
The Roles of DNA Demethylases in Triple-Negative Breast Cancer


Abstract
:1. Triple-Negative Breast Cancer
2. Epigenetics and Breast Cancer
2.1. DNA Methyltransferases
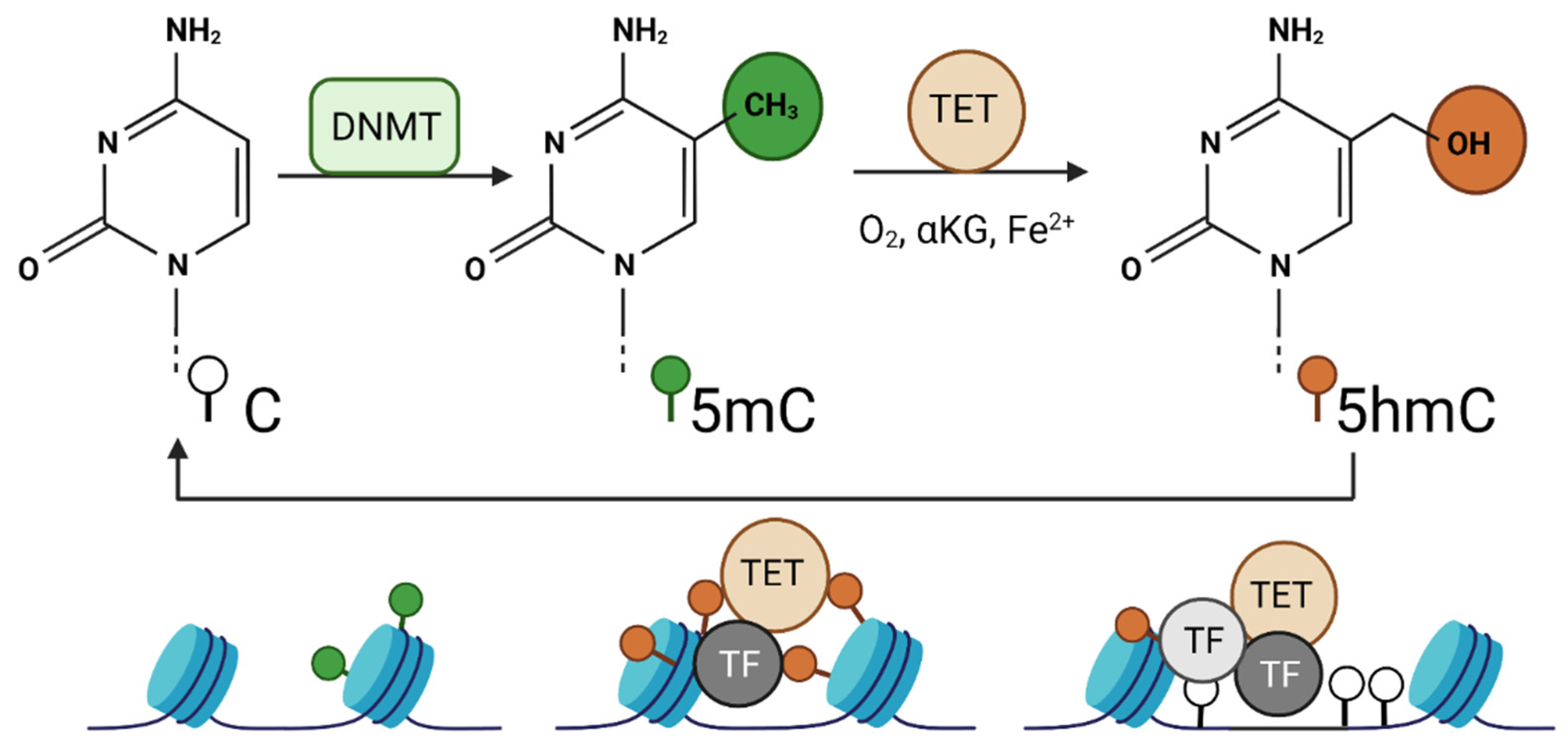
2.2. DNA Demethylases
2.3. DNA Methylation in TNBC
3. Are TET Enzymes Tumor-Suppressors or Oncogenes?
3.1. Tumor Suppressor Activity of TETs
3.2. Oncogenic Activity of TETs
3.3. Oncogenic Activity of TET1 in TNBC
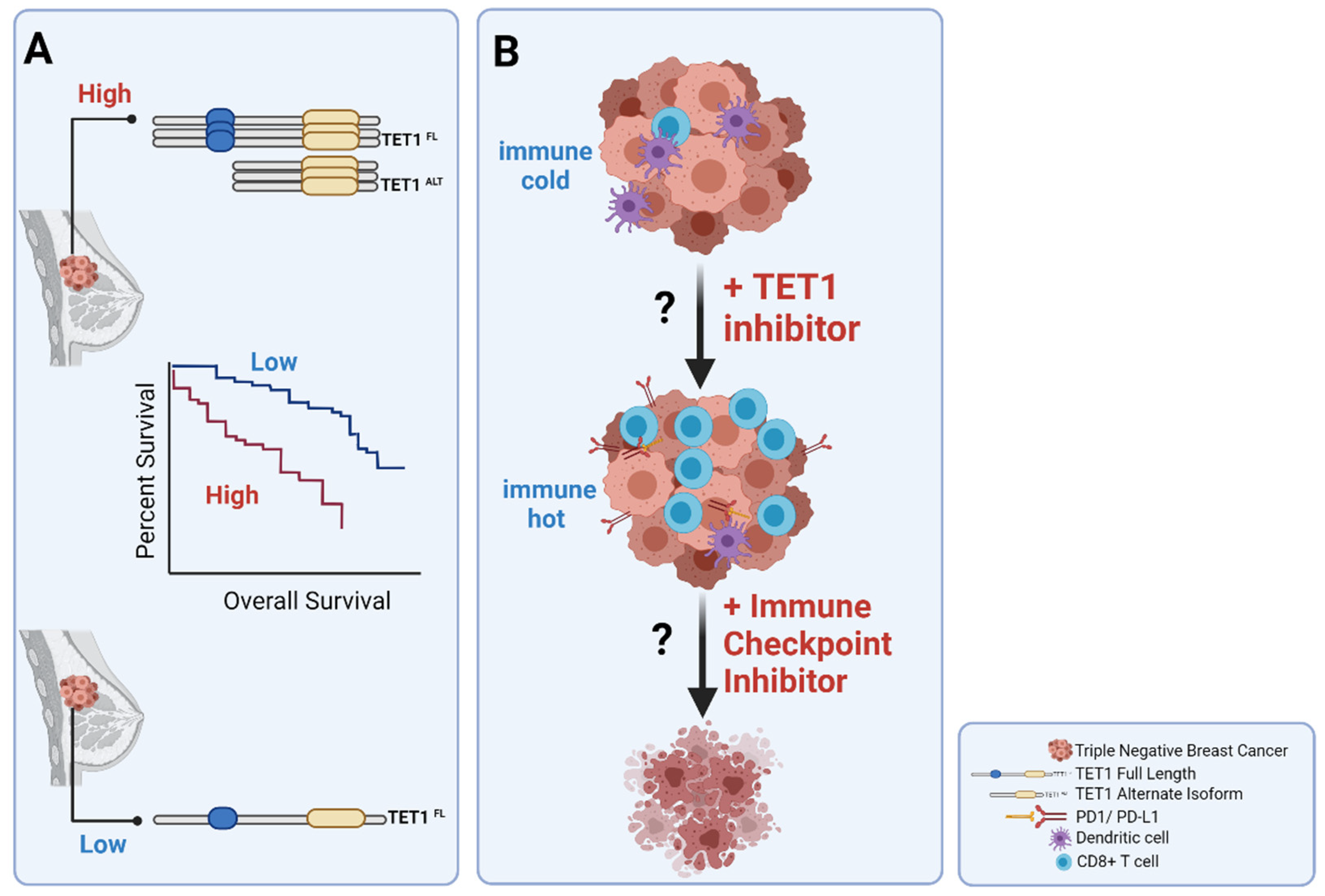
3.4. TET1ALT Is a Novel Isoform of TET1
3.5. Clinical Implications of TET1 in TNBCs
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marra, A.; Trapani, D.; Viale, G.; Criscitiello, C.; Curigliano, G. Practical classification of triple-negative breast cancer: Intratumoral heterogeneity, mechanisms of drug resistance, and novel therapies. NPJ Breast Cancer 2020, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Temian, D.C.; Pop, L.A.; Irimie, A.I.; Berindan-Neagoe, I. The Epigenetics of Triple-Negative and Basal-Like Breast Cancer: Current Knowledge. J. Breast Cancer 2018, 21, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Phillips, L.; Gill, A.J.; Baxter, R.C. Novel Prognostic Markers in Triple-Negative Breast Cancer Discovered by MALDI-Mass Spectrometry Imaging. Front. Oncol. 2019, 9, 379. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2019, 51, 411–412. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [Green Version]
- Network, C.G.A. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Stefansson, O.A.; Moran, S.; Gomez, A.; Sayols, S.; Arribas-Jorba, C.; Sandoval, J.; Hilmarsdottir, H.; Ólafsdóttir, E.; Tryggvadottir, L.; Jonasson, J.G.; et al. A DNA methylation-based definition of biologically distinct breast cancer subtypes. Mol. Oncol. 2014, 9, 555–568. [Google Scholar] [CrossRef]
- Lawrence, R.T.; Perez, E.M.; Hernández, D.; Miller, C.P.; Haas, K.M.; Irie, H.Y.; Lee, S.-I.; Blau, C.A.; Villén, J. The Proteomic Landscape of Triple-Negative Breast Cancer. Cell Rep. 2015, 11, 990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosok, M.; Alli-Shaik, A.; Bay, B.H.; Gunaratne, J. Comprehensive Proteomic Characterization Reveals Subclass-Specific Molecular Aberrations within Triple-negative Breast Cancer. iScience 2020, 23, 100868. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Piedras, I.; Vartak, D.; Sharma, M.; Pandey, S.; Casas, L.; Molehin, D.; Rasha, F.; Fokar, M.; Nichols, J.; Almodovar, S.; et al. Identification of Novel MeCP2 Cancer-Associated Target Genes and Post-Translational Modifications. Front. Oncol. 2020, 10, 576362. [Google Scholar] [CrossRef]
- Singh, M.; Bacolla, A.; Chaudhary, S.; Hunt, C.R.; Pandita, S.; Chauhan, R.; Gupta, A.; Tainer, J.A.; Pandita, T.K. Histone Acetyltransferase MOF Orchestrates Outcomes at the Crossroad of Oncogenesis, DNA Damage Response, Proliferation, and Stem Cell Development. Mol. Cell Biol. 2020, 40. [Google Scholar] [CrossRef] [PubMed]
- Berger, L.; Kolben, T.; Meister, S.; Kolben, T.M.; Schmoeckel, E.; Mayr, D.; Mahner, S.; Jeschke, U.; Ditsch, N.; Beyer, S. Expression of H3K4me3 and H3K9ac in breast cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 2017–2027. [Google Scholar] [CrossRef]
- Mollah, S.A.; Subramaniam, S. Histone Signatures Predict Therapeutic Efficacy in Breast Cancer. IEEE Open J. Eng. Med. Biol. 2020, 1, 74–82. [Google Scholar] [CrossRef]
- Gao, B.; Liu, X.; Li, Z.; Zhao, L.; Pan, Y. Overexpression of EZH2/NSD2 Histone Methyltransferase Axis Predicts Poor Prognosis and Accelerates Tumor Progression in Triple-Negative Breast Cancer. Front. Oncol. 2020, 10, 600514. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Feng, J.; Li, L.; Lin, L.; Ji, J.; Lin, C.; Liu, L.; Zhang, N.; Duan, D.; Li, Z.; et al. Arginine methylation-dependent LSD1 stability promotes invasion and metastasis of breast cancer. EMBO Rep. 2020, 21, e48597. [Google Scholar] [CrossRef]
- Søkilde, R.; Persson, H.; Ehinger, A.; Pirona, A.C.; Fernö, M.; Hegardt, C.; Larsson, C.; Loman, N.; Malmberg, M.; Rydén, L.; et al. Refinement of breast cancer molecular classification by miRNA expression profiles. BMC Genom. 2019, 20, 503. [Google Scholar] [CrossRef] [Green Version]
- Mansoori, B.; Silvestris, N.; Mohammadi, A.; Khaze, V.; Baghbani, E.; Mokhtarzadeh, A.; Shanehbandi, D.; Derakhshani, A.; Duijf, P.H.G.; Baradaran, B. miR-34a and miR-200c Have an Additive Tumor-Suppressive Effect on Breast Cancer Cells and Patient Prognosis. Genes 2021, 12, 267. [Google Scholar] [CrossRef] [PubMed]
- Koleckova, M.; Ehrmann, J.; Bouchal, J.; Janikova, M.; Brisudova, A.; Srovnal, J.; Staffova, K.; Svoboda, M.; Slaby, O.; Radova, L.; et al. Epithelial to mesenchymal transition and microRNA expression are associated with spindle and apocrine cell morphology in triple-negative breast cancer. Sci. Rep. 2021, 11, 5145. [Google Scholar] [CrossRef]
- Zhao, H.; Liu, X.; Yu, L.; Lin, S.; Zhang, C.; Xu, H.; Leng, Z.; Huang, W.; Lei, J.; Li, T.; et al. Comprehensive landscape of epigenetic-dysregulated lncRNAs reveals a profound role of enhancers in carcinogenesis in BC subtypes. Mol. Ther. Nucleic Acids 2021, 23, 667–681. [Google Scholar] [CrossRef]
- Sun, M.; Gadad, S.S.; Kim, D.S.; Kraus, W.L. Discovery, Annotation, and Functional Analysis of Long Noncoding RNAs Controlling Cell-Cycle Gene Expression and Proliferation in Breast Cancer Cells. Mol. Cell 2015, 59, 698–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.M.; Brane, A.C.; Tollefsbol, T.O. MicroRNAs and Epigenetics Strategies to Reverse Breast Cancer. Cells 2019, 8, 1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Gutierrez, A.D.; De León, D.C.; Millan-Catalan, O.; Coronel-Hernandez, J.; Campos-Parra, A.D.; Porras-Reyes, F.; Exayana-Alderete, A.; López-Camarillo, C.; Jacobo-Herrera, N.J.; Ramos-Payan, R.; et al. Identification of miRNA Master Regulators in Breast Cancer. Cells 2020, 9, 1610. [Google Scholar] [CrossRef]
- Hermann, A.; Goyal, R.; Jeltsch, A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem. 2004, 279, 48350–48359. [Google Scholar] [CrossRef] [Green Version]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [Green Version]
- Raynal, N.J.; Si, J.; Taby, R.F.; Gharibyan, V.; Ahmed, S.; Jelinek, J.; Estécio, M.R.H.; Issa, J.-P.J. DNA methylation does not stably lock gene expression but instead serves as a molecular mark for gene silencing memory. Cancer Res. 2012, 72, 1170–1181. [Google Scholar] [CrossRef] [Green Version]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Issa, J.P.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Rauscher, G.H.; Kresovich, J.K.; Poulin, M.; Yan, L.; Macias, V.; Mahmoud, A.M.; Al-Alem, U.; Kajdacsy-Balla, A.; Wiley, E.L.; Tonetti, D.; et al. Exploring DNA methylation changes in promoter, intragenic, and intergenic regions as early and late events in breast cancer formation. BMC Cancer 2015, 15, 816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef] [Green Version]
- Fang, F.; Turcan, S.; Rimner, A.; Kaufman, A.; Giri, D.; Morris, L.G.T.; Shen, R.; Seshan, V.; Mo, Q.; Heguy, A.; et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci. Transl. Med. 2011, 3, 75ra25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Issa, J.J.; Kropf, P. DNA Hypomethylating Drugs in Cancer Therapy. Cold Spring Harb. Perspect. Med. 2017, 7, a026948. [Google Scholar] [CrossRef] [Green Version]
- Issa, J.P.; Kantarjian, H.M. Targeting DNA methylation. Clin. Cancer Res. 2009, 15, 3938–3946. [Google Scholar] [CrossRef] [Green Version]
- Issa, J.-P.J.; Roboz, G.; Rizzieri, D.; Jabbour, E.; Stock, W.; O’Connell, C.; Yee, K.; Tibes, R.; Griffiths, E.A.; Walsh, K.; et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: A multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 2015, 16, 1099–1110. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.M.; Roboz, G.J.; Kropf, P.L.; Yee, K.W.L.; O’Connell, C.L.; Tibes, R.; Walsh, K.J.; Podoltsev, N.A.; Griffiths, E.A.; Jabbour, E.; et al. Guadecitabine (SGI-110) in treatment-naive patients with acute myeloid leukaemia: Phase 2 results from a multicentre, randomised, phase 1/2 trial. Lancet Oncol. 2017, 18, 1317–1326. [Google Scholar] [CrossRef]
- Roboz, G.J.; Kantarjian, H.M.; Yee, K.W.L.; Kropf, P.L.; O’Connell, C.L.; Griffiths, E.A.; Stock, W.; Daver, N.G.; Jabbour, E.; Ritchie, E.K.; et al. Dose, schedule, safety, and efficacy of guadecitabine in relapsed or refractory acute myeloid leukemia. Cancer 2018, 124, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.; Kelly, A.D.; Kropf, P.; Fung, H.; Jelinek, J.; Su, X.Y.; Roboz, G.J.; Kantarjian, H.M.; Azab, M.; Issa, J.-P.J. Genomic and epigenomic predictors of response to guadecitabine in relapsed/refractory acute myelogenous leukemia. Clin. Epigenet. 2019, 11, 106. [Google Scholar] [CrossRef]
- Yu, J.; Xie, T.; Wang, Z.; Wang, X.; Zeng, S.; Kang, Y.; Hou, T. DNA methyltransferases: Emerging targets for the discovery of inhibitors as potent anticancer drugs. Drug Discov. Today 2019, 24, 2323–2331. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loenarz, C.; Schofield, C.J. Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem. Sci. 2011, 36, 7–18. [Google Scholar] [CrossRef]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [Green Version]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [Green Version]
- Ko, M.; An, J.; Bandukwala, H.S.; Chavez, L.; Äijö, T.; Pastor, W.A.; Segal, M.F.; Li, H.; Koh, K.P.; Lähdesmäki, H.; et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature 2013, 497, 122–126. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, X.; Clark, E.; Mulcahey, M.; Huang, S.; Shi, Y.G. TET1 is a DNA-binding protein that modulates DNA methylation and gene transcription via hydroxylation of 5-methylcytosine. Cell Res. 2010, 20, 1390–1393. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Xu, C.; Kato, A.; Tempel, W.; Abreu, J.G.; Bian, C.; Hu, Y.; Hu, D.; Zhao, B.; Cerovina, T.; et al. Tet3 CXXC domain and dioxygenase activity cooperatively regulate key genes for Xenopus eye and neural development. Cell 2012, 151, 1200–1213. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Lu, Y.; Jelinek, J.; Liang, S.; Estecio, M.R.; Barton, M.C.; Issa, J.-P.J. TET1 is a maintenance DNA demethylase that prevents methylation spreading in differentiated cells. Nucleic Acids Res. 2014, 42, 6956–6971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, L.M.; Tahiliani, M.; Rao, A.; Aravind, L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle 2009, 8, 1698–1710. [Google Scholar] [CrossRef] [Green Version]
- Teif, V.B.; Beshnova, D.A.; Vainshtein, Y.; Marth, C.; Mallm, J.-P.; Höfer, T.; Rippe, K. Nucleosome repositioning links DNA (de)methylation and differential CTCF binding during stem cell development. Genome Res. 2014, 24, 1285–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulholland, C.B.; Nishiyama, A.; Ryan, J.; Nakamura, R.; Yiğit, M.; Glück, I.M.; Trummer, C.; Qin, W.; Bartoschek, M.D.; Traube, F.R.; et al. Author Correction: Recent evolution of a TET-controlled and DPPA3/STELLA-driven pathway of passive DNA demethylation in mammals. Nat. Commun. 2020, 11, 6443. [Google Scholar] [CrossRef] [PubMed]
- Neri, F.; Incarnato, D.; Krepelova, A.; Rapelli, S.; Pagnani, A.; Zecchina, R.; Parlato, C.; Oliviero, S. Genome-wide analysis identifies a functional association of Tet1 and Polycomb repressive complex 2 in mouse embryonic stem cells. Genome Biol. 2013, 14, R91. [Google Scholar] [CrossRef] [Green Version]
- Szyf, M. DNA methylation signatures for breast cancer classification and prognosis. Genome Med. 2012, 4, 26. [Google Scholar] [CrossRef]
- Stirzaker, C.; Zotenko, E.; Song, J.Z.; Qu, W.; Nair, S.S.; Locke, W.; Stone, A.; Armstong, N.J.; Robinson, M.D.; Dobrovic, A.; et al. Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value. Nat. Commun. 2015, 6, 5899. [Google Scholar] [CrossRef] [Green Version]
- Good, C.R.; Madzo, J.; Patel, B.; Maegawa, S.; Engel, N.; Jelinek, J.; Issa, J.-P.J. A novel isoform of TET1 that lacks a CXXC domain is overexpressed in cancer. Nucleic Acids Res. 2017, 45, 8269–8281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russler-Germain, D.; Spencer, D.H.; Young, M.A.; Lamprecht, T.L.; Miller, C.A.; Fulton, R.; Meyer, M.R.; Erdmann-Gilmore, P.; Townsend, R.R.; Wilson, R.K.; et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell 2014, 25, 442–454. [Google Scholar] [CrossRef] [Green Version]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2011, 44, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, J.K.; Dawlaty, M.M.; Verma, A.; Maitra, A. Roles and Regulations of TET Enzymes in Solid Tumors. Trends Cancer 2021, 7, 635–646. [Google Scholar] [CrossRef]
- Neri, F.; Dettori, D.; Incarnato, D.; Krepelova, A.; Rapelli, S.; Maldotti, M.; Parlato, C.; Paliogiannis, P.; Oliviero, S. TET1 is a tumour suppressor that inhibits colon cancer growth by derepressing inhibitors of the WNT pathway. Oncogene 2015, 34, 4168–4176. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.-H.; Peng, K.-L.; Kang, M.-L.; Chen, Y.-R.; Yang, Y.-C.; Tsai, C.-H.; Chu, C.-S.; Jeng, Y.-M.; Chen, Y.-T.; Lin, F.-M.; et al. TET1 suppresses cancer invasion by activating the tissue inhibitors of metalloproteinases. Cell Rep. 2012, 2, 568–579. [Google Scholar] [CrossRef] [Green Version]
- Nickerson, M.L.; Das, S.; Im, K.M.; Turan, S.; Berndt, S.I.; Li, H.; Lou, H.; Brodie, S.A.; Billaud, J.N.; Zhang, T.; et al. TET2 binds the androgen receptor and loss is associated with prostate cancer. Oncogene 2017, 36, 2172–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Huang, R.; Chan, M.W.; Yan, P.S.; Huang, T.; Wu, R.; Rahmanto, Y.S.; Su, P.; Weng, Y.; Chou, J.; et al. TET1 reprograms the epithelial ovarian cancer epigenome and reveals casein kinase 2α as a therapeutic target. J. Pathol. 2019, 248, 363–376. [Google Scholar] [CrossRef]
- Filipczak, P.T.; Leng, S.; Tellez, C.S.; Do, K.C.; Grimes, M.J.; Thomas, C.L.; Walton-Filipczak, S.R.; Picchi, M.A.; Belinsky, S.A. p53-Suppressed Oncogene TET1 Prevents Cellular Aging in Lung Cancer. Cancer Res. 2019, 79, 1758–1768. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Liu, Y.; Bai, F.; Zhang, J.-Y.; Ma, S.-H.; Liu, J.; Xu, Z.-D.; Zhu, H.-G.; Ling, Z.-Q.; Ye, D.; et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene 2013, 32, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Good, C.R.; Panjarian, S.; Kelly, A.D.; Madzo, J.; Patel, B.; Jelinek, J.; Issa, J.-P.J. TET1-Mediated Hypomethylation Activates Oncogenic Signaling in Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 4126–4137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Li, C.; Mao, H.; Du, Z.; Chan, W.Y.; Murray, P.; Luo, B.; Chan, A.T.C.; Mok, T.; Chan, F.K.; et al. Epigenetic inactivation of the CpG demethylase TET1 as a DNA methylation feedback loop in human cancers. Sci. Rep. 2016, 6, 26591. [Google Scholar] [CrossRef] [PubMed]
- Ke, Q.; Wang, K.; Fan, M.; Li, M.; Luo, G.; Wang, D. Prognostic role of high TET1 expression in patients with solid tumors: A meta-analysis. Medicine 2020, 99, e22863. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Qi, J.; Xiong, J.; Jiang, L.; Cui, D.; He, J.; Chen, P.; Li, L.; Wu, C.; Ma, T.; et al. Epigenetic Co-Deregulation of EZH2/TET1 is a Senescence-Countering, Actionable Vulnerability in Triple-Negative Breast Cancer. Theranostics 2019, 9, 761–777. [Google Scholar] [CrossRef]
- Yang, L.; Yu, S.J.; Hong, Q.; Yang, Y.; Shao, Z.M. Reduced Expression of TET1, TET2, TET3 and TDG mRNAs Are Associated with Poor Prognosis of Patients with Early Breast Cancer. PLoS ONE 2015, 10, e0133896. [Google Scholar] [CrossRef]
- Atlante, S.; Visintin, A.; Marini, E.; Savoia, M.; Dianzani, C.; Giorgis, M.; Sürün, D.; Maione, F.; Schnütgen, F.; Farsetti, A.; et al. α-ketoglutarate dehydrogenase inhibition counteracts breast cancer-associated lung metastasis. Cell Death Dis. 2018, 9, 756. [Google Scholar] [CrossRef] [Green Version]
- Bao, B.; Teslow, E.A.; Mitrea, C.; Boerner, J.L.; Dyson, G.; Bollig-Fischer, A. Role of TET1 and 5hmC in an Obesity-Linked Pathway Driving Cancer Stem Cells in Triple-Negative Breast Cancer. Mol. Cancer Res. 2020, 18, 1803–1814. [Google Scholar] [CrossRef]
- Teslow, E.A.; Mitrea, C.; Bao, B.; Mohammad, R.M.; Polin, L.A.; Dyson, G.; Purrington, K.S.; Bollig-Fischer, A. Obesity-induced MBD2_v2 expression promotes tumor-initiating triple-negative breast cancer stem cells. Mol. Oncol. 2019, 13, 894–908. [Google Scholar] [CrossRef]
- Wang, H.; An, X.; Yu, H.; Zhang, S.; Tang, B.; Zhang, X.; Li, Z. MiR-29b/TET1/ZEB2 signaling axis regulates metastatic properties and epithelial-mesenchymal transition in breast cancer cells. Oncotarget 2017, 8, 102119–102133. [Google Scholar] [CrossRef]
- Chen, N.; Zhao, G.; Yan, X.; Lv, Z.; Yin, H.; Zhang, S.; Song, W.; Li, X.; Li, L.; Du, Z.; et al. A novel FLI1 exonic circular RNA promotes metastasis in breast cancer by coordinately regulating TET1 and DNMT1. Genome Biol. 2018, 19, 218. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ozark, P.A.; Smith, E.; Zhao, Z.; Marshall, S.A.; Rendleman, E.J.; Piunti, A.; Ryan, C.; Whelan, A.L.; Helmin, K.A.; et al. TET2 coactivates gene expression through demethylation of enhancers. Sci. Adv. 2018, 4, eaau6986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.M.; Oh, M.H.; Go, J.H.; Han, K.; Choi, S.Y. Molecular subtypes of triple-negative breast cancer: Understanding of subtype categories and clinical implication. Genes Genom. 2020, 42, 1381–1387. [Google Scholar] [CrossRef] [PubMed]
- Collignon, E.; Canale, A.; Al Wardi, C.; Bizet, M.; Calonne, E.; Dedeurwaerder, S.; Garaud, S.; Naveaux, C.; Barham, W.; Wilson, A.; et al. Immunity drives TET1 regulation in cancer through NF-κB. Sci. Adv. 2018, 4, eaap7309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, C.; Qin, T.; Barton, M.C.; Jelinek, J.; Issa, J.P. Minimal role of base excision repair in TET-induced global DNA demethylation in HEK293T cells. Epigenetics 2015, 10, 1006–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Xia, W.; Wang, Q.; Towers, A.J.; Chen, J.; Gao, R.; Zhang, Y.; Yen, C.-A.; Lee, A.Y.; Li, Y.; et al. Isoform Switch of TET1 Regulates DNA Demethylation and Mouse Development. Mol. Cell 2016, 64, 1062–1073. [Google Scholar] [CrossRef] [Green Version]
- Chua, G.N.L.; Wassarman, K.L.; Sun, H.; Alp, J.A.; Jarczyk, E.I.; Kuzio, N.J.; Bennett, M.J.; Malachowsky, B.G.; Kruse, M.; Kennedy, A.J. Cytosine-Based TET Enzyme Inhibitors. ACS Med. Chem. Lett. 2019, 10, 180–185. [Google Scholar] [CrossRef]
- Singh, A.K.; Zhao, B.; Liu, X.; Wang, X.; Li, H.; Qin, H.; Wu, X.; Ma, Y.; Horne, D.; Yu, X. Selective targeting of TET catalytic domain promotes somatic cell reprogramming. Proc. Natl. Acad. Sci. USA 2020, 117, 3621–3626. [Google Scholar] [CrossRef]
- Wu, H.-X.; Chen, Y.-X.; Wang, Z.-X.; Zhao, Q.; He, M.-M.; Wang, Y.-N.; Wang, F.; Xu, R.-H. Alteration in TET1 as potential biomarker for immune checkpoint blockade in multiple cancers. J. Immunother. Cancer 2019, 7, 264. [Google Scholar] [CrossRef]
- Hock, H. A complex Polycomb issue: The two faces of EZH2 in cancer. Genes Dev. 2012, 26, 751–755. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Chemical Name | Generic Name | Mechanism | Drug Status |
|---|---|---|---|
| 5-Azacytidine | Azacitidine | Cytosine analog | FDA- and EMA-approved for treatment of MDS |
| 5-Aza-2-deoxycytidine | Decitabine | Cytosine analog | FDA-approved for treatment of MDS and EMA-approved for treatment of AML |
| SGI-110 | Guadecitabine | Cytosine analog | Phase III clinical trial in AML |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panjarian, S.; Issa, J.-P.J. The Roles of DNA Demethylases in Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 628. https://doi.org/10.3390/ph14070628
Panjarian S, Issa J-PJ. The Roles of DNA Demethylases in Triple-Negative Breast Cancer. Pharmaceuticals. 2021; 14(7):628. https://doi.org/10.3390/ph14070628
Chicago/Turabian StylePanjarian, Shoghag, and Jean-Pierre J. Issa. 2021. "The Roles of DNA Demethylases in Triple-Negative Breast Cancer" Pharmaceuticals 14, no. 7: 628. https://doi.org/10.3390/ph14070628
APA StylePanjarian, S., & Issa, J.-P. J. (2021). The Roles of DNA Demethylases in Triple-Negative Breast Cancer. Pharmaceuticals, 14(7), 628. https://doi.org/10.3390/ph14070628