
Repurposing Drugs to Treat Heart and Brain Illness


 , and
, and 
Abstract
1. Introduction
1.1. Technology to Repurpose Drugs
1.2. Heart and Brain Interconnectedness in Disease
2. Targeting Endothelial Dysfunction in Cardiovascular Disease
2.1. Colchicine
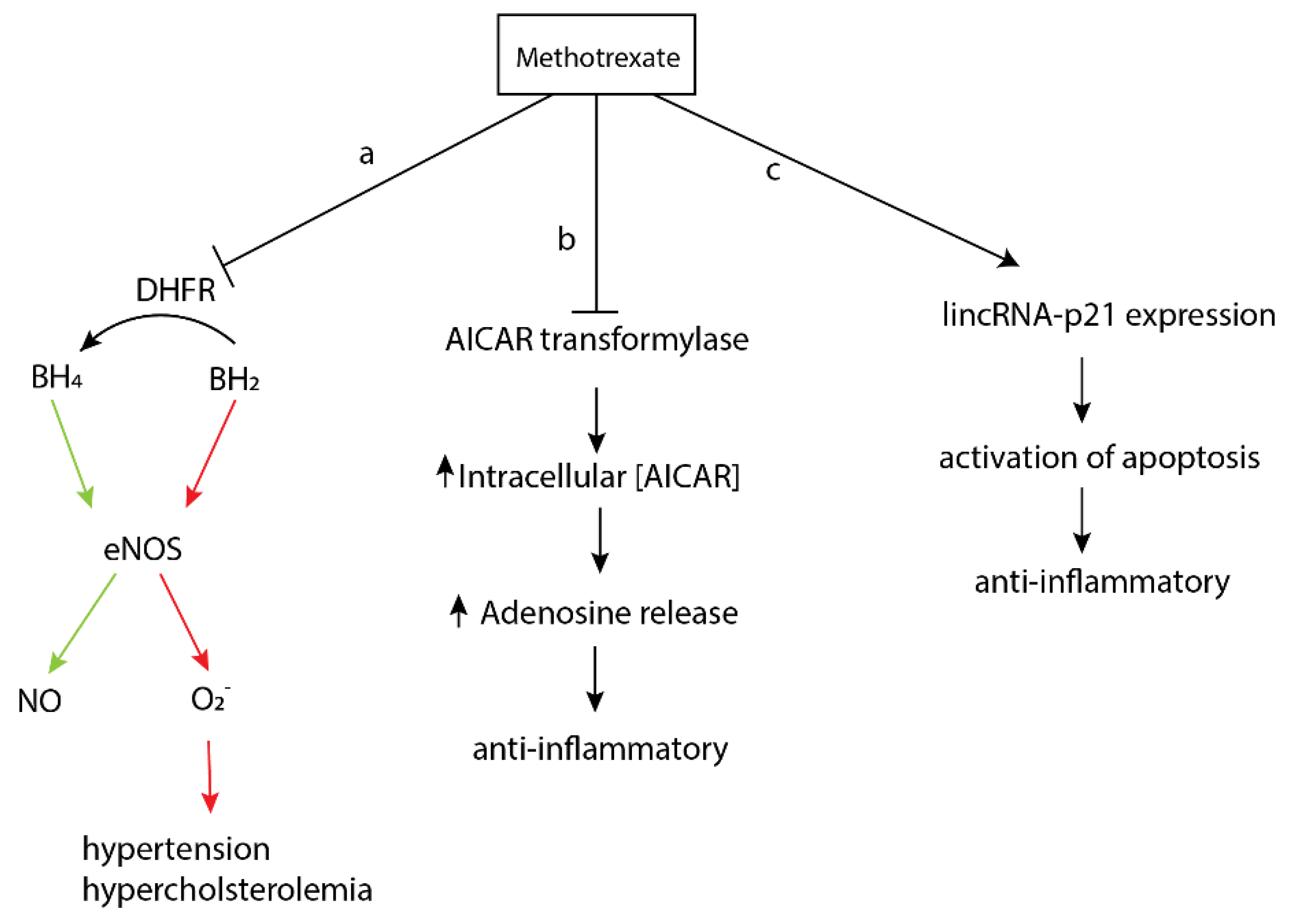
2.2. Methotrexate
2.3. Tocilizumab
3. Targeting Pulmonary Arterial Hypertension
3.1. Anakinra
3.2. Tadalafil
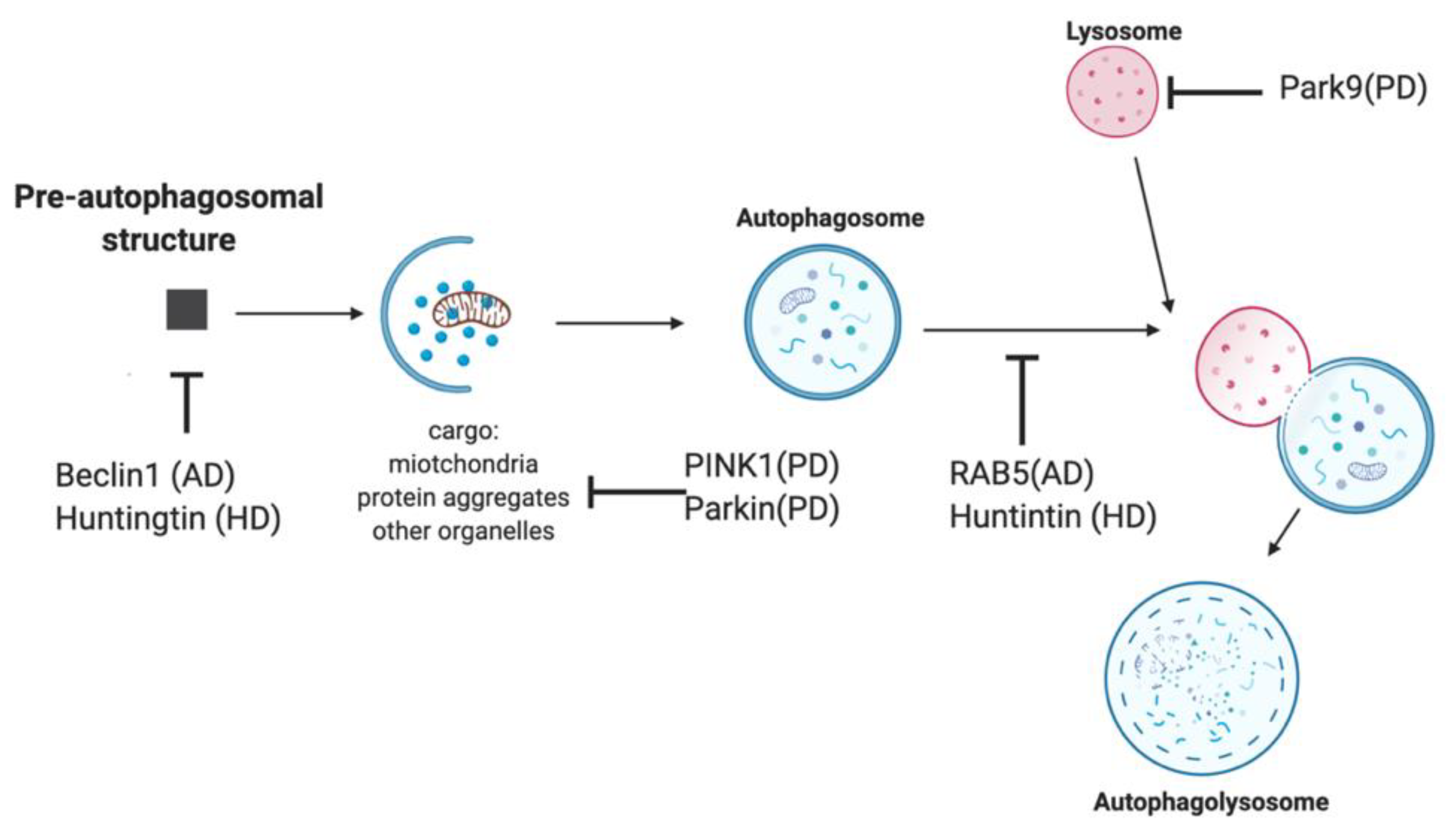
4. Modulating Autophagy in Neurodegeneration
4.1. Felodipine
4.2. Lonafarnib
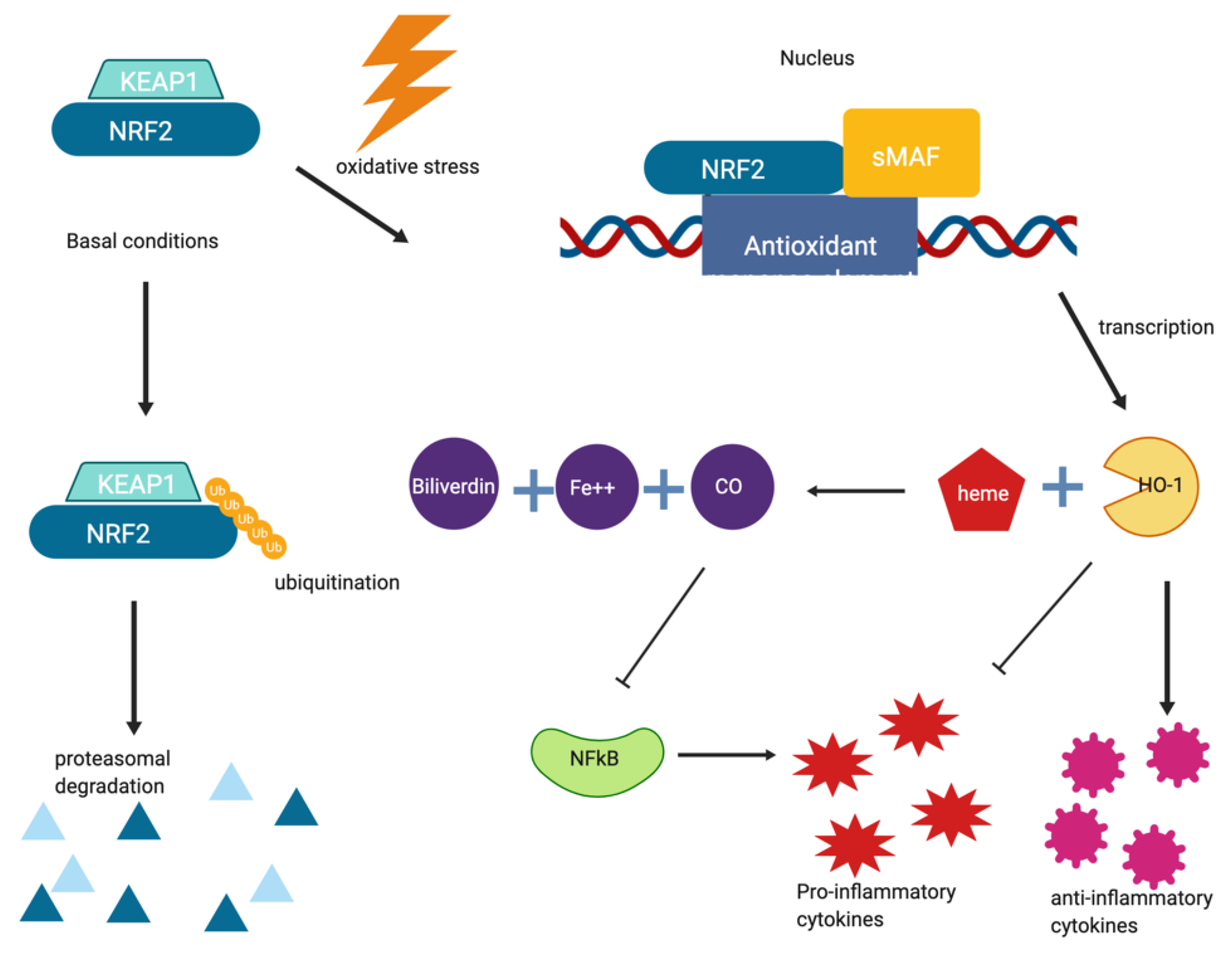
5. Inflammation and NRF2 as a Target in Neurodegeneration
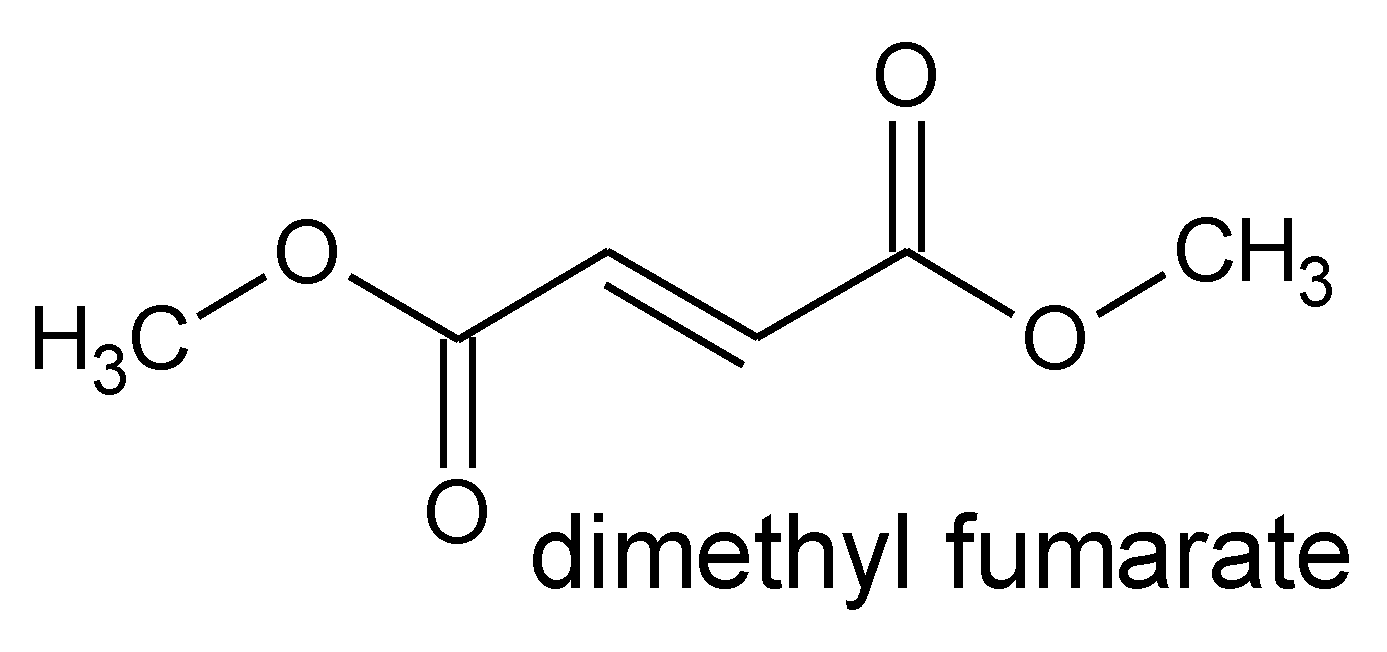
5.1. Dimethyl Fumarate
5.2. Exemestane
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 2012, 11, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Gns, H.; Saraswathy, G.R.; Murahari, M.; Krishnamurthy, M. An update on Drug Repurposing: Re-written saga of the drug’s fate. Biomed. Pharmacother. 2019, 110, 700–716. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Hubsher, G.; Haider, M.; Okun, M. Amantadine: The journey from fighting flu to treating Parkinson disease. Neurology 2012, 78, 1096–1099. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Griffin, W.; Li, Y.; Li, X.; Rousseau, J.F.; Ding, Y.; Song, M.; Lu, W. Understanding Drug Repurposing from the Perspective of Biomedical Entities and Their Evolution: Bibliographic Research Using Aspirin. JMIR Med. Inform. 2020, 8, e16739. [Google Scholar] [CrossRef]
- Colombo, D.; Ammirati, E. Cyclosporine in transplantation—A history of converging timelines. J. Boil. Regul. Homeost. Agents 2012, 25, 493–504. [Google Scholar]
- Varothai, S.; Bergfeld, W.F. Androgenetic Alopecia: An Evidence-Based Treatment Update. Am. J. Clin. Dermatol. 2014, 15, 217–230. [Google Scholar] [CrossRef]
- Ross, D.M.; Hughes, T.P. Cancer treatment with kinase inhibitors: What have we learnt from imatinib? Br. J. Cancer 2004, 90, 12–19. [Google Scholar] [CrossRef][Green Version]
- Cao, C.; Moult, J. GWAS and drug targets. BMC Genom. 2014, 15, S5. [Google Scholar] [CrossRef]
- Rudrapal, M.; Khairnar, S.J.; Jadhav, A.G. Drug Repurposing (DR): An emerging approach in drug discovery. In Drug Repurposing—Hypothesis, Molecular Aspects and Therapeutic Applications; IntechOpen: London, UK, 2020; p. 13. [Google Scholar]
- King, M.D.; Long, T.; Pfalmer, D.L.; Andersen, T.L.; McDougal, O.M. SPIDR: Small-molecule peptide-influenced drug repurposing. BMC Bioinform. 2018, 19, 138. [Google Scholar] [CrossRef]
- Campillos, M.; Kuhn, M.; Gavin, A.-C.; Jensen, L.J.; Bork, P. Drug Target Identification Using Side-Effect Similarity. Science 2008, 321, 263–266. [Google Scholar] [CrossRef]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Nabirotchkin, S.; Peluffo, A.E.; Rinaudo, P.; Yu, J.; Hajj, R.; Cohen, D. Next-generation drug repurposing using human genetics and network biology. Curr. Opin. Pharmacol. 2020, 51, 78–92. [Google Scholar] [CrossRef]
- Miller, I.N.; Cronin-Golomb, A. Gender differences in Parkinson’s disease: Clinical characteristics and cognition. Mov. Disord. 2010, 25, 2695–2703. [Google Scholar] [CrossRef]
- Shu, L.; Blencowe, M.; Yang, X. Translating GWAS Findings to Novel Therapeutic Targets for Coronary Artery Disease. Front. Cardiovasc. Med. 2018, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bullock, C.; Cornia, N.; Jacob, R.; Remm, A.; Peavey, T.; Weekes, K.; Mallory, C.; Oxford, J.T.; McDougal, O.M.; Andersen, T.L. DockoMatic 2.0: High Throughput Inverse Virtual Screening and Homology Modeling. J. Chem. Inf. Model. 2013, 53, 2161–2170. [Google Scholar] [CrossRef]
- Reilly, M.P.; Lindgren, C.; He, J.; Ferguson, J.F.; Stylianou, I.M.; Mehta, N.N.; Burnett, M.S.; Devaney, J.M.; Knouff, C.W.; Thompson, J.R.; et al. Identification of ADAMTS7 as a novel locus for coronary atherosclerosis and association of ABO with myocardial infarction in the presence of coronary atherosclerosis: Two genome-wide association studies. Lancet 2011, 377, 383–392. [Google Scholar] [CrossRef]
- Marquart, L.A.; Turner, M.W.; McDougal, O.M. Qualitative Assay to Detect Dopamine Release by Ligand Action on Nicotinic Acetylcholine Receptors. Toxins 2019, 11, 682. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Jaghoori, M.M.; Bleijlevens, B.; Olabarriaga, S.D. 1001 Ways to run AutoDock Vina for virtual screening. J. Comput. Mol. Des. 2016, 30, 237–249. [Google Scholar] [CrossRef]
- Koes, D.R.; Camacho, C.J. ZINCPharmer: Pharmacophore search of the ZINC database. Nucleic Acids Res. 2012, 40, W409–W414. [Google Scholar] [CrossRef]
- Athauda, D.; Foltynie, T. Drug Repurposing in Parkinson’s Disease. CNS Drugs 2018, 32, 747–761. [Google Scholar] [CrossRef]
- Zhang, X.; Che, C. Drug Repurposing for Parkinson’s Disease by Integrating Knowledge Graph Completion Model and Knowledge Fusion of Medical Literature. Futur. Internet 2021, 13, 14. [Google Scholar] [CrossRef]
- Gelosa, P.; Castiglioni, L.; Camera, M.; Sironi, L. Repurposing of drugs approved for cardiovascular diseases: Opportunity or mirage? Biochem. Pharmacol. 2020, 177, 113895. [Google Scholar] [CrossRef]
- Mao, X.-Y. Drug repurposing in neurological diseases: Opportunities and challenges. In Drug Repurposing—Hypothesis, Molecular Aspects and Therapeutic Applications; IntechOpen: London, UK, 2020; Volume 32, pp. 137–144. [Google Scholar]
- Gumina, G.; Virga, K.G. Recent Advances in Drug Repurposing for Parkinson’s Disease. Curr. Med. Chem. 2019, 26, 5340–5362. [Google Scholar] [CrossRef]
- Ballard, C.; Aarsland, D.; Cummings, J.; O’Brien, J.; Mills, R.; Molinuevo, J.L.; Fladby, T.; Williams, G.; Doherty, P.; Corbett, A.; et al. Drug repositioning and repurposing for Alzheimer disease. Nat. Rev. Neurol. 2020, 16, 661–673. [Google Scholar] [CrossRef]
- Shakkour, Z.; Issa, H.; Ismail, H.; Ashekyan, O.; Habashy, K.J.; Nasrallah, L.; Jourdi, H.; Hamade, E.; Mondello, S.; Sabra, M.; et al. Drug Repurposing: Promises of Edaravone Target Drug in Traumatic Brain Injury. Curr. Med. Chem. 2021, 28, 2369–2391. [Google Scholar] [CrossRef] [PubMed]
- Durães, F.; Pinto, M.; Sousa, E. Old Drugs as New Treatments for Neurodegenerative Diseases. Pharmaceuticals 2018, 11, 44. [Google Scholar] [CrossRef]
- Mangoni, A.A.; Tommasi, S.; Zinellu, A.; Sotgia, S.; Carru, C.; Piga, M.; Erre, G.L. Repurposing existing drugs for cardiovascular risk management: A focus on methotrexate. Drugs Context 2018, 7, 1–12. [Google Scholar] [CrossRef]
- Rodriguez, S.; Hug, C.; Todorov, P.; Moret, N.; Boswell, S.A.; Evans, K.; Zhou, G.; Johnson, N.T.; Hyman, B.T.; Sorger, P.K.; et al. Machine learning identifies candidates for drug repurposing in Alzheimer’s disease. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Ion, G.N.D.; Mihai, D.P.; Lupascu, G.; Nitulescu, G.M. Application of molecular framework-based data-mining method in the search for beta-secretase 1 inhibitors through drug repurposing. J. Biomol. Struct. Dyn. 2018, 37, 3674–3685. [Google Scholar] [CrossRef]
- Pessetto, Z.Y.; Ma, Y.; Hirst, J.J.; von Mehren, M.; Weir, S.J.; Godwin, A.K. Drug Repurposing Identifies a Synergistic Combination Therapy with Imatinib Mesylate for Gastrointestinal Stromal Tumor. Mol. Cancer Ther. 2014, 13, 2276–2287. [Google Scholar] [CrossRef]
- Murphy, S.L.; Xu, J.; Kochanek, K.D.; Arias, E. Mortality in the United States 2017; NCHS Data Brief; National Center for Health Statistics, U.S. Department of Health & Human Services: Hyattsville, MD, USA, 2018.
- Pinkaew, D.; Fujise, K. Fortilin: A Potential Target for the Prevention and Treatment of Human Diseases. Adv. Clin. Chem. 2017, 82, 265–300. [Google Scholar] [CrossRef] [PubMed]
- Feletou, M. The Endothelium: Part 1: Multiple Functions of the Endothelial Cells-Focus on Endothelium-Derived Vasoactive Mediators; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2011. [Google Scholar]
- Sakurada, M.; Yoshioka, N.; Kuse, A.; Nakagawa, K.; Morichika, M.; Takahashi, M.; Kondo, T.; Asano, M.; Ueno, Y. Rapid identification of Gloriosa superba and Colchicum autumnale by melting curve analysis: Application to a suicide case involving massive ingestion of G. superba. Int. J. Leg. Med. 2019, 133, 1065–1073. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis is an inflammatory disease. Am. Hearth J. 1999, 138, S419–S420. [Google Scholar] [CrossRef]
- Libby, P.; Hansson, G.K. Inflammation and Immunity in Diseases of the Arterial Tree. Circ. Res. 2015, 116, 307–311. [Google Scholar] [CrossRef]
- Davies, M.J. Stability and Instability: Two Faces of Coronary Atherosclerosis. Circulation 1996, 94, 2013–2020. [Google Scholar] [CrossRef]
- Verma, S.; Szmitko, P.E.; Ridker, P.M. C-reactive protein comes of age. Nat. Clin. Pr. Neurol. 2005, 2, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Nidorf, S.M.; Thompson, P.L. Why Colchicine Should Be Considered for Secondary Prevention of Atherosclerosis: An Overview. Clin. Ther. 2019, 41, 41–48. [Google Scholar] [CrossRef]
- Fiolet, A.T.; Nidorf, S.M.; Mosterd, A.; Cornel, J.H. Colchicine in Stable Coronary Artery Disease. Clin. Ther. 2019, 41, 30–40. [Google Scholar] [CrossRef]
- Leung, Y.Y.; Hui, L.L.Y.; Kraus, V.B. Colchicine—Update on mechanisms of action and therapeutic uses. Semin. Arthritis Rheum. 2015, 45, 341–350. [Google Scholar] [CrossRef]
- Redelinghuys, P.; Brown, G.D. Inhibitory C-type lectin receptors in myeloid cells. Immunol. Lett. 2011, 136, 1–12. [Google Scholar] [CrossRef]
- Brennan, K.; Zheng, J. Interleukin 8. In xPharm: The Comprehensive Pharmacology Reference; Elsevier: Amsterdam, The Netherlands, 2007; pp. 1–4. [Google Scholar]
- Weinblatt, M.E. Methotrexate in Rheumatoid Arthritis: A Quarter Century of Development. Trans. Am. Clin. Clim. Assoc. 2013, 124, 16–25. [Google Scholar]
- Kinder, A.J.; Hassell, A.B.; Brand, J.; Brownfield, A.; Grove, M.; Shadforth, M.F. The treatment of inflammatory arthritis with methotrexate in clinical practice: Treatment duration and incidence of adverse drug reactions. Rheumatology 2005, 44, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Aquilante, C.L.; Niemi, M.; Gong, L.; Altman, R.B.; Klein, T.E. PharmGKB summary. Pharm. Genom. 2013, 23, 721–728. [Google Scholar] [CrossRef]
- Cronstein, B.N.; Aune, T.M. Methotrexate and its mechanisms of action in inflammatory arthritis. Nat. Rev. Rheumatol. 2020, 16, 145–154. [Google Scholar] [CrossRef]
- Sankrityayan, H.; Majumdar, A.S. Curcumin and folic acid abrogated methotrexate induced vascular endothelial dysfunction. Can. J. Physiol. Pharmacol. 2016, 94, 89–96. [Google Scholar] [CrossRef]
- Panja, S.; Khatua, D.K.; Halder, M. Simultaneous Binding of Folic Acid and Methotrexate to Human Serum Albumin: Insights into the Structural Changes of Protein and the Location and Competitive Displacement of Drugs. ACS Omega 2018, 3, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, M.J.; Hale, A.B.; Channon, K. Dihydrofolate reductase protects endothelial nitric oxide synthase from uncoupling in tetrahydrobiopterin deficiency. Free Radic. Biol. Med. 2011, 50, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Yan, Z.; Ding, S.; Xu, K.; Wang, L. Endothelial Injury, an Intriguing Effect of Methotrexate and Cyclophosphamide During Hematopoietic Stem Cell Transplantation in Mice. Transplant. Proc. 2008, 40, 2670–2673. [Google Scholar] [CrossRef]
- Matei, V.; Rodríguez-Vilarrupla, A.; Deulofeu, R.; Colomer, D.; Fernandez, M.; Bosch, J.; Garcia-Pagán, J.-C. The eNOS cofactor tetrahydrobiopterin improves endothelial dysfunction in livers of rats with CCl4 cirrhosis. Hepatology 2006, 44, 44–52. [Google Scholar] [CrossRef]
- Gualtierotti, R.; Ingegnoli, F.; Boscolo, M.; Griffini, S.; Grovetti, E.; Cugno, M. Tocilizumab Effects on Coagulation Factor XIII in Patients with Rheumatoid Arthritis. Adv. Ther. 2019, 36, 3494–3502. [Google Scholar] [CrossRef]
- Ruiz-Limón, P.; Ortega, R.; de la Rosa, I.A.; Aguilera, M.C. Ábalos; Sanchez, C.P.; Gomez, Y.J.; Peralbo-Santaella, E.; Font, P.; Ruiz-Vilches, D.; Ferrín, G.; et al. Tocilizumab improves the proatherothrombotic profile of rheumatoid arthritis patients modulating endothelial dysfunction, NETosis, and inflammation. Transl. Res. 2017, 183, 87–103. [Google Scholar] [CrossRef]
- Williams, K.; Stern, M.P.; Freeman, G.L. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum. 2001, 44, 2737–2745. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Gibbs, J.S.R. The changing landscape of pulmonary arterial hypertension and implications for patient care. Eur. Respir. Rev. 2014, 23, 450–457. [Google Scholar] [CrossRef]
- Gaine, S.P.; Rubin, L.J. Primary pulmonary hypertension. Lancet 1998, 352, 719–725. [Google Scholar] [CrossRef]
- Sitbon, O.; Noordegraaf, A.V. Epoprostenol and pulmonary arterial hypertension: 20 years of clinical experience. Eur. Respir. Rev. 2017, 26, 160055. [Google Scholar] [CrossRef] [PubMed]
- Leeper, B.; Powell, B. Pulmonary arterial hypertension. Nurs. Crit. Care 2019, 14, 14–22. [Google Scholar] [CrossRef]
- Grinnan, D.; Trankle, C.; Andruska, A.; Bloom, B.; Spiekerkoetter, E.F.; Grinnan, D. Drug repositioning in pulmonary arterial hypertension: Challenges and opportunities. Pulm. Circ. 2019, 9. [Google Scholar] [CrossRef]
- Trankle, C.R.; Canada, J.M.; Kadariya, D.; Markley, R.; de Chazal, H.M.; Pinson, J.; Fox, A.; van Tassell, B.W.; Abbate, A.; Grinnan, D. IL-1 Blockade Reduces Inflammation in Pulmonary Arterial Hypertension and Right Ventricular Failure: A Single-Arm, Open-Label, Phase IB/II Pilot Study. Am. J. Respir. Crit. Care Med. 2019, 199, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Parpaleix, A.; Amsellem, V.; Houssaini, A.; Abid, S.; Breau, M.; Marcos, E.; Sawaki, D.; Delcroix, M.; Quarck, R.; Maillard, A.; et al. Role of interleukin-1 receptor 1/MyD88 signalling in the development and progression of pulmonary hypertension. Eur. Respir. J. 2016, 48, 470–483. [Google Scholar] [CrossRef]
- Fisher, A. FDA Approves Adcirca (tadalafil) Tablets for the Treatment of Pulmonary Arterial Hypertension. Available online: https://www.drugs.com/newdrugs/fda-approves-adcirca-tadalafil-pulmonary-arterial-hypertension-1366.html (accessed on 12 November 2019).
- Daugan, A.; Grondin, P.; Ruault, C.; Gouville, A.-C.L.M.D.; Coste, H.; Linget, J.M.; Kirilovsky, J.; Hyafil, A.F.; Labaudinière, R. The Discovery of Tadalafil: A Novel and Highly Selective PDE5 Inhibitor. 2: 2,3,6,7,12,12a-hexahydropyrazino[1‘,2‘:1,6]pyrido[3,4-b]indole-1,4-dione Analogues. J. Med. Chem. 2003, 46, 4533–4542. [Google Scholar] [CrossRef]
- Kotera, J.; Fujishige, K.; Omori, K. Immunohistochemical Localization of cGMP-binding cGMP-specific Phosphodiesterase (PDE5) in Rat Tissues. J. Histochem. Cytochem. 2000, 48, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Brundage, B.H.; Ghofrani, A.; Oudiz, R.J.; Simonneau, G.; Safdar, Z.; Shapiro, S.; White, R.J.; Chan, M.; Beardsworth, A.; et al. Tadalafil Therapy for Pulmonary Arterial Hypertension. Circulation 2009, 119, 2894–2903. [Google Scholar] [CrossRef]
- Amor, S.; Puentes, F.; Baker, D.; van der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef]
- Bredesen, D.E.; Rao, R.V.; Mehlen, P. Cell death in the nervous system. Nature 2006, 443, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nat. Cell Biol. 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Pan, W. The Treatment Strategies for Neurodegenerative Diseases by Integrative Medicine. Integr. Med. Int. 2015, 1, 223–225. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M. Autophagy in neurodegenerative diseases: From pathogenic dysfunction to therapeutic modulation. Semin. Cell Dev. Biol. 2015, 40, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Plendil NDA 19834/S009. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/98/19834S009_PLENDIL_APPROV.PDF (accessed on 10 December 2020).
- Tian, X.; Gala, U.; Zhang, Y.; Shang, W.; Jaiswal, S.N.; di Ronza, A.; Jaiswal, M.; Yamamoto, S.; Sandoval, H.; DuRaine, L.; et al. A voltage-gated calcium channel regulates lysosomal fusion with endosomes and autophagosomes and is required for neuronal homeostasis. PLoS Biol. 2015, 13, e1002103. [Google Scholar] [CrossRef]
- Siddiqi, F.H.; Menzies, F.M.; Lopez, A.; Stamatakou, E.; Karabiyik, C.; Ureshino, R.; Ricketts, T.; Jimenez-Sanchez, M.; Esteban, M.A.; Lai, L.; et al. Felodipine induces autophagy in mouse brains with pharmacokinetics amenable to repurposing. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Alto, P.; Biopharmaceuticals, E. Eiger Announces Breakthrough Therapy Designation Granted by FDA for Lonafarnib in Progeria and Progeroid Laminopathies. Survival Benefit in Children with Progeria Published in JAMA 2018. NDA Filing Planned in 2019. Available online: https://www.eigerbio.com/press_releases/eiger-announces-breakthrough-therapy-designation-granted-by-fda-for-lonafarnib-in-progeria-and-progeroid-laminopathies/ (accessed on 13 June 2021).
- Sinha, J.K.; Ghosh, S.; Raghunath, M. Progeria: A rare genetic premature ageing disorder. Indian J. Med. Res. 2014, 139, 667–674. [Google Scholar]
- Pan, J.; Song, E.; Cheng, C.; Lee, M.-H.; Yeung, S.-C.J. Farnesyltransferase inhibitors-induced autophagy: Alternative mechanisms? Autophagy 2009, 5, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, I.; Luna, G.; Rauch, J.N.; Reis, S.A.; Giroux, M.; Karch, C.M.; Boctor, D.; Sibih, Y.E.; Storm, N.J.; Diaz, A.; et al. A farnesyltransferase inhibitor activates lysosomes and reduces tau pathology in mice with tauopathy. Sci. Transl. Med. 2019, 11, eaat3005. [Google Scholar] [CrossRef]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Balak, D.M. Fumaric Acid Esters in the Management of Psoriasis. Psoriasis Targets Ther. 2015, 5, 9–23. [Google Scholar] [CrossRef]
- Linker, R.A.; Haghikia, A. Dimethyl fumarate in multiple sclerosis: Latest developments, evidence and place in therapy. Ther. Adv. Chronic Dis. 2016, 7, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Bomprezzi, R. Dimethyl fumarate in the treatment of relapsing–remitting multiple sclerosis: An overview. Ther. Adv. Neurol. Disord. 2015, 8, 20–30. [Google Scholar] [CrossRef]
- Tecfidera Drug Approval Package. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204063Orig1s000TOC.cfm (accessed on 10 December 2020).
- Lastres-Becker, I.; García-Yagüe, Á.J.; Scannevin, R.H.; Casarejos, M.J.; Kügler, S.; Rábano, A.; Cuadrado, A. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy against Synucleinopathy in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef]
- Bento-Pereira, C.; Dinkova-Kostova, A.T. Activation of transcription factor Nrf2 to counteract mitochondrial dysfunction in Parkinson’s disease. Med. Res. Rev. 2021, 41, 785–802. [Google Scholar] [CrossRef] [PubMed]
- Braley, T.J.; Huber, P.A.K.; Segal, B.; Kaplish, N.; Saban, B.R.; Washnock-Schmid, J.M.; Chervin, R.D. A randomized, subject and rater-blinded, placebo-controlled trial of dimethyl fumarate for obstructive sleep apnea. Sleep 2018, 41, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Aromasin Drug Approval Package. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/99/20-753_Aromasin.cfm (accessed on 10 December 2020).
- Miller, W.R.; Bartlett, J.; Brodie, A.M.H.; Brueggemeier, R.W.; di Salle, E.; Lonning, P.E.; Llombart, A.; Maass, N.; Maudelonde, T.; Sasano, H.; et al. Aromatase Inhibitors: Are There Differences between Steroidal and Nonsteroidal Aromatase Inhibitors and Do They Matter? Oncologist 2008, 13, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Son, H.J.; Han, S.H.; Lee, J.A.; Shin, E.J.; Hwang, O. Potential repositioning of exemestane as a neuroprotective agent for Parkinson’s disease. Free Radic. Res. 2017, 51, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Ishikawa, T.; Hozumi, Y.; Ikeda, M.; Iwata, H.; Yamashita, H.; Toyama, T.; Chishima, T.; Saji, S.; Yamamoto-Ibusuki, M.; et al. Randomized controlled trial of toremifene 120 mg compared with exemestane 25 mg after prior treatment with a non-steroidal aromatase inhibitor in postmenopausal women with hormone receptor-positive metastatic breast cancer. BMC Cancer 2013, 13, 239. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.Y.; Kim, J.H.; Moon, M.K.; Han, S.-H.; Yeon, S.K.; Choi, J.W.; Jang, B.K.; Song, H.J.; Kang, Y.G.; Kim, J.W.; et al. Discovery of Vinyl Sulfones as a Novel Class of Neuroprotective Agents toward Parkinson’s Disease Therapy. J. Med. Chem. 2014, 57, 1473–1487. [Google Scholar] [CrossRef]
- Liu, H.; Talalay, P. Relevance of anti-inflammatory and antioxidant activities of exemestane and synergism with sulforaphane for disease prevention. Proc. Natl. Acad. Sci. USA 2013, 110, 19065–19070. [Google Scholar] [CrossRef]
- Sandberg, M.; Patil, J.; D’Angelo, B.; Weber, S.G.; Mallard, C. NRF2-regulation in brain health and disease: Implication of cerebral inflammation. Neuropharmacology 2014, 79, 298–306. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Original Purpose | Repurposed Use | Reference |
|---|---|---|---|
| Amantadine | Parkinson’s disease | Influenza A | [4] |
| Acetylsalicylic acid | Inflammation, pain relief | Antiplatelet | [5] |
| Cyclosporine | Rheumatoid arthritis | Transplant rejection, psoriasis, chronic dry eye | [6] |
| Minoxidil | Hypertension | Alopecia | [7] |
| Imatinib | Chronic myelogous leukemia | Gastrointestinal stromal tumor | [8] |
| Drug or Target | Target Pathology | Research Method | Reference |
|---|---|---|---|
| kinase inhibitors | Alzheimer’s disease | machine learning | [32] |
| dopaminergic agonists | Parkinson’s disease | machine learning | [24] |
| beta-secretase 1 inhibitors | Alzheimer’s diseases | data mining | [33] |
| GIST-T1 cells | Gastrointestinal stromal tumor | High-throughput synergy screening | [34] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cantrell, M.S.; Soto-Avellaneda, A.; Wall, J.D.; Ajeti, A.D.; Morrison, B.E.; Warner, L.R.; McDougal, O.M. Repurposing Drugs to Treat Heart and Brain Illness. Pharmaceuticals 2021, 14, 573. https://doi.org/10.3390/ph14060573
Cantrell MS, Soto-Avellaneda A, Wall JD, Ajeti AD, Morrison BE, Warner LR, McDougal OM. Repurposing Drugs to Treat Heart and Brain Illness. Pharmaceuticals. 2021; 14(6):573. https://doi.org/10.3390/ph14060573
Chicago/Turabian StyleCantrell, Maranda S., Alejandro Soto-Avellaneda, Jackson D. Wall, Aaron D. Ajeti, Brad E. Morrison, Lisa R. Warner, and Owen M. McDougal. 2021. "Repurposing Drugs to Treat Heart and Brain Illness" Pharmaceuticals 14, no. 6: 573. https://doi.org/10.3390/ph14060573
APA StyleCantrell, M. S., Soto-Avellaneda, A., Wall, J. D., Ajeti, A. D., Morrison, B. E., Warner, L. R., & McDougal, O. M. (2021). Repurposing Drugs to Treat Heart and Brain Illness. Pharmaceuticals, 14(6), 573. https://doi.org/10.3390/ph14060573