
Development and Validation of an RP-HPLC-PDA Method for Determination of Paracetamol, Caffeine and Tramadol Hydrochloride in Pharmaceutical Formulations

 , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
2.1. Preliminary Studies
2.2. Analytical Method Optimization
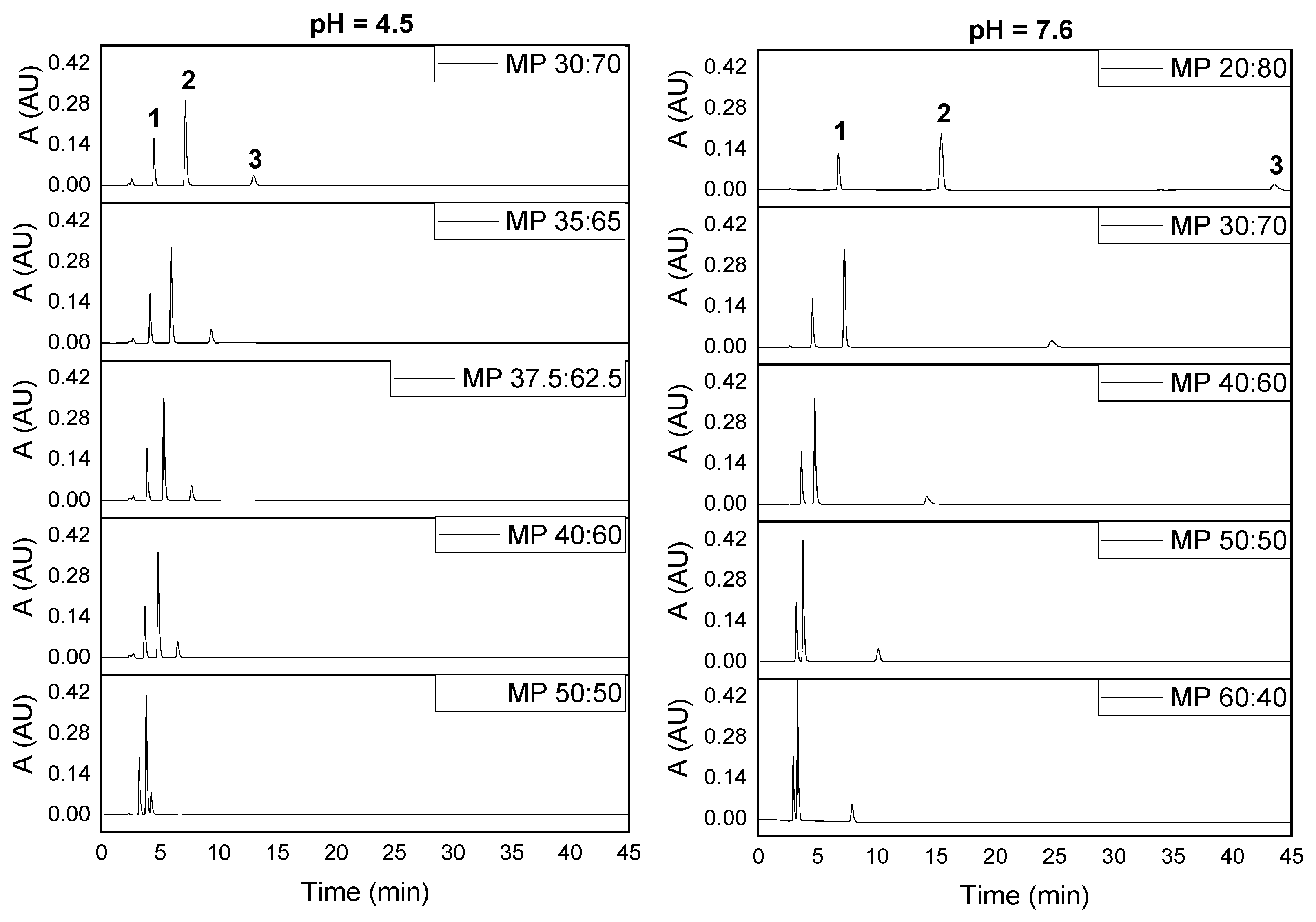
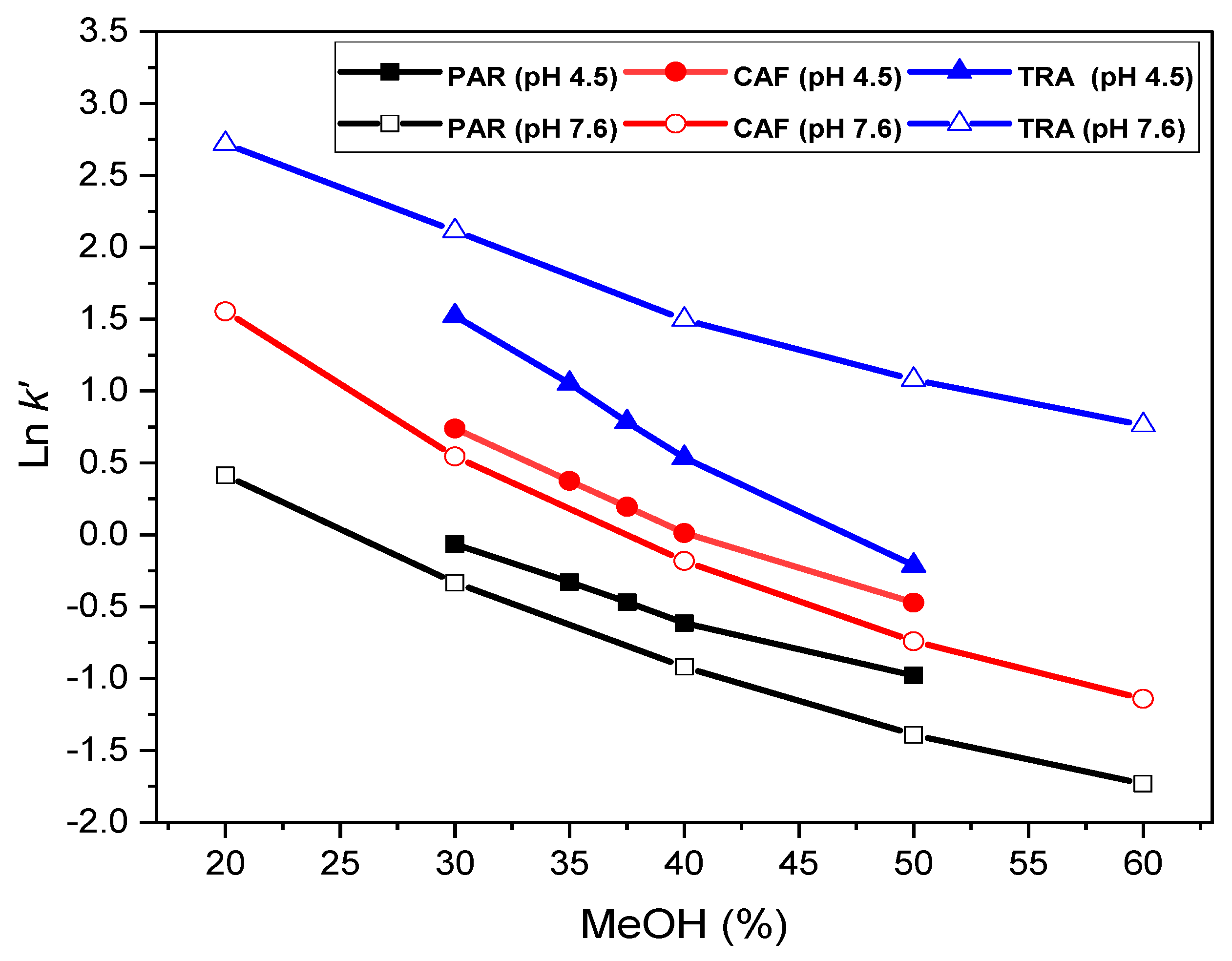
2.2.1. Effect of the Mobile Phase Composition
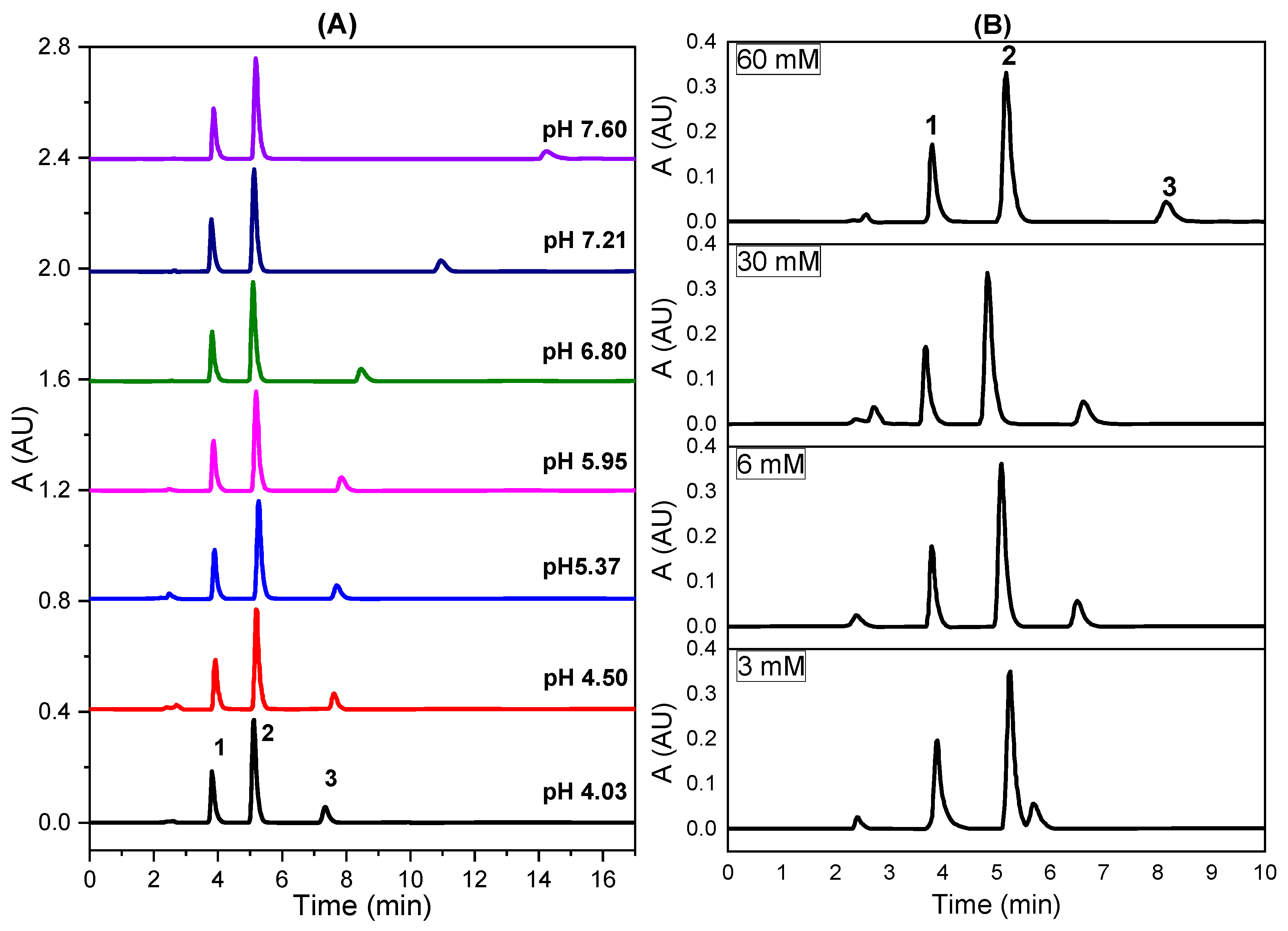
2.2.2. Effect of pH of the Mobile Phase at 40% (v/v) MeOH
2.2.3. Effect of pH of the Phosphate Concentration
2.2.4. Effect of the Flow Rate of the Mobile Phase
2.3. Validation of the Analytical Method
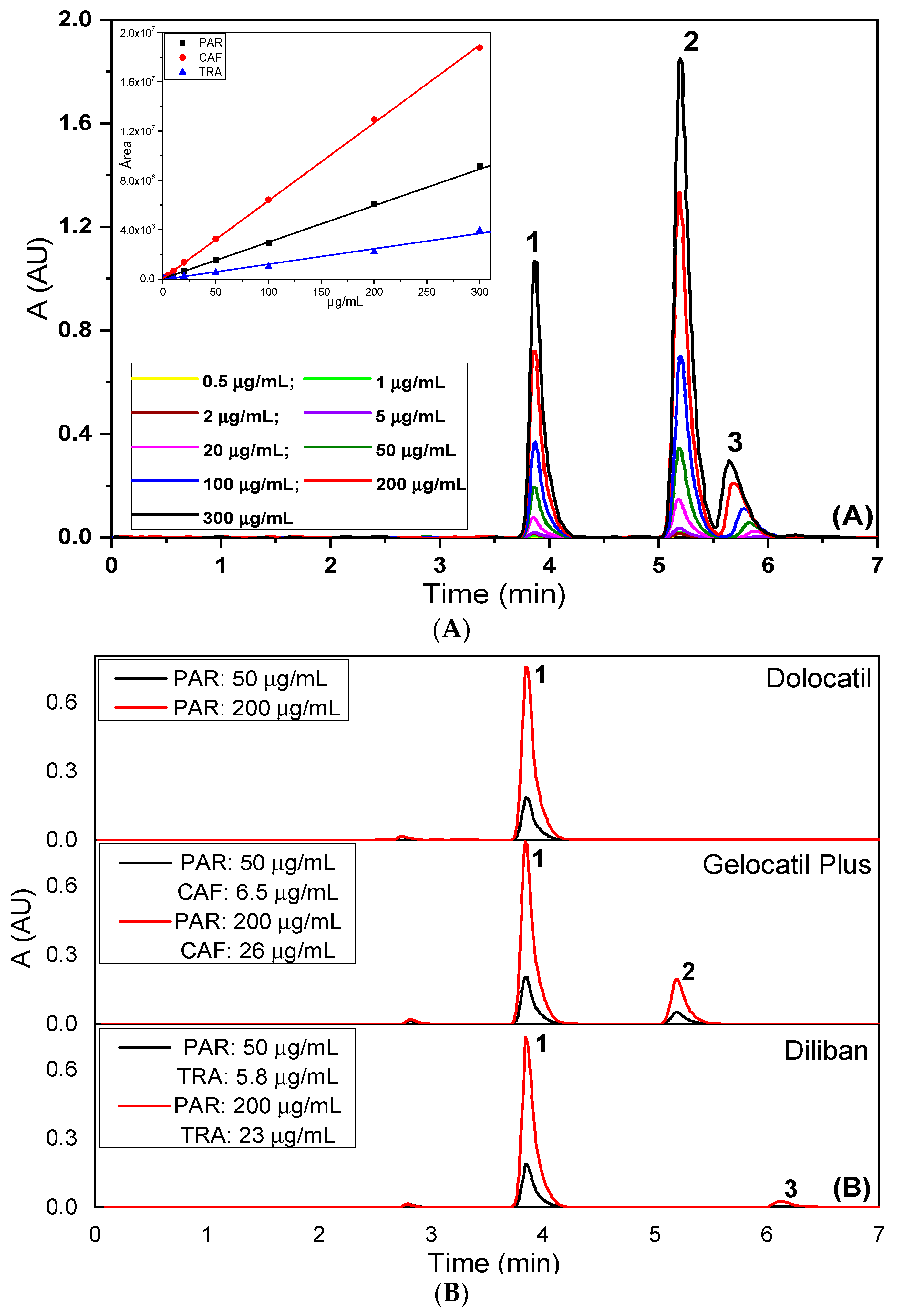
2.3.1. Linearity Range
2.3.2. Accuracy
2.3.3. Precision
2.3.4. Limit of Detection (LOD)
2.3.5. Limit of Quantitation (LOQ)
2.3.6. Robustness/Ruggedness
2.3.7. Specificity/Stability
2.4. Structural Characteristics of the Analytes
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Instrumentation
3.3. Chromatographic Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siddiqui, M.R.; AlOthman, Z.A.; Rahman, N. Analytical techniques in pharmaceutical analysis: A review. Arab. J. Chem. 2017, 10, S1409–S1421. [Google Scholar] [CrossRef]
- Khorshed, A.A.; Khairy, M.; Banks, C.E. Electrochemical determination of antihypertensive drugs by employing costless and portable unmodified screen-printed electrodes. Talanta 2019, 198, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Wu, Y.; Yang, J.; Chen, S.; Pu, Y.; Deng, H. Simultaneous Determination of Cortisol, Cortisone, and Multiple Illicit Drugs in Hair among Female Drug Addicts with LC-MS/MS. Molecules 2021, 26, 516. [Google Scholar] [CrossRef]
- González Peña, O.I.; López Zavala, M.Á.; Cabral Ruelas, H. Pharmaceuticals Market, Consumption Trends and Disease Incidence Are Not Driving the Pharmaceutical Research on Water and Wastewater. Int. J. Environ. Res. Public Health 2021, 18, 2532. [Google Scholar] [CrossRef]
- Turcios, A.E.; Hielscher, M.; Duarte, B.; Fonseca, V.F.; Caçador, I.; Papenbrock, J. Screening of Emerging Pollutants (EPs) in Estuarine Water and Phytoremediation Capacity of Tripolium pannonicum under Controlled Conditions. Int. J. Environ. Res. Public Health 2021, 18, 943. [Google Scholar] [CrossRef] [PubMed]
- Terzopoulou, Z.; Papageorgiou, M.; Kyzas, G.Z.; Bikiaris, D.N.; Lambropoulou, D.A. Preparation of molecularly imprinted solid-phase microextraction fiber for the selective removal and extraction of the antiviral drug abacavir in environmental and biological matrices. Anal. Chim. Acta 2016, 913, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Graham, G.G.; Davies, M.J.; Day, R.O.; Mohamudally, A.; Scott, K.F. The modern pharmacology of paracetamol: Therapeutic actions, mechanism of action, metabolism, toxicity and recent pharmacological findings. Inflammopharmacology 2013, 21, 201–232. [Google Scholar] [CrossRef] [PubMed]
- Schug, S.A.; Goddard, C. Recent advances in the pharmacological management of acute and chronic pain. Ann. Palliat. Med. 2014, 3, 263–275. [Google Scholar]
- Jendrzejewska, I.; Goryczka, T.; Pietrasik, E.; Klimontko, J.; Jampilek, J. X-ray and Thermal Analysis of Selected Drugs Containing Acetaminophen. Molecules 2020, 25, 5909. [Google Scholar] [CrossRef]
- Battu, P.R.; Reddy, M.S. RP-HPLC method for simultaneous estimation of paracetamol and ibuprofen in tablets. Asian J. Res. Chem. 2009, 2, 70–72. [Google Scholar]
- Călinescu, O.; Badea, I.A.; Vlădescu, L.; Meltzer, V.; Pincu, E. HPLC separation of acetaminophen and its impurities using a mixed-mode reversed-phase/cation exchange stationary phase. J. Chromatogr. Sci. 2012, 50, 335–342. [Google Scholar] [CrossRef]
- Afkhami, A.; Khoshsafar, H.; Bagheri, H.; Madrakian, T. Facile simultaneous electrochemical determination of codeine and acetaminophen in pharmaceutical samples and biological fluids by graphene–CoFe2O4 nanocomposite modified carbon paste electrode. Sens. Actuators B Chem. 2014, 203, 909–918. [Google Scholar] [CrossRef]
- Maslarska, V.; Tencheva, J. Simultaneous determination of paracetamol and codeine phosphate in pharmaceutical preparation by RP-HPLC. Int. J. Pharm. Pharm. Sci. 2013, 5, 417–419. [Google Scholar]
- Altun, M.L.; Ceyhan, T.; Kartal, M.; Atay, T.; Özdemir, N.; Cevheroğlu, Ş. LC method for the analysis of acetylsalicylic acid, caffeine and codeine phosphate in pharmaceutical preparations. J. Pharm. Biomed. Anal. 2001, 25, 93–101. [Google Scholar] [CrossRef]
- Takahashi, F.; Kobayashi, M.; Kobayashi, A.; Kobayashi, K.; Asamura, H. High-Frequency Heating Extraction Method for Sensitive Drug Analysis in Human Nails. Molecules 2018, 23, 3231. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.R.; Maleque, M.; Shihan, M.H. Development and validation of a simple RP-HPLC method for determination of caffeine in pharmaceutical dosage forms. Asian J. Pharm. Anal. 2012, 2, 1–4. [Google Scholar]
- Küçük, A.; Kadıoğlu, Y. Determination of tramadol hydrochloride in ampoule dosage forms by using UV spectrophotometric and HPLC-DAD methods in methanol and water media. Farmaco 2005, 60, 163–169. [Google Scholar] [CrossRef]
- Nobilis, M.; Pastera, J.; Anzenbacher, P.; Svoboda, D.; Kopecký, J.; Perlík, F. High-performance liquid chromatographic determination of tramadol in human plasma. J. Chromatogr. B Biomed. Sci. Appl. 1996, 681, 177–183. [Google Scholar] [CrossRef]
- Abdulbari, M.M.; Ihsan, M.S. Simultaneous determination and validation of chlorpheniramine maleate, acetaminophen, phenylpropanolamine hydrochloride and caffeine in tablet dosage form by using reverse phase high performance liquid chromatography (RP-HPLC). Int. J. Pharm. Pharm. Sci. 2013, 5 (Suppl. 3), 666–670. [Google Scholar]
- Altun, M.L. HPLC method for the analysis of paracetamol, caffeine and dipyrone. Turk. J. Chem. 2002, 26, 521–528. [Google Scholar]
- Franeta, J.T.; Agbaba, D.; Eric, S.; Pavkov, S.; Aleksic, M.; Vladimirov, S. HPLC assay of acetylsalicylic acid, paracetamol, caffeine and phenobarbital in tablets. Farmaco 2002, 57, 709–713. [Google Scholar] [CrossRef]
- Cunha, R.R.; Chaves, S.C.; Ribeiro, M.M.A.C.; Torres, L.M.F.C.; Muñoz, R.A.A.; DosSantos, W.T.P.; Richter, E.M. Simultaneous determination of caffeine, paracetamol, and ibuprofen in pharmaceutical formulations by high-performance liquid chromatography with UV detection and by capillary electrophoresis with conductivity detection. J. Sep. Sci. 2015, 38, 1657–1662. [Google Scholar] [CrossRef] [PubMed]
- Bandelwar, R.; Nikam, A.; Sawant, S. Analytical Method Development and Validation of Phenylepherine Hydrochloride, Chlorpheniramine Maleate, Paracetamol and Caffeine in Bulk Drug and Tablet Dosage Form by RP-HPLC. Indo Am. J. Pharm. Res. 2013, 3, 4330. [Google Scholar]
- Sultan, M. Simultaneous HPLC determination and validation of amphetamine, methamphetamine, caffeine, paracetamol and theophylline in illicit seized tablets. Int. J. Pharm. Pharm. Sci. 2014, 6, 294–298. [Google Scholar]
- Belal, T.; Awad, T.; Clark, R. Determination of paracetamol and tramadol hydrochloride in pharmaceutical mixture using HPLC and GC-MS. J. Chromatogr. Sci. 2009, 47, 849–854. [Google Scholar] [CrossRef]
- Ghorbani-Bidkorbeh, F.; Shahrokhian, S.; Mohammadi, A.; Dinarvand, R. Simultaneous voltammetric determination of tramadol and acetaminophen using carbon nanoparticles modified glassy carbon electrode. Electrochim. Acta 2010, 55, 2752–2759. [Google Scholar] [CrossRef]
- Kamble, R.M.; Singh, S.G. Stability-indicating RP-HPLC method for analysis of paracetamol and tramadol in a pharmaceutical dosage form. E J. Chem. 2012, 9, 1347–1356. [Google Scholar] [CrossRef]
- Gaikwad, H.; Borde, S.; Suralkar, M.; Choudhari, V.; Kuchekar, B. Development and validation of RP-HPLC method for simultaneous determination of tramadol hydrochloride, paracetamol and dicyclomine hydrochloride by using design of experiment software (DOE). Int. J. Pharm. Sci. 2014, 4, 792–801. [Google Scholar]
- Eisele, A.P.P.; Clausen, D.N.; Tarley, C.R.T.; Dall’Antonia, L.H.; Sartori, E.R. Simultaneous square-wave voltammetric determination of paracetamol, caffeine and orphenadrine in pharmaceutical formulations using a cathodically pretreated boron-doped diamond electrode. Electroanalysis 2013, 25, 1734–1741. [Google Scholar] [CrossRef]
- Nyansa, M.M.S.; Fiawoyife, P.D.; Mireku-Gyimah, N.A.; Addotey, J.N.A. Stability-Indicating HPLC Method for the Simultaneous Determination of Paracetamol and Tramadol Hydrochloride in Fixed-Dose Combination Tablets. J. Biomed. Sci. Eng. 2017, 5, 41–47. [Google Scholar] [CrossRef]
- Chandra, P.; Rathore, A.S.; Lohidasan, S.; Mahadik, K.R. Application of HPLC for the simultaneous determination of aceclofenac, paracetamol and tramadol hydrochloride in pharmaceutical dosage form. Sci. Pharm. 2012, 80, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Morelato, M.; Beavis, A.; Kirkbride, P.; Roux, C. Forensic applications of desorption electrospray ionisation mass spectrometry (DESI-MS). Forensic Sci. Int. 2013, 226, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Habibollahi, S.; Tavakkoli, N.; Nasirian, V.; Khani, H. Determination of tramadol by dispersive liquid–liquid microextraction combined with GC–MS. J. Chromatogr. Sci. 2015, 53, 655–661. [Google Scholar] [CrossRef]
- Grapp, M.; Maurer, H.H.; Desel, H. Systematic forensic toxicological analysis by GC-MS in serum using automated mass spectral deconvolution and identification system. Drug Test. Anal. 2016, 8, 816–825. [Google Scholar] [CrossRef]
- Engin, C.; Yilmaz, S.; Saglikoglu, G.; Yagmur, S.; Sadikoglu, M. Electroanalytical investigation of paracetamol on glassy carbon electrode by voltammetry. Int. J. Electrochem. Sci. 2015, 10, 1916–1925. [Google Scholar]
- Atty, S.A.; Ibrahim, A.H.; Ibrahim, H.; Abdelzaher, A.M.; Abdel-Raoof, A.M.; Fouad, F.A. Simultaneous voltammetric detection of anti-depressant drug, sertraline HCl and paracetamol in biological fluid at CNT-cesium modified electrode in micellar media. Microchem. J. 2020, 159, 105524. [Google Scholar] [CrossRef]
- Mulyasuryani, A.; Tjahjanto, R.T.; Andawiyah, R. Simultaneous Voltammetric Detection of Acetaminophen and Caffeine Base on Cassava Starch—Fe3O4 Nanoparticles Modified Glassy Carbon Electrode. Chemosensors 2019, 7, 49. [Google Scholar] [CrossRef]
- Chitravathi, S.; Munichandraiah, N. Voltammetric determination of paracetamol, tramadol and caffeine using poly (Nile blue) modified glassy carbon electrode. J. Electroanal. Chem. 2016, 764, 93–103. [Google Scholar] [CrossRef]
- Nobilis, M.; Kopecký, J.; Květina, J.; Chladek, J.; Svoboda, Z.; Voříšek, V.; Perlík, F.; Pour, M.; Kuneš, J. High-performance liquid chromatographic determination of tramadol and its O-desmethylated metabolite in blood plasma: Application to a bioequivalence study in humans. J. Chromatogr. A 2002, 949, 11–22. [Google Scholar] [CrossRef]
- Balcan, M.; Anghel, D.F.; Donescu, D.; Galatanu, A. Effect of the mobile phase composition on the retention behavior of polydisperse ethoxylated nonylphenols in reversed-phase high performance liquid chromatography. Rev. Roum. Chim. 2006, 51, 299. [Google Scholar]
- Molnár, I. Searching for robust HPLC methods–Csaba Horváth and the solvophobic theory. Chromatographia 2005, 62, s7–s17. [Google Scholar] [CrossRef]
- McCalley, D.V. Influence of organic solvent modifier and solvent strenght on peak shape of some basic compounds in high-performance liquid chromatography using a reversed-phase column. J. Chromatogr. A 1995, 708, 185–194. [Google Scholar] [CrossRef]
- Molnar, I.; Horváth, C. Reverse-phase chromatography of polar biological substances: Separation of catechol compounds by high-performance liquid chromatography. Clin. Chem. 1976, 22, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Snyder, L.R.; Quarry, M.A.; Glajch, J.L. Solvent-strength selectivity in reversed-phase HPLC. Chromatographia 1987, 24, 33–44. [Google Scholar] [CrossRef]
- Valkó, K.; Snyder, L.R.; Glajch, J.L. Retention in reversed-phase liquid chromatography as a function of mobile-phase composition. J. Chromatogr. A 1993, 656, 501–520. [Google Scholar] [CrossRef]
- Rived, F.; Canals, I.; Bosch, E.; Rosés, M. Acidity in methanol–water. Anal. Chim. Acta 2001, 439, 315–333. [Google Scholar] [CrossRef]
- Burns, D.T.; Danzer, K.; Townshend, A. A Tutorial Discussion of the use of the terms “Robust” and “Rugged” and the Associated Characteristics of “Robustness” and “Ruggedness” as used in Descriptions of Analytical Procedures. J. Assoc. Public Anal. 2009, 37, 40–60. [Google Scholar]
- Marques, R.R.N.; Sampaio, M.J.; Carrapiço, P.M.; Silva, C.G.; Morales-Torres, S.; Dražić, G.; Faria, J.L.; Silva, A.M.T. Photocatalytic degradation of caffeine: Developing solutions for emerging pollutants. Catal. Today 2013, 209, 108–115. [Google Scholar] [CrossRef]
- Vogna, D.; Marotta, R.; Napolitano, A.; D’Ischia, M. Advanced Oxidation Chemistry of Paracetamol. UV/H2O2-Induced Hydroxylation/Degradation Pathways and 15N-Aided Inventory of Nitrogenous Breakdown Products. J. Org. Chem. 2002, 67, 6143–6151. [Google Scholar] [CrossRef]
- Rosal, R.; Rodríguez, A.; Perdigón-Melón, J.A.; Petre, A.; García-Calvo, E.; Gómez, M.J.; Agüera, A.; Fernández-Alba, A.R. Degradation of caffeine and identification of the transformation products generated by ozonation. Chemosphere 2009, 74, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Benotti, M.J.; Brownawell, B.J. Microbial degradation of pharmaceuticals in estuarine and coastal seawater. Environ. Pollut. 2009, 157, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Rúa-Gómez, P.C.; Püttmann, W. Degradation of lidocaine, tramadol, venlafaxine and the metabolites O-desmethyltramadol and O-desmethylvenlafaxine in surface waters. Chemosphere 2013, 90, 1952–1959. [Google Scholar] [CrossRef] [PubMed]
- Rosés, M.; Canals, I.; Allemann, H.; Siigur, K.; Bosch, E. Retention of ionizable compounds on HPLC. 2. Effect of pH, ionic strength, and mobile phase composition on the retention of weak acids. Anal. Chem. 1996, 68, 4094–4100. [Google Scholar] [CrossRef] [PubMed]
- Meloun, M.; Syrový, T.; Vrána, A. The thermodynamic dissociation constants of losartan, paracetamol, phenylephrine and quinine by the regression analysis of spectrophotometric data. Anal. Chim. Acta 2005, 533, 97–110. [Google Scholar] [CrossRef]
- Chahiyan, H.; Gharib, F.; Farajtabar, A. Thermodynamic studies on solubility and protonation constant of acetaminophen at different ionic strengths and various temperatures. J. Mol. Liq. 2014, 199, 137–142. [Google Scholar] [CrossRef]
- Fuguet, E.; Ràfols, C.; Bosch, E.; Rosés, M. Chromatographic hydrophobicity index: pH profile for polyprotic compounds. J. Chromatogr. A 2009, 1216, 7798–7805. [Google Scholar] [CrossRef]
- Sudha, C.; Srinivasan, K. Nucleation control and separation of paracetamol polymorphs through swift cooling crystallization process. J. Cryst. Growth 2014, 401, 248–251. [Google Scholar] [CrossRef]
- ICH Harmonised Tripartie Guideline. Validation of Analytical Procedures: Text and Methodology Q2(R1); ICH: Geneva, Switzerland, 2005; Volume 1. [Google Scholar]
- Aboushady, D.; Parr, M.K.; Hanafi, R.S. Quality-by-Design Is a Tool for Quality Assurance in the Assessment of Enantioseparation of a Model Active Pharmaceutical Ingredient. Pharmaceuticals 2020, 13, 364. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH | Coefficient | Paracetamol | R2 | Caffeine | R2 | Tramadol | R2 |
|---|---|---|---|---|---|---|---|
| Linear fit: Ln k’ = a + m B | |||||||
| 7.6 | a | 1.350 ± 0.190 | 0.9721 | 2.700 ± 0.300 | 0.9597 | 3.620 ± 0.170 | 0.9755 |
| m | −0.054 ± 0.005 | −0.067 ± 0.007 | −0.050 ± 0.004 | ||||
| 4.5 | a | 1.260 ± 0.120 | 0.9818 | 2.478 ± 0.165 | 0.9805 | 4.070 ± 0.162 | 0.9909 |
| m | −0.046 ± 0.003 | −0.060 ± 0.004 | −0.087 ± 0.004 | ||||
| Quadratic fit: Ln k’ = a + m B + c B2 | |||||||
| 7.6 | a | 2.272 ± 0.040 | 0.9999 | 4.070 ± 0.110 | 0.9993 | 4.390 ± 0.140 | 0.9979 |
| m | −0.106 ± 0.002 | −0.146 ± 0.006 | −0.094 ± 0.008 | ||||
| c | 0.001 ± 0.000 | 0.001 ± 0.000 | 0.001 ± 0.000 | ||||
| 4.5 | a | 2.400 ± 0.400 | 0.9967 | 4.200 ± 0.300 | 0.9986 | 5.600 ± 0.500 | 0.9980 |
| m | −0.106 ± 0.016 | −0.146 ± 0.014 | −0.167 ± 0.024 | ||||
| c | 0.001 ± 0.000 | 0.001 ± 0.000 | 0.001 ± 0.000 | ||||
| Parameters | Paracetamol (40:60) (PDA Detection) | Caffeine (40:60) (PDA Detection) | Tramadol (40:60) | |
|---|---|---|---|---|
| (PDA Detection) | (Fl Detection) | |||
| Linear Range (μg/mL) | 0.8–270 | 0.4–250 | 1.0–300 | 0.2–40 |
| R2 | 0.9987 | 0.9998 | 0.9999 | 0.9999 |
| Accuracy (%) | 98.47–99.85 | 99.97–100.08 | 101.10–101.10 | 99.96–100.91 |
| RSD (%) (overall) | 3.45 | 3.92 | 3.16 | 2.94 |
| LOD (μg/mL) | 0.2 | 0.1 | 0.3 | 0.1 |
| LOQ (μg/mL) | 0.8 | 0.4 | 1.0 | 0.2 |
| Robustness/Ruggedness (%) | 3.9 | 4.1 | 3.6 | 3.2 |
| Specificity (%) | ≥95 | ≥95 | ≥95 | ≥95 |
| Stability (%) | ≤5 | ≤5 | ≤5 | ≤5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, F.J.; Rodríguez-Cordero, A.; López, R.; Robles, L.C.; Aller, A.J. Development and Validation of an RP-HPLC-PDA Method for Determination of Paracetamol, Caffeine and Tramadol Hydrochloride in Pharmaceutical Formulations. Pharmaceuticals 2021, 14, 466. https://doi.org/10.3390/ph14050466
Pereira FJ, Rodríguez-Cordero A, López R, Robles LC, Aller AJ. Development and Validation of an RP-HPLC-PDA Method for Determination of Paracetamol, Caffeine and Tramadol Hydrochloride in Pharmaceutical Formulations. Pharmaceuticals. 2021; 14(5):466. https://doi.org/10.3390/ph14050466
Chicago/Turabian StylePereira, Fernando J., Aida Rodríguez-Cordero, Roberto López, Luis C. Robles, and A. Javier Aller. 2021. "Development and Validation of an RP-HPLC-PDA Method for Determination of Paracetamol, Caffeine and Tramadol Hydrochloride in Pharmaceutical Formulations" Pharmaceuticals 14, no. 5: 466. https://doi.org/10.3390/ph14050466
APA StylePereira, F. J., Rodríguez-Cordero, A., López, R., Robles, L. C., & Aller, A. J. (2021). Development and Validation of an RP-HPLC-PDA Method for Determination of Paracetamol, Caffeine and Tramadol Hydrochloride in Pharmaceutical Formulations. Pharmaceuticals, 14(5), 466. https://doi.org/10.3390/ph14050466