.J.; Yang, D.; Wisinski, K.B.; Salgia, R.; et al. Nuclear Epidermal Growth Factor Receptor Is a Functional Molecular Target in Triple-Negative Breast Cancer. Mol. Cancer 2014, 13, 1356–1368. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Makino, K.; Xia, W.; Matin, A.; Wen, Y.; Kwong, K.Y.; Bourguignon, L.; Hung, M.-C. Nuclear Localization of EGF Receptor and Its Potential New Role as a Transcription Factor. Nat. Cell Biol. 2001, 3, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.-Y.; Tseng, J.T.; Lee, Y.-C.; Xia, W.; Wang, Y.-N.; Wu, M.-L.; Chuang, Y.-H.; Lai, C.-H.; Chang, W.-C. Nuclear Epidermal Growth Factor Receptor (EGFR) Interacts with Signal Transducer and Activator of Transcription 5 (STAT5) in Activating Aurora-A Gene Expression. Nucleic Acids Res. 2008, 36, 4337–4351. [Google Scholar] [CrossRef] [PubMed]
- Liccardi, G.; Hartley, J.A.; Hochhauser, D. EGFR Nuclear Translocation Modulates DNA Repair Following Cisplatin and Ionizing Radiation Treatment. Cancer Res. 2011, 71, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Reddy, Y.V.R.; Ding, Q.; Lees-Miller, S.P.; Meek, K.; Ramsden, D.A. Non-Homologous End Joining Requires That the DNA-PK Complex Undergo an Autophosphorylation-Dependent Rearrangement at DNA Ends. J. Biol. Chem. 2004, 279, 39408–39413. [Google Scholar] [CrossRef]
- Dittmann, K.; Mayer, C.; Rodemann, H.-P. Inhibition of Radiation-Induced EGFR Nuclear Import by C225 (Cetuximab) Suppresses DNA-PK Activity. Radiother. Oncol. 2005, 76, 157–161. [Google Scholar] [CrossRef]
- Friedmann, B.J.; Caplin, M.; Savic, B.; Shah, T.; Lord, C.J.; Ashworth, A.; Hartley, J.A.; Hochhauser, D. Interaction of the Epidermal Growth Factor Receptor and the DNA-Dependent Protein Kinase Pathway Following Gefitinib Treatment. Mol. Cancer 2006, 5, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.-C.; Ciou, S.-C.; Cheng, C.-M.; Wang, L.-H.; Hong, J.-H.; Jheng, M.-Y.; Ling, S.-T.; Lin, Y.-W. Involvement of Rad51 in Cytotoxicity Induced by Epidermal Growth Factor Receptor Inhibitor (Gefitinib, Iressa R) and Chemotherapeutic Agents in Human Lung Cancer Cells. Carcinogenesis 2008, 29, 1448–1458. [Google Scholar] [CrossRef]
- Li, L.; Wang, H.; Yang, E.S.; Arteaga, C.L.; Xia, F. Erlotinib Attenuates Homologous Recombinational Repair of Chromosomal Breaks in Human Breast Cancer Cells. Cancer Res. 2008, 68, 9141–9146. [Google Scholar] [CrossRef]
- Prat, M.; Oltolina, F.; Basilico, C. Monoclonal Antibodies against the MET/HGF Receptor and Its Ligand: Multitask Tools with Applications from Basic Research to Therapy. Biomedicines 2014, 2, 359–383. [Google Scholar] [CrossRef]
- Deb, T.B.; Zuo, A.H.; Barndt, R.J.; Sengupta, S.; Jankovic, R.; Johnson, M.D. Pnck Overexpression in HER-2 Gene-Amplified Breast Cancer Causes Trastuzumab Resistance through a Paradoxical PTEN-Mediated Process. Breast Cancer Res. Treat. 2015, 150, 347–361. [Google Scholar] [CrossRef]
- Riese, D.J., II. Ligand-Based Receptor Tyrosine Kinase Partial Agonists: New Paradigm for Cancer Drug Discovery? Expert Opin. Drug Dis. 2011, 6, 185–193. [Google Scholar] [CrossRef]
- Gurdal, H.; Tuglu, M.M.; Bostanabad, S.Y.; Dalkiliç, B. Partial Agonistic Effect of Cetuximab on Epidermal Growth Factor Receptor and Src Kinase Activation in Triple-Negative Breast Cancer Cell Lines. Int. J. Oncol. 2019, 54, 1345–1356. [Google Scholar] [CrossRef]
- Scott, G.K.; Dodson, J.M.; Montgomery, P.A.; Johnson, R.M.; Sarup, J.C.; Wong, W.L.; Ullrich, A.; Shepard, H.M.; Benz, C.C. P185HER2 Signal Transduction in Breast Cancer Cells. J. Biol. Chem. 1991, 266, 14300–14305. [Google Scholar] [CrossRef]
- Nagata, Y.; Lan, K.-H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. PTEN Activation Contributes to Tumor Inhibition by Trastuzumab, and Loss of PTEN Predicts Trastuzumab Resistance in Patients. Cancer Cell 2004, 6, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Okamoto, I.; Okabe, T.; Iwasa, T.; Satoh, T.; Nishio, K.; Fukuoka, M.; Nakagawa, K. Matuzumab and Cetuximab Activate the Epidermal Growth Factor Receptor but Fail to Trigger Downstream Signaling by Akt or Erk. Int. J. Cancer 2008, 122, 1530–1538. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.P.; Wolf-Yadlin, A.; Sevecka, M.; Grenier, J.K.; Root, D.E.; Lauffenburger, D.A.; MacBeath, G. Receptor Tyrosine Kinases Fall into Distinct Classes Based on Their Inferred Signaling Networks. Sci. Signal. 2013, 6, ra58. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread Potential for Growth-Factor-Driven Resistance to Anticancer Kinase Inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Wood, G.E.; Hockings, H.; Hilton, D.M.; Kermorgant, S. The Role of MET in Chemotherapy Resistance. Oncogene 2021, 40, 1927–1941. [Google Scholar] [CrossRef]
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y. Advances in Studies of Tyrosine Kinase Inhibitors and Their Acquired Resistance. Mol. Cancer 2018, 17, 36. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Choi, J.; Seo, J.; Song, J.; Lee, S.E.; Kwon, M.J.; Kwon, M.J.; Kundu, J.; Jung, K.; Oh, E.; et al. MET Is a Potential Target for Use in Combination Therapy with EGFR Inhibition in Triple-negative/Basal-like Breast Cancer. Int. J. Cancer 2014, 134, 2424–2436. [Google Scholar] [CrossRef]
- Ponzo, M.G.; Lesurf, R.; Petkiewicz, S.; O’Malley, F.P.; Pinnaduwage, D.; Andrulis, I.L.; Bull, S.B.; Chughtai, N.; Zuo, D.; Souleimanova, M.; et al. Met Induces Mammary Tumors with Diverse Histologies and Is Associated with Poor Outcome and Human Basal Breast Cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 12903–12908. [Google Scholar] [CrossRef]
- Mueller, K.L.; Madden, J.M.; Zoratti, G.L.; Kuperwasser, C.; List, K.; Boerner, J.L. Fibroblast-Secreted Hepatocyte Growth Factor Mediates Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Resistance in Triple-Negative Breast Cancers through Paracrine Activation of Met. Breast Cancer Res. 2012, 14, R104. [Google Scholar] [CrossRef]
- Sohn, J.; Liu, S.; Parinyanitikul, N.; Lee, J.; Hortobagyi, G.N.; Mills, G.B.; Ueno, N.T.; Gonzalez-Angulo, A.M. CMET Activation and EGFR-Directed Therapy Resistance in Triple-Negative Breast Cancer. J. Cancer 2014, 5, 745–753. [Google Scholar] [CrossRef]
- Kang, H.J.; Yi, Y.W.; Hong, Y.B.; Kim, H.J.; Jang, Y.-J.; Seong, Y.-S.; Bae, I. HER2 Confers Drug Resistance of Human Breast Cancer Cells through Activation of NRF2 by Direct Interaction. Sci. Rep. 2014, 4, 7201. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.W.; Oh, S. Comparative Analysis of NRF2-Responsive Gene Expression in AcPC-1 Pancreatic Cancer Cell Line. Genes Genom. 2015, 37, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Jura, N.; Shan, Y.; Cao, X.; Shaw, D.E.; Kuriyan, J. Structural Analysis of the Catalytically Inactive Kinase Domain of the Human EGF Receptor 3. Proc. Natl. Acad. Sci. USA 2009, 106, 21608–21613. [Google Scholar] [CrossRef]
- Sergina, N.V.; Rausch, M.; Wang, D.; Blair, J.; Hann, B.; Shokat, K.M.; Moasser, M.M. Escape from HER-Family Tyrosine Kinase Inhibitor Therapy by the Kinase-Inactive HER3. Nature 2007, 445, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Huang, S.; Kruser, T.J.; Nechrebecki, M.M.; Armstrong, E.A.; Benavente, S.; Gondi, V.; Hsu, K.-T.; Harari, P.M. Mechanisms of Acquired Resistance to Cetuximab: Role of HER (ErbB) Family Members. Oncogene 2008, 27, 3944–3956. [Google Scholar] [CrossRef]
- Tao, J.J.; Castel, P.; Radosevic-Robin, N.; Elkabets, M.; Auricchio, N.; Aceto, N.; Weitsman, G.; Barber, P.; Vojnovic, B.; Ellis, H.; et al. Antagonism of EGFR and HER3 Enhances the Response to Inhibitors of the PI3K-Akt Pathway in Triple-Negative Breast Cancer. Sci. Signal. 2014, 7, ra29. [Google Scholar] [CrossRef]
- Chakrabarty, A.; Sánchez, V.; Kuba, M.G.; Rinehart, C.; Arteaga, C.L. Feedback Upregulation of HER3 (ErbB3) Expression and Activity Attenuates Antitumor Effect of PI3K Inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 2718–2723. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Müller, A.-K.; Kothari, C.; Panayotopoulou, E.; Kedan, A.; Selitrennik, M.; Mills, G.B.; Nguyen, L.K.; Shin, S.; Karn, T.; et al. Targeting of PYK2 Synergizes with EGFR Antagonists in Basal-like TNBC and Circumvents HER3-Associated Resistance via the NEDD4–NDRG1 Axis. Cancer Res. 2017, 77, 86–99. [Google Scholar] [CrossRef]
- Ortiz-Cuaran, S.; Scheffler, M.; Plenker, D.; Dahmen, L.; Scheel, A.H.; Fernandez-Cuesta, L.; Meder, L.; Lovly, C.M.; Persigehl, T.; Merkelbach-Bruse, S.; et al. Heterogeneous Mechanisms of Primary and Acquired Resistance to Third-Generation EGFR Inhibitors. Clin. Cancer Res. 2016, 22, 4837–4847. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Yamada, T.; Wang, R.; Tanimura, K.; Adachi, Y.; Nishiyama, A.; Tanimoto, A.; Takeuchi, S.; Araujo, L.H.; Boroni, M.; et al. AXL Confers Intrinsic Resistance to Osimertinib and Advances the Emergence of Tolerant Cells. Nat. Commun. 2019, 10, 259. [Google Scholar] [CrossRef]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL Kinase Causes Resistance to EGFR-Targeted Therapy in Lung Cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An Epithelial–Mesenchymal Transition Gene Signature Predicts Resistance to EGFR and PI3K Inhibitors and Identifies Axl as a Therapeutic Target for Overcoming EGFR Inhibitor Resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.S.; Miller, M.A.; Gertler, F.B.; Lauffenburger, D.A. The Receptor AXL Diversifies EGFR Signaling and Limits the Response to EGFR-Targeted Inhibitors in Triple-Negative Breast Cancer Cells. Sci. Signal. 2013, 6, ra66. [Google Scholar] [CrossRef]
- Lev, S. Targeted Therapy and Drug Resistance in Triple-Negative Breast Cancer: The EGFR Axis. Biochem. Soc. Trans. 2020, 48, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.S.; Whittle, M.C.; Nakamura, K.; Abell, A.N.; Midland, A.A.; Zawistowski, J.S.; Johnson, N.L.; Granger, D.A.; Jordan, N.V.; Darr, D.B.; et al. Dynamic Reprogramming of the Kinome in Response to Targeted MEK Inhibition in Triple-Negative Breast Cancer. Cell 2012, 149, 307–321. [Google Scholar] [CrossRef]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-Dependent Phosphorylation Pathways Regulate Myc Protein Stability. Gene Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.W.; Kang, H.J.; Kim, H.J.; Hwang, J.S.; Wang, A.; Bae, I. Inhibition of Constitutively Activated Phosphoinositide 3-kinase/AKT Pathway Enhances Antitumor Activity of Chemotherapeutic Agents in Breast Cancer Susceptibility Gene 1-defective Breast Cancer Cells. Mol. Carcinog. 2013, 52, 667–675. [Google Scholar] [CrossRef]
- Yi, Y.W.; Kang, H.J.; Bae, E.J.; Oh, S.; Seong, Y.-S.; Bae, I. β-TrCP1 Degradation Is a Novel Action Mechanism of PI3K/MTOR Inhibitors in Triple-Negative Breast Cancer Cells. Exp. Mol. Med. 2015, 47, e143. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Yi, Y.W.; Kang, H.J.; Zhang, L.; Kim, H.J.; Kong, Y.; Liu, Y.; Wang, K.; Kong, H.-S.; Grindrod, S.; et al. Novel Carbazole Inhibits Phospho-STAT3 through Induction of Protein–Tyrosine Phosphatase PTPN6. J. Med. Chem. 2014, 57, 6342–6353. [Google Scholar] [CrossRef]
- Kang, H.J.; Yi, Y.W.; Hou, S.-J.; Kim, H.J.; Kong, Y.; Bae, I.; Brown, M.L. Disruption of STAT3-DNMT1 Interaction by SH-I-14 Induces Re-Expression of Tumor Suppressor Genes and Inhibits Growth of Triple-Negative Breast Tumor. Oncotarget 2014, 5, 83457–83468. [Google Scholar] [CrossRef] [PubMed]
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive Ras in Developmental Disorders and Cancer. Nat. Rev. Cancer 2007, 7, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; O’Brien, C.; Boyd, Z.; Cavet, G.; Guerrero, S.; Jung, K.; Januario, T.; Savage, H.; Punnoose, E.; Truong, T.; et al. In Vivo Antitumor Activity of MEK and Phosphatidylinositol 3-Kinase Inhibitors in Basal-Like Breast Cancer Models. Clin. Cancer Res. 2009, 15, 4649–4664. [Google Scholar] [CrossRef]
- Giltnane, J.M.; Balko, J.M. Rationale for Targeting the Ras/MAPK Pathway in Triple-Negative Breast Cancer. Discov. Med. 2014, 17, 275–283. [Google Scholar] [PubMed]
- Paranjape, T.; Heneghan, H.; Lindner, R.; Keane, F.K.; Hoffman, A.; Hollestelle, A.; Dorairaj, J.; Geyda, K.; Pelletier, C.; Nallur, S.; et al. A 3′-Untranslated Region KRAS Variant and Triple-Negative Breast Cancer: A Case-Control and Genetic Analysis. Lancet Oncol. 2011, 12, 377–386. [Google Scholar] [CrossRef]
- Tokumaru, Y.; Oshi, M.; Katsuta, E.; Yan, L.; Satyananda, V.; Matsuhashi, N.; Futamura, M.; Akao, Y.; Yoshida, K.; Takabe, K. KRAS Signaling Enriched Triple Negative Breast Cancer Is Associated with Favorable Tumor Immune Microenvironment and Better Survival. Am. J. Cancer Res. 2020, 10, 897–907. [Google Scholar]
- Popov, N.; Schülein, C.; Jaenicke, L.A.; Eilers, M. Ubiquitylation of the Amino Terminus of Myc by SCFβ-TrCP Antagonizes SCFFbw7-Mediated Turnover. Nat. Cell Biol. 2010, 12, 973–981. [Google Scholar] [CrossRef]
- Horiuchi, D.; Camarda, R.; Zhou, A.Y.; Yau, C.; Momcilovic, O.; Balakrishnan, S.; Corella, A.N.; Eyob, H.; Kessenbrock, K.; Lawson, D.A.; et al. PIM1 Kinase Inhibition as a Targeted Therapy against Triple-Negative Breast Tumors with Elevated MYC Expression. Nat. Med. 2016, 22, 1321–1329. [Google Scholar] [CrossRef]
- Lee, K.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sánchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647.e7. [Google Scholar] [CrossRef] [PubMed]
- Carey, J.P.; Karakas, C.; Bui, T.; Chen, X.; Vijayaraghavan, S.; Zhao, Y.; Wang, J.; Mikule, K.; Litton, J.K.; Hunt, K.K.; et al. Synthetic Lethality of PARP Inhibitors in Combination with MYC Blockade Is Independent of BRCA Status in Triple Negative Breast Cancer. Cancer Res. 2017, 78, 742–757. [Google Scholar] [CrossRef]
- Kipreos, E.T.; Pagano, M. The F-Box Protein Family. Genome Biol. 2000, 1. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, P.; Inuzuka, H.; Wei, W. Roles of F-Box Proteins in Cancer. Nat. Rev. Cancer 2014, 14, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.W.; Liu, Y.; Tron, A.E.; Inuzuka, H.; Wei, W. SCF and APC E3 Ubiquitin Ligases in Tumorigenesis; Springer Briefs in Cancer Research 2014; Springer: Berlin/Heidelberg, Germany, 2014; pp. 15–45. [Google Scholar] [CrossRef]
- Duan, S.; Skaar, J.R.; Kuchay, S.; Toschi, A.; Kanarek, N.; Ben-Neriah, Y.; Pagano, M. MTOR Generates an Auto-Amplification Loop by Triggering the ΒTrCP- and CK1α-Dependent Degradation of DEPTOR. Mol. Cell 2011, 44, 317–324. [Google Scholar] [CrossRef]
- Gao, D.; Inuzuka, H.; Tan, M.-K.M.; Fukushima, H.; Locasale, J.W.; Liu, P.; Wan, L.; Zhai, B.; Chin, Y.R.; Shaik, S.; et al. MTOR Drives Its Own Activation via SCFβTrCP-Dependent Degradation of the MTOR Inhibitor DEPTOR. Mol. Cell 2011, 44, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xiong, X.; Sun, Y. DEPTOR, an MTOR Inhibitor, Is a Physiological Substrate of SCFβTrCP E3 Ubiquitin Ligase and Regulates Survival and Autophagy. Mol. Cell 2011, 44, 304–316. [Google Scholar] [CrossRef]
- Karakas, B.; Bachman, K.E.; Park, B.H. Mutation of the PIK3CA Oncogene in Human Cancers. Br. J. Cancer 2006, 94, 455–459. [Google Scholar] [CrossRef]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The Clonal and Mutational Evolution Spectrum of Primary Triple-Negative Breast Cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef]
- Cossu-Rocca, P.; Orrù, S.; Muroni, M.R.; Sanges, F.; Sotgiu, G.; Ena, S.; Pira, G.; Murgia, L.; Manca, A.; Uras, M.G.; et al. Analysis of PIK3CA Mutations and Activation Pathways in Triple Negative Breast Cancer. PLoS ONE 2015, 10, e0141763. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive Molecular Portraits of Human Breast Tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Maehama, T.; Dixon, J.E. The Tumor Suppressor, PTEN/MMAC1, Dephosphorylates the Lipid Second Messenger, Phosphatidylinositol 3,4,5-Trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor Suppressor and Metabolic Regulator. Front. Endocrinol. 2018, 9, 338. [Google Scholar] [CrossRef]
- Hu, H.; Zhu, J.; Zhong, Y.; Geng, R.; Ji, Y.; Guan, Q.; Hong, C.; Wei, Y.; Min, N.; Qi, A.; et al. PIK3CA Mutation Confers Resistance to Chemotherapy in Triple-Negative Breast Cancer by Inhibiting Apoptosis and Activating the PI3K/AKT/MTOR Signaling Pathway. Ann. Transl. Med. 2021, 9, 410. [Google Scholar] [CrossRef]
- Young, C.D.; Zimmerman, L.J.; Hoshino, D.; Formisano, L.; Hanker, A.B.; Gatza, M.L.; Morrison, M.M.; Moore, P.D.; Whitwell, C.A.; Dave, B.; et al. Activating PIK3CA Mutations Induce an Epidermal Growth Factor Receptor (EGFR)/Extracellular Signal-Regulated Kinase (ERK) Paracrine Signaling Axis in Basal-like Breast Cancer. Mol. Cell Proteom. 2015, 14, 1959–1976. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.R.; Yoshida, T.; Marusyk, A.; Beck, A.H.; Polyak, K.; Toker, A. Targeting Akt3 Signaling in Triple-Negative Breast Cancer. Cancer Res. 2014, 74, 964–973. [Google Scholar] [CrossRef]
- Fan, Y.; Dutta, J.; Gupta, N.; Fan, G.; Gélinas, C. Regulation of Programmed Cell Death by NF-κB and its Role in Tumorigenesis and Therapy. In Programmed Cell Death in Cancer Progression and Therapy; Khosravi-Far, R., White, E., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 223–250. ISBN 9781402065538. [Google Scholar]
- Chaturvedi, M.M.; Sung, B.; Yadav, V.R.; Kannappan, R.; Aggarwal, B.B. NF-ΚB Addiction and Its Role in Cancer: ‘One Size Does Not Fit All’. Oncogene 2011, 30, 1615–1630. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.-W. NF-ΚB in Cancer: From Innocent Bystander to Major Culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. The IκB Kinase—a Bridge between Inflammation and Cancer. Cell Res. 2008, 18, 334–342. [Google Scholar] [CrossRef]
- De, S.; Dermawan, J.K.T.; Stark, G.R. EGF Receptor Uses SOS1 to Drive Constitutive Activation of NFκB in Cancer Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11721–11726. [Google Scholar] [CrossRef]
- Sethi, G.; Ahn, K.S.; Chaturvedi, M.M.; Aggarwal, B.B. Epidermal Growth Factor (EGF) Activates Nuclear Factor-ΚB through IκBα Kinase-Independent but EGF Receptor-Kinase Dependent Tyrosine 42 Phosphorylation of IκBα. Oncogene 2007, 26, 7324–7332. [Google Scholar] [CrossRef] [PubMed]
- Häussler, U.; von Wichert, G.; Schmid, R.M.; Keller, F.; Schneider, G. Epidermal Growth Factor Activates Nuclear Factor-ΚB in Human Proximal Tubule Cells. Am. J. Physiol. Ren. 2005, 289, F808–F815. [Google Scholar] [CrossRef] [PubMed]
- Alberti, C.; Pinciroli, P.; Valeri, B.; Ferri, R.; Ditto, A.; Umezawa, K.; Sensi, M.; Canevari, S.; Tomassetti, A. Ligand-Dependent EGFR Activation Induces the Co-Expression of IL-6 and PAI-1 via the NFkB Pathway in Advanced-Stage Epithelial Ovarian Cancer. Oncogene 2012, 31, 4139–4149. [Google Scholar] [CrossRef]
- Sun, L.; Carpenter, G. Epidermal Growth Factor Activation of NF-ΚB Is Mediated through IκBα Degradation and Intracellular Free Calcium. Oncogene 1998, 16, 2095–2102. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Deyama, Y.; Deyama, A.; Okitsu, M.; Yoshimura, Y.; Suzuki, K. Epidermal Growth Factor Receptor-Mediated Expression of NF-ΚB Transcription Factor in Osteoblastic MC3T3-E1 Cells Cultured under a Low-Calcium Environment. Life Sci. 1998, 62, 1623–1627. [Google Scholar] [CrossRef]
- Obata, H.; Biro, S.; Arima, N.; Kaieda, H.; Kihara, T.; Eto, H.; Miyata, M.; Tanaka, H. NF-ΚB Is Induced in the Nuclei of Cultured Rat Aortic Smooth Muscle Cells by Stimulation of Various Growth Factors. Biochem. Bioph Res. Commun. 1996, 224, 27–32. [Google Scholar] [CrossRef]
- Biswas, D.K.; Cruz, A.P.; Gansberger, E.; Pardee, A.B. Epidermal Growth Factor-Induced Nuclear Factor ΚB Activation: A Major Pathway of Cell-Cycle Progression in Estrogen-Receptor Negative Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2000, 97, 8542–8547. [Google Scholar] [CrossRef]
- Hardbower, D.M.; Singh, K.; Asim, M.; Verriere, T.G.; Olivares-Villagómez, D.; Barry, D.P.; Allaman, M.M.; Washington, M.K.; Peek, R.M.; Piazuelo, M.B.; et al. EGFR Regulates Macrophage Activation and Function in Bacterial Infection. J. Clin. Invest. 2016, 126, 3296–3312. [Google Scholar] [CrossRef]
- Shostak, K.; Zhang, X.; Hubert, P.; Göktuna, S.I.; Jiang, Z.; Klevernic, I.; Hildebrand, J.; Roncarati, P.; Hennuy, B.; Ladang, A.; et al. NF-ΚB-Induced KIAA1199 Promotes Survival through EGFR Signalling. Nat. Commun. 2014, 5, 5232. [Google Scholar] [CrossRef]
- Nottingham, L.K.; Yan, C.H.; Yang, X.; Si, H.; Coupar, J.; Bian, Y.; Cheng, T.-F.; Allen, C.; Arun, P.; Gius, D.; et al. Aberrant IKKα and IKKβ Cooperatively Activate NF-ΚB and Induce EGFR/AP1 Signaling to Promote Survival and Migration of Head and Neck Cancer. Oncogene 2014, 33, 1135–1147. [Google Scholar] [CrossRef]
- Kung, C.-P.; Raab-Traub, N. Epstein-Barr Virus Latent Membrane Protein 1 Modulates Distinctive NF-ΚB Pathways through C-Terminus-Activating Region 1 To Regulate Epidermal Growth Factor Receptor Expression ▿. J. Virol. 2010, 84, 6605–6614. [Google Scholar] [CrossRef]
- Li, Z.; Yang, Z.; Passaniti, A.; Lapidus, R.G.; Liu, X.; Cullen, K.J.; Dan, H.C. A Positive Feedback Loop Involving EGFR/Akt/MTORC1 and IKK/NF-KB Regulates Head and Neck Squamous Cell Carcinoma Proliferation. Oncotarget 2015, 7, 31892–31906. [Google Scholar] [CrossRef]
- Makhov, P.; Naito, S.; Haifler, M.; Kutikov, A.; Boumber, Y.; Uzzo, R.G.; Kolenko, V.M. The Convergent Roles of NF-ΚB and ER Stress in Sunitinib-Mediated Expression of pro-Tumorigenic Cytokines and Refractory Phenotype in Renal Cell Carcinoma. Cell Death Dis. 2018, 9, 374. [Google Scholar] [CrossRef] [PubMed]
- Day, E.K.; Sosale, N.G.; Xiao, A.; Zhong, Q.; Purow, B.; Lazzara, M.J. Glioblastoma Cell Resistance to EGFR and MET Inhibition Can Be Overcome via Blockade of FGFR-SPRY2 Bypass Signaling. Cell Rep. 2020, 30, 3383–3396.e7. [Google Scholar] [CrossRef] [PubMed]
- Bivona, T.G.; Hieronymus, H.; Parker, J.; Chang, K.; Taron, M.; Rosell, R.; Moonsamy, P.; Dahlman, K.; Miller, V.A.; Costa, C.; et al. FAS and NF-ΚB Signalling Modulate Dependence of Lung Cancers on Mutant EGFR. Nature 2011, 471, 523–526. [Google Scholar] [CrossRef]
- Tian, R.; Li, Y.; Gao, M. Shikonin Causes Cell-Cycle Arrest and Induces Apoptosis by Regulating the EGFR–NF-ΚB Signalling Pathway in Human Epidermoid Carcinoma A431 Cells. Biosci. Rep. 2015, 35, e00189. [Google Scholar] [CrossRef]
- Galvani, E.; Sun, J.; Leon, L.G.; Sciarrillo, R.; Narayan, R.S.; Sjin, R.T.T.; Lee, K.; Ohashi, K.; Heideman, D.A.M.; Alfieri, R.R.; et al. NF-ΚB Drives Acquired Resistance to a Novel Mutant-Selective EGFR Inhibitor. Oncotarget 2015, 6, 42717–42732. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, M.; Maeda, T.; Mehrotra, N.; Jin, C.; Alam, M.; Bouillez, A.; Hata, T.; Tagde, A.; Keating, A.; Kharbanda, S.; et al. Targeting MUC1-C Suppresses BCL2A1 in Triple-Negative Breast Cancer. Signal. Transduct. Target. 2018, 3, 13. [Google Scholar] [CrossRef]
- Ahmad, R.; Raina, D.; Joshi, M.D.; Kawano, T.; Ren, J.; Kharbanda, S.; Kufe, D. MUC1-C Oncoprotein Functions as a Direct Activator of the Nuclear Factor-ΚB P65 Transcription Factor. Cancer Res. 2009, 69, 7013–7021. [Google Scholar] [CrossRef] [PubMed]
- Kufe, D.W. MUC1-C Oncoprotein as a Target in Breast Cancer: Activation of Signaling Pathways and Therapeutic Approaches. Oncogene 2013, 32, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Siroy, A.; Abdul-Karim, F.W.; Miedler, J.; Fong, N.; Fu, P.; Gilmore, H.; Baar, J. MUC1 Is Expressed at High Frequency in Early-Stage Basal-like Triple-Negative Breast Cancer. Hum. Pathol. 2013, 44, 2159–2166. [Google Scholar] [CrossRef] [PubMed]
- Llona-Minguez, S.; Baiget, J.; Mackay, S.P. Small-Molecule Inhibitors of IκB Kinase (IKK) and IKK-Related Kinases. Pharm Pat. Anal. 2013, 2, 481–498. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chao, L.; Li, X.; Ma, G.; Chen, L.; Zang, Y.; Zhou, G. Elevated Expression of Phosphorylated C-Jun NH2-Terminal Kinase in Basal-like and “Triple-Negative” Breast Cancers. Hum. Pathol 2010, 41, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Kaoud, T.S.; Edupuganti, R.; Zhang, T.; Kogawa, T.; Zhao, Y.; Chauhan, G.B.; Giannoukos, D.N.; Qi, Y.; Tripathy, D.; et al. C-Jun N-Terminal Kinase Promotes Stem Cell Phenotype in Triple-Negative Breast Cancer through Upregulation of Notch1 via Activation of c-Jun. Oncogene 2017, 36, 2599–2608. [Google Scholar] [CrossRef] [PubMed]
- Gee, J.M.W.; Barroso, A.F.; Ellis, I.O.; Robertson, J.F.R.; Nicholson, R.I. Biological and Clinical Associations of C-jun Activation in Human Breast Cancer. Int. J. Cancer 2000, 89, 177–186. [Google Scholar] [CrossRef]
- Vleugel, M.M.; Greijer, A.E.; Bos, R.; van der Wall, E.; van Diest, P.J. C-Jun Activation Is Associated with Proliferation and Angiogenesis in Invasive Breast Cancer. Hum. Pathol. 2006, 37, 668–674. [Google Scholar] [CrossRef]
- Manole, S.; Richards, E.J.; Meyer, A.S. JNK Pathway Activation Modulates Acquired Resistance to EGFR/HER2–Targeted Therapies. Cancer Res. 2016, 76, 5219–5228. [Google Scholar] [CrossRef]
- Han, J.; Jeon, M.; Shin, I.; Kim, S. Elevated STC-1 Augments the Invasiveness of Triple-Negative Breast Cancer Cells through Activation of the JNK/c-Jun Signaling Pathway. Oncol. Rep. 2016, 36, 1764–1771. [Google Scholar] [CrossRef]
- Ebelt, N.D.; Kaoud, T.S.; Edupuganti, R.; Ravenstein, S.V.; Dalby, K.N.; Van, C.L. A C-Jun N-Terminal Kinase Inhibitor, JNK-IN-8, Sensitizes Triple Negative Breast Cancer Cells to Lapatinib. Oncotarget 2017, 5, 104894–104912. [Google Scholar] [CrossRef]
- Giuli, M.V.; Giuliani, E.; Screpanti, I.; Bellavia, D.; Checquolo, S. Notch Signaling Activation as a Hallmark for Triple-Negative Breast Cancer Subtype. J. Oncol. 2019, 2019, 1–15. [Google Scholar] [CrossRef]
- BeLow, M.; Osipo, C. Notch Signaling in Breast Cancer: A Role in Drug Resistance. Cells 2020, 9, 2204. [Google Scholar] [CrossRef]
- Diluvio, G.; Gaudio, F.D.; Giuli, M.V.; Franciosa, G.; Giuliani, E.; Palermo, R.; Besharat, Z.M.; Pignataro, M.G.; Vacca, A.; d’Amati, G.; et al. NOTCH3 Inactivation Increases Triple Negative Breast Cancer Sensitivity to Gefitinib by Promoting EGFR Tyrosine Dephosphorylation and Its Intracellular Arrest. Oncogenesis 2018, 7, 42. [Google Scholar] [CrossRef]
- Oren, M.; Rotter, V. Mutant P53 Gain-of-Function in Cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar] [CrossRef]
- Maslon, M.M.; Hupp, T.R. Drug Discovery and Mutant P53. Trends Cell Biol. 2010, 20, 542–555. [Google Scholar] [CrossRef]
- Goh, A.M.; Coffill, C.R.; Lane, D.P. The Role of Mutant P53 in Human Cancer. J. Pathol. 2011, 223, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H.; Norman, J.C. P53 and Its Mutants in Tumor Cell Migration and Invasion. J. Cell Biol. 2011, 192, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Ortiz, E.; de la Cruz-López, K.G.; Becerril-Rico, J.; Sarabia-Sánchez, M.A.; Ortiz-Sánchez, E.; García-Carrancá, A. Mutant P53 Gain-of-Function: Role in Cancer Development, Progression, and Therapeutic Approaches. Front. Cell Dev. Biol. 2021, 8, 607670. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.W.; Kang, H.J.; Kim, H.J.; Kong, Y.; Brown, M.M.; Bae, I. Targeting Mutant P53 by a SIRT1 Activator YK-3-237 Inhibits the Proliferation of Triple-Negative Breast Cancer Cells. Oncotarget 2013, 4, 984–994. [Google Scholar] [CrossRef] [PubMed]
- Darb-Esfahani, S.; Denkert, C.; Stenzinger, A.; Salat, C.; Sinn, B.; Schem, C.; Endris, V.; Klare, P.; Schmitt, W.; Blohmer, J.-U.; et al. Role of TP53 Mutations in Triple Negative and HER2-Positive Breast Cancer Treated with Neoadjuvant Anthracycline/Taxane-Based Chemotherapy. Oncotarget 2016, 7, 67686–67698. [Google Scholar] [CrossRef]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The Role of P53 in Cancer Drug Resistance and Targeted Chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef]
- Coradini, D.; Biganzoli, E.; Ardoino, I.; Ambrogi, F.; Boracchi, P.; Demicheli, R.; Daidone, M.G.; Moliterni, A. P53 Status Identifies Triple-Negative Breast Cancer Patients Who Do Not Respond to Adjuvant Chemotherapy. Breast 2015, 24, 294–297. [Google Scholar] [CrossRef]
- Shapira, I.; Lee, A.; Vora, R.; Budman, D.R. P53 Mutations in Triple Negative Breast Cancer Upregulate Endosomal Recycling of Epidermal Growth Factor Receptor (EGFR) Increasing Its Oncogenic Potency. Crit Rev. Oncol. Hematol. 2013, 88, 284–292. [Google Scholar] [CrossRef]
- Neilsen, P.M.; Noll, J.E.; Mattiske, S.; Bracken, C.P.; Gregory, P.A.; Schulz, R.B.; Lim, S.P.; Kumar, R.; Suetani, R.J.; Goodall, G.J.; et al. Mutant P53 Drives Invasion in Breast Tumors through Up-Regulation of MiR-155. Oncogene 2013, 32, 2992–3000. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cheng, B.; Miao, L.; Mei, Y.; Wu, M. Mutant P53-R273H Gains New Function in Sustained Activation of EGFR Signaling via Suppressing MiR-27a Expression. Cell Death Dis. 2013, 4, e574. [Google Scholar] [CrossRef]
- Steinman, R.M.; Mellman, I.S.; Muller, W.A.; Cohn, Z.A. Endocytosis and the Recycling of Plasma Membrane. J. Cell Biol. 1983, 96, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.W.; Lee, J.H.; Kim, S.-Y.; Pack, C.-G.; Ha, D.H.; Park, S.R.; Youn, J.; Cho, B.S. Advances in Analysis of Biodistribution of Exosomes by Molecular Imaging. Int. J. Mol. Sci. 2020, 21, 665. [Google Scholar] [CrossRef]
- Ha, D.H.; Kim, H.; Lee, J.; Kwon, H.H.; Park, G.-H.; Yang, S.H.; Jung, J.Y.; Choi, H.; Lee, J.H.; Sung, S.; et al. Mesenchymal Stem/Stromal Cell-Derived Exosomes for Immunomodulatory Therapeutics and Skin Regeneration. Cells 2020, 9, 1157. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.D.; Donaldson, J.G. Pathways and Mechanisms of Endocytic Recycling. Nat. Rev. Mol. Cell Bio 2009, 10, 597–608. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant P53 Drives Invasion by Promoting Integrin Recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Caswell, P.T.; Chan, M.; Lindsay, A.J.; McCaffrey, M.W.; Boettiger, D.; Norman, J.C. Rab-Coupling Protein Coordinates Recycling of A5β1 Integrin and EGFR1 to Promote Cell Migration in 3D Microenvironments. J. Cell Biol. 2008, 183, 143–155. [Google Scholar] [CrossRef]
- Shahar, N.; Larisch, S. Inhibiting the Inhibitors: Targeting Anti-Apoptotic Proteins in Cancer and Therapy Resistance. Drug Resist. Updates 2020, 52, 100712. [Google Scholar] [CrossRef]
- Giménez-Bonafé, P.; Tortosa, A.; Pérez-Tomás, R. Overcoming Drug Resistance by Enhancing Apoptosis of Tumor Cells. Curr. Cancer Drug Targets 2009, 9, 320–340. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.; Johnston, P.; Longley, D. Anti-Apoptotic Mechanisms of Drug Resistance in Cancer. Curr. Cancer Drug Targets 2009, 9, 307–319. [Google Scholar] [CrossRef]
- Goodwin, C.M.; Rossanese, O.W.; Olejniczak, E.T.; Fesik, S.W. Myeloid Cell Leukemia-1 Is an Important Apoptotic Survival Factor in Triple-Negative Breast Cancer. Cell Death Differ. 2015, 22, 2098–2106. [Google Scholar] [CrossRef] [PubMed]
- Ozretic, P.; Alvir, I.; Sarcevic, B.; Vujaskovic, Z.; Rendic-Miocevic, Z.; Roguljic, A.; Beketic-Oreskovic, L. Apoptosis Regulator Bcl-2 Is an Independent Prognostic Marker for Worse Overall Survival in Triple-Negative Breast Cancer Patients. Int. J. Biol. Markers 2017, 33, 109–115. [Google Scholar] [CrossRef]
- Zoeller, J.J.; Vagodny, A.; Daniels, V.W.; Taneja, K.; Tan, B.Y.; DeRose, Y.S.; Fujita, M.; Welm, A.L.; Letai, A.; Leverson, J.D.; et al. Navitoclax Enhances the Effectiveness of EGFR-Targeted Antibody-Drug Conjugates in PDX Models of EGFR-Expressing Triple-Negative Breast Cancer. Breast Cancer Res. 2020, 22, 132. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, M.; Suzuki, Y.; Alam, M.; Hinohara, K.; Hasegawa, M.; Jin, C.; Kharbanda, S.; Kufe, D. MUC1-C Stabilizes MCL-1 in the Oxidative Stress Response of Triple-Negative Breast Cancer Cells to BCL-2 Inhibitors. Sci. Rep. 2016, 6, 26643. [Google Scholar] [CrossRef]
- Cruz-Gordillo, P.; Honeywell, M.E.; Harper, N.W.; Leete, T.; Lee, M.J. ELP-Dependent Expression of MCL1 Promotes Resistance to EGFR Inhibition in Triple-Negative Breast Cancer Cells. Sci. Signal. 2020, 13, eabb9820. [Google Scholar] [CrossRef]
- Chen, M.J.; Dixon, J.E.; Manning, G. Genomics and Evolution of Protein Phosphatases. Sci. Signal. 2017, 10, eaag1796. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Aceto, N.; Meerbrey, K.L.; Kessler, J.D.; Zhou, C.; Migliaccio, I.; Nguyen, D.X.; Pavlova, N.N.; Botero, M.; Huang, J.; et al. Activation of Multiple Proto-Oncogenic Tyrosine Kinases in Breast Cancer via Loss of the PTPN12 Phosphatase. Cell 2011, 144, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.A.; Adamopoulos, C.; Karoulia, Z.; Wu, X.; Sachidanandam, R.; Aaronson, S.A.; Poulikakos, P.I. SHP2 Drives Adaptive Resistance to ERK Signaling Inhibition in Molecularly Defined Subsets of ERK-Dependent Tumors. Cell Rep. 2019, 26, 65–78.e5. [Google Scholar] [CrossRef]
- Matalkah, F.; Martin, E.; Zhao, H.; Agazie, Y.M. SHP2 Acts Both Upstream and Downstream of Multiple Receptor Tyrosine Kinases to Promote Basal-like and Triple-Negative Breast Cancer. Breast Cancer Res. 2016, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Libring, S.; Ruddraraju, K.V.; Miao, J.; Solorio, L.; Zhang, Z.-Y.; Wendt, M.K. SHP2 Is a Multifunctional Therapeutic Target in Drug Resistant Metastatic Breast Cancer. Oncogene 2020, 39, 7166–7180. [Google Scholar] [CrossRef]
- Song, Z.-D.; Wang, M.-J.; Ge, Y.; Chen, X.-P.; Xu, Z.-Y.; Sun, Y.; Xiong, X.-F. Tyrosine Phosphatase SHP2 Inhibitors in Tumor-Targeted Therapies. Acta Pharm. Sin. B 2020, 11, 13–29. [Google Scholar] [CrossRef]
- Yuan, X.; Bu, H.; Zhou, J.; Yang, C.-Y.; Zhang, H. Recent Advances of SHP2 Inhibitors in Cancer Therapy: Current Development and Clinical Application. J. Med. Chem. 2020, 63, 11368–11396. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.L. HSP90 and the Chaperoning of Cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P.; Le Breton, L. Hsp90: Breaking the Symmetry. Mol. Cell 2015, 58, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Röhl, A.; Rohrberg, J.; Buchner, J. The Chaperone Hsp90: Changing Partners for Demanding Clients. Trends Biochem. Sci. 2013, 38, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Sankhala, K.K.; Mita, M.M.; Mita, A.C.; Takimoto, C.H. Heat Shock Proteins: A Potential Anticancer Target. Curr. Drug Targets 2011, 12, 2001–2008. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-S.; Jang, C.-Y.; Kim, H.D.; Lee, J.Y.; Ahn, B.-Y.; Kim, J. Interaction of Hsp90 with Ribosomal Proteins Protects from Ubiquitination and Proteasome-Dependent Degradation. Mol. Biol. Cell 2006, 17, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Mumin, N.H.; Drobnitzky, N.; Patel, A.; Lourenco, L.M.; Cahill, F.F.; Jiang, Y.; Kong, A.; Ryan, A.J. Overcoming Acquired Resistance to HSP90 Inhibition by Targeting JAK-STAT Signalling in Triple-Negative Breast Cancer. BMC Cancer 2019, 19, 102. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. Identification, Cloning, and Expression of Human Estrogen Receptor-A36, a Novel Variant of Human Estrogen Receptor-A66. Biochem. Biophys Res. Commun 2005, 336, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, C.; Konan, H.-P.; Guerquin, M.-J.; Chesnel, A.; Livera, G.; Romancer, M.L.; Dumond, H. The Role of ERα36 in Development and Tumor Malignancy. Int. J. Mol. Sci. 2020, 21, 4116. [Google Scholar] [CrossRef] [PubMed]
- Mahboobifard, F.; Dargahi, L.; Jorjani, M.; Tehrani, F.R.; Pourgholami, M.H. The Role of ERα36 in Cell Type-Specific Functions of Estrogen and Cancer Development. Pharm. Res. 2021, 163, 105307. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. A Variant of Estrogen Receptor-α, HER-A36: Transduction of Estrogen- and Antiestrogen-Dependent Membrane-Initiated Mitogenic Signaling. Proc. Natl Acad. Sci. USA 2006, 103, 9063–9068. [Google Scholar] [CrossRef]
- Zhang, X.T.; Kang, L.G.; Ding, L.; Vranic, S.; Gatalica, Z.; Wang, Z.-Y. A Positive Feedback Loop of ER-A36/EGFR Promotes Malignant Growth of ER-Negative Breast Cancer Cells. Oncogene 2011, 30, 770–780. [Google Scholar] [CrossRef]
- Jordan, V.C. Tamoxifen (ICI46,474) as a Targeted Therapy to Treat and Prevent Breast Cancer. Br. J. Pharm. 2006, 147, S269–S276. [Google Scholar] [CrossRef]
- Wang, Q.; Jiang, J.; Ying, G.; Xie, X.-Q.; Zhang, X.; Xu, W.; Zhang, X.; Song, E.; Bu, H.; Ping, Y.-F.; et al. Tamoxifen Enhances Stemness and Promotes Metastasis of ERα36+ Breast Cancer by Upregulating ALDH1A1 in Cancer Cells. Cell Res. 2018, 28, 336–358. [Google Scholar] [CrossRef]
- Xu, X.; Wang, Q.; He, Y.; Ding, L.; Zhong, F.; Ou, Y.; Shen, Y.; Liu, H.; He, S. ADP-Ribosylation Factor 1 (ARF1) Takes Part in Cell Proliferation and Cell Adhesion-Mediated Drug Resistance (CAM-DR). Ann. Hematol. 2017, 96, 847–858. [Google Scholar] [CrossRef]
- Lang, L.; Shay, C.; Zhao, X.; Teng, Y. Combined Targeting of Arf1 and Ras Potentiates Anticancer Activity for Prostate Cancer Therapeutics. J. Exp. Clin. Cancer Res. 2017, 36, 112. [Google Scholar] [CrossRef]
- Gu, G.; Chen, Y.; Duan, C.; Zhou, L.; Chen, C.; Chen, J.; Cheng, J.; Shi, N.; Jin, Y.; Xi, Q.; et al. Overexpression of ARF1 Is Associated with Cell Proliferation and Migration through PI3K Signal Pathway in Ovarian Cancer. Oncol. Rep. 2017, 37, 1511–1520. [Google Scholar] [CrossRef]
- Luchsinger, C.; Aguilar, M.; Burgos, P.V.; Ehrenfeld, P.; Mardones, G.A. Functional Disruption of the Golgi Apparatus Protein ARF1 Sensitizes MDA-MB-231 Breast Cancer Cells to the Antitumor Drugs Actinomycin D and Vinblastine through ERK and AKT Signaling. PLoS ONE 2018, 13, e0195401. [Google Scholar] [CrossRef]
- Vo-Hoang, Y.; Paiva, S.; He, L.; Estaran, S.; Teng, Y. Design and Synthesis of Arf1-Targeting γ-Dipeptides as Potential Agents against Head and Neck Squamous Cell Carcinoma. Cells 2020, 9, 286. [Google Scholar] [CrossRef]
- Gillingham, A.K.; Munro, S. The Small G Proteins of the Arf Family and Their Regulators. Annu. Rev. Cell Dev. Biol. 2007, 23, 579–611. [Google Scholar] [CrossRef]
- Casalou, C.; Ferreira, A.; Barral, D.C. The Role of ARF Family Proteins and Their Regulators and Effectors in Cancer Progression: A Therapeutic Perspective. Front. Cell Dev. Biol. 2020, 8, 217. [Google Scholar] [CrossRef]
- Haines, E.; Schlienger, S.; Claing, A. The Small GTPase ADP-Ribosylation Factor 1 Mediates the Sensitivity of Triple Negative Breast Cancer Cells to EGFR Tyrosine Kinase Inhibitors. Cancer Biol. 2015, 16, 1535–1547. [Google Scholar] [CrossRef] [PubMed]
- Sauer, H.; Wartenberg, M.; Hescheler, J. Reactive Oxygen Species as Intracellular Messengers During Cell Growth and Differentiation. Cell Physiol. Biochem. 2001, 11, 173–186. [Google Scholar] [CrossRef]
- Cui, Q.; Wang, J.-Q.; Assaraf, Y.G.; Ren, L.; Gupta, P.; Wei, L.; Ashby, C.R.; Yang, D.-H.; Chen, Z.-S. Modulating ROS to Overcome Multidrug Resistance in Cancer. Drug Resist. Updates 2018, 41, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.-S.; Chang, J.-H.; Hung, W.-Y.; Yang, Y.-C.; Chien, M.-H. The Interplay of Reactive Oxygen Species and the Epidermal Growth Factor Receptor in Tumor Progression and Drug Resistance. J. Exp. Clin. Cancer Res. 2018, 37, 61. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, V.; He, J. Reactive Oxygen Species, Metabolic Plasticity, and Drug Resistance in Cancer. Int. J. Mol. Sci. 2020, 21, 3412. [Google Scholar] [CrossRef]
- Chen, C.-H.; Cheng, T.-H.; Lin, H.; Shih, N.-L.; Chen, Y.-L.; Chen, Y.-S.; Cheng, C.-F.; Lian, W.-S.; Meng, T.-C.; Chiu, W.-T.; et al. Reactive Oxygen Species Generation Is Involved in Epidermal Growth Factor Receptor Transactivation through the Transient Oxidization of Src Homology 2-Containing Tyrosine Phosphatase in Endothelin-1 Signaling Pathway in Rat Cardiac Fibroblasts. Mol. Pharm. 2006, 69, 1347–1355. [Google Scholar] [CrossRef]
- Gallatin, W.M.; Weissman, I.L.; Butcher, E.C. A Cell-Surface Molecule Involved in Organ-Specific Homing of Lymphocytes. Nature 1983, 304, 30–34. [Google Scholar] [CrossRef]
- Lesley, J.; Hyman, R.; Kincade, P.W. CD44 and Its Interaction with Extracellular Matrix. Adv. Immunol. 1993, 54, 271–335. [Google Scholar] [CrossRef]
- Peach, R.; Hollenbaugh, D.; Stamenkovic, I.; Aruffo, A. Identification of Hyaluronic Acid Binding Sites in the Extracellular Domain of CD44. J. Cell Biol. 1993, 122, 257–264. [Google Scholar] [CrossRef]
- Faassen, A.E.; Schrager, J.A.; Klein, D.J.; Oegema, T.R.; Couchman, J.R.; McCarthy, J.B. A Cell Surface Chondroitin Sulfate Proteoglycan, Immunologically Related to CD44, Is Involved in Type I Collagen-Mediated Melanoma Cell Motility and Invasion. J. Cell Biol. 1992, 116, 521–531. [Google Scholar] [CrossRef]
- Knutson, J.R.; Iida, J.; Fields, G.B.; McCarthy, J.B. CD44/Chondroitin Sulfate Proteoglycan and Alpha 2 Beta 1 Integrin Mediate Human Melanoma Cell Migration on Type IV Collagen and Invasion of Basement Membranes. Mol. Biol. Cell 1996, 7, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Dimitroff, C.J.; Lee, J.Y.; Rafii, S.; Fuhlbrigge, R.C.; Sackstein, R. CD44 Is a Major E-Selectin Ligand on Human Hematopoietic Progenitor Cells. J. Cell Biol. 2001, 153, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Hidalgo, A.; Chang, J.; Peired, A.; Frenette, P.S. CD44 Is a Physiological E-Selectin Ligand on Neutrophils. J. Exp. Med. 2005, 201, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Jalkanen, S.; Jalkanen, M. Lymphocyte CD44 Binds the COOH-Terminal Heparin-Binding Domain of Fibronectin. J. Cell Biol. 1992, 116, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.W.; Gunja-Smith, Z.; Iida, N.; Zhu, H.B.; Young, L.J.T.; Muller, W.J.; Cardiff, R.D. CD44v3,8–10 Is Involved in Cytoskeleton-mediated Tumor Cell Migration and Matrix Metalloproteinase (MMP-9) Association in Metastatic Breast Cancer Cells. J. Cell Physiol. 1998, 176, 206–215. [Google Scholar] [CrossRef]
- Weber, G.F.; Ashkar, S.; Glimcher, M.J.; Cantor, H. Receptor-Ligand Interaction Between CD44 and Osteopontin (Eta-1). Science 1996, 271, 509–512. [Google Scholar] [CrossRef]
- Haynes, B.F.; Telen, M.J.; Hale, L.P.; Denning, S.M. CD44—A Molecule Involved in Leukocyte Adherence and T-Cell Activation. Immunol. Today 1989, 10, 423–428. [Google Scholar] [CrossRef]
- Fukazawa, H.; Yoshida, K.; Ichinohasama, R.; Sawai, T.; Hiromatsu, Y.; Mori, K.; Kikuchi, K.; Aizawa, Y.; Abe, K.; Wall, J.R. Expression of the Hermes-1 (CD44) and ICAM-1 (CD54) Molecule on the Surface of Thyroid Cells from Patients with Graves’ Disease. Thyroid 1993, 3, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, T.; Komada, Y. Homing-Associated Cell Adhesion Molecule (H-CAM/CD44) on Human CD34+ Hematopoietic Progenitor Cells. Leuk. Lymphoma 2000, 40, 25–37. [Google Scholar] [CrossRef]
- Joensuu, H.; Ristamäki, R.; Klemi, P.; Jalkanen, S. Lymphocyte Homing Receptor (CD44) Expression Is Associated with Poor Prognosis in Gastrointestinal Lymphoma. Br. J. Cancer 1993, 68, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Raso-Barnett, L.; Banky, B.; Barbai, T.; Becsagh, P.; Timar, J.; Raso, E. Demonstration of a Melanoma-Specific CD44 Alternative Splicing Pattern That Remains Qualitatively Stable, but Shows Quantitative Changes during Tumour Progression. PLoS ONE 2013, 8, e53883. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The Biology and Role of CD44 in Cancer Progression: Therapeutic Implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef]
- Lee, J.H.; Ha, D.H.; Go, H.; Youn, J.; Kim, H.; Jin, R.C.; Miller, R.B.; Kim, D.; Cho, B.S.; Yi, Y.W. Reproducible Large-Scale Isolation of Exosomes from Adipose Tissue-Derived Mesenchymal Stem/Stromal Cells and Their Application in Acute Kidney Injury. Int. J. Mol. Sci. 2020, 21, 4774. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.-O.; Ha, D.H.; Kim, J.O.; Crumrine, D.A.; Meyer, J.M.; Wakefield, J.S.; Lee, Y.; Kim, B.; Kim, S.; Kim, H.; et al. Exosomes from Human Adipose Tissue-Derived Mesenchymal Stem Cells Promote Epidermal Barrier Repair by Inducing de Novo Synthesis of Ceramides in Atopic Dermatitis. Cells 2020, 9, 680. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.S.; Lee, J.; Won, Y.; Duncan, D.I.; Jin, R.C.; Lee, J.; Kwon, H.H.; Park, G.-H.; Yang, S.H.; Park, B.C.; et al. Skin Brightening Efficacy of Exosomes Derived from Human Adipose Tissue-Derived Stem/Stromal Cells: A Prospective, Split-Face, Randomized Placebo-Controlled Study. Cosmetics 2020, 7, 90. [Google Scholar] [CrossRef]
- Xu, H.; Niu, M.; Yuan, X.; Wu, K.; Liu, A. CD44 as a Tumor Biomarker and Therapeutic Target. Exp. Hematol. Oncol. 2020, 9, 36. [Google Scholar] [CrossRef]
- Jaggupilli, A.; Elkord, E. Significance of CD44 and CD24 as Cancer Stem Cell Markers: An Enduring Ambiguity. Clin. Dev. Immunol. 2012, 2012, 708036. [Google Scholar] [CrossRef]
- Yin, J.; Zhang, H.; Wu, X.; Zhang, Y.; Li, J.; Shen, J.; Zhao, Y.; Xiao, Z.; Lu, L.; Huang, C.; et al. CD44 Inhibition Attenuates EGFR Signaling and Enhances Cisplatin Sensitivity in Human EGFR Wild-Type Non-Small-Cell Lung Cancer Cells. Int. J. Mol. Med. 2020, 45, 1783–1792. [Google Scholar] [CrossRef]
- Fu, W.; Sun, H.; Zhao, Y.; Chen, M.; Yang, L.; Yang, X.; Jin, W. Targeted Delivery of CD44s-SiRNA by ScFv Overcomes de Novo Resistance to Cetuximab in Triple Negative Breast Cancer. Mol. Immunol. 2018, 99, 124–133. [Google Scholar] [CrossRef]
- Anborgh, P.H.; Lee, D.J.; Stam, P.F.; Tuck, A.B.; Chambers, A.F. Role of Osteopontin as a Predictive Biomarker for Anti-EGFR Therapy in Triple-Negative Breast Cancer. Expert Opin. Targets 2018, 22, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Tuck, A.B.; Hota, C.; Wilson, S.M.; Chambers, A.F. Osteopontin-Induced Migration of Human Mammary Epithelial Cells Involves Activation of EGF Receptor and Multiple Signal Transduction Pathways. Oncogene 2003, 22, 1198–1205. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Singhal, H.; Bautista, D.S.; Tonkin, K.S.; O’Malley, F.P.; Tuck, A.B.; Chambers, A.F.; Harris, J.F. Elevated Plasma Osteopontin in Metastatic Breast Cancer Associated with Increased Tumor Burden and Decreased Survival. Clin. Cancer Res. 1997, 3, 605–611. [Google Scholar] [PubMed]
- Tuck, A.B.; O’Malley, F.P.; Singhal, H.; Harris, J.F.; Tonkin, K.S.; Kerkvliet, N.; Saad, Z.; Doig, G.S.; Chambers, A.F. Osteopontin Expression in a Group of Lymph Node Negative Breast Cancer Patients. Int. J. Cancer 1998, 79, 502–508. [Google Scholar] [CrossRef]
- Bramwell, V.H.C.; Doig, G.S.; Tuck, A.B.; Wilson, S.M.; Tonkin, K.S.; Tomiak, A.; Perera, F.; Vandenberg, T.A.; Chambers, A.F. Serial Plasma Osteopontin Levels Have Prognostic Value in Metastatic Breast Cancer. Clin. Cancer Res. 2006, 12, 3337–3343. [Google Scholar] [CrossRef] [PubMed]
- Anborgh, P.H.; Caria, L.B.; Chambers, A.F.; Tuck, A.B.; Stitt, L.W.; Brackstone, M. Role of Plasma Osteopontin as a Biomarker in Locally Advanced Breast Cancer. Am. J. Transl. Res. 2015, 7, 723–732. [Google Scholar] [PubMed]
- Xu, Y.-Y.; Zhang, Y.-Y.; Lu, W.-F.; Mi, Y.-J.; Chen, Y.-Q. Prognostic Value of Osteopontin Expression in Breast Cancer: A Meta-Analysis. Mol. Clin. Oncol. 2015, 3, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bi, J.; Zhu, H.; Shi, M.; Zeng, X. ANXA2 Could Act as a Moderator of EGFR-Directed Therapy Resistance in Triple Negative Breast Cancer. Biosci. Biotechnol. Biochem. 2018, 82, 1–9. [Google Scholar] [CrossRef]
- Christensen, M.V.; Høgdall, C.K.; Jochumsen, K.M.; Høgdall, E.V.S. Annexin A2 and Cancer: A Systematic Review. Int. J. Oncol. 2018, 52, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, Z.; Niu, R.; Wang, L. Crucial Role of Anxa2 in Cancer Progression: Highlights on Its Novel Regulatory Mechanism. Cancer Biol. Med. 2019, 16, 671–687. [Google Scholar] [CrossRef] [PubMed]
- Shetty, P.K.; Thamake, S.I.; Biswas, S.; Johansson, S.L.; Vishwanatha, J.K. Reciprocal Regulation of Annexin A2 and EGFR with Her-2 in Her-2 Negative and Herceptin-Resistant Breast Cancer. PLoS ONE 2012, 7, e44299. [Google Scholar] [CrossRef]
- De Graauw, M.; Cao, L.; Winkel, L.; van Miltenburg, M.H.A.M.; le Dévédec, S.E.; Klop, M.; Yan, K.; Pont, C.; Rogkoti, V.-M.; Tijsma, A.; et al. Annexin A2 Depletion Delays EGFR Endocytic Trafficking via Cofilin Activation and Enhances EGFR Signaling and Metastasis Formation. Oncogene 2014, 33, 2610–2619. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy Revisited: A Conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, Y.; Li, Y.; Yang, L.; Ma, Y.; Peng, X.; Yang, S.; Liu, J.; Li, H. Autophagy: A Novel Mechanism of Chemoresistance in Cancers. Biomed. Pharm. 2019, 119, 109415. [Google Scholar] [CrossRef]
- Chang, H.; Zou, Z. Targeting Autophagy to Overcome Drug Resistance: Further Developments. J. Hematol. Oncol. 2020, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- Dragowska, W.H.; Weppler, S.A.; Wang, J.C.; Wong, L.Y.; Kapanen, A.I.; Rawji, J.S.; Warburton, C.; Qadir, M.A.; Donohue, E.; Roberge, M.; et al. Induction of Autophagy Is an Early Response to Gefitinib and a Potential Therapeutic Target in Breast Cancer. PLoS ONE 2013, 8, e76503. [Google Scholar] [CrossRef]
- Li, X.; Fan, Z. The Epidermal Growth Factor Receptor Antibody Cetuximab Induces Autophagy in Cancer Cells by Downregulating HIF-1α and Bcl-2 and Activating the Beclin 1/HVps34 Complex. Cancer Res. 2010, 70, 5942–5952. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zou, Z.; Becker, N.; Anderson, M.; Sumpter, R.; Xiao, G.; Kinch, L.; Koduru, P.; Christudass, C.S.; Veltri, R.W.; et al. EGFR-Mediated Beclin 1 Phosphorylation in Autophagy Suppression, Tumor Progression, and Tumor Chemoresistance. Cell 2013, 154, 1269–1284. [Google Scholar] [CrossRef] [PubMed]
- Jutten, B.; Rouschop, K. EGFR Signaling and Autophagy Dependence for Growth, Survival, and Therapy Resistance. Cell Cycle 2014, 13, 42–51. [Google Scholar] [CrossRef]
- Fung, C.; Chen, X.; Grandis, J.R.; Duvvuri, U. EGFR Tyrosine Kinase Inhibition Induces Autophagy in Cancer Cells. Cancer Biol. 2012, 13, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Thapa, N.; Sun, Y.; Anderson, R.A. A Kinase-Independent Role for EGF Receptor in Autophagy Initiation. Cell 2015, 160, 145–160. [Google Scholar] [CrossRef]
- Rontogianni, S.; Iskit, S.; van Doorn, S.; Peeper, D.S.; Altelaar, M. Combined EGFR and ROCK Inhibition in Triple-Negative Breast Cancer Leads to Cell Death Via Impaired Autophagic Flux. Mol. Cell Proteom. 2020, 19, 261–277. [Google Scholar] [CrossRef]
- Jones, V.S.; Huang, R.-Y.; Chen, L.-P.; Chen, Z.-S.; Fu, L.; Huang, R.-P. Cytokines in Cancer Drug Resistance: Cues to New Therapeutic Strategies. Biochim. Biophys. Acta 2016, 1865, 255–265. [Google Scholar] [CrossRef]
- Chen, W.; Qin, Y.; Liu, S. Cytokines, Breast Cancer Stem Cells (BCSCs) and Chemoresistance. Clin. Transl. Med. 2018, 7, 27. [Google Scholar] [CrossRef]
- Jena, B.C.; Mandal, M. The Emerging Roles of Exosomes in Anti-Cancer Drug Resistance and Tumor Progression: An Insight towards Tumor-Microenvironment Interaction. Biochim. Biophys. Acta 2021, 1875, 188488. [Google Scholar] [CrossRef]
- Namee, N.M.; O’Driscoll, L. Extracellular Vesicles and Anti-Cancer Drug Resistance. Biochim. Biophys. Acta 2018, 1870, 123–136. [Google Scholar] [CrossRef]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour Micro-Environment Elicits Innate Resistance to RAF Inhibitors through HGF Secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Rickel, E.A.; Siegel, L.A.; Yoon, B.-R.P.; Rottman, J.B.; Kugler, D.G.; Swart, D.A.; Anders, P.M.; Tocker, J.E.; Comeau, M.R.; Budelsky, A.L. Identification of Functional Roles for Both IL-17RB and IL-17RA in Mediating IL-25-Induced Activities. J. Immunol. 2008, 181, 4299–4310. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.M.; Lee, Y.-H.; Shi, Y.; Wang, X.; Angkasekwinai, P.; Nallaparaju, K.C.; Flaherty, S.; Chang, S.H.; Watarai, H.; Dong, C. Interleukin-17B Antagonizes Interleukin-25-Mediated Mucosal Inflammation. Immunity 2015, 42, 692–703. [Google Scholar] [CrossRef]
- Merrouche, Y.; Fabre, J.; Cure, H.; Garbar, C.; Fuselier, C.; Bastid, J.; Antonicelli, F.; Al-Daccak, R.; Bensussan, A.; Giustiniani, J. IL-17E Synergizes with EGF and Confers in Vitro Resistance to EGFR-Targeted Therapies in TNBC Cells. Oncotarget 2016, 7, 53350–53361. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The Biology, Function, and Biomedical Applications of Exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Bai, X.; Ni, J.; Zhang, H.; Duan, W.; Graham, P.; Li, Y. Exosomes and Breast Cancer Drug Resistance. Cell Death Dis. 2020, 11, 987. [Google Scholar] [CrossRef] [PubMed]
- Goh, C.Y.; Wyse, C.; Ho, M.; O’Beirne, E.; Howard, J.; Lindsay, S.; Kelly, P.; Higgins, M.; McCann, A. Exosomes in Triple Negative Breast Cancer: Garbage Disposals or Trojan Horses? Cancer Lett. 2020, 473, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, M.; Merkelbach-Bruse, S.; Bos, M.; Fassunke, J.; Gardizi, M.; Michels, S.; Groneck, L.; Schultheis, A.M.; Malchers, F.; Leenders, F.; et al. Spatial Tumor Heterogeneity in Lung Cancer with Acquired Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitor Resistance: Targeting High-Level MET-Amplification and EGFR T790M Mutation Occurring at Different Sites in the Same Patient. J. Thorac. Oncol. 2015, 10, e40–e43. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nishio, M.; Horiike, A.; Murakami, H.; Yamamoto, N.; Kaneda, H.; Nakagawa, K.; Horinouchi, H.; Nagashima, M.; Sekiguchi, M.; Tamura, T. Phase I Study of the HER3-Targeted Antibody Patritumab (U3-1287) Combined with Erlotinib in Japanese Patients with Non-Small Cell Lung Cancer. Lung Cancer 2015, 88, 275–281. [Google Scholar] [CrossRef]
- Reckamp, K.L.; Koczywas, M.; Cristea, M.C.; Dowell, J.E.; Wang, H.; Gardner, B.K.; Milne, G.L.; Figlin, R.A.; Fishbein, M.C.; Elashoff, R.M.; et al. Randomized Phase 2 Trial of Erlotinib in Combination with High-dose Celecoxib or Placebo in Patients with Advanced Non-small Cell Lung Cancer. Cancer 2015, 121, 3298–3306. [Google Scholar] [CrossRef]
- Lee, J.Y.; Sun, J.-M.; Lim, S.H.; Kim, H.S.; Yoo, K.H.; Jung, K.S.; Song, H.-N.; Ku, B.M.; Koh, J.; Bae, Y.-H.; et al. A Phase Ib/II Study of Afatinib in Combination with Nimotuzumab in Non–Small Cell Lung Cancer Patients with Acquired Resistance to Gefitinib or Erlotinib. Clin. Cancer Res. 2016, 22, 2139–2145. [Google Scholar] [CrossRef] [PubMed]
- Zwitter, M.; Rajer, M.; Stanic, K.; Vrankar, M.; Doma, A.; Cuderman, A.; Grmek, M.; Kern, I.; Kovac, V. Intercalated Chemotherapy and Erlotinib for Non-Small Cell Lung Cancer (NSCLC) with Activating Epidermal Growth Factor Receptor (EGFR) Mutations. Cancer Biol. 2016, 17, 1–7. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rosell, R.; Dafni, U.; Felip, E.; Curioni-Fontecedro, A.; Gautschi, O.; Peters, S.; Massutí, B.; Palmero, R.; Aix, S.P.; Carcereny, E.; et al. Erlotinib and Bevacizumab in Patients with Advanced Non-Small-Cell Lung Cancer and Activating EGFR Mutations (BELIEF): An International, Multicentre, Single-Arm, Phase 2 Trial. Lancet Respir. Med. 2017, 5, 435–444. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Zhang, L.; Kim, D.-W.; Liu, X.; Lee, D.H.; Yang, J.C.-H.; Ahn, M.-J.; Vansteenkiste, J.F.; Su, W.-C.; Felip, E.; et al. Phase Ib/II Study of Capmatinib (INC280) Plus Gefitinib After Failure of Epidermal Growth Factor Receptor (EGFR) Inhibitor Therapy in Patients with EGFR-Mutated, MET Factor–Dysregulated Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 3101–3109. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.; Katakami, N.; Kaji, R.; Yokoyama, T.; Kaneda, T.; Tamiya, M.; Inoue, T.; Kimura, H.; Yano, Y.; Tamura, D.; et al. Afatinib plus Bevacizumab Combination after Acquired Resistance to EGFR Tyrosine Kinase Inhibitors in EGFR-mutant Non–Small Cell Lung Cancer: Multicenter, Single-arm, Phase 2 Trial (ABC Study). Cancer 2018, 124, 3830–3838. [Google Scholar] [CrossRef] [PubMed]
- Uchibori, K.; Satouchi, M.; Sueoka-Aragane, N.; Urata, Y.; Sato, A.; Imamura, F.; Inoue, T.; Tachihara, M.; Kobayashi, K.; Katakami, N.; et al. Phase II Trial of Gefitinib plus Pemetrexed after Relapse Using First-Line Gefitinib in Patients with Non-Small Cell Lung Cancer Harboring EGFR Gene Mutations. Lung Cancer 2018, 124, 65–70. [Google Scholar] [CrossRef]
- Park, J.S.; Hong, M.H.; Chun, Y.J.; Kim, H.R.; Cho, B.C. A Phase Ib Study of the Combination of Afatinib and Ruxolitinib in EGFR Mutant NSCLC with Progression on EGFR-TKIs. Lung Cancer 2019, 134, 46–51. [Google Scholar] [CrossRef]
- Yang, J.C.-H.; Cheng, Y.; Murakami, H.; Yang, P.-C.; He, J.; Nakagawa, K.; Kang, J.H.; Kim, J.-H.; Hozak, R.R.; Nguyen, T.S.; et al. A Randomized Phase 2 Study of Gefitinib with or without Pemetrexed as First-Line Treatment in Nonsquamous NSCLC with EGFR Mutation: Final Overall Survival and Biomarker Analysis. J. Thorac. Oncol. 2020, 15, 91–100. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Cheng, Y.; Zhou, J.; Lu, S.; Zhang, Y.; Zhao, J.; Kim, D.-W.; Soo, R.A.; Kim, S.-W.; Pan, H.; et al. Tepotinib plus Gefitinib in Patients with EGFR-Mutant Non-Small-Cell Lung Cancer with MET Overexpression or MET Amplification and Acquired Resistance to Previous EGFR Inhibitor (INSIGHT Study): An Open-Label, Phase 1b/2, Multicentre, Randomised Trial. Lancet Respir. Med. 2020, 8, 1132–1143. [Google Scholar] [CrossRef]
- Sequist, L.V.; Han, J.-Y.; Ahn, M.-J.; Cho, B.C.; Yu, H.; Kim, S.-W.; Yang, J.C.-H.; Lee, J.S.; Su, W.-C.; Kowalski, D.; et al. Osimertinib plus Savolitinib in Patients with EGFR Mutation-Positive, MET-Amplified, Non-Small-Cell Lung Cancer after Progression on EGFR Tyrosine Kinase Inhibitors: Interim Results from a Multicentre, Open-Label, Phase 1b Study. Lancet Oncol. 2020, 21, 373–386. [Google Scholar] [CrossRef]
- Matar, P.; Rojo, F.; Cassia, R.; Moreno-Bueno, G.; Cosimo, S.D.; Tabernero, J.; Guzmán, M.; Rodriguez, S.; Arribas, J.; Palacios, J.; et al. Combined Epidermal Growth Factor Receptor Targeting with the Tyrosine Kinase Inhibitor Gefitinib (ZD1839) and the Monoclonal Antibody Cetuximab (IMC-C225) Superiority Over Single-Agent Receptor Targeting. Clin. Cancer Res. 2004, 10, 6487–6501. [Google Scholar] [CrossRef]
- Oliveras-Ferraros, C.; Vazquez-Martin, A.; López-Bonet, E.; Martín-Castillo, B.; Barco, S.D.; Brunet, J.; Menendez, J.A. Growth and Molecular Interactions of the Anti-EGFR Antibody Cetuximab and the DNA Cross-Linking Agent Cisplatin in Gefitinib-Resistant MDA-MB-468 Cells: New Prospects in the Treatment of Triple-Negative/Basal-like Breast Cancer. Int. J. Oncol. 2008, 33, 1165–1176. [Google Scholar] [CrossRef]
- Corkery, B.; Crown, J.; Clynes, M.; O’Donovan, N. Epidermal Growth Factor Receptor as a Potential Therapeutic Target in Triple-Negative Breast Cancer. Ann. Oncol. 2009, 20, 862–867. [Google Scholar] [CrossRef]
- Edwards, S.R.; Wandless, T.J. The Rapamycin-Binding Domain of the Protein Kinase Mammalian Target of Rapamycin Is a Destabilizing Domain. J. Biol. Chem. 2007, 282, 13395–13401. [Google Scholar] [CrossRef]
- Liu, T.; Yacoub, R.; Taliaferro-Smith, L.D.; Sun, S.-Y.; Graham, T.R.; Dolan, R.; Lobo, C.; Tighiouart, M.; Yang, L.; Adams, A.; et al. Combinatorial Effects of Lapatinib and Rapamycin in Triple-Negative Breast Cancer Cells. Mol. Cancer 2011, 10, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Roberti, M.P.; Barrio, M.M.; Bravo, A.I.; Rocca, Y.S.; Arriaga, J.M.; Bianchini, M.; Mordoh, J.; Levy, E.M. IL-15 and IL-2 Increase Cetuximab-Mediated Cellular Cytotoxicity against Triple Negative Breast Cancer Cell Lines Expressing EGFR. Breast Cancer Res. Treat. 2011, 130, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Roberti, M.P.; Rocca, Y.S.; Amat, M.; Pampena, M.B.; Loza, J.; Coló, F.; Fabiano, V.; Loza, C.M.; Arriaga, J.M.; Bianchini, M.; et al. IL-2- or IL-15-Activated NK Cells Enhance Cetuximab-Mediated Activity against Triple-Negative Breast Cancer in Xenografts and in Breast Cancer Patients. Breast Cancer Res. Treat. 2012, 136, 659–671. [Google Scholar] [CrossRef]
- Donawho, C.K.; Luo, Y.; Luo, Y.; Penning, T.D.; Bauch, J.L.; Bouska, J.J.; Bontcheva-Diaz, V.D.; Cox, B.F.; DeWeese, T.L.; Dillehay, L.E.; et al. ABT-888, an Orally Active Poly(ADP-Ribose) Polymerase Inhibitor That Potentiates DNA-Damaging Agents in Preclinical Tumor Models. Clin. Cancer Res. 2007, 13, 2728–2737. [Google Scholar] [CrossRef] [PubMed]
- Nowsheen, S.; Cooper, T.; Stanley, J.A.; Yang, E.S. Synthetic Lethal Interactions between EGFR and PARP Inhibition in Human Triple Negative Breast Cancer Cells. PLoS ONE 2012, 7, e46614. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.R.; Mueller, K.L.; Terlecky, L.J.; Krentz, K.A.; Bollig-Fischer, A.; Terlecky, S.R.; Boerner, J.L. A Targeted Enzyme Approach to Sensitization of Tyrosine Kinase Inhibitor-Resistant Breast Cancer Cells. Exp. Cell Res. 2012, 318, 2014–2021. [Google Scholar] [CrossRef]
- O’Hare, T.; Walters, D.K.; Stoffregen, E.P.; Jia, T.; Manley, P.W.; Mestan, J.; Cowan-Jacob, S.W.; Lee, F.Y.; Heinrich, M.C.; Deininger, M.W.N.; et al. In Vitro Activity of Bcr-Abl Inhibitors AMN107 and BMS-354825 against Clinically Relevant Imatinib-Resistant Abl Kinase Domain Mutants. Cancer Res. 2005, 65, 4500–4505. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.P.; Lee, F.Y.; Luo, R.; Jiang, Y.; Donker, M.; Akin, C. Dasatinib (BMS-354825) Inhibits KITD816V, an Imatinib-Resistant Activating Mutation That Triggers Neoplastic Growth in Most Patients with Systemic Mastocytosis. Blood 2006, 108, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.M.H.; Mueller, K.; Gartner, E.; Boerner, J. Dasatinib Is Synergistic with Cetuximab and Cisplatin in Triple-Negative Breast Cancer Cells. J. Surg Res. 2013, 185, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Gonzalez, B.; Feldman, M.E.; Zunder, E.R.; Goldenberg, D.D.; Williams, O.; Loewith, R.; Stokoe, D.; Balla, A.; Toth, B.; et al. A Pharmacological Map of the PI3-K Family Defines a Role for P110α in Insulin Signaling. Cell 2006, 125, 733–747. [Google Scholar] [CrossRef]
- Yi, Y.W.; Hong, W.; Kang, H.J.; Kim, H.J.; Zhao, W.; Wang, A.; Seong, Y.; Bae, I. Inhibition of the PI3K/AKT Pathway Potentiates Cytotoxicity of EGFR Kinase Inhibitors in Triple-negative Breast Cancer Cells. J. Cell Mol. Med. 2013, 17, 648–656. [Google Scholar] [CrossRef]
- Adams, J.; Palombella, V.J.; Sausville, E.A.; Johnson, J.; Destree, A.; Lazarus, D.D.; Maas, J.; Pien, C.S.; Prakash, S.; Elliott, P.J. Proteasome Inhibitors: A Novel Class of Potent and Effective Antitumor Agents. Cancer Res. 1999, 59, 2615–2622. [Google Scholar] [PubMed]
- Chen, Y.-J.; Yeh, M.-H.; Yu, M.-C.; Wei, Y.-L.; Chen, W.-S.; Chen, J.-Y.; Shih, C.-Y.; Tu, C.-Y.; Chen, C.-H.; Hsia, T.-C.; et al. Lapatinib–Induced NF-KappaB Activation Sensitizes Triple-Negative Breast Cancer Cells to Proteasome Inhibitors. Breast Cancer Res. 2013, 15, R108. [Google Scholar] [CrossRef]
- Ferraro, D.A.; Gaborit, N.; Maron, R.; Cohen-Dvashi, H.; Porat, Z.; Pareja, F.; Lavi, S.; Lindzen, M.; Ben-Chetrit, N.; Sela, M.; et al. Inhibition of Triple-Negative Breast Cancer Models by Combinations of Antibodies to EGFR. Proc. Natl. Acad. Sci. USA 2013, 110, 1815–1820. [Google Scholar] [CrossRef]
- Christensen, J.G.; Schreck, R.; Burrows, J.; Kuruganti, P.; Chan, E.; Le, P.; Chen, J.; Wang, X.; Ruslim, L.; Blake, R.; et al. A Selective Small Molecule Inhibitor of C-Met Kinase Inhibits c-Met-Dependent Phenotypes in Vitro and Exhibits Cytoreductive Antitumor Activity in Vivo. Cancer Res. 2003, 63, 7345–7355. [Google Scholar] [PubMed]
- Bladt, F.; Faden, B.; Friese-Hamim, M.; Knuehl, C.; Wilm, C.; Fittschen, C.; Grädler, U.; Meyring, M.; Dorsch, D.; Jaehrling, F.; et al. EMD 1214063 and EMD 1204831 Constitute a New Class of Potent and Highly Selective C-Met Inhibitors. Am. Assoc. Cancer Res. 2013, 19, 2941–2951. [Google Scholar] [CrossRef]
- Saura, C.; Roda, D.; Roselló, S.; Oliveira, M.; Macarulla, T.; Pérez-Fidalgo, J.A.; Morales-Barrera, R.; Sanchis-García, J.M.; Musib, L.; Budha, N.; et al. A First-in-Human Phase I Study of the ATP-Competitive AKT Inhibitor Ipatasertib Demonstrates Robust and Safe Targeting of AKT in Patients with Solid Tumors. Cancer Discov. 2017, 7, 102–113. [Google Scholar] [CrossRef]
- Folkes, A.J.; Ahmadi, K.; Alderton, W.K.; Alix, S.; Baker, S.J.; Box, G.; Chuckowree, I.S.; Clarke, P.A.; Depledge, P.; Eccles, S.A.; et al. The Identification of 2-(1 H -Indazol-4-Yl)-6-(4-Methanesulfonyl-Piperazin-1-Ylmethyl)-4-Morpholin-4-Yl-Thieno[3,2- d ]Pyrimidine (GDC-0941) as a Potent, Selective, Orally Bioavailable Inhibitor of Class I PI3 Kinase for the Treatment of Cancer. J. Med. Chem. 2008, 51, 5522–5532. [Google Scholar] [CrossRef]
- Shor, B.; Zhang, W.-G.; Toral-Barza, L.; Lucas, J.; Abraham, R.T.; Gibbons, J.J.; Yu, K. A New Pharmacologic Action of CCI-779 Involves FKBP12-Independent Inhibition of MTOR Kinase Activity and Profound Repression of Global Protein Synthesis. Cancer Res. 2008, 68, 2934–2943. [Google Scholar] [CrossRef]
- Madden, J.M.; Mueller, K.L.; Bollig-Fischer, A.; Stemmer, P.; Mattingly, R.R.; Boerner, J.L. Abrogating Phosphorylation of EIF4B Is Required for EGFR and MTOR Inhibitor Synergy in Triple-Negative Breast Cancer. Breast Cancer Res. Treat. 2014, 147, 283–293. [Google Scholar] [CrossRef][Green Version]
- French, K.J.; Schrecengost, R.S.; Lee, B.D.; Zhuang, Y.; Smith, S.N.; Eberly, J.L.; Yun, J.K.; Smith, C.D. Discovery and Evaluation of Inhibitors of Human Sphingosine Kinase. Cancer Res. 2003, 63, 5962–5969. [Google Scholar]
- Martin, J.L.; de Silva, H.C.; Lin, M.Z.; Scott, C.D.; Baxter, R.C. Inhibition of Insulin-like Growth Factor–Binding Protein-3 Signaling through Sphingosine Kinase-1 Sensitizes Triple-Negative Breast Cancer Cells to EGF Receptor Blockade. Mol. Cancer 2014, 13, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Le, P.; Liang, C.; Chan, J.; Kiewlich, D.; Miller, T.; Harris, D.; Sun, L.; Rice, A.; Vasile, S.; et al. Potent and Selective Inhibitors of the Met [Hepatocyte Growthfactor/Scatter Factor (HGF/SF) Receptor] Tyrosine Kinaseblock HGF/SF-Induced Tumor Cell Growthand Invasion. Mol. Cancer 2003, 2, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Soo, K.C.; Chow, P.K.H.; Tran, E. Targeted Inhibition of the Extracellular Signal-Regulated Kinase Kinase Pathway with AZD6244 (ARRY-142886) in the Treatment of Hepatocellular Carcinoma. Mol. Cancer 2007, 6, 138–146. [Google Scholar] [CrossRef]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Maiello, M.R.; D’Alessio, A.; Bevilacqua, S.; Gallo, M.; Normanno, N.; Luca, A.D. EGFR and MEK Blockade in Triple Negative Breast Cancer Cells. J. Cell Biochem. 2015, 116, 2778–2785. [Google Scholar] [CrossRef]
- Randazzo, P.A.; Yang, Y.C.; Rulka, C.; Kahn, R.A. Activation of ADP-Ribosylation Factor by Golgi Membranes. Evidence for a Brefeldin A- and Protease-Sensitive Activating Factor on Golgi Membranes. J. Biol. Chem. 1993, 268, 9555–9563. [Google Scholar] [CrossRef]
- Donaldson, J.G.; Finazzi, D.; Klausner, R.D. Brefeldin A Inhibits Golgi Membrane-Catalysed Exchange of Guanine Nucleotide onto ARF Protein. Nature 1992, 360, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Griffith, D.J.; Druker, B.J.; Wait, C.L.; Ott, K.A.; Zigler, A.J. Inhibition of C-Kit Receptor Tyrosine Kinase Activity by STI 571, a Selective Tyrosine Kinase Inhibitor. Blood 2000, 96, 925–932. [Google Scholar] [CrossRef]
- Wang, Y.-L.; Overstreet, A.-M.; Chen, M.-S.; Wang, J.; Zhao, H.-J.; Ho, P.-C.; Smith, M.; Wang, S.-C. Combined Inhibition of EGFR and C-ABL Suppresses the Growth of Triple-Negative Breast Cancer Growth through Inhibition of HOTAIR. Oncotarget 2015, 6, 11150–11161. [Google Scholar] [CrossRef]
- Wang, X.; Song, H.; Yu, Q.; Liu, Q.; Wang, L.; Liu, Z.; Yu, Z. Ad-P53 Enhances the Sensitivity of Triple-Negative Breast Cancer MDA-MB-468 Cells to the EGFR Inhibitor Gefitinib. Oncol. Rep. 2015, 33, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Engstrom, L.D.; Aranda, R.; Lee, M.; Tovar, E.A.; Essenburg, C.J.; Madaj, Z.; Chiang, H.; Briere, D.; Hallin, J.; Lopez-Casas, P.P.; et al. Glesatinib Exhibits Antitumor Activity in Lung Cancer Models and Patients Harboring MET Exon 14 Mutations and Overcomes Mutation-Mediated Resistance to Type I MET Inhibitors in Nonclinical Models. Clin. Cancer Res. 2017, 23, 6661–6672. [Google Scholar] [CrossRef]
- Linklater, E.S.; Tovar, E.A.; Essenburg, C.J.; Turner, L.; Madaj, Z.; Winn, M.E.; Melnik, M.K.; Korkaya, H.; Maroun, C.R.; Christensen, J.G.; et al. Targeting MET and EGFR Crosstalk Signaling in Triple-Negative Breast Cancers. Oncotarget 2016, 7, 69903–69915. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.Y.; Li, Q.; Lee, J.H.; Arango, M.E.; McDonnell, S.R.; Yamazaki, S.; Koudriakova, T.B.; Alton, G.; Cui, J.J.; Kung, P.-P.; et al. An Orally Available Small-Molecule Inhibitor of c-Met, PF-2341066, Exhibits Cytoreductive Antitumor Efficacy through Antiproliferative and Antiangiogenic Mechanisms. Cancer Res. 2007, 67, 4408–4417. [Google Scholar] [CrossRef]
- Zou, H.Y.; Li, Q.; Engstrom, L.D.; West, M.; Appleman, V.; Wong, K.A.; McTigue, M.; Deng, Y.-L.; Liu, W.; Brooun, A.; et al. PF-06463922 Is a Potent and Selective next-Generation ROS1/ALK Inhibitor Capable of Blocking Crizotinib-Resistant ROS1 Mutations. Proc. Natl. Acad. Sci. USA 2015, 112, 3493–3498. [Google Scholar] [CrossRef]
- Guerrab, A.E.; Bamdad, M.; Kwiatkowski, F.; Bignon, Y.-J.; Penault-Llorca, F.; Aubel, C. Anti-EGFR Monoclonal Antibodies and EGFR Tyrosine Kinase Inhibitors as Combination Therapy for Triple-Negative Breast Cancer. Oncotarget 2016, 5, 73618–73637. [Google Scholar] [CrossRef] [PubMed]
- Doe, C.; Bentley, R.; Behm, D.J.; Lafferty, R.; Stavenger, R.; Jung, D.; Bamford, M.; Panchal, T.; Grygielko, E.; Wright, L.L.; et al. Novel Rho Kinase Inhibitors with Anti-Inflammatory and Vasodilatory Activities. J. Pharm. Exp. 2007, 320, 89–98. [Google Scholar] [CrossRef]
- Iskit, S.; Lieftink, C.; Halonen, P.; Shahrabi, A.; Possik, P.A.; Beijersbergen, R.L.; Peeper, D.S. Integrated in Vivo Genetic and Pharmacologic Screening Identifies Co-Inhibition of EGRF and ROCK as a Potential Treatment Regimen for Triple-Negative Breast Cancer. Oncotarget 2016, 7, 42859–42872. [Google Scholar] [CrossRef]
- Tanei, T.; Choi, D.S.; Rodriguez, A.A.; Liang, D.H.; Dobrolecki, L.; Ghosh, M.; Landis, M.D.; Chang, J.C. Antitumor Activity of Cetuximab in Combination with Ixabepilone on Triple Negative Breast Cancer Stem Cells. Breast Cancer Res. 2016, 18, 6. [Google Scholar] [CrossRef]
- Guerrab, A.E.; Bamdad, M.; Bignon, Y.; Penault-Llorca, F.; Aubel, C. Anti-EGFR Monoclonal Antibodies Enhance Sensitivity to DNA-damaging Agents in BRCA1-mutated and PTEN-wild-type Triple-negative Breast Cancer Cells. Mol. Carcinog. 2017, 56, 1383–1394. [Google Scholar] [CrossRef]
- Zhang, T.; Inesta-Vaquera, F.; Niepel, M.; Zhang, J.; Ficarro, S.B.; Machleidt, T.; Xie, T.; Marto, J.A.; Kim, N.; Sim, T.; et al. Discovery of Potent and Selective Covalent Inhibitors of JNK. Chem. Biol. 2012, 19, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Slack-Davis, J.K.; Martin, K.H.; Tilghman, R.W.; Iwanicki, M.; Ung, E.J.; Autry, C.; Luzzio, M.J.; Cooper, B.; Kath, J.C.; Roberts, W.G.; et al. Cellular Characterization of a Novel Focal Adhesion Kinase Inhibitor. J. Biol. Chem. 2007, 282, 14845–14852. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal Adhesion Kinase: In Command and Control of Cell Motility. Nat. Rev. Mol. Cell Bio. 2005, 6, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Mistry, A.; Chang, J.S.; Cunningham, D.; Griffor, M.; Bonnette, P.C.; Wang, H.; Chrunyk, B.A.; Aspnes, G.E.; Walker, D.P.; et al. Structural Characterization of Proline-Rich Tyrosine Kinase 2 (PYK2) Reveals a Unique (DFG-out) Conformation and Enables Inhibitor Design. J. Biol. Chem. 2009, 284, 13193–13201. [Google Scholar] [CrossRef]
- Rolin, J.; Sand, K.L.; Knudsen, E.; Maghazachi, A.A. FTY720 and SEW2871 Reverse the Inhibitory Effect of S1P on Natural Killer Cell Mediated Lysis of K562 Tumor Cells and Dendritic Cells but Not on Cytokine Release. Cancer Immunol. Immunother. 2010, 59, 575–586. [Google Scholar] [CrossRef]
- Martin, J.L.; Julovi, S.M.; Lin, M.Z.; de Silva, H.C.; Boyle, F.M.; Baxter, R.C. Inhibition of Basal-like Breast Cancer Growth by FTY720 in Combination with Epidermal Growth Factor Receptor Kinase Blockade. Breast Cancer Res. 2017, 19, 90. [Google Scholar] [CrossRef]
- Miller, S.; Tavshanjian, B.; Oleksy, A.; Perisic, O.; Houseman, B.T.; Shokat, K.M.; Williams, R.L. Shaping Development of Autophagy Inhibitors with the Structure of the Lipid Kinase Vps34. Science 2010, 327, 1638–1642. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; He, K.; Ma, Q.; Yu, Q.; Liu, C.; Ndege, I.; Wang, X.; Yu, Z. Autophagy Inhibitor Facilitates Gefitinib Sensitivity in Vitro and in Vivo by Activating Mitochondrial Apoptosis in Triple Negative Breast Cancer. PLoS ONE 2017, 12, e0177694. [Google Scholar] [CrossRef] [PubMed]
- Al-Fifi, Z.I.A.; Marshall, S.L.; Hyde, D.; Anstee, J.H.; Bowler, K. Characterization of ATPases of Apical Membrane Fractions from Locusta Migratoria Malpighian Tubules. Insect Biochem. Mol. Biol. 1998, 28, 201–211. [Google Scholar] [CrossRef]
- Bao, B.; Mitrea, C.; Wijesinghe, P.; Marchetti, L.; Girsch, E.; Farr, R.L.; Boerner, J.L.; Mohammad, R.; Dyson, G.; Terlecky, S.R.; et al. Treating Triple Negative Breast Cancer Cells with Erlotinib plus a Select Antioxidant Overcomes Drug Resistance by Targeting Cancer Cell Heterogeneity. Sci. Rep. 2017, 7, 44125. [Google Scholar] [CrossRef]
- Colvin, T.A.; Gabai, V.L.; Gong, J.; Calderwood, S.K.; Li, H.; Gummuluru, S.; Matchuk, O.N.; Smirnova, S.G.; Orlova, N.V.; Zamulaeva, I.A.; et al. Hsp70–Bag3 Interactions Regulate Cancer-Related Signaling Networks. Cancer Res. 2014, 74, 4731–4740. [Google Scholar] [CrossRef]
- Shields, S.; Conroy, E.; O’Grady, T.; McGoldrick, A.; Connor, K.; Ward, M.P.; Useckaite, Z.; Dempsey, E.; Reilly, R.; Fan, Y.; et al. BAG3 Promotes Tumour Cell Proliferation by Regulating EGFR Signal Transduction Pathways in Triple Negative Breast Cancer. Oncotarget 2018, 9, 15673–15690. [Google Scholar] [CrossRef]
- Yap, T.A.; Yan, L.; Patnaik, A.; Fearen, I.; Olmos, D.; Papadopoulos, K.; Baird, R.D.; Delgado, L.; Taylor, A.; Lupinacci, L.; et al. First-in-Man Clinical Trial of the Oral Pan-AKT Inhibitor MK-2206 in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2011, 29, 4688–4695. [Google Scholar] [CrossRef]
- You, K.S.; Yi, Y.W.; Kwak, S.-J.; Seong, Y.-S. Inhibition of RPTOR Overcomes Resistance to EGFR Inhibition in Triple-Negative Breast Cancer Cells. Int. J. Oncol. 2018, 52, 828–840. [Google Scholar] [CrossRef]
- Qian, F.; Engst, S.; Yamaguchi, K.; Yu, P.; Won, K.-A.; Mock, L.; Lou, T.; Tan, J.; Li, C.; Tam, D.; et al. Inhibition of Tumor Cell Growth, Invasion, and Metastasis by EXEL-2880 (XL880, GSK1363089), a Novel Inhibitor of HGF and VEGF Receptor Tyrosine Kinases. Cancer Res. 2009, 69, 8009–8016. [Google Scholar] [CrossRef] [PubMed]
- Simiczyjew, A.; Dratkiewicz, E.; Troys, M.V.; Ampe, C.; Styczeń, I.; Nowak, D. Combination of EGFR Inhibitor Lapatinib and MET Inhibitor Foretinib Inhibits Migration of Triple Negative Breast Cancer Cell Lines. Cancers 2018, 10, 335. [Google Scholar] [CrossRef]
- Shin, S.-Y.; Müller, A.-K.; Verma, N.; Lev, S.; Nguyen, L.K. Systems Modelling of the EGFR-PYK2-c-Met Interaction Network Predicts and Prioritizes Synergistic Drug Combinations for Triple-Negative Breast Cancer. PLoS Comput. Biol. 2018, 14, e1006192. [Google Scholar] [CrossRef]
- Montagnoli, A.; Valsasina, B.; Croci, V.; Menichincheri, M.; Rainoldi, S.; Marchesi, V.; Tibolla, M.; Tenca, P.; Brotherton, D.; Albanese, C.; et al. A Cdc7 Kinase Inhibitor Restricts Initiation of DNA Replication and Has Antitumor Activity. Nat. Chem. Biol. 2008, 4, 357–365. [Google Scholar] [CrossRef]
- Fleisher, B.; Mody, H.; Werkman, C.; Ait-Oudhia, S. Chloroquine Sensitizes MDA-MB-231 Cells to Osimertinib through Autophagy–Apoptosis Crosstalk Pathway. Breast Cancer Targets 2019, 11, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.; Trachet, E.; Albassam, M.; Zheng, X.; Leopold, W.R.; Pryer, N.K.; et al. Specific Inhibition of Cyclin-Dependent Kinase 4/6 by PD 0332991 and Associated Antitumor Activity in Human Tumor Xenografts. Mol. Cancer 2004, 3, 1427–1437. [Google Scholar]
- Foidart, P.; Yip, C.; Radermacher, J.; Lienard, M.; Blacher, S.; Montero-Ruiz, L.; Maquoi, E.; Montaudon, E.; Chateau-Joubert, S.; Collignon, J.; et al. Expression of MT4-MMP, EGFR and RB in Triple Negative Breast Cancer Strongly Sensitizes Tumors to Erlotinib and Palbociclib Combination Therapy. Clin. Cancer Res. 2019, 25, 1838–1850. [Google Scholar] [CrossRef]
- Sedrani, R.; Cottens, S.; Kallen, J.; Schuler, W. Chemical Modification of Rapamycin: The Discovery of SDZ RAD. Transplant. P 1998, 30, 2192–2194. [Google Scholar] [CrossRef]
- He, J.; McLaughlin, R.P.; van der Noord, V.; Foekens, J.A.; Martens, J.W.M.; van Westen, G.; Zhang, Y.; van de Water, B. Multi-Targeted Kinase Inhibition Alleviates MTOR Inhibitor Resistance in Triple-Negative Breast Cancer. Breast Cancer Res. Treat. 2019, 178, 263–274. [Google Scholar] [CrossRef]
- Hanke, J.H.; Gardner, J.P.; Dow, R.L.; Changelian, P.S.; Brissette, W.H.; Weringer, E.J.; Pollok, B.A.; Connelly, P.A. Discovery of a Novel, Potent, and Src Family-Selective Tyrosine Kinase Inhibitor Study of Lck- and Fynt-Dependent T Cell Activation. J. Biol. Chem. 1996, 271, 695–701. [Google Scholar] [CrossRef]
- Karni, R.; Mizrachi, S.; Reiss-Sklan, E.; Gazit, A.; Livnah, O.; Levitzki, A. The Pp60c-Src Inhibitor PP1 Is Non-competitive against ATP. Febs Lett. 2003, 537, 47–52. [Google Scholar] [CrossRef]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A Potent and Orally Bioavailable Bcl-2 Family Inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [PubMed]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Toumelin-Braizat, G.L.; Chanrion, M.; Kelly, G.L.; Gong, J.-N.; Moujalled, D.M.; et al. The MCL1 Inhibitor S63845 Is Tolerable and Effective in Diverse Cancer Models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef]
- Shen, Y.; Rehman, F.L.; Feng, Y.; Boshuizen, J.; Bajrami, I.; Elliott, R.; Wang, B.; Lord, C.J.; Post, L.E.; Ashworth, A. BMN 673, a Novel and Highly Potent PARP1/2 Inhibitor for the Treatment of Human Cancers with DNA Repair Deficiency. Clin. Cancer Res. 2013, 19, 5003–5015. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.-Y.; Yam, C.; Chen, M.-K.; Chan, L.-C.; Xiao, M.; Wei, Y.-K.; Yamaguchi, H.; Lee, P.-C.; Han, Y.; Nie, L.; et al. Blocking C-Met and EGFR Reverses Acquired Resistance of PARP Inhibitors in Triple-Negative Breast Cancer. Am. J. Cancer Res. 2020, 10, 648–661. [Google Scholar] [PubMed]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting Transcription Regulation in Cancer with a Covalent CDK7 Inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef]
- McDermott, M.S.J.; Sharko, A.C.; Munie, J.; Kassler, S.; Melendez, T.; Lim, C.; Broude, E.V. CDK7 Inhibition Is Effective in All the Subtypes of Breast Cancer: Determinants of Response and Synergy with EGFR Inhibition. Cells 2020, 9, 638. [Google Scholar] [CrossRef] [PubMed]
- Juliá, E.P.; Mordoh, J.; Levy, E.M. Cetuximab and IL-15 Promote NK and Dendritic Cell Activation In Vitro in Triple Negative Breast Cancer. Cells 2020, 9, 1573. [Google Scholar] [CrossRef] [PubMed]
- McKnight, B.N.; Kim, S.; Boerner, J.L.; Viola, N.T. Cetuximab PET Delineated Changes in Cellular Distribution of EGFR upon Dasatinib Treatment in Triple Negative Breast Cancer. Breast Cancer Res. 2020, 22, 37. [Google Scholar] [CrossRef]
- Canonici, A.; Browne, A.L.; Ibrahim, M.F.K.; Fanning, K.P.; Roche, S.; Conlon, N.T.; O’Neill, F.; Meiller, J.; Cremona, M.; Morgan, C.; et al. Combined Targeting EGFR and SRC as a Potential Novel Therapeutic Approach for the Treatment of Triple Negative Breast Cancer. Adv. Med. Oncol. 2020, 12, 1758835919897546. [Google Scholar] [CrossRef]
- Guerrab, A.E.; Bamdad, M.; Bignon, Y.-J.; Penault-Llorca, F.; Aubel, C. Co-Targeting EGFR and MTOR with Gefitinib and Everolimus in Triple-Negative Breast Cancer Cells. Sci. Rep. 2020, 10, 6367. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Kakefuda, R.; Tajima, N.; Sowa, Y.; Sakai, T. Antitumor Activities of JTP-74057 (GSK1120212), a Novel MEK1/2 Inhibitor, on Colorectal Cancer Cell Lines in Vitro and in Vivo. Int. J. Oncol. 2011, 39, 23–31. [Google Scholar] [CrossRef]
- Ahmad, S.; He, Q.; Williams, K.P.; Scott, J.E. Identification of a Triple Drug Combination That Is Synergistically Cytotoxic for Triple-Negative Breast Cancer Cells Using a Novel Combination Discovery Approach. SLAS Discov. 2020, 25, 923–938. [Google Scholar] [CrossRef] [PubMed]
- Rau, A.; Lieb, W.S.; Seifert, O.; Honer, J.; Birnstock, D.; Richter, F.; Aschmoneit, N.; Olayioye, M.A.; Kontermann, R.E. Inhibition of Tumor Cell Growth and Cancer Stem Cell Expansion by a Bispecific Antibody Targeting EGFR and HER3. Mol. Cancer 2020, 19, 1474–1485. [Google Scholar] [CrossRef]
- Reddy, T.P.; Choi, D.S.; Anselme, A.C.; Qian, W.; Chen, W.; Lantto, J.; Horak, I.D.; Kragh, M.; Chang, J.C.; Rosato, R.R. Simultaneous Targeting of HER Family Pro-Survival Signaling with Pan-HER Antibody Mixture Is Highly Effective in TNBC: A Preclinical Trial with PDXs. Breast Cancer Res. 2020, 22, 48. [Google Scholar] [CrossRef]
- Caratelli, S.; Arriga, R.; Sconocchia, T.; Ottaviani, A.; Lanzilli, G.; Pastore, D.; Cenciarelli, C.; Venditti, A.; Principe, M.I.D.; Lauro, D.; et al. In Vitro Elimination of Epidermal Growth Factor Receptor-overexpressing Cancer Cells by CD32A-chimeric Receptor T Cells in Combination with Cetuximab or Panitumumab. Int. J. Cancer 2020, 146, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Takeuchi, M.; Kinoyama, I.; Minematsu, T.; Shirasuna, K.; Matsuhisa, A.; Kita, A.; Tominaga, F.; Yamanaka, K.; Kudoh, M.; et al. YM155, a Novel Small-Molecule Survivin Suppressant, Induces Regression of Established Human Hormone-Refractory Prostate Tumor Xenografts. Cancer Res. 2007, 67, 8014–8021. [Google Scholar] [CrossRef] [PubMed]
- Turner, T.H.; Alzubi, M.A.; Harrell, J.C. Identification of Synergistic Drug Combinations Using Breast Cancer Patient-Derived Xenografts. Sci. Rep. 2020, 10, 1493. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Qi, X.-W.; Liu, X.-Z.; Yang, Z.-Y.; Cai, R.-L.; Cui, H.-J.; Chen, L.; Yu, S.-C. Icaritin Enhances the Efficacy of Cetuximab against Triple-Negative Breast Cancer Cells. Oncol. Lett. 2020, 19, 3950–3958. [Google Scholar] [CrossRef]
- Zecchin, D.; Moore, C.; Michailidis, F.; Horswell, S.; Rana, S.; Howell, M.; Downward, J. Combined Targeting of G Protein-coupled Receptor and EGF Receptor Signaling Overcomes Resistance to PI3K Pathway Inhibitors in PTEN-null Triple Negative Breast Cancer. EMBO Mol. Med. 2020, 12, e11987. [Google Scholar] [CrossRef]
- Barlaam, B.; Cosulich, S.; Degorce, S.; Fitzek, M.; Green, S.; Hancox, U.; der Brempt, C.L.; Lohmann, J.-J.; Maudet, M.; Morgentin, R.; et al. Discovery of (R)-8-(1-(3,5-Difluorophenylamino)Ethyl)-N,N-dimethyl-2-Morpholino-4-Oxo-4H-chromene-6-Carboxamide (AZD8186): A Potent and Selective Inhibitor of PI3Kβ and PI3Kδ for the Treatment of PTEN-Deficient Cancers. J. Med. Chem. 2015, 58, 943–962. [Google Scholar] [CrossRef]
- Xu, W.; Song, C.; Wang, X.; Li, Y.; Bai, X.; Liang, X.; Wu, J.; Liu, J. Downregulation of MiR-155-5p Enhances the Anti-Tumor Effect of Cetuximab on Triple-Negative Breast Cancer Cells via Inducing Cell Apoptosis and Pyroptosis. Aging 2021, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Grimshaw, K.M.; Hunter, L.-J.K.; Yap, T.A.; Heaton, S.P.; Walton, M.I.; Woodhead, S.J.; Fazal, L.; Reule, M.; Davies, T.G.; Seavers, L.C.; et al. AT7867 Is a Potent and Oral Inhibitor of AKT and P70 S6 Kinase That Induces Pharmacodynamic Changes and Inhibits Human Tumor Xenograft Growth. Mol. Cancer 2010, 9, 1100–1110. [Google Scholar] [CrossRef]
- Barrett, S.D.; Bridges, A.J.; Dudley, D.T.; Saltiel, A.R.; Fergus, J.H.; Flamme, C.M.; Delaney, A.M.; Kaufman, M.; LePage, S.; Leopold, W.R.; et al. The Discovery of the Benzhydroxamate MEK Inhibitors CI-1040 and PD 0325901. Bioorg. Med. Chem. Lett. 2008, 18, 6501–6504. [Google Scholar] [CrossRef] [PubMed]
- Koepke, J.I.; Nakrieko, K.; Wood, C.S.; Boucher, K.K.; Terlecky, L.J.; Walton, P.A.; Terlecky, S.R. Restoration of Peroxisomal Catalase Import in a Model of Human Cellular Aging. Traffic 2007, 8, 1590–1600. [Google Scholar] [CrossRef] [PubMed]
- Carles, F.; Bourg, S.; Meyer, C.; Bonnet, P. PKIDB: A Curated, Annotated and Updated Database of Protein Kinase Inhibitors in Clinical Trials. Molecules 2018, 23, 908. [Google Scholar] [CrossRef] [PubMed]
- Bournez, C.; Carles, F.; Peyrat, G.; Aci-Sèche, S.; Bourg, S.; Meyer, C.; Bonnet, P. Comparative Assessment of Protein Kinase Inhibitors in Public Databases and in PKIDB. Molecules 2020, 25, 3226. [Google Scholar] [CrossRef] [PubMed]
- Anido, J.; Matar, P.; Albanell, J.; Guzmán, M.; Rojo, F.; Arribas, J.; Averbuch, S.; Baselga, J. ZD1839, a Specific Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitor, Induces the Formation of Inactive EGFR/HER2 and EGFR/HER3 Heterodimers and Prevents Heregulin Signaling in HER2-Overexpressing Breast Cancer Cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 1274–1283. [Google Scholar]
- Senichkin, V.V.; Streletskaia, A.Y.; Gorbunova, A.S.; Zhivotovsky, B.; Kopeina, G.S. Saga of Mcl-1: Regulation from Transcription to Degradation. Cell Death Differ. 2020, 27, 405–419. [Google Scholar] [CrossRef]
- Deng, M.; Wang, J.; Chen, Y.; Zhang, L.; Liu, D. Combination of SF1126 and Gefitinib Induces Apoptosis of Triple-Negative Breast Cancer Cells through the PI3K/AKT–MTOR Pathway. Anti Cancer Drug 2015, 26, 422–427. [Google Scholar] [CrossRef]
- De, P.; Dey, N.; Terakedis, B.; Bergsagel, P.L.; Li, Z.H.; Mahadevan, D.; Garlich, J.R.; Trudel, S.; Makale, M.T.; Durden, D.L. An Integrin-Targeted, Pan-Isoform, Phosphoinositide-3 Kinase Inhibitor, SF1126, Has Activity against Multiple Myeloma in Vivo. Cancer Chemoth. Pharm. 2013, 71, 867–881. [Google Scholar] [CrossRef]
- Heitman, J.; Movva, N.; Hall, M. Targets for Cell Cycle Arrest by the Immunosuppressant Rapamycin in Yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. MTOR Interacts with Raptor to Form a Nutrient-Sensitive Complex That Signals to the Cell Growth Machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Polunovsky, V.A.; Rosenwald, I.B.; Tan, A.T.; White, J.; Chiang, L.; Sonenberg, N.; Bitterman, P.B. Translational Control of Programmed Cell Death: Eukaryotic Translation Initiation Factor 4E Blocks Apoptosis in Growth-Factor-Restricted Fibroblasts with Physiologically Expressed or Deregulated Myc. Mol. Cell Biol. 1996, 16, 6573–6581. [Google Scholar] [CrossRef] [PubMed]
- Wendel, H.-G.; Silva, R.L.A.; Malina, A.; Mills, J.R.; Zhu, H.; Ueda, T.; Watanabe-Fukunaga, R.; Fukunaga, R.; Teruya-Feldstein, J.; Pelletier, J.; et al. Dissecting EIF4E Action in Tumorigenesis. Gene Dev. 2007, 21, 3232–3237. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Zheng, X.S. Toward Rapamycin Analog (Rapalog)-Based Precision Cancer Therapy. Acta Pharm. Sin. 2015, 36, 1163–1169. [Google Scholar] [CrossRef]
- Salmond, R.J.; Emery, J.; Okkenhaug, K.; Zamoyska, R. MAPK, Phosphatidylinositol 3-Kinase, and Mammalian Target of Rapamycin Pathways Converge at the Level of Ribosomal Protein S6 Phosphorylation to Control Metabolic Signaling in CD8 T Cells. J. Immunol. 2009, 183, 7388–7397. [Google Scholar] [CrossRef]
- Shahbazian, D.; Roux, P.P.; Mieulet, V.; Cohen, M.S.; Raught, B.; Taunton, J.; Hershey, J.W.; Blenis, J.; Pende, M.; Sonenberg, N. The MTOR/PI3K and MAPK Pathways Converge on EIF4B to Control Its Phosphorylation and Activity. EMBO J. 2006, 25, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Traxler, P.; Allegrini, P.R.; Brandt, R.; Brueggen, J.; Cozens, R.; Fabbro, D.; Grosios, K.; Lane, H.A.; McSheehy, P.; Mestan, J.; et al. AEE788 A Dual Family Epidermal Growth Factor Receptor/ErbB2 and Vascular Endothelial Growth Factor Receptor Tyrosine Kinase Inhibitor with Antitumor and Antiangiogenic Activity. Cancer Res. 2004, 64, 4931–4941. [Google Scholar] [CrossRef]
- Keraite, I.; Alvarez-Garcia, V.; Garcia-Murillas, I.; Beaney, M.; Turner, N.C.; Bartos, C.; Oikonomidou, O.; Kersaudy-Kerhoas, M.; Leslie, N.R. PIK3CA Mutation Enrichment and Quantitation from Blood and Tissue. Sci. Rep. 2020, 10, 17082. [Google Scholar] [CrossRef] [PubMed]
- Fayette, J.; Wirth, L.; Oprean, C.; Udrea, A.; Jimeno, A.; Rischin, D.; Nutting, C.; Harari, P.M.; Csoszi, T.; Cernea, D.; et al. Randomized Phase II Study of Duligotuzumab (MEHD7945A) vs. Cetuximab in Squamous Cell Carcinoma of the Head and Neck (MEHGAN Study). Front. Oncol. 2016, 6, 232. [Google Scholar] [CrossRef]
- Hill, A.G.; Findlay, M.; Burge, M.; Jackson, C.; Alfonso, P.G.; Samuel, L.; Ganju, V.; Karthaus, M.; Amatu, A.; Jeffery, M.; et al. Phase II Study of the Dual EGFR/HER3 Inhibitor Duligotuzumab (MEHD7945A) vs. Cetuximab in Combination with FOLFIRI in RAS Wild-Type Metastatic Colorectal Cancer. Clin. Cancer Res. 2018, 24, 2276–2284. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT Inhibition Relieves Feedback Suppression of Receptor Tyrosine Kinase Expression and Activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Serra, V.; Scaltriti, M.; Prudkin, L.; Eichhorn, P.J.A.; Ibrahim, Y.H.; Chandarlapaty, S.; Markman, B.; Rodriguez, O.; Guzman, M.; Rodriguez, S.; et al. PI3K Inhibition Results in Enhanced HER Signaling and Acquired ERK Dependency in HER2-Overexpressing Breast Cancer. Oncogene 2011, 30, 2547–2557. [Google Scholar] [CrossRef]
- Rodrik-Outmezguine, V.S.; Chandarlapaty, S.; Pagano, N.C.; Poulikakos, P.I.; Scaltriti, M.; Moskatel, E.; Baselga, J.; Guichard, S.; Rosen, N. MTOR Kinase Inhibition Causes Feedback-Dependent Biphasic Regulation of AKT Signaling. Cancer Discov. 2011, 1, 248–259. [Google Scholar] [CrossRef]
- Kane, R.C.; Farrell, A.T.; Sridhara, R.; Pazdur, R. United States Food and Drug Administration Approval Summary: Bortezomib for the Treatment of Progressive Multiple Myeloma after One Prior Therapy. Clin. Cancer Res. 2006, 12, 2955–2960. [Google Scholar] [CrossRef] [PubMed]
- Shahshahan, M.A.; Beckley, M.N.; Jazirehi, A.R. Potential Usage of Proteasome Inhibitor Bortezomib (Velcade, PS-341) in the Treatment of Metastatic Melanoma: Basic and Clinical Aspects. Am. J. Cancer Res. 2011, 1, 913–924. [Google Scholar] [PubMed]
- Nakshatri, H.; Bhat-Nakshatri, P.; Martin, D.A.; Goulet, R.J.; Sledge, G.W. Constitutive Activation of NF-KappaB during Progression of Breast Cancer to Hormone-Independent Growth. Mol. Cell Biol. 1997, 17, 3629–3639. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Gonzalez-Angulo, A.M.; Reuben, J.M.; Booser, D.J.; Pusztai, L.; Krishnamurthy, S.; Esseltine, D.; Stec, J.; Broglio, K.R.; Islam, R.; et al. Bortezomib (VELCADE®) in Metastatic Breast Cancer: Pharmacodynamics, Biological Effects, and Prediction of Clinical Benefits. Ann. Oncol. 2006, 17, 813–817. [Google Scholar] [CrossRef]
- Duong, H.-Q.; You, K.; Oh, S.; Kwak, S.-J.; Seong, Y.-S. Silencing of NRF2 Reduces the Expression of ALDH1A1 and ALDH3A1 and Sensitizes to 5-FU in Pancreatic Cancer Cells. Antioxidants 2017, 6, 52. [Google Scholar] [CrossRef]
- Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; Lahti, J.M.; Manning, G.; Morgan, D.O.; Tsai, L.-H.; Wolgemuth, D.J. Cyclin-Dependent Kinases: A Family Portrait. Nat. Cell Biol. 2009, 11, 1275–1276. [Google Scholar] [CrossRef]
- Roskoski, R. Cyclin-Dependent Protein Serine/Threonine Kinase Inhibitors as Anticancer Drugs. Pharm. Res. 2018, 139, 471–488. [Google Scholar] [CrossRef] [PubMed]
- Cassandri, M.; Fioravanti, R.; Pomella, S.; Valente, S.; Rotili, D.; Baldo, G.D.; Angelis, B.D.; Rota, R.; Mai, A. CDK9 as a Valuable Target in Cancer: From Natural Compounds Inhibitors to Current Treatment in Pediatric Soft Tissue Sarcomas. Front. Pharm. 2020, 11, 1230. [Google Scholar] [CrossRef]
- Chou, J.; Quigley, D.A.; Robinson, T.M.; Feng, F.Y.; Ashworth, A. Transcription-Associated Cyclin-Dependent Kinases as Targets and Biomarkers for Cancer Therapy. Cancer Discov. 2020, 10, 351–370. [Google Scholar] [CrossRef]
- Giacinti, C.; Giordano, A. RB and Cell Cycle Progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK Inhibitors: Cell Cycle Regulators and Beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Paye, A.; Truong, A.; Yip, C.; Cimino, J.; Blacher, S.; Munaut, C.; Cataldo, D.; Foidart, J.M.; Maquoi, E.; Collignon, J.; et al. EGFR Activation and Signaling in Cancer Cells Are Enhanced by the Membrane-Bound Metalloprotease MT4-MMP. Cancer Res. 2014, 74, 6758–6770. [Google Scholar] [CrossRef]
- Yip, C.; Foidart, P.; Somja, J.; Truong, A.; Lienard, M.; Feyereisen, E.; Schroeder, H.; Gofflot, S.; Donneau, A.-F.; Collignon, J.; et al. MT4-MMP and EGFR Expression Levels Are Key Biomarkers for Breast Cancer Patient Response to Chemotherapy and Erlotinib. Br. J. Cancer 2017, 116, 742–751. [Google Scholar] [CrossRef]
- Peng, J.; Zhu, Y.; Milton, J.T.; Price, D.H. Identification of Multiple Cyclin Subunits of Human P-TEFb. Gene Dev. 1998, 12, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Larochelle, S.; Amat, R.; Glover-Cutter, K.; Sansó, M.; Zhang, C.; Allen, J.J.; Shokat, K.M.; Bentley, D.L.; Fisher, R.P. Cyclin-Dependent Kinase Control of the Initiation-to-Elongation Switch of RNA Polymerase II. Nat. Struct Mol. Biol. 2012, 19, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Viladevall, L.; Amour, C.V.S.; Rosebrock, A.; Schneider, S.; Zhang, C.; Allen, J.J.; Shokat, K.M.; Schwer, B.; Leatherwood, J.K.; Fisher, R.P. TFIIH and P-TEFb Coordinate Transcription with Capping Enzyme Recruitment at Specific Genes in Fission Yeast. Mol. Cell 2009, 33, 738–751. [Google Scholar] [CrossRef]
- Yang, Z.; Yik, J.H.N.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for Stimulation of Transcriptional Elongation by the Bromodomain Protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef]
- Ahn, S.H.; Kim, M.; Buratowski, S. Phosphorylation of Serine 2 within the RNA Polymerase II C-Terminal Domain Couples Transcription and 3′ End Processing. Mol. Cell 2004, 13, 67–76. [Google Scholar] [CrossRef]
- Li, B.; Chonghaile, T.N.; Fan, Y.; Madden, S.F.; Klinger, R.; O’Connor, A.E.; Walsh, L.; O’Hurley, G.; Udupi, G.M.; Joseph, J.; et al. Therapeutic Rationale to Target Highly Expressed CDK7 Conferring Poor Outcomes in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 3834–3845. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; et al. CDK7-Dependent Transcriptional Addiction in Triple-Negative Breast Cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef]
- Sun, B.; Mason, S.; Wilson, R.C.; Hazard, S.E.; Wang, Y.; Fang, R.; Wang, Q.; Yeh, E.S.; Yang, M.; Roberts, T.M.; et al. Inhibition of the Transcriptional Kinase CDK7 Overcomes Therapeutic Resistance in HER2-Positive Breast Cancers. Oncogene 2020, 39, 50–63. [Google Scholar] [CrossRef]
- Labib, K.; Tercero, J.A.; Diffley, J.F.X. Uninterrupted MCM2-7 Function Required for DNA Replication Fork Progression. Science 2000, 288, 1643–1647. [Google Scholar] [CrossRef]
- Benedict, B.; van Harn, T.; Dekker, M.; Hermsen, S.; Kucukosmanoglu, A.; Pieters, W.; Delzenne-Goette, E.; Dorsman, J.C.; Petermann, E.; Foijer, F.; et al. Loss of P53 Suppresses Replication-Stress-Induced DNA Breakage in G1/S Checkpoint Deficient Cells. Elife 2018, 7, e37868. [Google Scholar] [CrossRef] [PubMed]
- Parsons, S.J.; Parsons, J.T. Src Family Kinases, Key Regulators of Signal Transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef]
- Boggon, T.J.; Eck, M.J. Structure and Regulation of Src Family Kinases. Oncogene 2004, 23, 7918–7927. [Google Scholar] [CrossRef]
- Lindauer, M.; Hochhaus, A. Small Molecules in Oncology. Recent Results Cancer Res. 2014, 201, 27–65. [Google Scholar] [CrossRef]
- Kim, D.-J.; Yi, Y.-W.; Kim, J.H. In Situ Monitoring of Bindings between Dasatinib and Its Target Protein Kinases Using Magnetic Nanoparticles in Live Cells. J. Am Chem. Soc. 2008, 130, 16466–16467. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long Non-Coding RNA HOTAIR Reprograms Chromatin State to Promote Cancer Metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, L.; Zhang, L.; Wang, Y.; Li, H.; Ren, X.; Wei, F.; Yu, W.; Liu, T.; Wang, X.; et al. Long Non-Coding RNA HOTAIR Promotes Tumor Cell Invasion and Metastasis by Recruiting EZH2 and Repressing E-Cadherin in Oral Squamous Cell Carcinoma. Int. J. Oncol. 2015, 46, 2586–2594. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Yao, L.; Zhang, Q.; Wang, F.; Mei, H.; Guo, X.; Huang, W. Long Noncoding RNA HOTAIR Promotes Metastasis of Renal Cell Carcinoma by Up-Regulating Histone H3K27 Demethylase JMJD3. Oncotarget 2014, 5, 19795–19802. [Google Scholar] [CrossRef]
- Yom, C.K.; Noh, D.-Y.; Kim, W.H.; Kim, H.S. Clinical Significance of High Focal Adhesion Kinase Gene Copy Number and Overexpression in Invasive Breast Cancer. Breast Cancer Res. Treat. 2011, 128, 647–655. [Google Scholar] [CrossRef]
- Boyd, Z.S.; Wu, Q.J.; O’Brien, C.; Spoerke, J.; Savage, H.; Fielder, P.J.; Amler, L.; Yan, Y.; Lackner, M.R. Proteomic Analysis of Breast Cancer Molecular Subtypes and Biomarkers of Response to Targeted Kinase Inhibitors Using Reverse-Phase Protein Microarrays. Mol. Cancer 2008, 7, 3695–3706. [Google Scholar] [CrossRef]
- Glenisson, M.; Vacher, S.; Callens, C.; Susini, A.; Cizeron-Clairac, G.; Scodan, R.L.; Meseure, D.; Lerebours, F.; Spyratos, F.; Lidereau, R.; et al. Identification of New Candidate Therapeutic Target Genes in Triple-Negative Breast Cancer. Genes Cancer 2012, 3, 63–70. [Google Scholar] [CrossRef][Green Version]
- Sun, J.; Zhang, D.; Bae, D.-H.; Sahni, S.; Jansson, P.; Zheng, Y.; Zhao, Q.; Yue, F.; Zheng, M.; Kovacevic, Z.; et al. Metastasis Suppressor, NDRG1, Mediates Its Activity through Signaling Pathways and Molecular Motors. Carcinogenesis 2013, 34, 1943–1954. [Google Scholar] [CrossRef]
- Ono-Saito, N.; Niki, I.; Hidaka, H. H-Series Protein Kinase Inhibitors and Potential Clinical Applications. Pharm. Ther. 1999, 82, 123–131. [Google Scholar] [CrossRef]
- Zhao, J.; Zhou, D.; Guo, J.; Ren, Z.; Zhou, L.; Wang, S.; Xu, B.; Wang, R. Effect of Fasudil Hydrochloride, a Protein Kinase Inhibitor, on Cerebral Vasospasm and Delayed Cerebral Ischemic Symptoms After Aneurysmal Subarachnoid Hemorrhage. Neurol. Med. Chir 2006, 46, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lin, X.; Aske, J.C.; Ye, P.; Williams, C.; Abramovitz, M.; Leyland-Jones, B.R. Dasatinib Attenuates Overexpression of Src Signaling Induced by the Combination Treatment of Veliparib plus Carboplatin in Triple-Negative Breast Cancer. Cancer Chemoth. Pharm. 2019, 84, 1241–1256. [Google Scholar] [CrossRef] [PubMed]
- Tryfonopoulos, D.; Walsh, S.; Collins, D.M.; Flanagan, L.; Quinn, C.; Corkery, B.; McDermott, E.W.; Evoy, D.; Pierce, A.; O’Donovan, N.; et al. Src: A Potential Target for the Treatment of Triple-Negative Breast Cancer. Ann. Oncol. 2011, 22, 2234–2240. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Wali, V.B.; Cheng, C.J.; Inukai, S.; Booth, C.J.; Agarwal, S.; Rimm, D.L.; Győrffy, B.; Santarpia, L.; Pusztai, L.; et al. MiR-34a Silences c-SRC to Attenuate Tumor Growth in Triple-Negative Breast Cancer. Cancer Res. 2016, 76, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.-L.; Zhang, J.; Li, P.-Z.; Lang, R.-G.; Li, W.-D.; Sun, H.; Liu, F.-F.; Guo, X.-J.; Gu, F.; Fu, L. Dasatinib Inhibits C-Src Phosphorylation and Prevents the Proliferation of Triple-Negative Breast Cancer (TNBC) Cells Which Overexpress Syndecan-Binding Protein (SDCBP). PLoS ONE 2017, 12, e0171169. [Google Scholar] [CrossRef]
- Lou, L.; Yu, Z.; Wang, Y.; Wang, S.; Zhao, Y. C-Src Inhibitor Selectively Inhibits Triple-negative Breast Cancer Overexpressed Vimentin in Vitro and in Vivo. Cancer Sci. 2018, 109, 1648–1659. [Google Scholar] [CrossRef]
- Song, L.; Morris, M.; Bagui, T.; Lee, F.Y.; Jove, R.; Haura, E.B. Dasatinib (BMS-354825) Selectively Induces Apoptosis in Lung Cancer Cells Dependent on Epidermal Growth Factor Receptor Signaling for Survival. Cancer Res. 2006, 66, 5542–5548. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Wu, M.-H.; Wei, T.-T.; Chung, S.-H.; Chen, K.-F.; Cheng, A.-L.; Chen, C.-C. Degradation of Epidermal Growth Factor Receptor Mediates Dasatinib-Induced Apoptosis in Head and Neck Squamous Cell Carcinoma Cells. Neoplasia 2012, 14, 463–475. [Google Scholar] [CrossRef]
- Duong, H.-Q.; Yi, Y.W.; Kang, H.J.; Bae, I.; Jang, Y.-J.; Kwak, S.-J.; Seong, Y.-S. Combination of Dasatinib and Gemcitabine Reduces the ALDH1A1 Expression and the Proliferation of Gemcitabine-Resistant Pancreatic Cancer MIA PaCa-2 Cells. Int. J. Oncol. 2014, 44, 2132–2138. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.; Dean, C.A.; Giacomantonio, C.A.; Lee, P.W.K. Aldehyde Dehydrogenase: Its Role as a Cancer Stem Cell Marker Comes down to the Specific Isoform. Cell Cycle 2011, 10, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Gay, C.M.; Byers, L.A.; O’Connor, M.J.; Yap, T.A. PARP Inhibitors: Extending Benefit Beyond BRCA-Mutant Cancers. Clin. Cancer Res. 2019, 25, 3759–3771. [Google Scholar] [CrossRef] [PubMed]
- Keung, M.Y.; Wu, Y.; Badar, F.; Vadgama, J.V. Response of Breast Cancer Cells to PARP Inhibitors Is Independent of BRCA Status. J. Clin. Med. 2020, 9, 940. [Google Scholar] [CrossRef]
- Elstrodt, F.; Hollestelle, A.; Nagel, J.H.A.; Gorin, M.; Wasielewski, M.; van den Ouweland, A.; Merajver, S.D.; Ethier, S.P.; Schutte, M. BRCA1 Mutation Analysis of 41 Human Breast Cancer Cell Lines Reveals Three New Deleterious Mutants. Cancer Res. 2006, 66, 41–45. [Google Scholar] [CrossRef]
- Kumaraswamy, E.; Wendt, K.L.; Augustine, L.A.; Stecklein, S.R.; Sibala, E.C.; Li, D.; Gunewardena, S.; Jensen, R.A. BRCA1 Regulation of Epidermal Growth Factor Receptor (EGFR) Expression in Human Breast Cancer Cells Involves MicroRNA-146a and Is Critical for Its Tumor Suppressor Function. Oncogene 2015, 34, 4333–4346. [Google Scholar] [CrossRef]
- Yi, Y.W.; Park, J.-S.; Kwak, S.-J.; Seong, Y.-S. Co-Treatment with BEZ235 Enhances Sensitivity of BRCA1-Negative Breast Cancer Cells to Olaparib. Anticancer Res. 2015, 35, 3829–3838. [Google Scholar]
- Reilly, E.B.; Phillips, A.C.; Buchanan, F.G.; Kingsbury, G.; Zhang, Y.; Meulbroek, J.A.; Cole, T.B.; DeVries, P.J.; Falls, H.D.; Beam, C.; et al. Characterization of ABT-806, a Humanized Tumor-Specific Anti-EGFR Monoclonal Antibody. Mol. Cancer 2015, 14, 1141–1151. [Google Scholar] [CrossRef]
- Phillips, A.C.; Boghaert, E.R.; Vaidya, K.S.; Mitten, M.J.; Norvell, S.; Falls, H.D.; DeVries, P.J.; Cheng, D.; Meulbroek, J.A.; Buchanan, F.G.; et al. ABT-414, an Antibody–Drug Conjugate Targeting a Tumor-Selective EGFR Epitope. Mol. Cancer 2016, 15, 661–669. [Google Scholar] [CrossRef]
- Anderson, M.G.; Falls, H.D.; Mitten, M.J.; Oleksijew, A.; Vaidya, K.S.; Boghaert, E.R.; Gao, W.; Palma, J.P.; Cao, D.; Chia, P.-L.; et al. Targeting Multiple EGFR-Expressing Tumors with a Highly Potent Tumor-Selective Antibody–Drug Conjugate. Mol. Cancer 2020, 19, 2117–2125. [Google Scholar] [CrossRef] [PubMed]
- Guillermet-Guibert, J.; Davenne, L.; Pchejetski, D.; Saint-Laurent, N.; Brizuela, L.; Guilbeau-Frugier, C.; Delisle, M.-B.; Cuvillier, O.; Susini, C.; Bousquet, C. Targeting the Sphingolipid Metabolism to Defeat Pancreatic Cancer Cell Resistance to the Chemotherapeutic Gemcitabine Drug. Mol. Cancer 2009, 8, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Baran, Y.; Salas, A.; Senkal, C.E.; Gunduz, U.; Bielawski, J.; Obeid, L.M.; Ogretmen, B. Alterations of Ceramide/Sphingosine 1-Phosphate Rheostat Involved in the Regulation of Resistance to Imatinib-Induced Apoptosis in K562 Human Chronic Myeloid Leukemia Cells. J. Biol. Chem. 2007, 282, 10922–10934. [Google Scholar] [CrossRef] [PubMed]
- Sukocheva, O.; Wang, L.; Verrier, E.; Vadas, M.A.; Xia, P. Restoring Endocrine Response in Breast Cancer Cells by Inhibition of the Sphingosine Kinase-1 Signaling Pathway. Endocrinology 2009, 150, 4484–4492. [Google Scholar] [CrossRef] [PubMed]
- Madhunapantula, S.V.; Hengst, J.; Gowda, R.; Fox, T.E.; Yun, J.K.; Robertson, G.P. Targeting Sphingosine Kinase-1 to Inhibit Melanoma. Pigm. Cell Melanoma R 2012, 25, 259–274. [Google Scholar] [CrossRef]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; de Peredo, A.G.; et al. Resistance of Melanoma to Immune Checkpoint Inhibitors Is Overcome by Targeting the Sphingosine Kinase-1. Nat. Commun. 2020, 11, 437. [Google Scholar] [CrossRef]
- Ren, X.; Su, C. Sphingosine Kinase 1 Contributes to Doxorubicin Resistance and Glycolysis in Osteosarcoma. Mol. Med. Rep. 2020, 22, 2183–2190. [Google Scholar] [CrossRef]
- Maczis, M.A.; Maceyka, M.; Waters, M.R.; Newton, J.; Singh, M.; Rigsby, M.F.; Turner, T.H.; Alzubi, M.A.; Harrell, J.C.; Milstien, S.; et al. Sphingosine Kinase 1 Activation by Estrogen Receptor A36 Contributes to Tamoxifen Resistance in Breast Cancer. J. Lipid Res. 2018, 59, 2297–2307. [Google Scholar] [CrossRef]
- Xu, Y.; Dong, B.; Wang, J.; Zhang, J.; Xue, W.; Huang, Y. Sphingosine Kinase 1 Overexpression Contributes to Sunitinib Resistance in Clear Cell Renal Cell Carcinoma. Oncoimmunology 2018, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wu, Q.; Zhou, X.; Huang, J. SphK1 Functions Downstream of IGF-1 to Modulate IGF-1-Induced EMT, Migration and Paclitaxel Resistance of A549 Cells: A Preliminary in Vitro Study. J. Cancer 2019, 10, 4264–4269. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liang, Y.; Chang, W.; Hu, B.; Zhang, Y. Triple Negative Breast Cancer Depends on Sphingosine Kinase 1 (SphK1)/Sphingosine-1-Phosphate (S1P)/Sphingosine 1-Phosphate Receptor 3 (S1PR3)/Notch Signaling for Metastasis. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 1912–1923. [Google Scholar] [CrossRef]
- Wang, X.; Sun, Y.; Peng, X.; Naqvi, S.M.A.S.; Yang, Y.; Zhang, J.; Chen, M.; Chen, Y.; Chen, H.; Yan, H.; et al. The Tumorigenic Effect of Sphingosine Kinase 1 and Its Potential Therapeutic Target. Cancer Control. 2020, 27, 1073274820976664. [Google Scholar] [CrossRef] [PubMed]
- Ruckhäberle, E.; Rody, A.; Engels, K.; Gaetje, R.; von Minckwitz, G.; Schiffmann, S.; Grösch, S.; Geisslinger, G.; Holtrich, U.; Karn, T.; et al. Microarray Analysis of Altered Sphingolipid Metabolism Reveals Prognostic Significance of Sphingosine Kinase 1 in Breast Cancer. Breast Cancer Res. Treat. 2008, 112, 41–52. [Google Scholar] [CrossRef]
- Lim, K.G.; Tonelli, F.; Li, Z.; Lu, X.; Bittman, R.; Pyne, S.; Pyne, N.J. FTY720 Analogues as Sphingosine Kinase 1 Inhibitors Enzyme Inhibition Kinetics, Allosterism, Proteasomal Degradation, And Actin Rearrangement In Mcf-7 Breast Cancer Cells. J. Biol. Chem. 2011, 286, 18633–18640. [Google Scholar] [CrossRef]
- Sharma, S.; Mathur, A.; Pradhan, S.; Singh, D.; Gupta, S. Fingolimod (FTY720): First Approved Oral Therapy for Multiple Sclerosis. J. Pharm. Pharm. 2011, 2, 49–51. [Google Scholar] [CrossRef]
- Xu, H.; Wu, K.; Tian, Y.; Liu, Q.; Han, N.; Yuan, X.; Zhang, L.; Wu, G.S.; Wu, K. CD44 Correlates with Clinicopathological Characteristics and Is Upregulated by EGFR in Breast Cancer. Int. J. Oncol. 2016, 49, 1343–1350. [Google Scholar] [CrossRef]
- Kawahara, S.; Otsuji, Y.; Nakamura, M.; Murakami, M.; Murate, T.; Matsunaga, T.; Kanoh, H.; Seishima, M.; Banno, Y.; Hara, A. Sphingosine Kinase 1 Plays a Role in the Upregulation of CD44 Expression through Extracellular Signal-Regulated Kinase Signaling in Human Colon Cancer Cells. Anti Cancer Drug 2013, 24, 473–483. [Google Scholar] [CrossRef]
- Collina, F.; Bonito, M.D.; Bergolis, V.L.; Laurentiis, M.D.; Vitagliano, C.; Cerrone, M.; Nuzzo, F.; Cantile, M.; Botti, G. Prognostic Value of Cancer Stem Cells Markers in Triple-Negative Breast Cancer. Biomed. Res. Int. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.Z.; Marzec, K.A.; Martin, J.L.; Baxter, R.C. The Role of Insulin-like Growth Factor Binding Protein-3 in the Breast Cancer Cell Response to DNA-Damaging Agents. Oncogene 2014, 33, 85–96. [Google Scholar] [CrossRef]
- Schnute, M.E.; McReynolds, M.D.; Kasten, T.; Yates, M.; Jerome, G.; Rains, J.W.; Hall, T.; Chrencik, J.; Kraus, M.; Cronin, C.N.; et al. Modulation of Cellular S1P Levels with a Novel, Potent and Specific Inhibitor of Sphingosine Kinase-1. Biochem. J. 2012, 444, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Peyroche, A.; Antonny, B.; Robineau, S.; Acker, J.; Cherfils, J.; Jackson, C.L. Brefeldin A Acts to Stabilize an Abortive ARF–GDP–Sec7 Domain Protein Complex Involvement of Specific Residues of the Sec7 Domain. Mol. Cell 1999, 3, 275–285. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent Advances in the Development of Protein–Protein Interactions Modulators: Mechanisms and Clinical Trials. Signal. Transduct. Target. 2020, 5, 213. [Google Scholar] [CrossRef]
- Takayama, S.; Xie, Z.; Reed, J.C. An Evolutionarily Conserved Family of Hsp70/Hsc70 Molecular Chaperone Regulators. J. Biol. Chem. 1999, 274, 781–786. [Google Scholar] [CrossRef]
- Rauch, J.N.; Zuiderweg, E.R.P.; Gestwicki, J.E. Non-Canonical Interactions between Heat Shock Cognate Protein 70 (Hsc70) and Bcl2-Associated Anthanogene (BAG) Co-Chaperones Are Important for Client Release. J. Biol. Chem. 2016, 291, 19848–19857. [Google Scholar] [CrossRef]
- Wang, J.; Lou, P.; Lesniewski, R.; Henkin, J. Paclitaxel at Ultra Low Concentrations Inhibits Angiogenesis without Affecting Cellular Microtubule Assembly. Anti Cancer Drug 2003, 14, 13–19. [Google Scholar] [CrossRef]
- Bollag, D.M.; McQueney, P.A.; Zhu, J.; Hensens, O.; Koupal, L.; Liesch, J.; Goetz, M.; Lazarides, E.; Woods, C.M. Epothilones, a New Class of Microtubule-Stabilizing Agents with a Taxol-like Mechanism of Action. Cancer Res. 1995, 55, 2325–2333. [Google Scholar]
- Lee, F.Y.; Borzilleri, R.; Fairchild, C.R.; Kim, S.H.; Long, B.H.; Reventos-Suarez, C.; Vite, G.D.; Rose, W.C.; Kramer, R.A. BMS-247550: A Novel Epothilone Analog with a Mode of Action Similar to Paclitaxel but Possessing Superior Antitumor Efficacy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2001, 7, 1429–1437. [Google Scholar]
- De Almeida, L.C.; Calil, F.A.; Machado-Neto, J.A.; Costa-Lotufo, L.V. DNA Damaging Agents and DNA Repair: From Carcinogenesis to Cancer Therapy. Cancer Genet. 2020, 252–253, 6–24. [Google Scholar] [CrossRef]
- Yimit, A.; Adebali, O.; Sancar, A.; Jiang, Y. Differential Damage and Repair of DNA-Adducts Induced by Anti-Cancer Drug Cisplatin across Mouse Organs. Nat. Commun 2019, 10, 309. [Google Scholar] [CrossRef]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular Advances and Pharmacologic Developments in Antitumor Activity and Cardiotoxicity. Pharm. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.; Ohashi, A.; Huang, Y.; Pandita, T.K.; Ludwig, T.; Powell, S.N.; Yang, Q. Negative Regulation of AKT Activation by BRCA1. Cancer Res. 2008, 68, 10040–10044. [Google Scholar] [CrossRef] [PubMed]
- Galmarini, C.M.; Mackey, J.R.; Dumontet, C. Nucleoside Analogues and Nucleobases in Cancer Treatment. Lancet Oncol. 2002, 3, 415–424. [Google Scholar] [CrossRef]
- Von der Maase, H.; Hansen, S.W.; Roberts, J.T.; Dogliotti, L.; Oliver, T.; Moore, M.J.; Bodrogi, I.; Albers, P.; Knuth, A.; Lippert, C.M.; et al. Gemcitabine and Cisplatin Versus Methotrexate, Vinblastine, Doxorubicin, and Cisplatin in Advanced or Metastatic Bladder Cancer: Results of a Large, Randomized, Multinational, Multicenter, Phase III Study. J. Clin. Oncol. 2000, 18, 3068–3077. [Google Scholar] [CrossRef] [PubMed]
- Albain, K.S.; Nag, S.M.; Calderillo-Ruiz, G.; Jordaan, J.P.; Llombart, A.C.; Pluzanska, A.; Rolski, J.; Melemed, A.S.; Reyes-Vidal, J.M.; Sekhon, J.S.; et al. Gemcitabine Plus Paclitaxel Versus Paclitaxel Monotherapy in Patients with Metastatic Breast Cancer and Prior Anthracycline Treatment. J. Clin. Oncol. 2008, 26, 3950–3957. [Google Scholar] [CrossRef] [PubMed]
- Sandler, A.B.; Nemunaitis, J.; Denham, C.; von Pawel, J.; Cormier, Y.; Gatzemeier, U.; Mattson, K.; Manegold, C.; Palmer, M.C.; Gregor, A.; et al. Phase III Trial of Gemcitabine Plus Cisplatin Versus Cisplatin Alone in Patients with Locally Advanced or Metastatic Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2000, 18, 122. [Google Scholar] [CrossRef] [PubMed]
- Mutch, D.G.; Orlando, M.; Goss, T.; Teneriello, M.G.; Gordon, A.N.; McMeekin, S.D.; Wang, Y.; Scribner, D.R.S., Jr.; Marciniack, M.; Naumann, R.W.; et al. Randomized Phase III Trial of Gemcitabine Compared with Pegylated Liposomal Doxorubicin in Patients with Platinum-Resistant Ovarian Cancer. J. Clin. Oncol. 2007, 25, 2811–2818. [Google Scholar] [CrossRef] [PubMed]
- Ferrandina, G.; Ludovisi, M.; Lorusso, D.; Pignata, S.; Breda, E.; Savarese, A.; Medico, P.D.; Scaltriti, L.; Katsaros, D.; Priolo, D.; et al. Phase III Trial of Gemcitabine Compared with Pegylated Liposomal Doxorubicin in Progressive or Recurrent Ovarian Cancer. J. Clin. Oncol. 2008, 26, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in Survival and Clinical Benefit with Gemcitabine as First-Line Therapy for Patients with Advanced Pancreas Cancer: A Randomized Trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef]
- Mini, E.; Nobili, S.; Caciagli, B.; Landini, I.; Mazzei, T. Cellular Pharmacology of Gemcitabine. Ann. Oncol. 2006, 17, v7–v12. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, V.; Plunkett, W. Modulatory Activity of 2′,2′-Difluorodeoxycytidine on the Phosphorylation and Cytotoxicity of Arabinosyl Nucleosides. Cancer Res. 1990, 50, 3675–3680. [Google Scholar] [PubMed]
- Heinemann, V.; Xu, Y.Z.; Chubb, S.; Sen, A.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Inhibition of Ribonucleotide Reduction in CCRF-CEM Cells by 2′,2′-Difluorodeoxycytidine. Mol. Pharm. 1990, 38, 567–572. [Google Scholar]
- Baker, C.H.; Banzon, J.; Bollinger, J.M.; Stubbe, J.; Samano, V.; Robins, M.J.; Lippert, B.; Jarvi, E.; Resvick, R. 2′-Deoxy-2′-Methylenecytidine and 2′-Deoxy-2′,2′-Difluorocytidine 5′-Diphosphates: Potent Mechanism-Based Inhibitors of Ribonucleotide Reductase. J. Med. Chem. 1991, 34, 1879–1884. [Google Scholar] [CrossRef]
- Honeywell, R.J.; van Haperen, V.W.T.R.; Veerman, G.; Smid, K.; Peters, G.J. Inhibition of Thymidylate Synthase by 2′,2′-Difluoro-2′-Deoxycytidine (Gemcitabine) and Its Metabolite 2′,2′-Difluoro-2′-Deoxyuridine. Int. J. Biochem. Cell Biol. 2015, 60, 73–81. [Google Scholar] [CrossRef]
- Holliger, P.; Prospero, T.; Winter, G. “Diabodies”: Small Bivalent and Bispecific Antibody Fragments. Proc. Natl. Acad. Sci. USA 1993, 90, 6444–6448. [Google Scholar] [CrossRef]
- Han, W.; Pan, H.; Chen, Y.; Sun, J.; Wang, Y.; Li, J.; Ge, W.; Feng, L.; Lin, X.; Wang, X.; et al. EGFR Tyrosine Kinase Inhibitors Activate Autophagy as a Cytoprotective Response in Human Lung Cancer Cells. PLoS ONE 2011, 6, e18691. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-Q.; Yu, Z.-Y.; Li, J.; Ouyang, X.-N. Gefitinib Induces Lung Cancer Cell Autophagy and Apoptosis via Blockade of the PI3K/AKT/MTOR Pathway. Oncol. Lett 2016, 12, 63–68. [Google Scholar] [CrossRef]
- Mah, L.-J.; El-Osta, A.; Karagiannis, T.C. ΓH2AX: A Sensitive Molecular Marker of DNA Damage and Repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Said, A.; Bock, S.; Lajqi, T.; Müller, G.; Weindl, G. Chloroquine Promotes IL-17 Production by CD4+ T Cells via P38-Dependent IL-23 Release by Monocyte-Derived Langerhans-like Cells. J. Immunol. 2014, 193, 6135–6143. [Google Scholar] [CrossRef]
- Hansen, T.E.; Johansen, T. Following Autophagy Step by Step. BMC Biol. 2011, 9, 39. [Google Scholar] [CrossRef]
- Bui, N.-L.-C.; Pandey, V.; Zhu, T.; Ma, L.; Basappa; Lobie, P.E. Bad Phosphorylation as a Target of Inhibition in Oncology. Cancer Lett. 2018, 415, 177–186. [Google Scholar] [CrossRef]
- Yang, X.-J.; Xi, Y.-M.; Li, Z.-J. Icaritin: A Novel Natural Candidate for Hematological Malignancies Therapy. Biomed. Res. Int. 2019, 2019, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zheng, N.; Dong, J.; Wang, X.; Liu, L.; Huang, J. Estrogen Receptor-A36 Is Involved in Icaritin Induced Growth Inhibition of Triple-Negative Breast Cancer Cells. J. Steroid Biochem. Mol. Biol. 2017, 171, 318–327. [Google Scholar] [CrossRef]
- Daley, J. Gene Therapy Arrives. Nature 2019, 576, S12–S13. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host Cell Death and Inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.-C.; Shao, F. Pore-Forming Activity and Structural Autoinhibition of the Gasdermin Family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy Drugs Induce Pyroptosis through Caspase-3 Cleavage of a Gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Jia, H.; Truica, C.I.; Wang, B.; Wang, Y.; Ren, X.; Harvey, H.A.; Song, J.; Yang, J.-M. Immunotherapy for Triple-Negative Breast Cancer: Existing Challenges and Exciting Prospects. Drug Resist. Update 2017, 32, 1–15. [Google Scholar] [CrossRef]
- Li, Z.; Qiu, Y.; Lu, W.; Jiang, Y.; Wang, J. Immunotherapeutic Interventions of Triple Negative Breast Cancer. J. Transl. Med. 2018, 16, 147. [Google Scholar] [CrossRef]
- Katz, H.; Alsharedi, M. Immunotherapy in Triple-Negative Breast Cancer. Med. Oncol. 2018, 35, 13. [Google Scholar] [CrossRef]
- Marra, A.; Viale, G.; Curigliano, G. Recent Advances in Triple Negative Breast Cancer: The Immunotherapy Era. BMC Med. 2019, 17, 90. [Google Scholar] [CrossRef]
- Wahby, S.; Fashoyin-Aje, L.; Osgood, C.L.; Cheng, J.; Fiero, M.H.; Zhang, L.; Tang, S.; Hamed, S.S.; Song, P.; Charlab, R.; et al. FDA Approval Summary: Accelerated Approval of Sacituzumab Govitecan-Hziy for Third-Line Treatment of Metastatic Triple-Negative Breast Cancer. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Bardia, A.; Mayer, I.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond, J.R.; O’Shaughnessy, J.; Moroose, R.L.; Santin, A.D.; Abramson, V.G.; et al. Sacituzumab Govitecan-Hziy in Refractory Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2019, 380, 741–751. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA Grants Regular Approval to Sacituzumab Govitecan for Triple-Negative Breast Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-regular-approval-sacituzumab-govitecan-triple-negative-breast-cancer#:~:text=On%20April%207%2C%202021%2C%20the,least%20one%20of%20them%20for (accessed on 13 April 2021).
- Larson, R.C.; Maus, M.V. Recent Advances and Discoveries in the Mechanisms and Functions of CAR T Cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity. Front. Immunol. 2017, 8, 533. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human Chimeric Antigen Receptor Macrophages for Cancer Immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T Cell Immunotherapy for Human Cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Bagley, S.J.; O’Rourke, D.M. Clinical Investigation of CAR T Cells for Solid Tumors: Lessons Learned and Future Directions. Pharm. Ther. 2020, 205, 107419. [Google Scholar] [CrossRef]
- Dees, S.; Ganesan, R.; Singh, S.; Grewal, I.S. Emerging CAR-T Cell Therapy for the Treatment of Triple-Negative Breast Cancer. Mol. Cancer 2020, 19, 2409–2421. [Google Scholar] [CrossRef] [PubMed]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S.; Daëron, M. Specificity and Affinity of Human Fcγ Receptors and Their Polymorphic Variants for Human IgG Subclasses. Blood 2009, 113, 3716–3725. [Google Scholar] [CrossRef]
- Schneider-Merck, T.; van Bueren, J.J.L.; Berger, S.; Rossen, K.; van Berkel, P.H.C.; Derer, S.; Beyer, T.; Lohse, S.; Bleeker, W.K.; Peipp, M.; et al. Human IgG2 Antibodies against Epidermal Growth Factor Receptor Effectively Trigger Antibody-Dependent Cellular Cytotoxicity but, in Contrast to IgG1, Only by Cells of Myeloid Lineage. J. Immunol. 2010, 184, 512–520. [Google Scholar] [CrossRef]
- Ramello, M.C.; Haura, E.B.; Abate-Daga, D. CAR-T Cells and Combination Therapies: What’s next in the Immunotherapy Revolution? Pharm. Res. 2018, 129, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, L.M.; Nassin, M.L.; Hadji, A.; LaBelle, J.L. Killing Two Cells with One Stone: Pharmacologic BCL-2 Family Targeting for Cancer Cell Death and Immune Modulation. Front. Pediatrics 2016, 4, 135. [Google Scholar] [CrossRef] [PubMed]
- Dinh, T.N.; Onea, A.S.; Jazirehi, A.R. Combination of Celecoxib (Celebrex®) and CD19 CAR-Redirected CTL Immunotherapy for the Treatment of B-Cell Non-Hodgkin’s Lymphomas. Am. J. Clin. Exp. Immunol. 2017, 6, 27–42. [Google Scholar]
- Joshi, S.; Durden, D.L. Combinatorial Approach to Improve Cancer Immunotherapy: Rational Drug Design Strategy to Simultaneously Hit Multiple Targets to Kill Tumor Cells and to Activate the Immune System. J. Oncol. 2019, 2019, 1–18. [Google Scholar] [CrossRef]
- He, X.; Xu, C. Immune Checkpoint Signaling and Cancer Immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced Expression of PD-1, a Novel Member of the Immunoglobulin Gene Superfamily, upon Programmed Cell Death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 Expression in Triple-Negative Breast Cancer. Cancer Immunol. 2014, 2, 361–370. [Google Scholar] [CrossRef]
- Narayan, P.; Wahby, S.; Gao, J.J.; Amiri-Kordestani, L.; Ibrahim, A.; Bloomquist, E.; Tang, S.; Xu, Y.; Liu, J.; Fu, W.; et al. FDA Approval Summary: Atezolizumab Plus Paclitaxel Protein-Bound for the Treatment of Patients with Advanced or Metastatic TNBC Whose Tumors Express PD-L1. Clin. Cancer Res. 2020, 26, 2284–2289. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA Grants Accelerated Approval to Pembrolizumab for Locally Recurrent Unresectable or Metastatic Triple Negative Breast Cancer. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-grants-accelerated-approval-pembrolizumab-locally-recurrent-unresectable-or-metastatic-triple (accessed on 13 April 2021).
- Boonstra, M.C.; Geus, S.W.L.D.; Prevoo, H.A.J.M.; Hawinkels, L.J.A.C.; Velde, C.J.H.V.D.; Kuppen, P.J.K.; Vahrmeijer, A.L.; Sier, C.F.M. Selecting Targets for Tumor Imaging: An Overview of Cancer-Associated Membrane Proteins. Biomark. Cancer 2016, 8, BIC.S38542. [Google Scholar] [CrossRef]
- Yu, B.; Tai, H.C.; Xue, W.; Lee, L.J.; Lee, R.J. Receptor-Targeted Nanocarriers for Therapeutic Delivery to Cancer. Mol. Membr. Biol. 2010, 27, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Park, K.-S.; Bandeira, E.; Shelke, G.V.; Lässer, C.; Lötvall, J. Enhancement of Therapeutic Potential of Mesenchymal Stem Cell-Derived Extracellular Vesicles. Stem Cell Res. 2019, 10, 288. [Google Scholar] [CrossRef]
- Yeo, R.W.Y.; Lai, R.C.; Zhang, B.; Tan, S.S.; Yin, Y.; Teh, B.J.; Lim, S.K. Mesenchymal Stem Cell: An Efficient Mass Producer of Exosomes for Drug Delivery. Adv. Drug Deliv. Rev. 2013, 65, 336–341. [Google Scholar] [CrossRef]
- Jain, A.; Singh, S.K.; Arya, S.K.; Kundu, S.C.; Kapoor, S. Protein Nanoparticles: Promising Platforms for Drug Delivery Applications. Acs Biomater. Sci. Eng 2018, 4, 3939–3961. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Choi, D.W.; Kim, H.N.; Park, C.G.; Lee, W.; Park, H.H. Protein-Based Nanoparticles as Drug Delivery Systems. Pharmaceutics 2020, 12, 604. [Google Scholar] [CrossRef] [PubMed]
- Kianfar, E. Protein Nanoparticles in Drug Delivery: Animal Protein, Plant Proteins and Protein Cages, Albumin Nanoparticles. J. Nanobiotechnol. 2021, 19, 159. [Google Scholar] [CrossRef]
- Imai, Y.; Leung, C.K.H.; Friesen, H.G.; Shiu, R.P.C. Epidermal Growth Factor Receptors and Effect of Epidermal Growth Factor on Growth of Human Breast Cancer Cells in Long-Term Tissue Culture. Cancer Res. 1982, 42, 4394–4398. [Google Scholar] [PubMed]
- Nestor, M.; Ekberg, T.; Dring, J.; van Dongen, G.A.M.S.; Wester, K.; Tolmachev, V.; Anniko, M. Quantification of CD44v6 and EGFR Expression in Head and Neck Squamous Cell Carcinomas Using a Single-Dose Radioimmunoassay. Tumor Biol. 2007, 28, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the Potential of Antibody–Drug Conjugates for Cancer Therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef]
- Biteghe, F.A.N.; Mungra, N.; Chalomie, N.E.T.; Ndong, J.D.L.C.; Engohang-Ndong, J.; Vignaux, G.; Padayachee, E.; Naran, K.; Barth, S. Advances in Epidermal Growth Factor Receptor Specific Immunotherapy: Lessons to Be Learned from Armed Antibodies. Oncotarget 2020, 11, 3531–3557. [Google Scholar] [CrossRef]
- Large, D.E.; Soucy, J.R.; Hebert, J.; Auguste, D.T. Advances in Receptor-Mediated, Tumor-Targeted Drug Delivery. Adv. Ther. 2019, 2, 1800091. [Google Scholar] [CrossRef]
- Kunjiappan, S.; Pavadai, P.; Vellaichamy, S.; Pandian, S.R.K.; Ravishankar, V.; Palanisamy, P.; Govindaraj, S.; Srinivasan, G.; Premanand, A.; Sankaranarayanan, M.; et al. Surface Receptor-mediated Targeted Drug Delivery Systems for Enhanced Cancer Treatment: A State-of-the-art Review. Drug Dev. Res. 2021, 82, 309–340. [Google Scholar] [CrossRef]
- Shu, D.; Li, H.; Shu, Y.; Xiong, G.; Carson, W.E.; Haque, F.; Xu, R.; Guo, P. Systemic Delivery of Anti-MiRNA for Suppression of Triple Negative Breast Cancer Utilizing RNA Nanotechnology. ACS Nano 2015, 9, 9731–9740. [Google Scholar] [CrossRef]
- Camorani, S.; Crescenzi, E.; Fedele, M.; Cerchia, L. Oligonucleotide Aptamers against Tyrosine Kinase Receptors: Prospect for Anticancer Applications. Biochim. Biophys. Acta Bba Rev. Cancer 2018, 1869, 263–277. [Google Scholar] [CrossRef]
- Guo, S.; Vieweger, M.; Zhang, K.; Yin, H.; Wang, H.; Li, X.; Li, S.; Hu, S.; Sparreboom, A.; Evers, B.M.; et al. Ultra-Thermostable RNA Nanoparticles for Solubilizing and High-Yield Loading of Paclitaxel for Breast Cancer Therapy. Nat. Commun. 2020, 11, 972. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Mao, H.; Wang, Y.A.; Cao, Z.; Peng, X.; Wang, X.; Duan, H.; Ni, C.; Yuan, Q.; Adams, G.; et al. Single Chain Epidermal Growth Factor Receptor Antibody Conjugated Nanoparticles for in Vivo Tumor Targeting and Imaging. Small 2009, 5, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Master, A.M.; Gupta, A.S. EGF Receptor-Targeted Nanocarriers for Enhanced Cancer Treatment. Nanomedicine 2012, 7, 1895–1906. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Ding, Z.; Yang, X.; Zhao, X.; Zhao, M.; Gao, L.; Chen, Q.; Xie, S.; Liu, A.; Yin, S.; et al. Nanobody: A Small Antibody with Big Implications for Tumor Therapeutic Strategy. Int. J. Nanomed. 2021, 16, 2337–2356. [Google Scholar] [CrossRef]
- Tiede, C.; Bedford, R.; Heseltine, S.J.; Smith, G.; Wijetunga, I.; Ross, R.; AlQallaf, D.; Roberts, A.P.; Balls, A.; Curd, A.; et al. Affimer Proteins Are Versatile and Renewable Affinity Reagents. Elife 2017, 6, e24903. [Google Scholar] [CrossRef]
- Feiner, R.C.; Kemker, I.; Krutzke, L.; Allmendinger, E.; Mandell, D.J.; Sewald, N.; Kochanek, S.; Müller, K.M. EGFR-Binding Peptides: From Computational Design towards Tumor-Targeting of Adeno-Associated Virus Capsids. Int. J. Mol. Sci. 2020, 21, 9535. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Selby, L.I.; Johnston, A.P.R.; Such, G.K. The Endosomal Escape of Nanoparticles: Toward More Efficient Cellular Delivery. Bioconjugate Chem. 2018, 30, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Cupic, K.I.; Rennick, J.J.; Johnston, A.P.; Such, G.K. Controlling Endosomal Escape Using Nanoparticle Composition: Current Progress and Future Perspectives. Nanomedicine 2019, 14, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Khan, J.M.; Haque, S. Strategies in the Design of Endosomolytic Agents for Facilitating Endosomal Escape in Nanoparticles. Biochimie 2019, 160, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Gholam, D.; Chebib, A.; Hauteville, D.; Bralet, M.-P.; Jasmin, C. Combined Paclitaxel and Cetuximab Achieved a Major Response on the Skin Metastases of a Patient with Epidermal Growth Factor Receptor-Positive, Estrogen Receptor-Negative, Progesterone Receptor-Negative and Human Epidermal Growth Factor Receptor-2-Negative (Triple-Negative) Breast Cancer. Anti Cancer Drug 2007, 18, 835–837. [Google Scholar] [CrossRef]
- Carey, L.A.; Rugo, H.S.; Marcom, P.K.; Mayer, E.L.; Esteva, F.J.; Ma, C.X.; Liu, M.C.; Storniolo, A.M.; Rimawi, M.F.; Forero-Torres, A.; et al. TBCRC 001: Randomized Phase II Study of Cetuximab in Combination with Carboplatin in Stage IV Triple-Negative Breast Cancer. J. Clin. Oncol. 2012, 30, 2615–2623. [Google Scholar] [CrossRef]
- Baselga, J.; Gómez, P.; Greil, R.; Braga, S.; Climent, M.A.; Wardley, A.M.; Kaufman, B.; Stemmer, S.M.; Pêgo, A.; Chan, A.; et al. Randomized Phase II Study of the Anti–Epidermal Growth Factor Receptor Monoclonal Antibody Cetuximab with Cisplatin Versus Cisplatin Alone in Patients with Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2013, 31, 2586–2592. [Google Scholar] [CrossRef]
- Nabholtz, J.M.; Chalabi, N.; Radosevic-Robin, N.; Dauplat, M.M.; Mouret-Reynier, M.A.; Praagh, I.V.; Servent, V.; Jacquin, J.; Benmammar, K.E.; Kullab, S.; et al. Multicentric Neoadjuvant Pilot Phase II Study of Cetuximab Combined with Docetaxel in Operable Triple Negative Breast Cancer. Int. J. Cancer 2016, 138, 2274–2280. [Google Scholar] [CrossRef] [PubMed]
- Trédan, O.; Campone, M.; Jassem, J.; Vyzula, R.; Coudert, B.; Pacilio, C.; Prausova, J.; Hardy-Bessard, A.-C.; Arance, A.; Mukhopadhyay, P.; et al. Ixabepilone Alone or with Cetuximab as First-Line Treatment for Advanced/Metastatic Triple-Negative Breast Cancer. Clin. Breast Cancer 2015, 15, 8–15. [Google Scholar] [CrossRef]
- Crozier, J.A.; Advani, P.P.; LaPlant, B.; Hobday, T.; Jaslowski, A.J.; Moreno-Aspitia, A.; Perez, E.A. N0436 (Alliance): A Phase II Trial of Irinotecan with Cetuximab in Patients with Metastatic Breast Cancer Previously Exposed to Anthracycline and/or Taxane-Containing Therapy. Clin. Breast Cancer 2016, 16, 23–30. [Google Scholar] [CrossRef]
- Nabholtz, J.M.; Abrial, C.; Mouret-Reynier, M.A.; Dauplat, M.M.; Weber, B.; Gligorov, J.; Forest, A.M.; Tredan, O.; Vanlemmens, L.; Petit, T.; et al. Multicentric Neoadjuvant Phase II Study of Panitumumab Combined with an Anthracycline/Taxane-Based Chemotherapy in Operable Triple-Negative Breast Cancer: Identification of Biologically Defined Signatures Predicting Treatment Impact. Ann. Oncol. 2014, 25, 1570–1577. [Google Scholar] [CrossRef] [PubMed]
- Cowherd, S.; Miller, L.D.; Melin, S.A.; Akman, S.; Isom, S.; Cole, J.; Pullikuth, A.; Lawrence, J.A. A Phase II Clinical Trial of Weekly Paclitaxel and Carboplatin in Combination with Panitumumab in Metastatic Triple Negative Breast Cancer. Cancer Biol. 2015, 16, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Patel, T.; Dreisbach, L.; McCleod, M.; Heim, W.J.; Hermann, R.C.; Paschold, E.; Iannotti, N.O.; Dakhil, S.; Gorton, S.; et al. Cetuximab and First-Line Taxane/Carboplatin Chemotherapy in Advanced Non–Small-Cell Lung Cancer: Results of the Randomized Multicenter Phase III Trial BMS099. J. Clin. Oncol. 2010, 28, 911–917. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J.A. Highlights in Metastatic Breast Cancer from the 2013 San Antonio Breast Cancer Symposium (SABCS). Clin. Adv. Hematol. Oncol. 2014, 12, 3–17. [Google Scholar]
- Pirker, R.; Pereira, J.R.; Szczesna, A.; von Pawel, J.; Krzakowski, M.; Ramlau, R.; Vynnychenko, I.; Park, K.; Yu, C.-T.; Ganul, V.; et al. Cetuximab plus Chemotherapy in Patients with Advanced Non-Small-Cell Lung Cancer (FLEX): An Open-Label Randomised Phase III Trial. Lancet 2009, 373, 1525–1531. [Google Scholar] [CrossRef]
- Matsuda, N.; Wang, X.; Lim, B.; Krishnamurthy, S.; Alvarez, R.H.; Willey, J.S.; Parker, C.A.; Song, J.; Shen, Y.; Hu, J.; et al. Safety and Efficacy of Panitumumab Plus Neoadjuvant Chemotherapy in Patients with Primary HER2-Negative Inflammatory Breast Cancer. JAMA Oncol. 2018, 4, 1207. [Google Scholar] [CrossRef] [PubMed]
- Yardley, D.A.; Ward, P.J.; Daniel, B.R.; Eakle, J.F.; Lamar, R.E.; Lane, C.M.; Hainsworth, J.D. Panitumumab, Gemcitabine, and Carboplatin as Treatment for Women with Metastatic Triple-Negative Breast Cancer: A Sarah Cannon Research Institute Phase II Trial. Clin. Breast Cancer 2016, 16, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Bernsdorf, M.; Ingvar, C.; Jörgensen, L.; Tuxen, M.K.; Jakobsen, E.H.; Saetersdal, A.; Kimper-Karl, M.L.; Kroman, N.; Balslev, E.; Ejlertsen, B. Effect of Adding Gefitinib to Neoadjuvant Chemotherapy in Estrogen Receptor Negative Early Breast Cancer in a Randomized Phase II Trial. Breast Cancer Res. Tr 2011, 126, 463–470. [Google Scholar] [CrossRef]
- Stringer-Reasor, E.M.; May, J.E.; Olariu, E.; Caterinicchia, V.; Li, Y.; Chen, D.; Manna, D.L.D.; Rocque, G.B.; Vaklavas, C.; Falkson, C.I.; et al. An Open-Label, Pilot Study of Veliparib and Lapatinib in Patients with Metastatic, Triple-Negative Breast Cancer. Breast Cancer Res. 2021, 23, 30. [Google Scholar] [CrossRef] [PubMed]
- Fenn, K.; Maurer, M.; Lee, S.M.; Crew, K.D.; Trivedi, M.S.; Accordino, M.K.; Hershman, D.L.; Kalinsky, K. Phase I Study of Erlotinib and Metformin in Metastatic Triple Negative Breast Cancer. Clin. Breast Cancer 2019, 20, 80–86. [Google Scholar] [CrossRef]


| Year | EGFRi | Combination Drug | TNBC Cells | Molecules Affected by Combination | Combination Effects | Ref | ||
|---|---|---|---|---|---|---|---|---|
| Name | Known Target (IC50, nM) | Level | Effects | |||||
| 2004 | Gefitinib | Cetuximab | EGFR | MDA-MB-468 | - | Cell | - Reduced cell proliferation | [364] |
| 2008 | Cetuximab | Cisplatin | DNA | MDA-MB-468 | Down: EGFR Up: BRCA1 | Cell | - Synergistically induced apoptotic cell death - Depleted EGFR protein | [365] |
| 2009 | Gefitinib | Carboplatin + Docetaxel | DNA Microtubule | BT20 HCC9137 | - | Cell | - Reduced cell viability - Induced G2/M cell cycle arrest | [366] |
| 2011 | Erlotinib | Gemcitabine | Antimetabolite | BT-549 MBA-MB-231 MDA-MB-468 | - | Cell | - Synergistically reduced cell viability | [11] |
| 2011 | Lapatinib | Sirolimus (Rapamycin) | mTOR (~0.1) [367] | MDA-MB-231 MDA-MB-468 | Down: p-AKT (S473), p-EGFR (Y1173), p-ERK1/2 (T202/Y204), p-S6 (S235/236) | Cell | - Reduced survival of MDA-MB-231 and MDA-MB-468 but only induced apoptosis of MDA-MB-468 with a concomitant increase of cleaved caspase 3 - Differentially affected p-eIF4E (S209) levels in MDA-MB-468 (down) versus in MDA-MB-231 (up) | [368] |
| MDA-MB-231 MDA-MB-468 | Down: Ki67 Up: Caspase-3 | Xeno 1 | - Inhibited growth of xenograft tumors of both cells with decreased Ki67 in both tumors and increased apoptosis in MDA-MB-468 tumor | |||||
| 2011 | Cetuximab | IL-2 or IL-15 | NK cells | IIB-BR-G IIB-BR-G MT | Up: IFN-γ from NK cells in a co-culture | Cell | - Increased ADCC by NK cells | [369] |
| 2012 | Cetuximab | IL-2 or IL-15 | NK cells | IIB-BR-G IIB-BR-G MT | Up: CD107a in NK cells in a co-culture | Cell Ex vivo Xeno | - Increased ADCC by NK cells - Reduced tumor volume in xenografts with an increase in the number of infiltrating NK cells in the tumors - No effect on tumor proliferation in xenografts | [370] |
| 2012 | Lapatinib | Veliparib (NSC 737664, ABT-888) | PARP2 (2.9), PARP1 (5.2) [371] | MDA-MB-231 MDA-MB-453 MDA-MB-468 | Down: nuclear BRCA1, nuclear EGFR Up: cleaved caspase-3, cleaved caspase-9, γ-H2AX | Cell | - Reduced colony formation - Induced apoptosis | [372] |
| MDA-MB-231 | - | Xeno | - Reduced tumor growth | |||||
| 2012 | Gefitinib | CAT-SKL 2 | - | MDA-MB-468 SUM149PT | Down: p-EGFR (Y1068) | Cell | - Reduced cell proliferation | [373] |
| 2013 | Cetuximab | Dasatinib + Cisplatin | Dasatinib: ABL1 (0.6), SRC (0.8) [374]; KITD816V (37), KIT (79) [375] | BT20 BT549 MDA-MB-231 SUM102PT SUM149PT SUM229PE | Down: p-AKT (S473), p-EGFR (Y845), p-EGFR (Y1068), p-EGFR (Y1173), p-ERK (T202/Y204) Up: cleaved caspase-9 | Cell | - Induced apoptosis - Reduced cell growth and migration | [376] |
| 2013 | Gefitinib | PI-103 | DNA-PK (2), PI3Kα (8), mTORC1 (20), PI3Kδ (48), mTORC2 (83), PI3Kβ (88), PI3Kγ (150) [377] | MDA-MB-468SUM149PT | Down: p-AKT (S473), BCL2, p-ERK (Y204.Y187), MCL1, XIAP Up: caspase-3/7 | Cell | - Reduced cell viability and induced apoptosis in the BL subtype (MDA-MB-468 and SUM149PT) but not in the MSL subtype (HS578T and MDA-MB-231) | [378] |
| 2013 | Lapatinib | Bortezomib (Velcade®, PS-341, LDP-341, LM341) | 20S proteasome (Ki = 0.6) [379] | HS578T MDA-MB-231 | Down: BCL2 Up: BAX, Cleaved Caspase-3, Cleaved PARP | Cell | - Reduced colony formation | [380] |
| MDA-MB-231 | Down: P65 Up: BAX | Xeno | - Reduced tumor growth | |||||
| 2013 | Panitumumab | mAb111 | EGFR | BT549 | Down: EGFR | Cell | - Reduced in vitro invasion of BT549- Reduced colony formation of HCC70 | [381] |
| HCC70 | - | Xeno | - Reduced tumor volume | |||||
| 2014 | Erlotinib | PHA-665752 | MET (9), RON (68), FLK1 (200) [382] | BT20 CRC029 MDA-MB-468 | - | Cell | - Reduced cell viability | [150] |
| 2014 | Gefitinib | Tepotinib (Tepmetko®, EMD1214063) | MET (4) [383] | MDA-MB-468 | Down: p-AKT (T308), p-ERK (T202/Y204), p-RPS6 (S235/236) | Cell | - Reduced growth and colony formation | [153] |
| 2014 | Duligotuzumab (MEHD7945A) | Ipatasertib (GDC-0068, RG7440) | AKT1 (5), AKT3 (8), AKT2 (18) [384] | HCC70 MDA-MB-468 | Down: p-EGFR (Y1068), p-ERK (T202/Y204), p-HER3 (Y1289), p-RPS6 (S240/244)Up: p-AKT (T308) | Cell | - Reduced cell proliferation | [159] |
| Pictilisib (GDC-0941, RG7321) | PI3Kα (3), PI3Kδ (3), PI3Kβ (33), PI3Kγ (75), mTOR (Ki = 0.58 μM) [385] | Down: p-AKT (S473), p-AKT (T308), p-EGFR (Y1068), p-ERK (T202/Y204), p-HER3 (Y1289), p-RPS6 (S240/244) | Xeno PDX | - Reduced tumor growth of HCC70 and PDX - Reduced Ki67 index in xenograft tumor | ||||
| 2014 | Gefitinib | Temsirolimus | mTOR (1760) [386] | BT20 MDA-MB-468 MDA-MB-231 | Down: p-eIF4B (S422) | Cell | - Reduced cell growth, colony formation, and cell viability - Reduced cap-dependent translation | [387] |
| 2014 | Gefitinib | SKI II (SphK-I2) | SPHK (500) [388] | HS578T MDA-MB-231 MDA-MB-436 MDA-MB-468 | - | Cell | - Reduced cell proliferation | [389] |
| Xeno | - Reduced the growth of MDA-BM-468 xenograft tumor | |||||||
| 2015 | Gefitinib | SU11274 | MET (10) [390] | HS578T MDA-MB-231 | Down: p-AKT (S473), RPS6, p-RPS6 (S235/236) | Cell | - Reduced cell viability and colony formation - No synergistic effect on cell cycle distribution | [15] |
| 2015 | Gefitinib | Selumetinib (AZD6244) | MEK1 (14) [391], MEK2 (KD = 530) [392] | HCC70 MDA-MB-231 MDA-MB-468 SUM149PT SUM159PT | Down: p-AKT (S473), p-ERK (T202/Y204) | Cell | - Induced G0/G1 cell cycle arrest and apoptosis in SUM149PT | [393] |
| 2015 | Gefitinib | Brefeldin A | ARF purified from bovine brain (2 μM) [394], ARF1 (10 μM) [395] | MDA-MB-231 | Down: p-AKT, EGFR, p-ERK1/2, HER2, p-SRC | Cell | - Induced cell death | [288] |
| 2015 | Lapatinib | Imatinib (Gleevec®) | PDGFR (100), KIT (100), ABL1 (600) [396] | HCC1806 MDA-MB-231 SUM159PT | Down: HOTAIR, β-catenin, MYC | Cell | - Reduced cell viability | [397] |
| MDA-MB-231 | - | Xeno | - Reduced tumor growth | |||||
| 2015 | Gefitinib | Ad-wtp53 | - | MDA-MB-468 | Down: p-AKT (S473) Up: cleaved caspase-3, caspase-9 | Cell | - Inhibited cell growth and colony formation - Induced apoptosis and G2/M arrest | [398] |
| Xeno | - Reduced tumor growth | |||||||
| 2016 | Erlotinib | Glesatinib (MGCD265) | DDR2 (1.9), AXL (5.3), MERTK (7.8), PDGFRα (14), VEGFR3 (FLT4) (23), FLT3 (31), MET (46), VEGFR2 (KDR) (66), PDGFRβ (69) [399] | Primary TNBC | Down: p-ERK1/2 (T202/Y204), p-RPS6 (S240/244) | PDX 3 | - Reduced tumor growth | [400] |
| Crizotinib (PF-02341066) | MET (11) [401], ROS1 (Ki < 0.025), ALK (Ki < 0.07), LTK (2.7), FER (3.3), FES (6), PTK2B (FAK2) (14), TNK2 (17), PTK2 (FAK) (17), TRKB (23), TRKA (24), TRKC (46), FRK (PTK5) (53), EGFRL858R/T790M (53), EGFRT790M (56) [402] | |||||||
| 2016 | Gefitinib or Erlotinib | Cetuximab or Panitumumab | EGFR | MDA-MB-468 SUM1315MO2 | - | Cell | - Induced G1 cell cycle arrest | [403] |
| 2016 | Gefitinib | GSK269962A | ROCK1 (1.6), ROCK2 (4), MSK1 (49), RSK1 (132) [404] | HCC1806 | - | Xeno | - Reduced tumor growth | [405] |
| MDA-MB-231 | Down: CDK2, Cyclin A, p27, p-RB (S807/811) | Cell | - Inhibited colony formation - Reduced S phase and increased G2 cell cycle arrest | |||||
| 2016 | Cetuximab | Ixabepilone | Microtubule | SUM159PT | Down: LC3B, p62 | Cell | - Reduced mammosphere formation efficiency | [406] |
| - | Xeno | - Reduced tumor growth | ||||||
| 2017 | Cetuximab or panitumumab | Cisplatin or epirubicin | SUM1315MO2 | - | Cell | - Induced G1 cell cycle arrest compared to a DNA-damaging agent alone that induced G2 cell cycle arrest | [407] | |
| 2017 | Lapatinib | JNK-IN-8 (JNK Inhibitor XVI) | JNK3 (1), JNK1 (4.7), JNK2 (18.7), KITV559D/T670I (56), KITV559D (92) [408] | HCC1569 MDA-MB-231 MDA-MB-436 | - | Cell | - Induced apoptosis | [231] |
| MDA-MB-231 | - | Xeno | - Reduced tumor growth | |||||
| 2017 | Erlotinib or gefitinib | PF573228 | FAK1 (4) [409] | BT20 HCC38 HCC1143 HCC1937 MDA-MB-468 | Down: p-AKT (S473), p-AKT (T308), HER3, p-S6K (T389), p-STAT3 (Y705) | Cell | - Reduced cell proliferation and colony formation | [410] |
| PF431396 | FAK1 (2), PYK2 (11) [411] | MDA-MB-468 | - | Xeno | - Reduced tumor growth | |||
| 2017 | Gefitinib | Fingolimod (Gilenya®, FTY720) | S1PR (0.033) [412] | HCC1806 MDA-MB-468 | Down: CD44, IGFBP3 | Cell | - Reduced cell proliferation | [413] |
| HCC1806 MDA-MB-468 | Down: p-EGFR (Y1068), Ki67 Up: cleaved caspase-3 | Xeno | - Inhibited tumor growth and increased mouse survival | |||||
| 4T1 | Syn 4 | |||||||
| 2017 | Gefitinib | 3-methyladenine (NSC 66389) | PIK3C3 (25 μM), PI3Kγ (60 μM) [414] | MDA-MB-231 MDA-MB0465 | Down: BCL2 Up: p-ATM (S1981), BAX, cleaved caspase-3, p-CHK1 (S345), p-CHK2 (T68), Cytochrome C, γ-H2AX | Cell | - Reduced cell viability and colony formation - Induced G0/G1 cell cycle arrest - Induced DNA damage - Promoted mitochondria-dependent apoptosis | [415] |
| Bafilomycin A1 | H+-ATPase (0.44) [416] | MDA-MB-468 | Up: cleaved caspase-3 | Xeno | - Reduced tumor growth | |||
| 2017 | Erlotinib | CAT-SKL | - | HCC70 MDA-MB-468 | - | Cell | - Reduced cell viability | [417] |
| (−)-epicatechin | ||||||||
| 2018 | Cetuximab | YM-1 | BAG3-HSP70 interaction (4.8 μM) [418] | BT549 MDA-MB-468 | - | Cell | - Reduced cell viability | [419] |
| 2018 | Gefitinib | MK-2206 | AKT1 (5), AKT2 (12), AKT3 (65) [420] | HS578T MDA-MB-231 | Down: p-mTOR (S2448), RPS6, p-RPS6 (S235/236), p-RPAS40 (T246), XIAP | Cell | - Reduced cell viability, proliferation, and colony formation - No synergistic effect on cell cycle distribution | [421] |
| 2018 | Lapatinib | Foretinib (GSK1363089, XL880) | MET (0.4), VEGFR2/KDR (0.86), TIE2 (1.1), VEGFR3/FLT4 (2.8), RON (3), FLT3 (3.6), PDGFRα (3.6), KIT (3.6), VEGFR1/FLT1 (6.8), PDGFRβ (9.6) [422] | BT549 MDA-MB-231 | Down: p-AKT (S473) | Cell | - Reduced cell viability and migration | [423] |
| 2018 | Gefitinib | Tepotinib (Tepmetko®, EMD1214063) | MET (4) [383] | MDA-MB-468 | Down: p-ERK1/2 (T202/Y204), p-STAT3 (Y705) | Cell | - Reduced cell viability and colony formation | [424] |
| PF431396 | FAK1 (2), PYK2 (11) [411] | |||||||
| 2019 | Erlotinib or Gefitinib or Lapatinib | PHA-767491 | CDC7 (10), CDK9 (34), GSK3β (220), CDK2 (240), CDK1 (250), CDK5 (460), MAPKAPK2/MK2 (470), PLK1 (980) [425] | BT549 HS578T SKBR7 | Down: p-AKT (S473), CDC7, CDK4, Cyclin D1, p-EGFR (T1173), pERK1/2 (T202/Y204), MCM2, p-MCM2 (S40/41), RB, p-RB (S780), POLR2A, p-POLR2A (S2/5), | Cell | - Reduced cell proliferation - Inhibited DNA replication and CDK9-mediated transcriptional elongation - Increased apoptosis and G2/M cell cycle arrest | [99] |
| 2019 | Osimertinib | Chloroquine (CHQ, Aralen) | - | MDA-MB-231 | Up: p-BAD (S112), caspase-3, LC3B-II | Cell | - Reduced cell viability | [426] |
| 2019 | Erlotinib | Palbociclib (Ibrance®, PD-0332991, LQQ) | CDK4/Cyclin D3 (9), CDK4/Cyclin D1 (11), CDK6/Cyclin D1 (15) [427] | HS578T-MMP17 5 MDA-MB-231-MMP17 MDA-MB-468-MMP17 | Down: p-EGFR (Y1068), p-RB (S807/811) | Cell | - Reduced cell proliferation | [428] |
| MDA-MB-231-MMP17PDX | Down: Ki67, p-RB (S807/811) | Xeno | - Reduced tumor growth - Reduced MMP17 and p-RB | |||||
| 2019 | AEE788 | Everolimus (RAD001) | mTOR (1.6–2.4) [429] | SUM149PT | Down: CDK4, Cyclin B1, Cyclin D1, p-mTOR (S2448) | Cell | - Reduced cell proliferation - Induced apoptosis | [430] |
| Sirolimus (Rapamycin) | mTOR (~0.1) [367] | |||||||
| Temsirolimus | mTOR (1760) [386] | |||||||
| 2019 | Cetuximab | PP2 (AG 1879, AGL 1879) | LCK (4), FYN (5), HCK (5), SRC (100) [431,432] | MDA-MB-231 MDA-MB-468 | Down: p-AKT (S473), p-EGFR (Y1173), p-IGF1R (Y1161), p-PI3K, p-SRC (Y416), p-VEGFR2 (Y1175) | Cell | - Reduced cell proliferation | [142] |
| 2020 | ABT-414 | Navitoclax (ABT-263) | BCL-xL (Ki ≤ 0.5), BCL2 (Ki ≤ 1), BCL2L2 (Ki ≤ 1) [433] | - | - | PDX | - Reduced PDX tumor growth in vivo | [258] |
| ABBV-321 | ||||||||
| 2020 | Erlotinib | S63845 | MCL1 (Kd = 0.19) [434] | BT20, H38, H1806, H1937, MDA-MB-231, MDA-MB-468, | - | Cell | - Reduced cell viability | [260] |
| 2020 | Gefitinib | Crizotinib + Talazoparib | Crizotinib: see above Talazoparib: PARP1 (0.57) [435] | PARPi-resistant SUM149PT | - | Cell | - Reduced cell viability | [436] |
| 2020 | Erlotinib | THZ1 | CDK7 (3.2) [437] | MDA-MB-231 | - | Cell | - Reduced cell proliferation | [438] |
| 2020 | Cetuximab | IL-15 | NK cells | IIB-BR-G | Up: CD25 and CD69 in NK cells; CD83 and CD86 in DCs; IFN-γ and TNF-α in co-culture supernatant | Cell | - Activated NK cell tumor killing - Stimulated maturation of DCs | [439] |
| 2020 | Cetuximab | Dasatinib | ABL1 (0.6), SRC (0.8) [374] KITD816V (37), KIT (79) [375] | MDA-MB-468 | Down: SRC, p-SRC (Y416) | Xeno | - Reduced tumor volume | [440] |
| 2020 | Gefitinib | GSK269962A | ROCK1 (1.6), ROCK2 (4), MSK1 (49), RSK1 (132) [404] | MDA-MB-231 | Down: p-RPS6 (S235/236) Up: p-AMPK (T172) | Cell | - Increased accumulation of autophagic vacuoles | [339] |
| CAL120HCC1806HS578T | Down: p-RPS6 (S235/236) Up: LC3-II | |||||||
| 2020 | Afatinib | Dasatinib | ABL1 (0.6), SRC (0.8) [374] KITD816V (37), KIT (79) [375] | BT20 HCC1937 HDQP1 | Down: p-EGFR (Y1068), p-SFKs (Y416), p-SRC (Y527)Up: cleaved caspase-7, p27 | Cell | - Reduced cell proliferation- Induced G1 cell cycle arrest | [441] |
| HCC1806 | Down: CDC42, p-EGFR (Y1068) | Xeno | - Reduced tumor growth | |||||
| 2020 | Gefitinib | Everolimus (RAD001) | mTOR (1.6–2.4) [429] | CAL-51 | Down: p-4E-BP1 (T37/46), Cyclin B1, Cyclin E1, p-mTOR (S2448) | Cell | - Reduced cell proliferation - Induced G1 cell cycle arrest and apoptosis | [442] |
| 2020 | Afatinib | Dasatinib + Trametinib (Mekinist®, JTP-74057, GSK1120212) | Dasatinib: see above Trametinib: MEK1 (0.92), MEK2 (1.8) [443] | BT20 MDA-MB-468 | - | Cell | - Reduced cell proliferation | [444] |
| 2020 | Bispecific antibody for EGFR (cetuximab) and HER3 (IgG 3-43) | EGFR (KD = 21), HER3 (KD = 19) [445] | MDA-MB-468 | Down: p-AKT (T308), p-EGFR (Y1068), p-ERK (T202/Y204), p-HER3 (Y1289) in FaDu, the hypopharyngeal carcinoma cell line | Cell | - Reduced cell proliferation and mammosphere formation | [445] | |
| Xeno | - Reduced tumor growth with concordant reduction of CSCs | |||||||
| 2020 | Pan-HER antibody mixture (combination of 3 sets of 2 antibodies each targeting non-overlapping epitopes of EGFR, HER2, and HER3) | - | 15 PDXs | Down: p-AKT (T308), EGFR, p-EGFR (Y1068), p-ERK1/2, p-FAK (Y397), HER3, p-HER3 (Y1289) | PDX | - Reduced tumor growth | [446] | |
| 2020 | Cetuximab or Panitumumab | CD32A-CR T cells | CD32A131R | MDA-MB-468 | Up: IFN-γ and TNF-α from CD32A131R-CR T cells | Cell | - Induced cancer cell killing | [447] |
| 2020 | Afatinib | YM155 (Sepantronium bromide) | BIRC56 gene expression (0.54) [448] | - | - | PDX | - Reduced PDX mammary tumor growth in vivo | [449] |
| 2020 | Cetuximab | Icaritin | - | MDA-MB-231 MDA-MB-436 | Cell | - Induced apoptosis- Reduced cell proliferation | [450] | |
| 2020 | Gefitinib, Erlotinib, or Lapatinib | MK-2206 | AKT1 (5), AKT2 (12), AKT3 (65) [420] | MDA-MB-468 | Down: p-AKT (S473), p-AKT (T308), p-EGFR (Y1068), p-ERK (T202/Y204), p-PRAS40 (T246), p-RPS6 (S235/236) | Cell | - Reduced cell proliferation (by gefitinib or lapatinib in the presence of MK-2206, AZD8186, or pictilisib) | [451] |
| AZD8186 | PI3Kβ (4), PI3Kδ (12), PI3Kα (35) [452] | Xeno | - Reduced tumor growth of HCC70 or MDA-MB-468 xenografts (by Erlotinib + AZD8186) | |||||
| Pictilisib (GDC-0941, RG7321) | PI3Kα (3), PI3Kδ (3), PI3Kβ (33), PI3Kγ (75), mTOR (Ki = 0.58 μM) [385] | |||||||
| 2021 | Cetuximab | miR-155-5p antagomir | miR-155-5p | MDA-MB-468 | Down: BCL2, p-EGFR Up: BAX, cleaved caspase-1, cleaved caspase-3, GSDME, IL-1β | Cell | - Reduced cell proliferation - Increased apoptosis and induced pyroptosis- Reduced migration and invasion | [453] |
| MDA-MB-468 | Down: p-EGFR, Ki67 Up: cleaved caspase-1, GSDME | Xeno | - Reduced tumor growth - Increased TUNEL-positive cells | |||||
| 2021 | Gefitinib | AT7867 + PD-0325901 (Mirdametinib) | AT7867: AKT2 (17), PKA (20), AKT1 (32), AKT3 (47), p70 S6K (85) [454] PD-0325901: MEK (0.33) [455] | HS578T MDA-MB-231 | Down: ERK1/2, GSK3β, p-GSK3β (S9), RPS6, p-RPS6 (S235/236), XIAP Up: cleaved caspase-3 | Cell | - Reduced cell proliferation and colony formation- Induced apoptotic cell death | [22] |
| Drug | Primary Target | Initial Year Approval | Company | Approved Indications 1 | EGFRi Tested |
|---|---|---|---|---|---|
| Crizotinib | ALK, ROS1, MET | 2011 | Pfizer | ∙ ALK -positive advanced NSCLC ∙ ROS1-positive NSCLC ∙ ALK-positive anaplastic large cell lymphoma | Erlotinib [400] |
| Dasatinib | BCR-ABL, SRC | 2006 | Bristol-Myers Squibb | ∙ CML ∙ Philadelphia chromosome (Ph)-positive ALL ∙ Ph-positive CML | Cetuximab [376,440] |
| Everolimus | mTOR | 2009 | Novartis | ∙ HER2-negative breast cancer, pancreatic neuroendocrine tumors, renal cell carcinoma, angiomyolipoma, subependymal giant cell astrocytoma | AEE788 [430] Gefitinib [442] |
| Imatinib | BCR-ABL | 2002 | Novartis | ∙ Rare gastrointestinal cancer ∙ ALL | Lapatinib [397] |
| Palbociclib | CDK4/6 | 2015 | Park Davis | ∙ ER- and HER2-positive breast cancer | Erlotinib [428] |
| Sirolimus | mTOR | 1999 | Wyeth | ∙ Kidney transplant, lymphangioleiomyomatosis | Lapatinib [368] |
| Selumetinib | MEK1/2 | 2020 | Astra Zeneca | ∙ Neurofibromatosis type 1 plexiform neurofibromas | Gefitinib [393] |
| Temsirolimus | mTOR | 2007 | Wyeth | ∙ Advanced kidney cancer | Gefitinib [387] |
| Tepotinib | MET | 2021 | EMD Serono | ∙ Metastatic NSCLC with METex14 skipping alterations | Gefitinib [153,424] |
| Trametinib | MEK1/2 | 2013 | GlaxoSmithKline | ∙ Melanoma | Afatinib (with Dasatinib) [444] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, K.S.; Yi, Y.W.; Cho, J.; Park, J.-S.; Seong, Y.-S. Potentiating Therapeutic Effects of Epidermal Growth Factor Receptor Inhibition in Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 589. https://doi.org/10.3390/ph14060589
You KS, Yi YW, Cho J, Park J-S, Seong Y-S. Potentiating Therapeutic Effects of Epidermal Growth Factor Receptor Inhibition in Triple-Negative Breast Cancer. Pharmaceuticals. 2021; 14(6):589. https://doi.org/10.3390/ph14060589
Chicago/Turabian StyleYou, Kyu Sic, Yong Weon Yi, Jeonghee Cho, Jeong-Soo Park, and Yeon-Sun Seong. 2021. "Potentiating Therapeutic Effects of Epidermal Growth Factor Receptor Inhibition in Triple-Negative Breast Cancer" Pharmaceuticals 14, no. 6: 589. https://doi.org/10.3390/ph14060589
APA StyleYou, K. S., Yi, Y. W., Cho, J., Park, J.-S., & Seong, Y.-S. (2021). Potentiating Therapeutic Effects of Epidermal Growth Factor Receptor Inhibition in Triple-Negative Breast Cancer. Pharmaceuticals, 14(6), 589. https://doi.org/10.3390/ph14060589