
Chemical Classes Presenting Novel Antituberculosis Agents Currently in Different Phases of Drug Development: A 2010–2020 Review



Abstract
1. Introduction
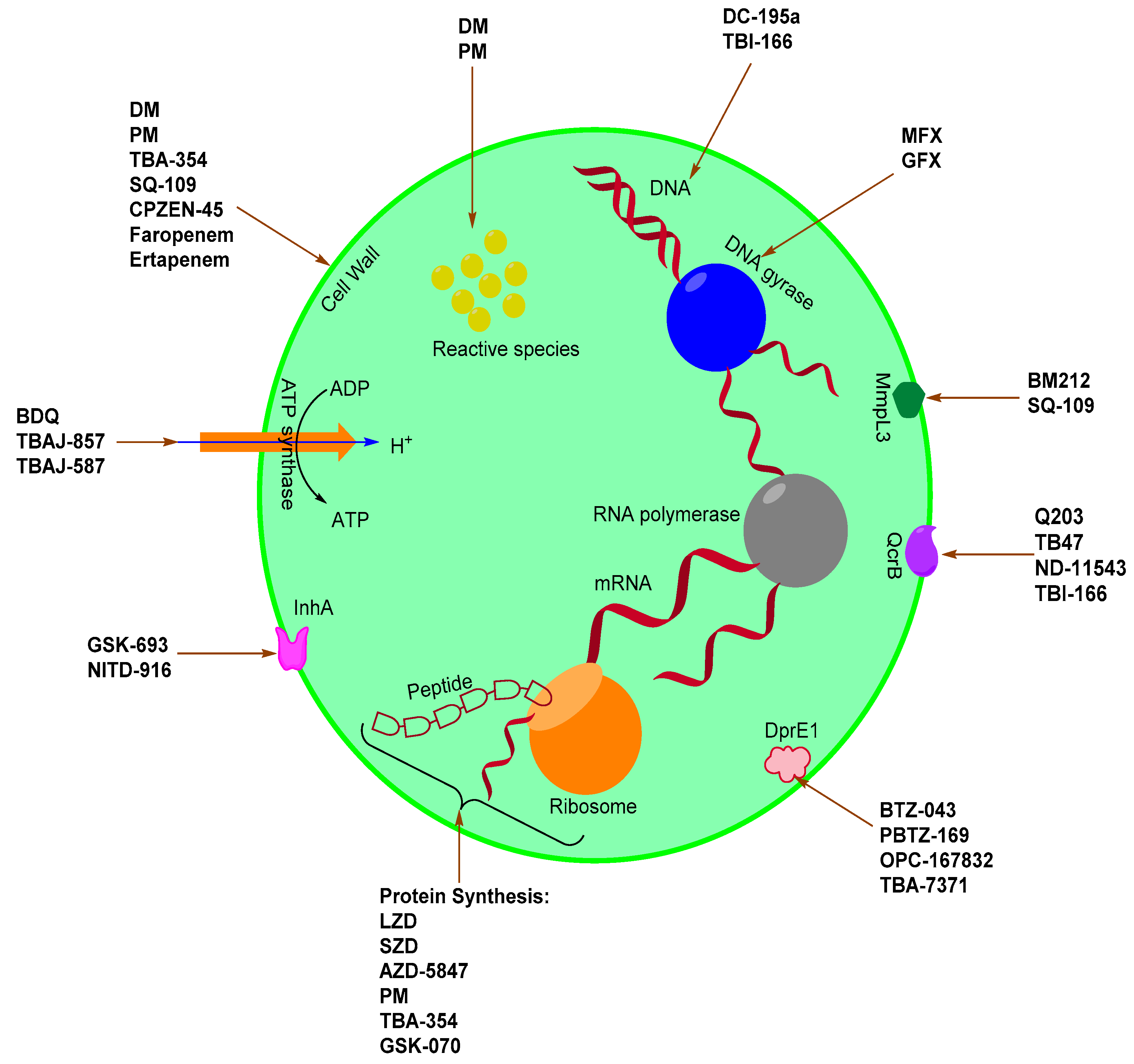
2. Novel Anti-Mtb Agents
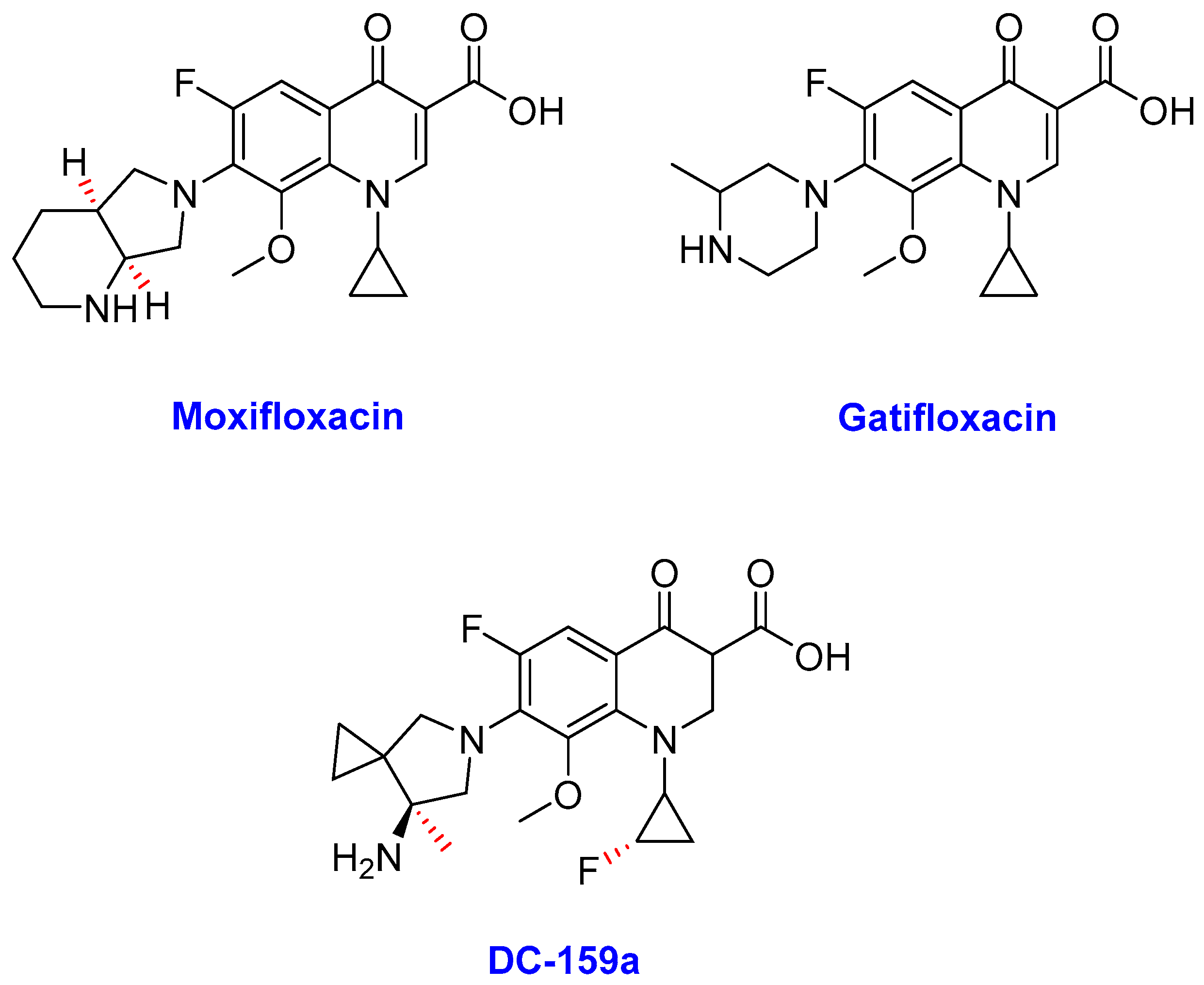
2.1. Quinolone Derivatives
Fluoroquinolones
- (i)
- Moxifloxacin
- (ii)
- Gatifloxacin
- (iii)
- DC-159a
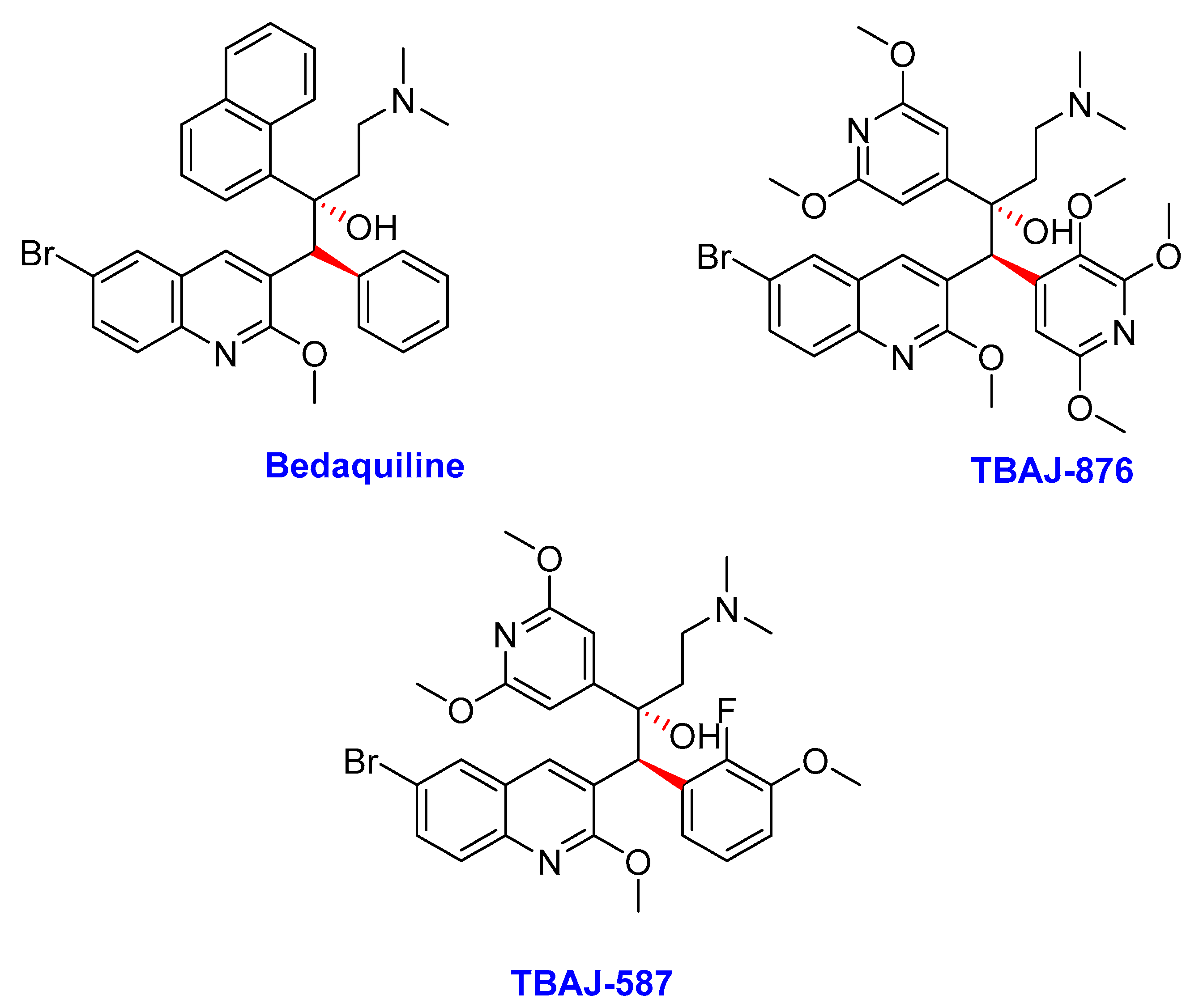
2.2. Diarylquinolines
- (i)
- Bedaquiline
- (ii)
- TBAJ-876
- (iii)
- TBAJ-587
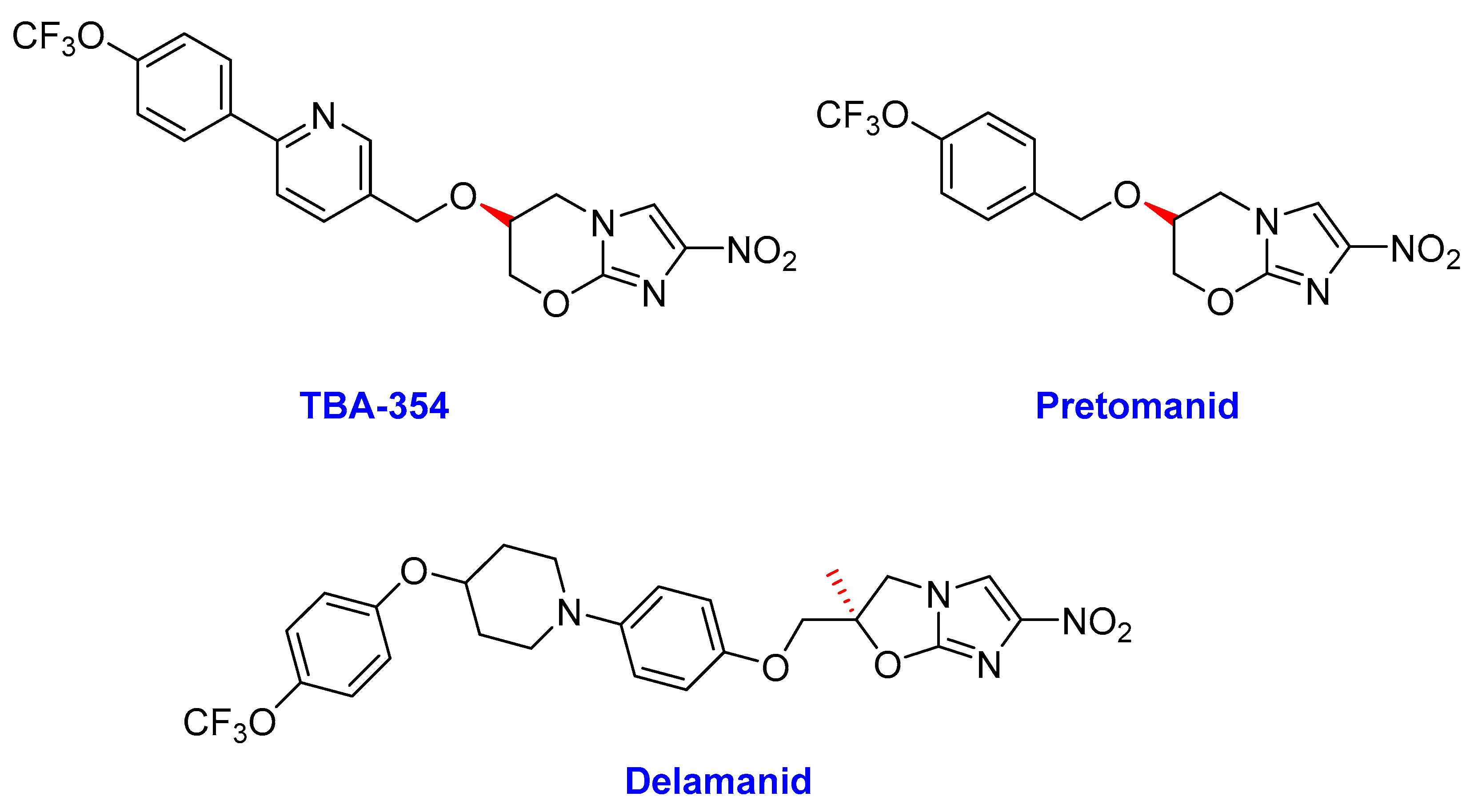
2.3. Nitroimidazoles
- (i)
- Delamanid
- (ii)
- Pretomanid (PD)
- (iii)
- TBA-354
2.4. Oxazolidinones
- (i)
- Linezolid (LZD)
- (ii)
- Sutezolid
- (iii)
- Posizolid (AZD-5847)
- (iv) Tedizolid (TZD)
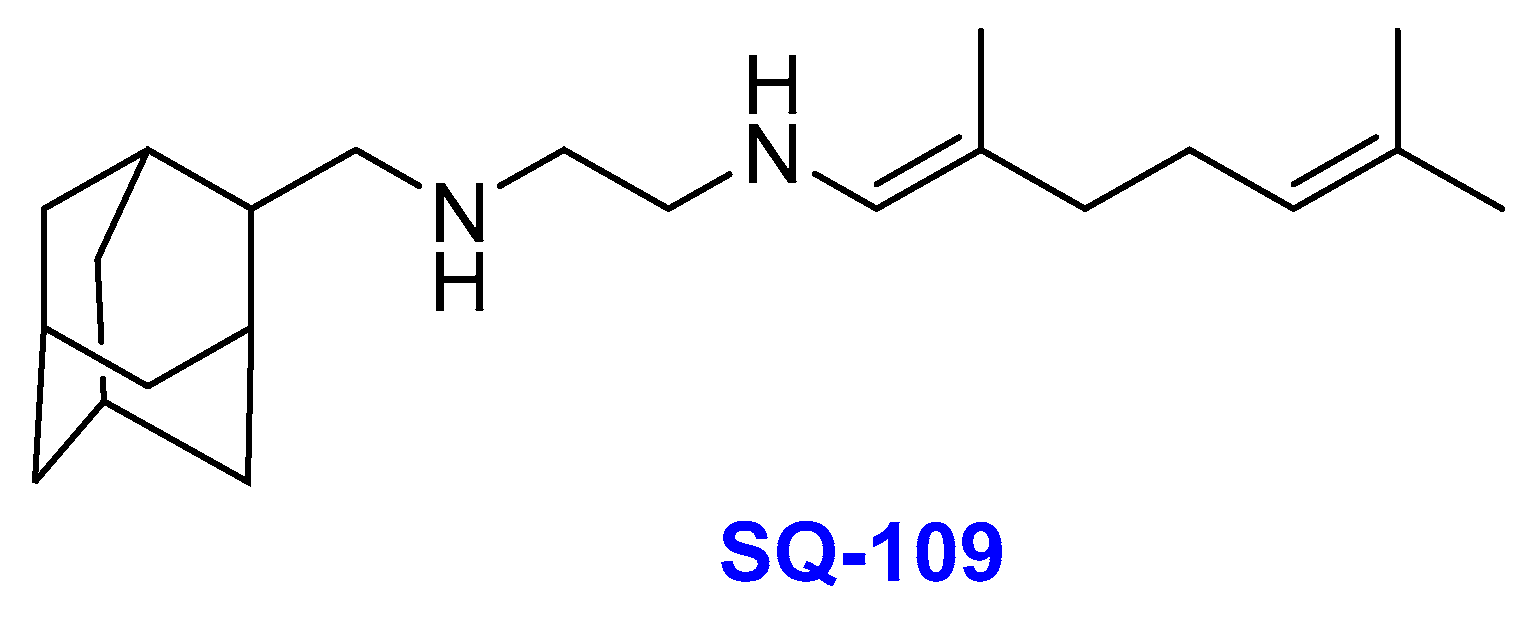
2.5. Ethylenediamines
- (i)
- SQ-109
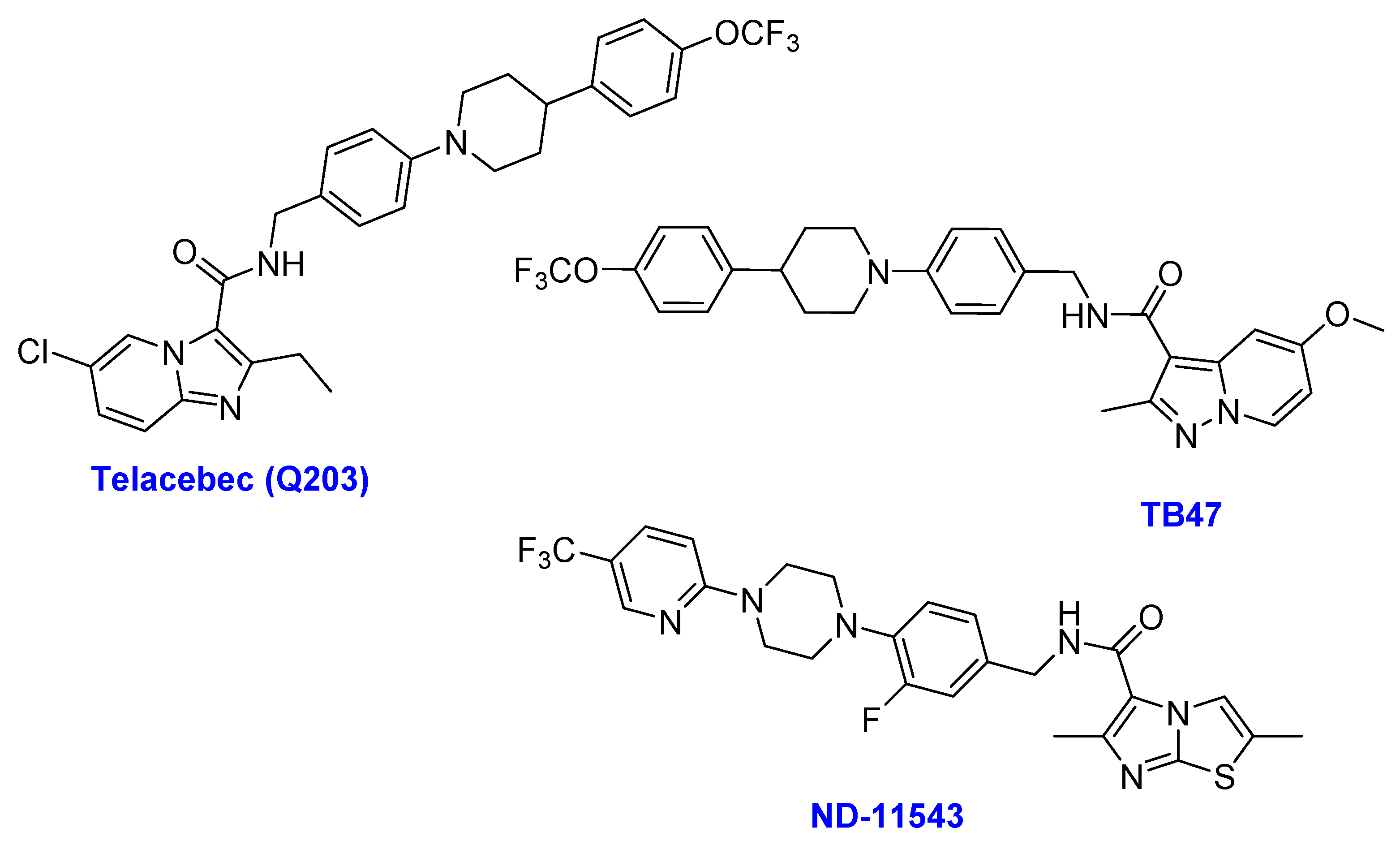
2.6. Imidazopyridine Amides
- (i)
- Telacebec (Q203)
- (ii)
- TB47
- (iii)
- ND-11543
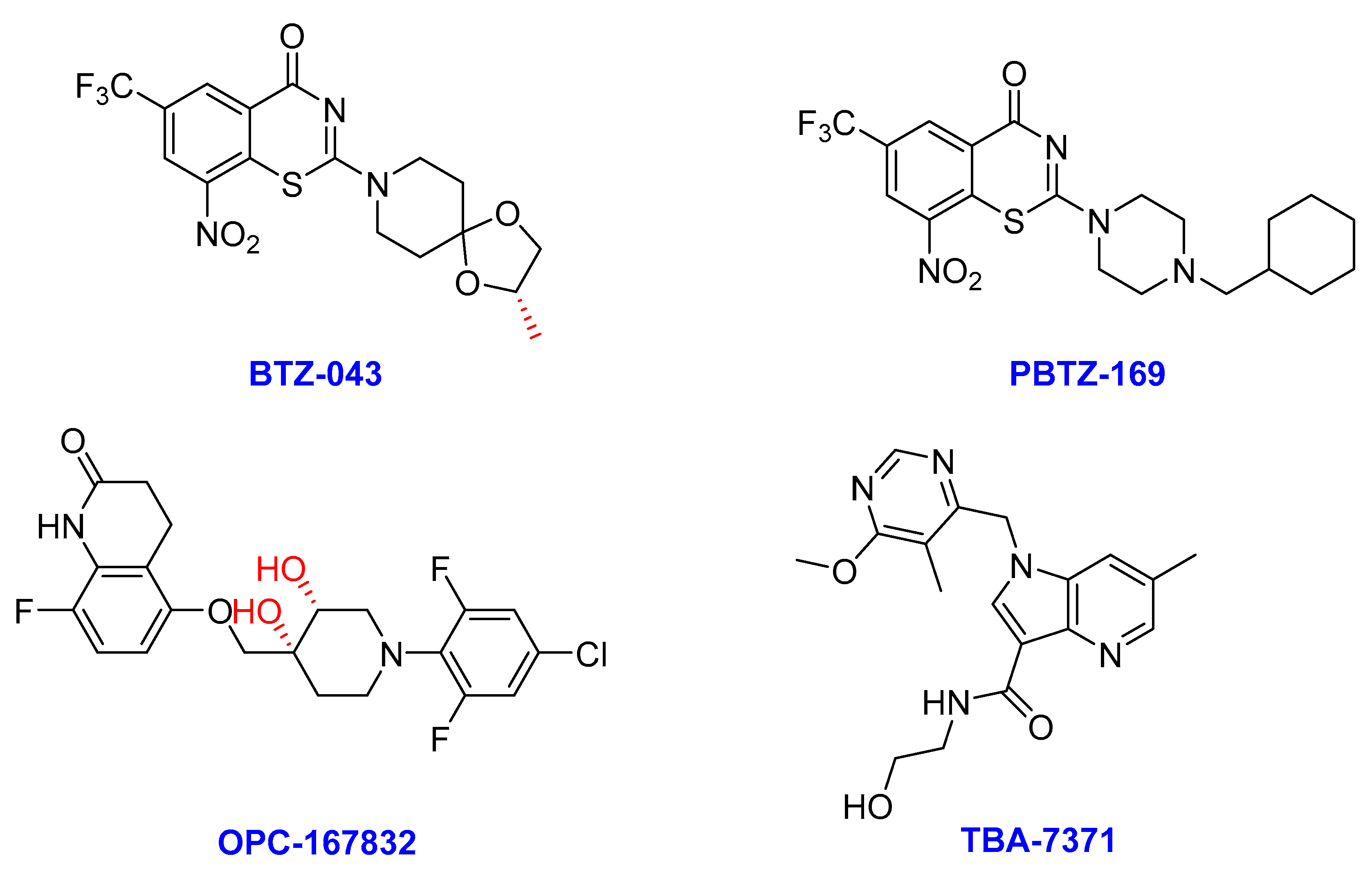
2.7. DprE1 Inhibitors
- (i)
- BTZ-043
- (ii)
- Macozinone (PBTZ-169)
- (iii)
- OPC-167832
- (iv) TBA-7371
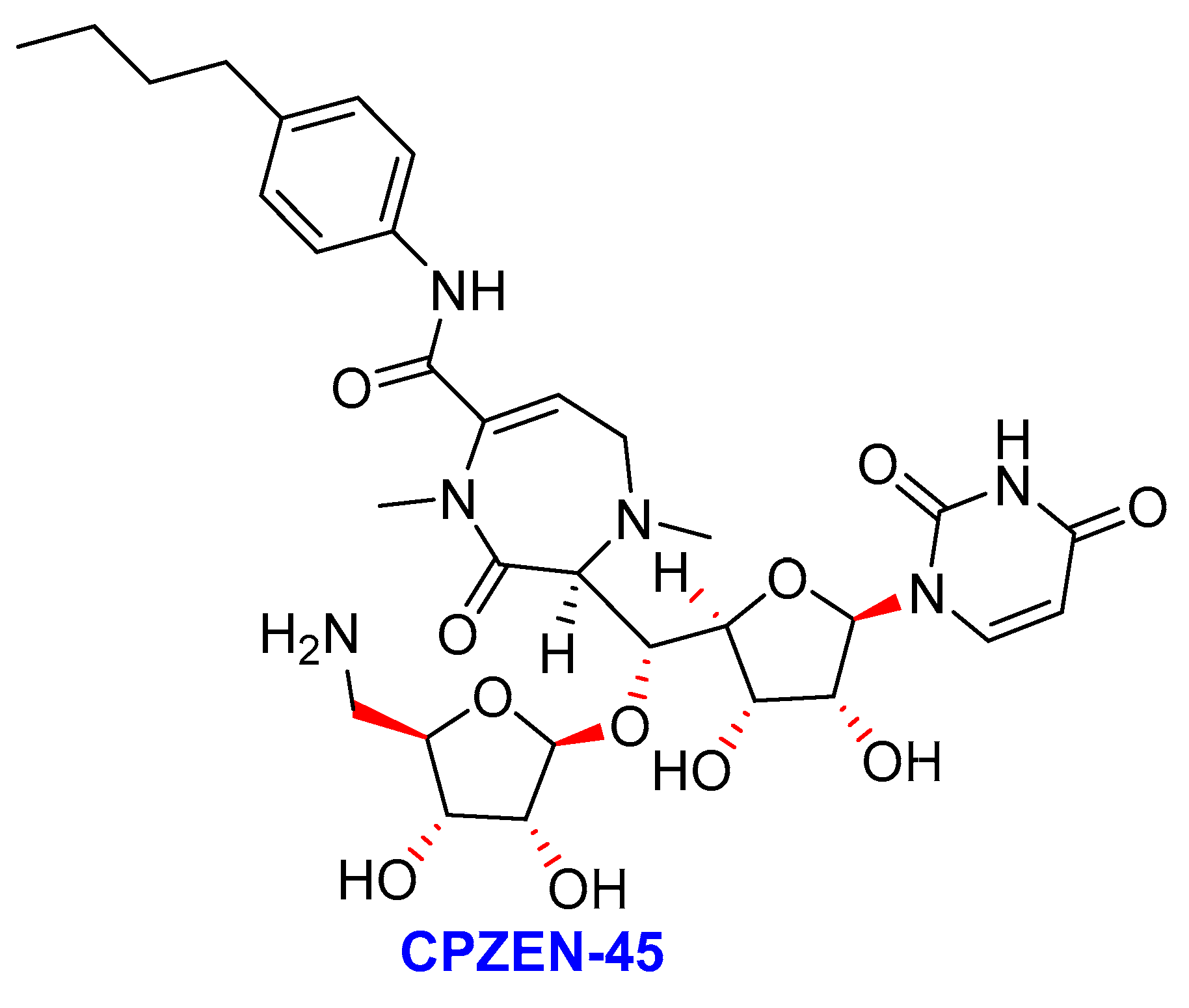
2.8. Caprazamycins
- (i)
- Caprezene-4-butylanilide (CPZEN-45)
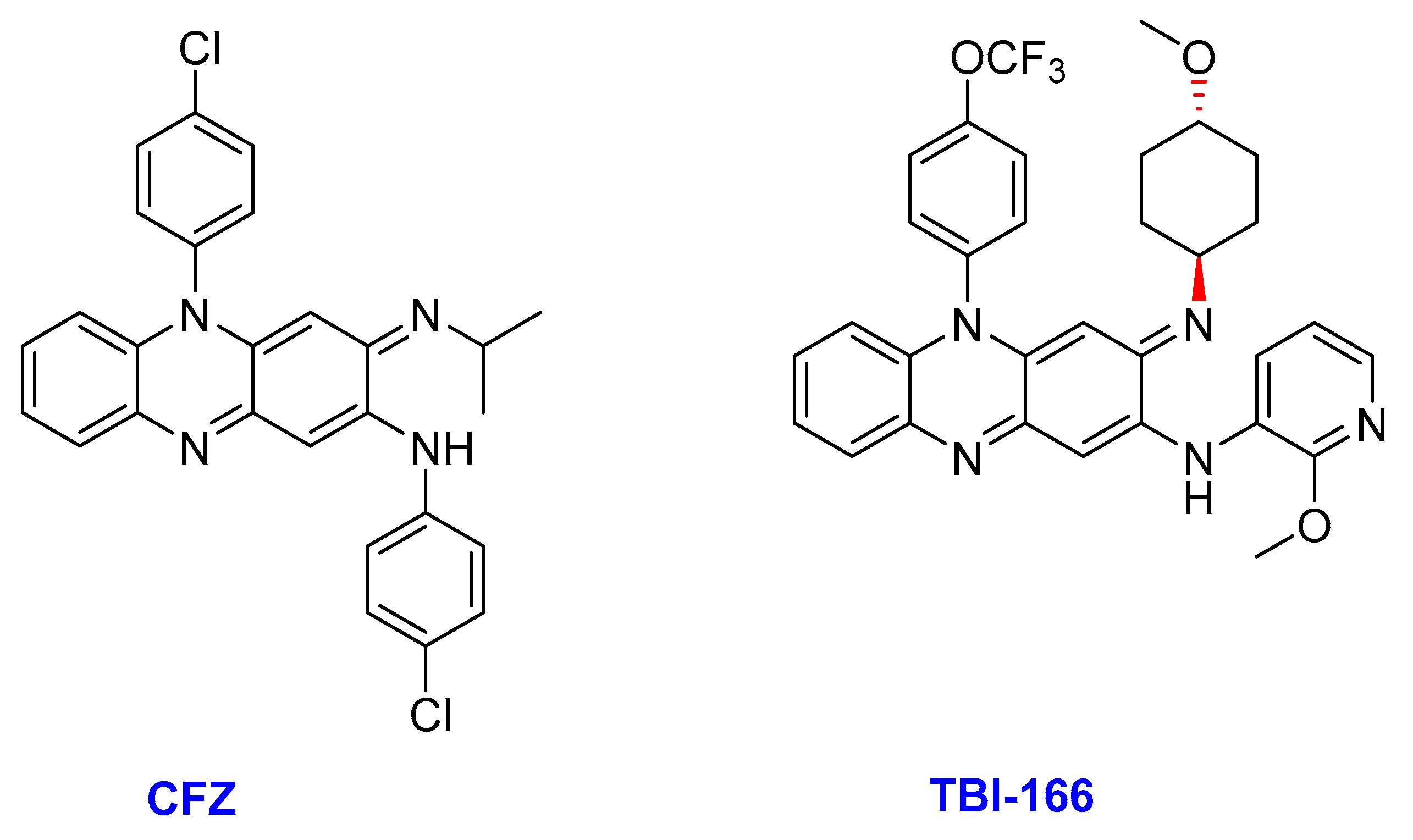
2.9. Riminophenazine
- (ii)
- TBI-166
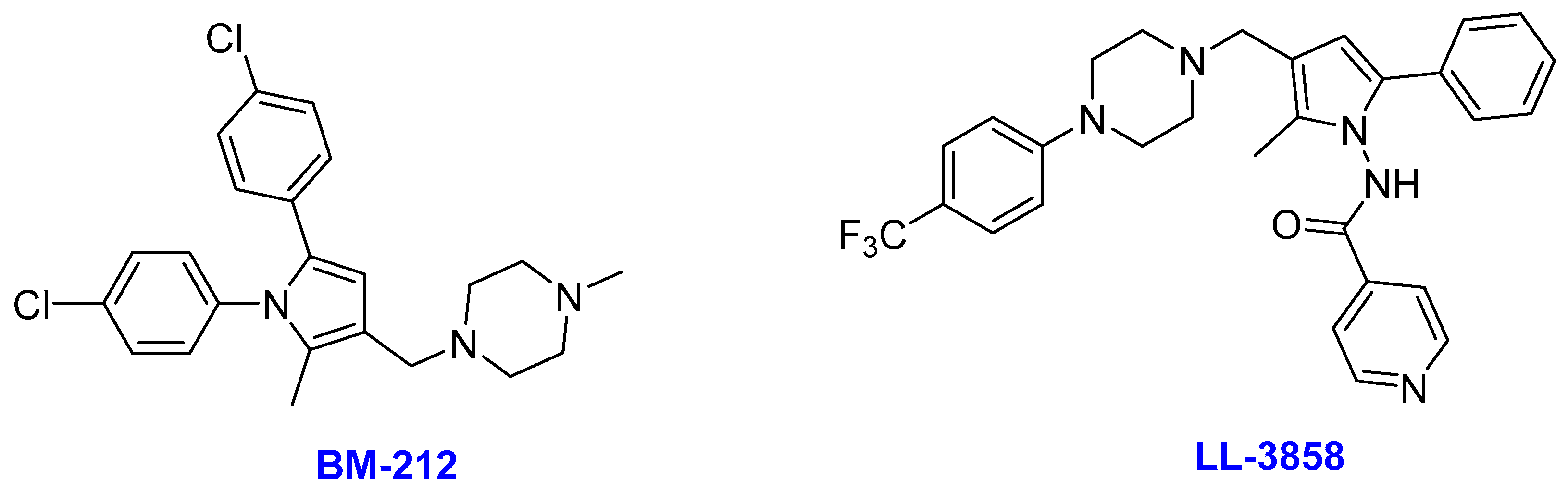
2.10. Pyrroles
- (i)
- BM212
- (ii)
- LL-3858
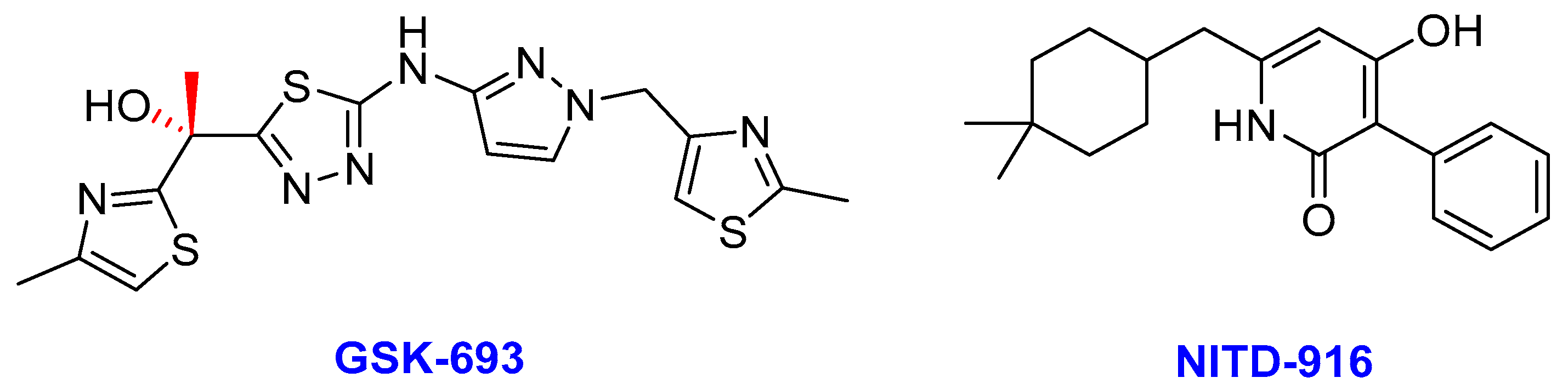
2.11. InhA Inhibitors
- (i)
- GSK-693
- (ii)
- NITD-916
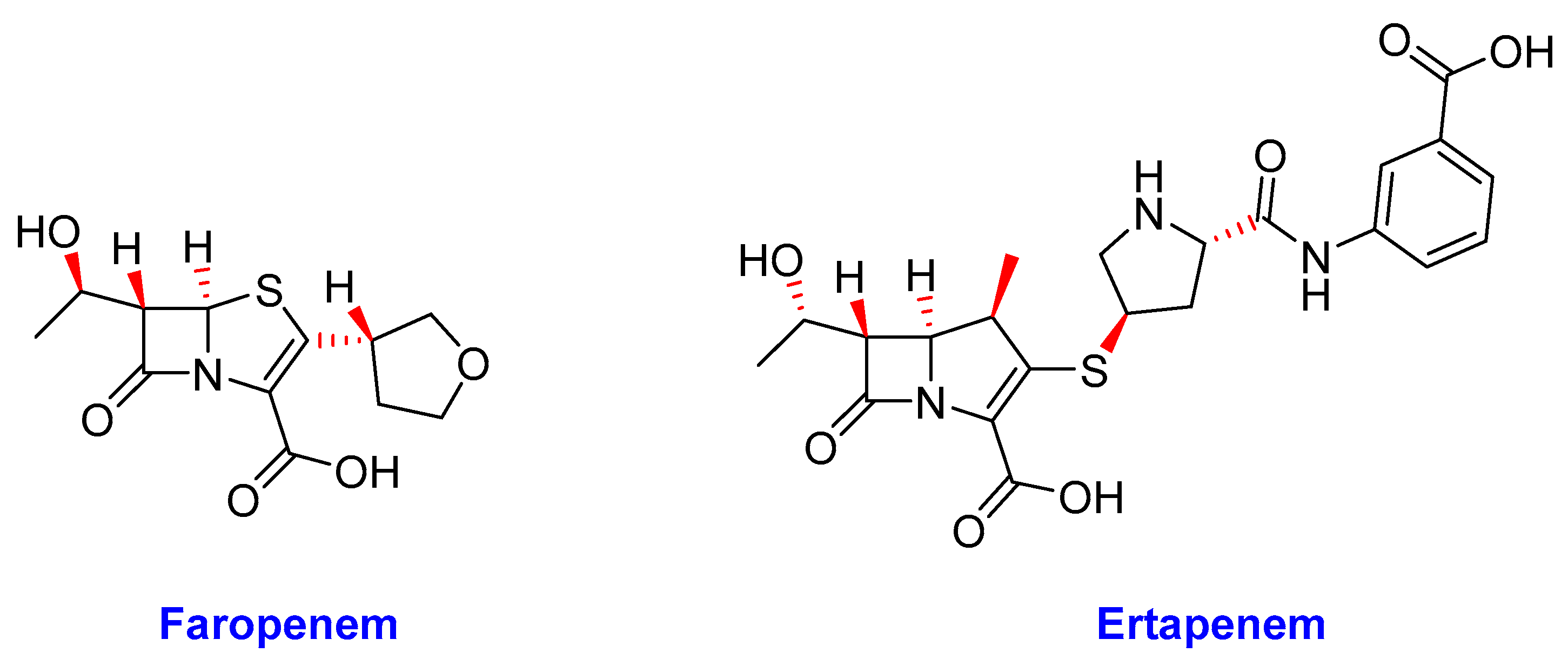
2.12. β-Lactams
- (i)
- Faropenem
- (ii)
- Ertapenem
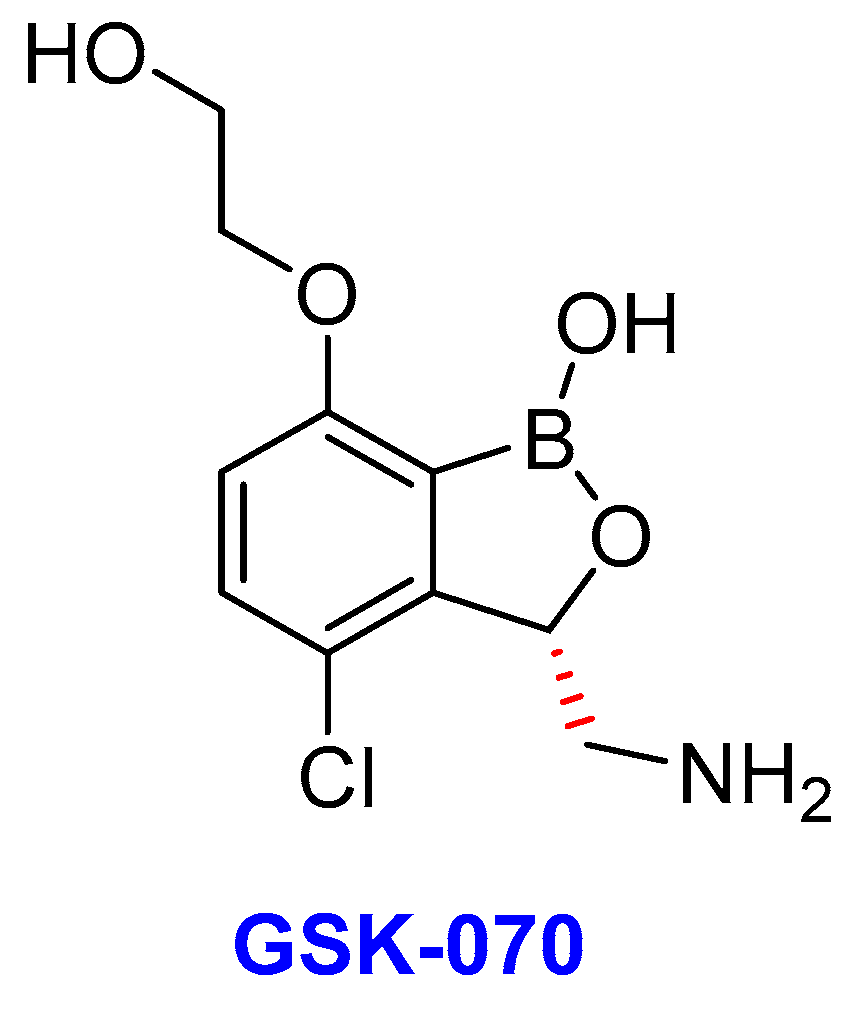
2.13. Oxoborates
- (i)
- GSK-3036656 (GSK-070)
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patil, J. Novel tubercular therapeutic agents: Need of the Day. Pharmacoepidemiol. Drug Saf. 2015, 4, e137. [Google Scholar]
- World Health Organization. Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-report-2019 (accessed on 18 June 2020).
- Brites, D.; Loiseau, C.; Menardo, F.; Borrell, S.; Boniotti, M.; Warren, R.; Dippenaar, A.; Parsons, S.D.; Beisel, C.; Behr, M.A.; et al. A new phylogenetic framework for the animal-adapted Mycobacterium tuberculosis complex. Front. Microbiol. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.C.; Brisse, S.; Brosch, R.; Fabre, M.; Omaïs, B.; Marmiesse, M.; Supply, P.; Vincent, V. Ancient origin and gene mosaicism of the progenitor of Mycobacterium tuberculosis. PLoS Pathog. 2005, 1, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Glaziou, P.; Sismanidis, C.; Floyd, K.; Raviglione, M. Global epidemiology of tuberculosis. Cold Spring Harb Perspect. Med. 2014, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.L.; Flynn, J.L. Understanding latent tuberculosis: A moving target. J. Immunol. 2010, 185, 15–22. [Google Scholar] [CrossRef]
- Heemskerk, D.; Caws, M.; Marais, B.; Farrar, J. Tuberculosis in Adults and Children; Springer: London, UK, 2015; pp. 1–71, Springer eBook collection. Available online: https://www.ncbi.nlm.nih.gov/books/NBK344402 (accessed on 12 May 2020).
- Moghadam, M.T.; Amirmozafari, N.; Shariati, A.; Hallajzadeh, M.; Mirkalantari, S.; Khoshbayan, A.; Jazi, F.M. How phages overcome the challenges of drug resistant bacteria in clinical infections. Infect. Drug Resist. 2020, 13, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Castañeda-Hernández, D.M.; Rodriguez-Morales, A.J. Epidemiological burden of tuberculosis in developing countries. In Current Topics in Public Health; Rodriguez-Morales, A.J., Ed.; Intech: Trujillo, Venezuela, 2013; pp. 317–340. [Google Scholar]
- Kawatsu, L.; Uchimura, K.; Izumi, K.; Ohkado, A.; Ishikawa, N. Profile of tuberculosis among the foreign-born population in Japan, 2007–2014. Western Pac. Surveill Response J. 2016, 7, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Lee, S.; Ishizuka, A.; Hinoshita, E.; Hori, H.; Ishibashi, N.; Komada, K.; Norizuki, M.; Katsuma, Y.; Akashi, H.; et al. Challenges and opportunities for eliminating tuberculosis–leveraging political momentum of the UN high-level meeting on tuberculosis. BMC Public Health 2019, 19, 1–7. [Google Scholar] [CrossRef]
- Kumar, K.; Kon, O.M. Diagnosis and treatment of tuberculosis: Latest developments and future priorities. Ann. Res. Hosp. 2017, 1, 1–15. [Google Scholar] [CrossRef]
- Patil, J. New theoretical background for tuberculosis treatment. J. Pharmacovigil. 2014, 2, 123–124. [Google Scholar] [CrossRef]
- Kanabus, A. Information about Tuberculosis. GHE: United Kingdom. 2020. Available online: https://www.tbfacts.org (accessed on 5 May 2020).
- Eker, B.; Ortmann, J.; Migliori, G.B.; Sotgiu, G.; Muetterlein, R.; Centis, R.; Hoffmann, H.; Kirsten, D.; Schaberg, T.; Ruesch-Gerdes, S.; et al. Multidrug-and extensively drug-resistant tuberculosis, Germany. Emerg. Infect. Dis. 2008, 14, 1700–1706. [Google Scholar] [CrossRef] [PubMed]
- World Health Organisation. Available online: https://www.who.int/tb/publications/2019/consolidated-guidelines-drug-resistant-TB-treatment/en/ (accessed on 25 May 2020).
- Sharma, S.K.; Dheda, K. What is new in the WHO consolidated guidelines on drug-resistant tuberculosis treatment? Indian J. Med. Res. 2019, 149, 309–312. [Google Scholar] [PubMed]
- Rendon, A.; Tiberi, S.; Scardigli, A.; D’Ambrosio, L.; Centis, R.; Caminero, J.A.; Migliori, G.B. Classification of drugs to treat multidrug-resistant tuberculosis (MDR-TB): Evidence and perspectives. J. Thorac. Dis. 2016, 8, 2666. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.S.; Challagundla, L.; Baugh, E.H.; Omar, S.V.; Mustaev, A.; Auld, S.C.; Shah, N.S.; Kreiswirth, B.N.; Brust, J.C.; Nelson, K.N.; et al. Pre-detection history of extensively drug-resistant tuberculosis in KwaZulu-Natal, South Africa. Proc. Natl. Acad. Sci. USA. 2019, 116, 23284–23291. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L. Antibiotic resistance mechanisms in M. tuberculosis: An update. Arch. Toxicol. 2016, 90, 1585–1604. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.; Wolff, K.A.; Nguyen, L. Molecular biology of drug resistance in Mycobacterium tuberculosis. In Pathogenesis of Mycobacterium Tuberculosis and Its Interaction with the Host Organism; Pieters, J., McKinney, J., Eds.; Springer: Heidelberg/Berlin, Germany, 2012; Volume 374, pp. 53–80. [Google Scholar]
- Arya, N.; Raut, M.K.; Tekale, S.G.; Shishoo, C.J.; Jain, K.S. Tuberculosis: New drug discovery pipelines. Austin J. Anal. Pharm. Chem. 2014, 1, 1–9. [Google Scholar]
- Poissy, J.; Aubry, A.; Fernandez, C.; Lott, M.-C.; Chauffour, A.; Jarlier, V.; Farinotti, R.; Veziris, N. Should moxifloxacin be used for the treatment of extensively drug-resistant tuberculosis? An answer from a murine model. Antimicrob. Agents Chemother. 2010, 54, 4765–4771. [Google Scholar] [CrossRef] [PubMed]
- Aubry, A.; Pan, X.-S.; Fisher, L.M.; Jarlier, V.; Cambau, E. Mycobacterium tuberculosis DNA gyrase: Interaction with quinolones and correlation with antimycobacterial drug activity. Antimicrob. Agents Chemother. 2004, 48, 1281–1288. [Google Scholar] [CrossRef]
- Madhusudan, K.; Ramesh, V.; Nagaraja, V. Molecular cloning of gyrA and gyrB genes of Mycobacterium tuberculosis: Analysis of nucleotide sequence. Int. J. Biochem. Mol. Biol. 1994, 33, 651–660. [Google Scholar]
- Gillespie, S.H. The role of moxifloxacin in tuberculosis therapy. Eur. Respir. J. 2016, 25, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Jindani, A.; Harrison, T.S.; Nunn, A.J.; Phillips, P.P.; Churchyard, G.J.; Charalambous, S.; Hatherill, M.; Geldenhuys, H.; McIlleron, H.M.; Zvada, S.P.; et al. High-dose rifapentine with moxifloxacin for pulmonary tuberculosis. N. Engl. J. Med. 2014, 371, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, A.; Naidoo, K.; McIlleron, H.; Essack, S.; Padayatchi, N. A review of moxifloxacin for the treatment of drug-susceptible tuberculosis. J. Clin. Pharmacol. 2017, 57, 1369–1386. [Google Scholar] [CrossRef]
- Dookie, N.; Sturm, A.W.; Moodley, P. Moxifloxacin resistance in the F15/LAM4/KZN extensively drug-resistant strain of Mycobacterium tuberculosis. Infect. Drug Resist. 2014, 7, 223–228. [Google Scholar] [PubMed]
- Pletz, M.W.R.; De Roux, A.; Roth, A.; Neumann, K.-H.; Mauch, H.; Lode, H. Early bactericidal activity of moxifloxacin in treatment of pulmonary tuberculosis: A prospective, randomized study. Antimicrob. Agents Chemother. 2004, 48, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.C.; Ruiz, M.; López, M.; Royo, G. In vitro activity of moxifloxacin, levofloxacin, gatifloxacin and linezolid against Mycobacterium tuberculosis. Int. J. Antimicrob. Agents 2002, 20, 464–467. [Google Scholar] [CrossRef]
- Balfour, J.A.B.; Wiseman, L.R. Moxifloxacin. Drugs 1999, 57, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Nijland, H.; Ruslami, R.; Suroto, A.J.; Burger, D.; Alisjahbana, B.; Van Crevel, R.; Aarnoutse, R.E. Rifampicin reduces plasma concentrations of moxifloxacin in patients with tuberculosis. Clin. Infect. Dis. 2007, 45, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Al Omari, M.M.; Jaafari, D.S.; Al-Sou’od, K.A.; Badwan, A.A. Moxifloxacin hydrochloride. Profiles Drug Subst. Excip. Relat. Methodol. 2014, 39, 299–431. [Google Scholar] [PubMed]
- Zvada, S.P.; Denti, P.; Sirgel, F.A.; Chigutsa, E.; Hatherill, M.; Charalambous, S.; Mungofa, S.; Wiesner, L.; Simonsson, U.S.; Jindani, A.; et al. Moxifloxacin population pharmacokinetics and model-based comparison of efficacy between moxifloxacin and ofloxacin in African patients. Antimicrob. Agents Chemother. 2014, 58, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.F.; Hsueh, P.-R.; Gillespie, S.H.; Blasi, F. Community-acquired pneumonia and tuberculosis: Differential diagnosis and the use of fluoroquinolones. Int. J. Infect. Dis. 2014, 18, 14–21. [Google Scholar] [CrossRef]
- Hofman, S.; Segers, M.; Ghimire, S.; Bolhuis, M.; Sturkenboom, M.; Van Soolingen, D.; Alffenaar, J.W. Emerging drugs and alternative possibilities in the treatment of tuberculosis. Expert Opin. Emerg. Drugs 2016, 21, 103–116. [Google Scholar] [CrossRef]
- Gajjar, D.A.; LaCreta, F.P.; Uderman, H.D.; Kollia, G.D.; Duncan, G.; Birkhofer, M.J.; Grasela, D.M. A dose-escalation study of the safety, tolerability, and pharmacokinetics of intravenous gatifloxacin in healthy adult men. Pharmacotherapy 2000, 20, 49–58. [Google Scholar] [CrossRef]
- Nakashima, M.; Uematsu, T.; Kosuge, K.; Kusajima, H.; Ooie, T.; Masuda, Y.; Ishida, R.Y.; Uchida, H.I. Single-and multiple-dose pharmacokinetics of AM-1155, a new 6-fluoro-8-methoxy quinolone, in humans. Antimicrob. Agents Chemother. 1995, 39, 2635–2640. [Google Scholar] [CrossRef]
- Deshpande, D.; Pasipanodya, J.G.; Srivastava, S.; Bendet, P.; Koeuth, T.; Bhavnani, S.M.; Ambrose, P.G.; Smythe, W.; McIlleron, H.; Thwaites, G.; et al. Gatifloxacin pharmacokinetics/pharmacodynamics–based optimal dosing for pulmonary and meningeal multidrug-resistant tuberculosis. Clin. Infect. Dis. 2018, 67, 274–283. [Google Scholar] [CrossRef]
- LaCreta, F.P.; Kaul, S.; Kollia, G.D.; Duncan, G.; Randall, D.M.; Grasela, D.M. Interchangeability of 400-mg intravenous and oral gatifloxacin in healthy adults. Pharmacotherapy 2000, 20, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Grasela, D.M. Clinical pharmacology of gatifloxacin, a new fluoroquinolone. Clin. Infect. Dis. 2000, 31, 51–58. [Google Scholar] [CrossRef]
- Mignot, A.; Guillaume, M.; Brault, M.; Gualano, V.; Millérioux, L.; Göhler, K.; Stahlberg, H.J. Multiple-dose pharmacokinetics and excretion balance of gatifloxacin, a new fluoroquinolone antibiotic, following oral administration to healthy Caucasian volunteers. Chemotherapy 2002, 48, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Ganguly, A.; Sunwoo, H. Current overview of anti-tuberculosis Drugs: Metabolism and toxicities. Mycobact. Dis. 2016, 6, 1–6. [Google Scholar] [CrossRef]
- Chiang, C.; Van Deun, A.; Rieder, H. Gatifloxacin for short, effective treatment of multidrug-resistant tuberculosis. Int. J. Tuberc. Lung. Dis. 2016, 20, 1143–1147. [Google Scholar] [CrossRef]
- Quan, D.; Nagalingam, G.; Payne, R.; Triccas, J.A. New tuberculosis drug leads from naturally occurring compounds. J. Infect. Dis. 2017, 56, 212–220. [Google Scholar] [CrossRef]
- Ahmad, Z.; Minkowski, A.; Peloquin, C.A.; Williams, K.N.; Mdluli, K.E.; Grosset, J.H.; Nuermberger, E.L. Activity of the fluoroquinolone DC-159a in the initial and continuation phases of treatment of murine tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 1781–1783. [Google Scholar] [CrossRef] [PubMed]
- Onodera, Y.; Hirata, T.; Hoshino, K.; Otani, T. DC-159a, a novel quinolone, showed high inhibitory activity against altered topoisomerases of Streptococcus pneumoniae and Mycobacterium tuberculosis. In Proceedings of the 47th Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL, USA, 17–20 September 2007; American Society for Microbiology: Washington, DC, USA; pp. F1–F2126. [Google Scholar]
- Asif, M.; Farhan, S.S. An overview on antitubercular activity profile of fluoroquinolone derivatives and their molecular hybridization. J. Med. Chem. 2020, 3, 145–153. [Google Scholar]
- Disratthakit, A.; Doi, N. In vitro activities of DC-159a, a novel fluoroquinolone, against Mycobacterium species. Antimicrob. Agents Chemother. 2010, 54, 2684–2686. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, J.; Disratthakit, A.; Maeda, S.; Doi, N. Characteristic resistance mechanism of Mycobacterium tuberculosis to DC-159a, a new respiratory quinolone. Antimicrob. Agents Chemother. 2011, 55, 3958–3960. [Google Scholar] [CrossRef]
- Villemagne, B.; Crauste, C.; Flipo, M.; Baulard, A.R.; Déprez, B.; Willand, N. Tuberculosis: The drug development pipeline at a glance. Eur. J. Med. Chem. 2012, 51, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mdluli, K.; Ma, Z. Mycobacterium tuberculosis DNA gyrase as a target for drug discovery. Infect. Disord. Drug Targets. 2007, 7, 159–168. [Google Scholar] [CrossRef]
- Schaaf, H.S.; Thee, S.; van der Laan, L.; Hesseling, A.C.; Garcia-Prats, A.J. Adverse effects of oral second-line antituberculosis drugs in children. Expert Opin. Drug Saf. 2016, 15, 1369–1381. [Google Scholar] [CrossRef]
- Koul, A.; Vranckx, L.; Dendouga, N.; Balemans, W.; Van den Wyngaert, I.; Vergauwen, K.; Göhlmann, H.W.; Willebrords, R.; Poncelet, A.; Guillemont, J.; et al. Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J. Biol. Chem. 2008, 283, 25273–25280. [Google Scholar] [CrossRef] [PubMed]
- Biuković, G.; Basak, S.; Manimekalai, M.S.S.; Rishikesan, S.; Roessle, M.; Dick, T.; Rao, S.P.; Hunke, C.; Grüber, G. Variations of subunit ε of the Mycobacterium tuberculosis F1Fo ATP synthase and a novel model for mechanism of action of the tuberculosis drug TMC207. Antimicrob. Agents Chemother. 2013, 57, 168–176. [Google Scholar] [CrossRef]
- Olaru, I.D.; von Groote-Bidlingmaier, F.; Heyckendorf, J.; Yew, W.W.; Lange, C.; Chang, K.C. Novel drugs against tuberculosis: A clinician’s perspective. Eur. Respir. J. 2015, 45, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Hards, K.; McMillan, D.G.; Schurig-Briccio, L.A.; Gennis, R.B.; Lill, H.; Bald, D.; Cook, G.M. Ionophoric effects of the antitubercular drug bedaquiline. Proc. Natl. Acad. Sci. USA 2018, 115, 7326–7331. [Google Scholar] [CrossRef] [PubMed]
- Matteelli, A.; Carvalho, A.C.; Dooley, K.E.; Kritski, A. TMC207: The first compound of a new class of potent anti-tuberculosis drugs. Future Microbiol. 2010, 5, 849–858. [Google Scholar] [CrossRef]
- Dooley, K.E.; Park, J.-G.; Swindells, S.; Allen, R.; Haas, D.W.; Cramer, Y.; Aweeka, F.; Wiggins, I.; Gupta, A.; Lizak, P.; et al. Safety, tolerability, and pharmacokinetic interactions of the antituberculous agent TMC207 (bedaquiline) with efavirenz in healthy volunteers: AIDS Clinical Trials Group Study A5267. J. Acquir. Immune Defic. Syndr. 2012, 59, 455–462. [Google Scholar] [CrossRef]
- Pethe, K.; Bifani, P.; Jang, J.; Kang, S.; Park, S.; Ahn, S.; Jiricek, J.; Jung, J.; Jeon, H.K.; Cechetto, J.; et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 2013, 19, 1157–1160. [Google Scholar] [CrossRef]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Göhlmann, H.W.; Neefs, J.-M.; Winkler, H.; Van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Gobedo, A.; Hwoldi, A.; Toma, A. Recent advances in the development of anti-tuberculosis drugs acting on multidrug-resistant strains: A review. Int. J. Pharm. Biol. Sci. 2015, 2, 1–18. [Google Scholar]
- Nguyen, T.; Cao, T.; Akkerman, O.; Tiberi, S.; Vu, D.; Alffenaar, J. Bedaquiline as part of combination therapy in adults with pulmonary multi-drug resistant tuberculosis. Expert Rev. Clin. Pharmacol. 2016, 9, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Van Heeswijk, R.P.G.; Dannemann, B.; Hoetelmans, R.M.W. Bedaquiline: A review of human pharmacokinetics and drug–drug interactions. Antimicrob. Agents Chemother. 2014, 69, 2310–2318. [Google Scholar] [CrossRef]
- Lounis, N.; Gevers, T.; Van Den Berg, J.; Andries, K. Impact of the interaction of R207910 with rifampin on the treatment of tuberculosis studied in the mouse model. Antimicrob. Agents Chemother. 2008, 52, 3568–3572. [Google Scholar] [CrossRef] [PubMed]
- Tiberi, S.; du Plessis, N.; Walzl, G.; Vjecha, M.J.; Rao, M.; Ntoumi, F.; Mfinanga, S.; Kapata, N.; Mwaba, P.; McHugh, T.D.; et al. Tuberculosis: Progress and advances in development of new drugs, treatment regimens, and host-directed therapies. Lancet Infect. Dis. 2018, 18, 183–198. [Google Scholar] [CrossRef]
- Fox, G.J.; Menzies, D. A review of the evidence for using bedaquiline (TMC207) to treat multi-drug resistant tuberculosis. Infect. Dis Ther. 2013, 2, 123–144. [Google Scholar] [CrossRef] [PubMed]
- Van Heeswijk, R.; Vandevoorde, A.; Meyvisch, P.; De Marez, T.; De Beule, K.; Mc Neeley, D.; Hoetelmans, R. The effect of lopinavir/ritonavir on the pharmacokinetics of TMC207, an investigational antimycobacterial agent. In Proceedings of the 18th International AIDS Conference, Viena, Austria, 18–23 July 2010. [Google Scholar]
- Svensson, E.M.; Aweeka, F.; Park, J.-G.; Marzan, F.; Dooley, K.E.; Karlsson, M.O. Model-based estimates of the effects of efavirenz on bedaquiline pharmacokinetics and suggested dose adjustments for patients coinfected with HIV and tuberculosis. Antimicrob. Agents Chemother. 2013, 57, 2780–2787. [Google Scholar] [CrossRef]
- Coyne, K.M.; Pozniak, A.L.; Lamorde, M.; Boffito, M. Pharmacology of second-line antituberculosis drugs and potential for interactions with antiretroviral agents. Aids 2009, 23, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, D.T.; Liu, J.; Lee, R.B.; Lee, R.E. New agents for the treatment of drug-resistant Mycobacterium tuberculosis. Adv. Drug Deliv. Rev. 2016, 102, 55–72. [Google Scholar] [CrossRef]
- Pontali, E.; Sotgiu, G.; Tiberi, S.; D’Ambrosio, L.; Centis, R.; Migliori, G.B. Cardiac safety of bedaquiline: A systematic and critical analysis of the evidence. Eur. Respir. J. 2017, 50, 1–10. [Google Scholar] [CrossRef]
- Sarathy, J.P.; Ragunathan, P.; Shin, J.; Cooper, C.B.; Upton, A.M.; Grüber, G.; Dick, T. TBAJ-876 retains bedaquiline’s activity against subunits c and ε of Mycobacterium tuberculosis F-ATP synthase. Antimicrob. Agents Chemother. 2019, 63, 1–11. [Google Scholar] [CrossRef]
- Sutherland, H.S.; Tong, A.S.; Choi, P.J.; Conole, D.; Blaser, A.; Franzblau, S.G.; Cooper, C.B.; Upton, A.M.; Lotlikar, M.U.; Denny, W.A.; et al. Structure-activity relationships for analogs of the tuberculosis drug bedaquiline with the naphthalene unit replaced by bicyclic heterocycles. Bioorg. Med. Chem. 2018, 26, 1797–1809. [Google Scholar] [CrossRef] [PubMed]
- Choi, P.J.; Sutherland, H.S.; Blaser, A.; Tong, A.S.; Cooper, C.B.; Upton, A.M.; Upton, A.M.; Palmer, B.D.; Denny, W.A. Synthetic studies to help elucidate the metabolism of the preclinical candidate TBAJ-876—A less toxic and more potent analogue of bedaquiline. Molecules 2020, 25, 1423. [Google Scholar] [CrossRef] [PubMed]
- Sarathy, J.P.; Gruber, G.; Dick, T. Re-understanding the mechanisms of action of the anti-mycobacterial drug bedaquiline. Antibiotics 2019, 8, 261. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, H.S.; Tong, A.S.; Choi, P.J.; Blaser, A.; Conole, D.; Franzblau, S.G.; Lotlikar, M.U.; Cooper, C.B.; Upton, A.M.; Denny, W.A.; et al. 3, 5-Dialkoxypyridine analogues of bedaquiline are potent antituberculosis agents with minimal inhibition of the hERG channel. Bioorg. Med. Chem. 2019, 27, 1292–1307. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, H.S.; Tong, A.S.; Choi, P.J.; Blaser, A.; Franzblau, S.G.; Cooper, C.B.; Upton, A.M.; Lotlikar, M.; Denny, W.A.; Palmer, B.D. Variations in the C-unit of bedaquiline provides analogues with improved biology and pharmacology. Bioorg. Med. Chem. 2020, 28, 1–13. [Google Scholar] [CrossRef]
- Kumar, D.; Negi, B.; Rawat, D.S. The anti-tuberculosis agents under development and the challenges ahead. Future Med. Chem. 2015, 7, 1981–2003. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.-S.; Jeong, B.-H.; Koh, W.-J. Delamanid when other anti-tuberculosis-treatment regimens failed due to resistance or tolerability. Expert Opin. Pharmacother. 2015, 16, 253–261. [Google Scholar] [CrossRef]
- Szumowski, J.D.; Lynch, J.B. Profile of delamanid for the treatment of multidrug-resistant tuberculosis. Drug Des. Devel. Ther. 2015, 9, 677–682. [Google Scholar] [PubMed]
- Karekar, S.R.; Marathe, P.A. Current status of delamanid in the management of MDR tuberculosis. J. Assoc. Physicians India 2018, 66, 72–75. [Google Scholar] [PubMed]
- Ryan, N.J.; Lo, J.H. Delamanid: First global approval. Drugs 2014, 74, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- TB Alliance. Available online: https://www.treatmentactiongroup.org/wp-content/uploads/2019/12/pipeline_tb_treatment_lm_final.pdf (accessed on 24 May 2020).
- Saliu, O.Y.; Crismale, C.; Schwander, S.K.; Wallis, R.S. Bactericidal activity of OPC-67683 against drug-tolerant Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2007, 60, 994–998. [Google Scholar] [CrossRef]
- Xavier, A.S.; Lakshmanan, M. Delamanid: A new armor in combating drug-resistant tuberculosis. J. Pharm. Pharm. 2014, 5, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A.; Scott, L.J. Delamanid: A review of its use in patients with multidrug-resistant tuberculosis. Drugs 2015, 75, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, Y.; Sasahara, K.; Yoda, N.; Mizuno, K.; Umehara, K. Delamanid does not inhibit or induce cytochrome p450 enzymes in vitro. Biol. Pharm. Bull. 2014, 37, 1727–1735. [Google Scholar] [CrossRef]
- Gler, M.T.; Skripconoka, V.; Sanchez-Garavito, E.; Xiao, H.; Cabrera-Rivero, J.L.; Vargas-Vasquez, D.E.; Gao, M.; Awad, M.; Park, S.K.; Shim, T.S.; et al. Delamanid for multidrug-resistant pulmonary tuberculosis. N. Engl. J. Med. 2012, 366, 2151–2160. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Accinelli, A.; Tejada, F.; Kharel, M. Drugs used in tuberculosis and leprosy. In Side Effects of Drugs Annual. 39; Elsevier: Amsterdam, The Netherlands, 2017; pp. 283–293. [Google Scholar]
- Mallikaarjun, S.; Wells, C.; Petersen, C.; Paccaly, A.; Shoaf, S.E.; Patil, S.; Geiter, L. Delamanid coadministered with antiretroviral drugs or antituberculosis drugs shows no clinically relevant drug-drug interactions in healthy subjects. Antimicrob. Agents Chemother. 2016, 60, 5976–5985. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lewis, J.M.; Sloan, D.J. The role of delamanid in the treatment of drug-resistant tuberculosis. Clin. Risk Manag. 2015, 11, 779–791. [Google Scholar]
- Tyagi, S.; Nuermberger, E.; Yoshimatsu, T.; Williams, K.; Rosenthal, I.; Lounis, N.; Bishai, W.; Grosset, J. Bactericidal activity of the nitroimidazopyran PA-824 in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2005, 49, 2289–2293. [Google Scholar] [CrossRef] [PubMed]
- Manjunatha, U.; Boshoff, H.I.; Barry, C.E. The mechanism of action of PA-824: Novel insights from transcriptional profiling. Commun. Integr. Biol. 2009, 2, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Haver, H.L.; Chua, A.; Ghode, P.; Lakshminarayana, S.B.; Singhal, A.; Mathema, B.; Wintjens, R.; Bifani, P. Mutations in genes for the F420 biosynthetic pathway and a nitroreductase enzyme are the primary resistance determinants in spontaneous in vitro-selected PA-824-resistant mutants of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 5316–5323. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.-P.; Bair, T.B.; Bae, Y.-M.; Daniels, L. Use of transposon Tn5367 mutagenesis and a nitroimidazopyran-based selection system to demonstrate a requirement for fbiA and fbiB in coenzyme F420 biosynthesis by Mycobacterium bovis BCG. J. Bacteriol. 2001, 183, 7058–7066. [Google Scholar] [CrossRef] [PubMed]
- Conradie, F.; Diacon, A.H.; Ngubane, N.; Howell, P.; Everitt, D.; Crook, A.M.; Mendel, C.M.; Egizi, E.; Moreira, J.; Timm, J.; et al. Treatment of highly drug-resistant pulmonary tuberculosis. N. Engl. J. Med. 2020, 382, 893–902. [Google Scholar] [CrossRef]
- TB Alliance. Available online: https://www.tballiance.org/pathway-potential-new-tb-treatments (accessed on 23 February 2021).
- Keam, S.J. Pretomanid: First Approval. Drugs. 2019, 79, 1797–1803. [Google Scholar] [CrossRef] [PubMed]
- Deb, U.; Biswas, S. Pretomanid: The latest USFDA-approved anti-tuberculosis drug. Indian J. Tuberc. 2020, 68, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wedajo, W.; Yamada, Y.; Dahlroth, S.-L.; Neo, J.J.-L.; Dick, T.; Chui, W.K. 1, 3, 5-triazaspiro [5.5] undeca-2, 4-dienes as selective Mycobacterium tuberculosis dihydrofolate reductase inhibitors with potent whole cell activity. Eur. J. Med. Chem. 2018, 144, 262–276. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Zhang, T.; Zong, Z.; Xue, Y.; Shang, Y.; Wang, F.; Huang, H.; Chu, N.; Pang, Y. Comparison of in vitro activity of the nitroimidazoles delamanid and pretomanid against multidrug-resistant and extensively drug-resistant tuberculosis. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1293–1296. [Google Scholar]
- Jia, L.; Noker, P.E.; Coward, L.; Gorman, G.S.; Protopopova, M.; Tomaszewski, J.E. Interspecies pharmacokinetics and in vitro metabolism of SQ109. Br. J. Pharmacol. 2006, 147, 476–485. [Google Scholar] [CrossRef]
- Clain, J.M.; Escalante, P. Novel treatments for drug-resistant tuberculosis. Clin. Med. Insights Ther. 2016, 8, 21–28. [Google Scholar] [CrossRef]
- Dawson, R.; Diacon, A.H.; Everitt, D.; van Niekerk, C.; Donald, P.R.; Burger, D.A.; Schall, R.; Spigelman, M.; Conradie, A.; Eisenach, K.; et al. Efficiency and safety of the combination of moxifloxacin, pretomanid (PA-824), and pyrazinamide during the first 8 weeks of antituberculosis treatment: A phase 2b, open-label, partly randomised trial in patients with drug-susceptible or drug-resistant pulmonary tuberculosis. Lancet 2015, 385, 1738–1747. [Google Scholar] [PubMed]
- Diacon, A.H.; Dawson, R.; du Bois, J.; Narunsky, K.; Venter, A.; Donald, P.R.; van Niekerk, C.; Erondu, N.; Ginsberg, A.M.; Becker, P.; et al. Phase II dose-ranging trial of the early bactericidal activity of PA-824. Antimicrob. Agents Chemother. 2012, 56, 3027–3031. [Google Scholar] [CrossRef]
- Salinger, D.H.; Subramoney, V.; Everitt, D.; Nedelman, J.R. Population pharmacokinetics of the antituberculosis agent pretomanid. Antimicrob. Agents Chemother. 2019, 63, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dooley, K.E.; Luetkemeyer, A.F.; Park, J.-G.; Allen, R.; Cramer, Y.; Murray, S.; Sutherland, D.; Aweeka, F.; Koletar, S.L.; Marzan, F.; et al. Phase I safety, pharmacokinetics, and pharmacogenetics study of the antituberculosis drug PA-824 with concomitant lopinavir-ritonavir, efavirenz, or rifampin. Antimicrob. Agents Chemother. 2014, 58, 5245–5252. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.-C.; Silverman, M.H.; Yu, X.Y.; Wright, G.; Brown, N. Discovery and development of DNA polymerase IIIC inhibitors to treat Gram-positive infections. Bioorg. Med. Chem. 2019, 15, 3209–3217. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, A.M. Drugs in Development for Tuberculosis. Drugs. 2010, 70, 2201–2214. [Google Scholar] [CrossRef] [PubMed]
- Conradie, F.; Diacon, A.; Everitt, D.; Mendel, C.; van Niekerk, C.; Howell, P. Sustained high rate of successful treatment outcomes: Interim results of 75 patients in the Nix-TB clinical study of pretomanid, bedaquiline and linezolid. In Proceedings of the 49th World Conference on Lung Health of the International Union Against Tuberculosis and Lung Disease, Hague, The Netherlands, 25 May 2018. [Google Scholar]
- Kmentova, I.; Sutherland, H.S.; Palmer, B.D.; Blaser, A.; Franzblau, S.G.; Wan, B.; Wang, Y.; Ma, Z.; Denny, W.A.; Thompson, A.M. Synthesis and structure− activity relationships of aza-and diazabiphenyl analogues of the antitubercular drug (6 S)-2-Nitro-6-{[4-(trifluoromethoxy) benzyl] oxy}-6, 7-dihydro-5 H-imidazo [2, 1-b][1, 3] oxazine (PA-824). J. Med. Chem. 2010, 53, 8421–8439. [Google Scholar] [CrossRef] [PubMed]
- Upton, A.; Cho, S.; Yang, T.; Kim, Y.; Wang, Y.; Lu, Y.; Wang, B.; Xu, J.; Mdluli, K.; Ma, Z.; et al. In vitro and in vivo activities of the nitroimidazole TBA-354 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 136–144. [Google Scholar] [CrossRef]
- Da Silva, P.B.; Campos, D.L.; Ribeiro, C.M.; da Silva, I.C.; Pavan, F.R. New antimycobacterial agents in the pre-clinical phase or beyond: Recent advances in patent literature (2001–2016). Expert Opin. Pat. 2017, 27, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Wallis, R.S.; Maeurer, M.; Mwaba, P.; Chakaya, J.; Rustomjee, R.; Migliori, G.B.; Marais, B.; Schito, M.; Churchyard, G.; Swaminathan, S.; et al. Tuberculosis—advances in development of new drugs, treatment regimens, host-directed therapies, and biomarkers. Lancet Infect. Dis. 2016, 16, 34–46. [Google Scholar] [CrossRef]
- TB Alliance. Available online: https://www.newtbdrugs.org/pipeline/compound/tba-354 (accessed on 23 February 2021).
- Ntshangase, S.; Shobo, A.; Kruger, H.G.; Asperger, A.; Niemeyer, D.; Arvidsson, P.I.; Govender, T.; Baijnath, S. The downfall of TBA-354-a possible explanation for its neurotoxicity via mass spectrometric imaging. Xenobiotica 2018, 48, 938–944. [Google Scholar] [CrossRef]
- Pandit, N.; Singla, R.K.; Shrivastava, B. Current updates on oxazolidinone and its significance. Int. J. Med. Chem. 2012, 2012, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Marchese, A.; Schito, G. The oxazolidinones as a new family of antimicrobial agent. Clin. Microbiol. Infect. 2001, 7, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Kanafani, Z.A.; Corey, G.R. Tedizolid (TR-701): A new oxazolidinone with enhanced potency. Expert Opin. Investig. Drugs 2012, 21, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Carena, A.A.; Stryjewski, M.E. Tedizolid (torezolid) for the treatment of complicated skin and skin structure infections. Expert Rev. Clin. Pharmacol. 2020, 13, 577–592. [Google Scholar] [CrossRef]
- Shorr, A.F.; Lodise, T.P.; Corey, G.R.; De Anda, C.; Fang, E.; Das, A.F.; Prokocimer, P. Analysis of the phase 3 establish trials of tedizolid versus linezolid in acute bacterial skin and skin structure infections. Antimicrob. Agents Chemother. 2015, 59, 864–871. [Google Scholar] [CrossRef]
- Alcalá, L.; Ruiz-Serrano, M.J.; Turégano, C.P.-F.; de Viedma, D.G.; Díaz-Infantes, M.; Marín-Arriaza, M.; Bouza, E. In vitro activities of linezolid against clinical isolates of Mycobacterium tuberculosis that are susceptible or resistant to first-line antituberculous drugs. Antimicrob. Agents Chemother. 2003, 47, 416–417. [Google Scholar] [CrossRef]
- Stalker, D.J.; Jungbluth, G.L.; Hopkins, N.K.; Batts, D.H. Pharmacokinetics and tolerance of single-and multiple-dose oral or intravenous linezolid, an oxazolidinone antibiotic, in healthy volunteers. J. Antimicrob. Chemother. 2003, 51, 1239–1246. [Google Scholar] [CrossRef]
- Tang, S.; Yao, L.; Hao, X.; Zhang, X.; Liu, G.; Liu, X.; Wu, M.; Zen, L.; Sun, H.; Liu, Y. Efficacy, safety and tolerability of linezolid for the treatment of XDR-TB: A study in China. Eur. Respir. J. 2015, 45, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, B.T.; Kaatz, G.; Rybak, M. Linezolid and other oxazolidinones. In Antimicrobial Therapy and Vaccines; Yu, L., Edwards, G., McKinnon, P.S., Peloquin, C., Morse, G.D., Eds.; Esun Technologies, LLC: Pittsburgh, PA, USA, 2005; pp. 223–241. [Google Scholar]
- Diacon, A.H.; De Jager, V.R.; Dawson, R.; Narunsky, K.; Vanker, N.; Burger, D.A.; Everitt, D.; Pappas, F.; Nedelman, J.; Mendel, C.M. Fourteen-day bactericidal activity, safety, and pharmacokinetics of linezolid in adults with drug-sensitive pulmonary tuberculosis. Antimicrob. Agents Chemother. 2020, 64, 2012–2019. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.N.; Brickner, S.J.; Stover, C.K.; Zhu, T.; Ogden, A.; Tasneen, R.; Tyagi, S.; Grosset, J.H.; Nuermberger, E.L. Addition of PNU-100480 to first-line drugs shortens the time needed to cure murine tuberculosis. Am. J. Respir. Crit. Care Med. 2009, 180, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Gualano, G.; Capone, S.; Matteelli, A.; Palmieri, F. New antituberculosis drugs: From clinical trial to programmatic use. Curr. Infect. Dis. Rep. 2016, 8, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Wallis, R.S.; Dawson, R.; Friedrich, S.O.; Venter, A.; Paige, D.; Zhu, T.; Silvia, A.; Gobey, J.; Ellery, C.; Zhang, Y.; et al. Mycobactericidal activity of sutezolid (PNU-100480) in sputum (EBA) and blood (WBA) of patients with pulmonary tuberculosis. PLoS ONE 2014, 9, e94462. [Google Scholar] [CrossRef] [PubMed]
- Dooley, K.E.; Kim, P.S.; Williams, S.D.; Hafner, R. TB and HIV therapeutics: Pharmacology research priorities. AIDS Res Treat 2012, 2012, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Louie, A.; Eichas, K.; Files, K.; Swift, M.; Bahniuk, N.; Brown, D.; Drusano, G.L. Activities of PNU-100480 (PNU 480) alone, PNU 480 plus its major metabolite PNU-101603 (PNU 1603) and PNU 480 plus PNU 1603 in combination with rifampin (RIF) against Mycobacterium tuberculosis: Comparison with linezolid. In Proceedings of the 51st Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL, USA, 17–20 September 2011. [Google Scholar]
- TB Alliance. Available online: https://www.tballiance.org/news/tb-alliance-sublicenses-promising-anti-tuberculosis-drug-medicines-patent-pool (accessed on 23 February 2021).
- Libardo, M.D.; Boshoff, H.I.; Barry, C.E., III. The present state of the tuberculosis drug development pipeline. Curr. Opin. Pharmacol. 2018, 42, 81–94. [Google Scholar] [CrossRef]
- Wallis, R.S.; Jakubiec, W.; Kumar, V.; Bedarida, G.; Silvia, A.; Paige, D.; Zhu, T.; Mitton-Fry, M.; Ladutko, L.; Campbell, S.; et al. Biomarker-assisted dose selection for safety and efficacy in early development of PNU-100480 for tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 567–574. [Google Scholar] [CrossRef]
- Zhu, T.; Friedrich, S.O.; Diacon, A.; Wallis, R.S. Population pharmacokinetic/pharmacodynamic analysis of the bactericidal activities of sutezolid (PNU-100480) and its major metabolite against intracellular Mycobacterium tuberculosis in ex vivo whole-blood cultures of patients with pulmonary tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 3306–3311. [Google Scholar] [CrossRef] [PubMed]
- Furin, J.J.; Du Bois, J.; van Brakel, E.; Chheng, P.; Venter, A.; Peloquin, C.A.; Alsultan, A.; Thiel, B.A.; Debanne, S.M.; Boom, W.H.; et al. Early bactericidal activity of AZD5847 in patients with pulmonary tuberculosis. Antimicrob. Agents Chemother. 2016, 60, 6591–6599. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sala, C.; Dhar, N.; Vocat, A.; Sambandamurthy, V.K.; Sharma, S.; Marriner, G.; Balasubramanian, V.; Cole, S.T. In vitro and in vivo activities of three oxazolidinones against nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 3217–3223. [Google Scholar] [CrossRef] [PubMed]
- Mistry, N.; Tolani, M.; Dholakia, Y. New drugs for tuberculosis. Drugs Future. 2015, 40, 39–56. [Google Scholar] [CrossRef]
- Balasubramanian, V.; Solapure, S.; Shandil, R.; Gaonkar, S.; Mahesh, K.; Reddy, J.; Deshpande, A.; Bharath, S.; Kumar, N.; Wright, L.; et al. Pharmacokinetic and pharmacodynamic evaluation of AZD5847 in a mouse model of tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 4185–4190. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kisgen, J.J.; Mansour, H.; Unger, N.R.; Childs, L.M. Tedizolid: A new oxazolidinone antimicrobial. Am. J. Health Syst Pharm. 2014, 71, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Zhanel, G.G.; Love, R.; Adam, H.; Golden, A.; Zelenitsky, S.; Schweizer, F.; Gorityala, B.; Lagacé-Wiens, P.R.; Rubinstein, E.; Walkty, A.; et al. Tedizolid: A novel oxazolidinone with potent activity against multidrug-resistant gram-positive pathogens. Drugs 2015, 75, 253–270. [Google Scholar] [CrossRef]
- Ong, V.; Flanagan, S.; Fang, E.; Dreskin, H.J.; Locke, J.B.; Bartizal, K.; Prokocimer, P. Absorption, Distribution, Metabolism, and Excretion of the novel antibacterial prodrug tedizolid phosphate. Drug Metab. Dispos. 2014, 42, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, S.; McKee, E.E.; Das, D.; Tulkens, P.M.; Hosako, H.; Fiedler-Kelly, J.; Passarell, J.; Radovsky, A.; Prokocimer, P. Nonclinical and pharmacokinetic assessments to evaluate the potential of tedizolid and linezolid to affect mitochondrial function. Antimicrob. Agents Chemother. 2015, 59, 178–185. [Google Scholar] [CrossRef]
- Fugitt, R.B.; Luckenbaugh, R.W. 3-(p-Alkylsulfonylphenyl) Oxazolidinone Derivatives as Antibacterial Agents. U.S. Patent 4340606, 20 July 1982. [Google Scholar]
- Michalska, K.; Karpiuk, I.; Król, M.; Tyski, S. Recent development of potent analogues of oxazolidinone antibacterial agents. Bioorg. Med. Chem. 2013, 21, 577–591. [Google Scholar] [CrossRef]
- Vera-Cabrera, L.; Castro-Garza, J.; Rendon, A.; Ocampo-Candiani, J.; Welsh, O.; Choi, S.H.; Blackwood, K.; Molina-Torres, C. In vitro susceptibility of mycobacterium tuberculosis clinical isolates to garenoxacin and DA-7867. Antimicrob. Agents Chemother. 2005, 49, 4351–4353. [Google Scholar] [CrossRef]
- Ruiz, P.; Causse, M.; Vaquero, M.; Casal, M. In vitro activity of tedizolid against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2019, 63, 1939-18. [Google Scholar] [CrossRef]
- Deshpande, D.; Srivastava, S.; Nuermberger, E.; Koeuth, T.; Martin, K.R.; Cirrincione, K.N.; Lee, P.S.; Gumbo, T. Multiparameter responses to tedizolid monotherapy and moxifloxacin combination therapy models of children with intracellular tuberculosis. Clin. Infect. Dis. 2018, 67 (Suppl. 3), 342–348. [Google Scholar] [CrossRef]
- Srivastava, S.; van Rijn, S.P.; Wessels, A.M.A.; Alffenaar, J.-W.C.; Gumbo, T. Susceptibility testing of antibiotics that degrade faster than the doubling time of slow-growing mycobacteria: Ertapenem sterilizing effect versus Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2016, 60, 3193–3195. [Google Scholar] [CrossRef]
- Hoffner, S.; Chan, M.M.; Chan, E.D.; Ordway, D. Drug discovery targeting drug-resistant nontuberculous mycobacteria. In Drug Discovery Targeting Drug-Resistant Bacteria, 4; Kesharwani, P., Chopra, S., Dasgupta, A., Eds.; Elsevier: London, UK, 2020; pp. 361–376. [Google Scholar]
- Kim, Y.; Kim, A.; Lee, S.; Choi, S.-H.; Lee, D.Y.; Song, J.-S.; Lee, H.; Jang, I.J.; Yu, K.S. Pharmacokinetics, safety, and tolerability of tedizolid phosphate after single-dose administration in healthy Korean male subjects. Clin. Ther. 2017, 39, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Eum, S.Y.; Kong, J.H.; Hong, M.S.; Lee, Y.J.; Kim, J.H.; Hwang, S.H.; Cho, S.N.; Via, L.E.; Barry, C.E., III. Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest 2010, 137, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Deshpande, D.; Nuermberger, E.; Lee, P.S.; Cirrincione, K.; Dheda, K.; Gumbo, T. The Sterilizing effect of intermittent tedizolid for pulmonary tuberculosis. Clin. Infect. Dis. 2018, 67 (Suppl. 3), 336–341. [Google Scholar] [CrossRef] [PubMed]
- Sacksteder, K.A.; Protopopova, M.; Barry, C.E.; Andries, K.; Nacy, C.A. Discovery and development of SQ109: A new antitubercular drug with a novel mechanism of action. Future Microbiol. 2012, 7, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.E.; Protopopova, M.; Crooks, E.; Slayden, R.A.; Terrot, M.; Barry, C.E. Combinatorial lead optimization of [1, 2]-diamines based on ethambutol as potential antituberculosis preclinical candidates. J. Comb. Chem. 2003, 5, 172–187. [Google Scholar] [CrossRef]
- Su, C.-C.; Klenotic, P.A.; Bolla, J.R.; Purdy, G.E.; Robinson, C.V.; Yu, E.W. MmpL3 is a lipid transporter that binds trehalose monomycolate and phosphatidylethanolamine. Proc. Natl. Acad. Sci. USA 2019, 116, 11241–11246. [Google Scholar] [CrossRef]
- Heinrich, N.; Dawson, R.; du Bois, J.; Narunsky, K.; Horwith, G.; Phipps, A.J.; Nacy, C.A.; Aarnoutse, R.E.; Boeree, M.J.; Gillespie, S.H.; et al. Early phase evaluation of SQ109 alone and in combination with rifampicin in pulmonary TB patients. J. Antimicrob. Chemother. 2015, 70, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Protopopova, M.; Hanrahan, C.; Nikonenko, B.; Samala, R.; Chen, P.; Gearhart, J.; Einck, L.; Nacy, C.A. Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1, 2-ethylenediamines. J. Antimicrob. Chemother. 2005, 56, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Nikonenko, B.V.; Protopopova, M.; Samala, R.; Einck, L.; Nacy, C.A. Drug therapy of experimental tuberculosis (TB): Improved outcome by combining SQ109, a new diamine antibiotic, with existing TB drugs. Antimicrob. Agents Chemother. 2007, 51, 1563–1565. [Google Scholar] [CrossRef] [PubMed]
- Boeree, M.J.; Heinrich, N.; Aarnoutse, R.; Diacon, A.H.; Dawson, R.; Rehal, S.; Kibiki, G.S.; Churchyard, G.; Sanne, I.; Ntinginya, N.E.; et al. High-dose rifampicin, moxifloxacin, and SQ109 for treating tuberculosis: A multi-arm, multi-stage randomised controlled trial. Lancet Infect. Dis. 2017, 17, 39–49. [Google Scholar] [CrossRef]
- Jia, L.; Tomaszewski, J.E.; Hanrahan, C.; Coward, L.; Noker, P.; Gorman, G.; Nikonenko, B.; Protopopova, M. Pharmacodynamics and pharmacokinetics of SQ109, a new diamine-based antitubercular drug. Br. J. Pharmacol. 2005, 144, 80–87. [Google Scholar] [CrossRef]
- Niemi, M.; Backman, J.T.; Fromm, M.F.; Neuvonen, P.J.; Kivistö, K.T. Pharmacokinetic interactions with rifampicin. Clin. Pharmacokinet. 2003, 42, 819–850. [Google Scholar] [CrossRef]
- Vilchèze, C. Mycobacterial Cell Wall: A Source of Successful Targets for Old and New Drugs. Appl. Sci. 2020, 10, 2278. [Google Scholar] [CrossRef]
- Working Group for New TB Drugs. Available online: https://www.newtbdrugs.org/pipeline/trials (accessed on 23 June 2020).
- Kang, S.; Kim, Y.M.; Jeon, H.; Park, S.; Seo, M.J.; Lee, S.; Park, D.; Nam, J.; Lee, S.; Nam, K.; et al. Synthesis and structure-activity relationships of novel fused ring analogues of Q203 as antitubercular agents. Eur. J. Med. Chem. 2017, 136, 420–427. [Google Scholar] [CrossRef]
- Lohrasbi, V.; Talebi, M.; Bialvaei, A.Z.; Fattorini, L.; Drancourt, M.; Heidary, M.; Darban-Sarokhalil, D. Trends in the discovery of new drugs for Mycobacterium tuberculosis therapy with a glance at resistance. Tuberculosis 2018, 109, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Matsoso, L.G.; Kana, B.D.; Crellin, P.K.; Lea-Smith, D.J.; Pelosi, A.; Powell, D.; Powell, D.; Dawes, S.S.; Rubin, H.; Coppel, R.L.; et al. Function of the cytochrome bc1-aa3 branch of the respiratory network in mycobacteria and network adaptation occurring in response to its disruption. J. Bacteriol. 2005, 187, 6300–6308. [Google Scholar] [CrossRef]
- Kang, S.; Kim, R.Y.; Seo, M.J.; Lee, S.; Kim, Y.M.; Seo, M.; Seo, J.J.; Ko, Y.; Choi, I.; Jang, J.; et al. Lead optimization of a novel series of imidazo [1, 2-a] pyridine amides leading to a clinical candidate (Q203) as a multi-and extensively-drug-resistant anti-tuberculosis agent. J. Med. Chem. 2014, 57, 5293–5305. [Google Scholar] [CrossRef]
- Tang, J.; Wang, B.; Wu, T.; Wan, J.; Tu, Z.; Njire, M.; Wan, B.; Franzblauc, S.G.; Zhang, T.; Lu, X.; et al. Design, synthesis, and biological evaluation of pyrazolo [1, 5-a] pyridine-3-carboxamides as novel antitubercular agents. ACS Medicinal Chem. Lett. 2015, 6, 814–818. [Google Scholar] [CrossRef]
- Lu, X.; Williams, Z.; Hards, K.; Tang, J.; Cheung, C.-Y.; Aung, H.L.; Wang, B.; Liu, Z.; Hu, X.; Lenaerts, A.; et al. Pyrazolo [1, 5-a] pyridine inhibitor of the respiratory cytochrome bcc complex for the treatment of drug-resistant tuberculosis. ACS Infect. Dis. 2018, 5, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chiwala, G.; Gao, Y.; Liu, Z.; Sapkota, S.; Lu, Z.; Guo, L.; Khan, S.A.; Zhong, N.; Zhang, T. TB47 and clofazimine form a highly synergistic sterilizing block in a second-line regimen for tuberculosis in mice. Biomed. Pharmacother. 2020, 131, 110782–110789. [Google Scholar] [CrossRef]
- Lu, X.; Tang, J.; Liu, Z.; Li, M.; Zhang, T.; Zhang, X.; Ding, K. Discovery of new chemical entities as potential leads against Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 2016, 26, 5916–5919. [Google Scholar] [CrossRef] [PubMed]
- Grosset, J.H.; Tyagi, S.; Almeida, D.V.; Converse, P.J.; Li, S.-Y.; Ammerman, N.C.; Bishai, W.R.; Enarson, D.; Trébucq, A. Assessment of clofazimine activity in a second-line regimen for tuberculosis in mice. Am. J. Respir. Crit. Care Med. 2013, 188, 608–612. [Google Scholar] [CrossRef]
- Moraski, G.C.; Seeger, N.; Miller, P.A.; Oliver, A.G.; Boshoff, H.I.; Cho, S.; Mulugeta, S.; Anderson, J.R.; Franzblau, S.G.; Miller, M.J. Arrival of imidazo [2, 1-b] thiazole-5-carboxamides: Potent anti-tuberculosis agents that target QcrB. ACS Infect. Dis. 2016, 2, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Moraski, G.C.; Deboosère, N.; Marshall, K.L.; Weaver, H.A.; Vandeputte, A.; Hastings, C.; Woolhiser, L.; Lenaerts, A.J.; Brodin, P.; Miller, M.J. Intracellular and in vivo evaluation of imidazo [2, 1-b] thiazole-5-carboxamide anti-tuberculosis compounds. PLoS ONE 2020, 15, e0227224. [Google Scholar] [CrossRef] [PubMed]
- Trefzer, C.; Škovierová, H.; Buroni, S.; Bobovská, A.; Nenci, S.; Molteni, E.; Pojer, F.; Pasca, M.R.; Makarov, V.; Cole, S.T. Benzothiazinones are suicide inhibitors of mycobacterial decaprenylphosphoryl-β-d-ribofuranose 2′-oxidase DprE1. J. Am. Chem. Soc. 2012, 134, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Shetye, G.S.; Franzblau, S.G. New tuberculosis drug targets, their inhibitors and potential therapeutic impact. Transl. Res. 2020, 220, 68–97. [Google Scholar] [CrossRef]
- Lechartier, B.; Hartkoorn, R.C.; Cole, S.T. In vitro combination studies of benzothiazinone lead compound BTZ043 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 5790–5793. [Google Scholar] [CrossRef]
- Makarov, V.; Manina, G.; Mikusova, K.; Möllmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 2009, 324, 801–804. [Google Scholar] [CrossRef]
- Gao, C.; Peng, C.; Shi, Y.; You, X.; Ran, K.; Xiong, L.; Ye, T.H.; Zhang, L.; Wang, N.; Zhu, Y.; et al. Benzothiazinethione is a potent preclinical candidate for the treatment of drug-resistant tuberculosis. Sci. Rep. 2016, 6, 1–9. [Google Scholar]
- Mahapatra, S.; Basu, J.; Brennan, P.; Crick, D.; Cole, S.; Eisenach, K.; McMurray, D.N.; Jacobs, W.R. Tuberculosis and the Tubercle Bacillus. Emerg. Infect. Dis. 2005, 11, 275–285. [Google Scholar]
- Makarov, V.; Lechartier, B.; Zhang, M.; Neres, J.; van der Sar, A.M.; Raadsen, S.A.; Hartkoorn, R.C.; Ryabova, O.B.; Vocat, A.; Decosterd, L.A.; et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Makarov, V.; Mikušová, K. Development of macozinone for TB treatment: An update. Appl. Sci. 2020, 10, 2269. [Google Scholar] [CrossRef]
- Shi, J.; Lu, J.; Zong, Z.; Huo, F.; Luo, J.; Liang, Q.; Li, Y.; Huang, H.; Pang, Y. In vitro activity of PBTZ169 against multiple Mycobacterium species. Antimicrob. Agents Chemother. 2018, 62, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lechartier, B.; Cole, S.T. Mode of action of clofazimine and combination therapy with benzothiazinones against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 4457–4463. [Google Scholar] [CrossRef] [PubMed]
- Mariandyshev, A.; Khokhlov, A.; Smerdin, S.; Shcherbakova, V.; Igumnova, O.; Ozerova, I.; Bolgarina, A.A.; Nikitina, N.A. The main results of clinical trials of the efficacy, safety and pharmacokinetics of the perspective anti-tuberculosis drug makozinone (PBTZ169). Ter. Arkh. 2020, 92, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Spaggiari, D.; Desfontaine, V.; Cruchon, S.; Guinchard, S.; Vocat, A.; Blattes, E.; Pitteloud, J.; Ciullini, L.; Bardinet, C.; Ivanyuk, A.; et al. Development and validation of a multiplex UHPLC-MS/MS method for the determination of the investigational antibiotic against multi-resistant tuberculosis macozinone (PBTZ169) and five active metabolites in human plasma. PLoS ONE 2019, 14, e0217139. [Google Scholar] [CrossRef]
- Kloss, F.; Krchnak, V.; Krchnakova, A.; Schieferdecker, S.; Dreisbach, J.; Krone, V.; Möllmann, U.; Hoelscher, M.; Miller, M.J. In vivo dearomatization of the potent antituberculosis agent BTZ043 via Meisenheimer complex formation. Angew. Chem. Int. 2017, 56, 2187–2191. [Google Scholar] [CrossRef]
- Yuan, T.; Sampson, N.S. Hit generation in TB drug discovery: From genome to granuloma. Chem. Rev. 2018, 118, 1887–1916. [Google Scholar] [CrossRef] [PubMed]
- Torfs, E.; Piller, T.; Cos, P.; Cappoen, D. Opportunities for overcoming Mycobacterium tuberculosis drug resistance: Emerging mycobacterial targets and host-directed therapy. Int. J. Mol. Sci. 2019, 20, 2868. [Google Scholar] [CrossRef]
- Riccardi, G.; Pasca, M.R.; Chiarelli, L.R.; Manina, G.; Mattevi, A.; Binda, C. The DprE1 enzyme, one of the most vulnerable targets of Mycobacterium tuberculosis. Appl. Microbiol. Biotechnol. 2013, 97, 8841–8848. [Google Scholar] [CrossRef] [PubMed]
- Panda, M.; Ramachandran, S.; Ramachandran, V.; Shirude, P.S.; Humnabadkar, V.; Nagalapur, K.; Sharma, S.; Kaur, P.; Guptha, S.; Narayan, A.; et al. Discovery of pyrazolopyridones as a novel class of noncovalent DprE1 inhibitor with potent anti-mycobacterial activity. J. Med. Chem. 2014, 57, 4761–4771. [Google Scholar] [CrossRef] [PubMed]
- Hariguchi, N.; Chen, X.; Hayashi, Y.; Kawano, Y.; Fujiwara, M.; Matsuba, M.; Shimizu, H.; Ohba, Y.; Nakamura, I.; Kitamoto, R.; et al. OPC-167832, a novel carbostyril derivative with potent anti-tuberculosis activity as a DprE1 inhibitor. Antimicrob. Agents Chemother. 2020, 64, 1–13. [Google Scholar] [CrossRef]
- Milano, A.; Pasca, M.R.; Provvedi, R.; Lucarelli, A.P.; Manina, G.; Ribeiro, A.L.dJ.L.; Manganelli, R.; Riccardi, G. Azole resistance in Mycobacterium tuberculosis is mediated by the MmpS5–MmpL5 efflux system. Tuberculosis 2009, 89, 84–90. [Google Scholar] [CrossRef]
- Heifets, L.B. Antimycobacterial drugs. Semin Respir. Infect. 1994, 9, 84–103. [Google Scholar]
- Shirude, P.S.; Shandil, R.; Sadler, C.; Naik, M.; Hosagrahara, V.; Hameed, S.; Shinde, V.; Bathula, C.; Humnabadkar, V.; Kumar, N.; et al. Azaindoles: Noncovalent DprE1 inhibitors from scaffold morphing efforts, kill Mycobacterium tuberculosis and are efficacious in vivo. J. Med. Chem. 2013, 56, 9701–9708. [Google Scholar] [CrossRef]
- Chatterji, M.; Shandil, R.; Manjunatha, M.; Solapure, S.; Ramachandran, V.; Kumar, N.; Saralaya, R.; Panduga, V.; Reddy, J.; Prabhakar, K.R.; et al. 1,4-Azaindole, a potential drug candidate for treatment of tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 5325–5331. [Google Scholar] [CrossRef]
- Shirude, P.S.; Shandil, R.K.; Manjunatha, M.; Sadler, C.; Panda, M.; Panduga, V.; Reddy, J.; Saralaya, R.; Nanduri, R.; Ambady, A.; et al. Lead optimization of 1,4-azaindoles as antimycobacterial agents. J. Med. Chem. 2014, 57, 5728–5737. [Google Scholar] [CrossRef]
- Igarashi, M.; Nakagawa, N.; Doi, N.; Hattori, S.; Naganawa, H.; Hamada, M. Caprazamycin B, a novel anti-tuberculosis antibiotic, from Streptomyces sp. J. Antibiot. Res. 2003, 56, 580–583. [Google Scholar] [CrossRef]
- Hirano, S.; Ichikawa, S.; Matsuda, A. Structure–activity relationship of truncated analogs of caprazamycins as potential anti-tuberculosis agents. Bioorg. Med. Chem. 2008, 16, 5123–5133. [Google Scholar] [CrossRef] [PubMed]
- Wiker, F.; Hauck, N.; Grond, S.; Gust, B. Caprazamycins: Biosynthesis and structure activity relationship studies. Int. J. Med. Microbiol. 2019, 309, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Bugg, T.D.; Lloyd, A.J.; Roper, D.I. Phospho-MurNAc-pentapeptide translocase (MraY) as a target for antibacterial agents and antibacterial proteins. Infect. Disord. Drug Targets 2006, 6, 85–106. [Google Scholar] [CrossRef]
- Nakamura, H.; Yoshida, T.; Tsukano, C.; Takemoto, Y. Synthesis of CPZEN-45: Construction of the 1, 4-diazepin-2-one core by the Cu-catalyzed intramolecular amidation of a vinyl iodide. Org. Lett. 2016, 18, 2300–2303. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, Y.; Hayashi, C.; Inoue, K.; Igarashi, M.; Takahashi, Y.; Pujari, V.; Crick, D.C.; Brennan, P.J.; Nomoto, A. Inhibition of the first step in synthesis of the mycobacterial cell wall core, catalyzed by the GlcNAc-1-phosphate transferase WecA, by the novel caprazamycin derivative CPZEN-45. J. Biol. Chem. 2013, 288, 30309–30319. [Google Scholar] [CrossRef]
- Takahashi, Y.; Igarashi, M.; Miyake, T.; Soutome, H.; Ishikawa, K.; Komatsuki, Y.; Koyama, Y.; Nakagawa, N.; Hattori, S.; Inoue, K.; et al. Novel semisynthetic antibiotics from caprazamycins A–G: Caprazene derivatives and their antibacterial activity. J. Antibiot. Res. 2013, 66, 171–178. [Google Scholar] [CrossRef]
- Pitner, R.A.; Durham, P.G.; Stewart, I.E.; Reed, S.G.; Cassell, G.H.; Hickey, A.J.; Carter, D. A Spray-dried combination of capreomycin and CPZEN-45 for inhaled tuberculosis therapy. J. Pharm. Sci. 2019, 108, 3302–3311. [Google Scholar] [CrossRef]
- Salomon, J.J.; Galeron, P.; Schulte, N.; Morow, P.R.; Severynse-Stevens, D.; Huwer, H.; Daum, N.; Lehr, C.M.; Hickey, A.J.; Ehrhardt, C. Biopharmaceutical in vitro characterization of CPZEN-45, a drug candidate for inhalation therapy of tuberculosis. Ther. Deliv. 2013, 4, 915–923. [Google Scholar] [CrossRef]
- Barry, V.C.; Belton, J.G.; Conalty, M.L.; Twomey, D. Anti-tubercular activity of oxidation products of substituted o-phenylene diamines. Nature 1948, 162, 622–623. [Google Scholar] [CrossRef] [PubMed]
- Barry, V.C.; Buggle, K.; Byrne, J.; Conalty, M.L.; Winder, F. Absorption, distribution and retention of the rimino-compounds in the experimental animal. Ir. J. Med. Sci. 1960, 35, 345–352. [Google Scholar] [CrossRef]
- Reddy, V.M.; O’ Sullivan, J.F.; Gangadharam, P.R.J. Antimycobacterial activities of riminophenazines. J. Antimicrob. Chemother. 1999, 43, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Bvumbi, M.V. Activity of riminophenazines against mycobacterium tuberculosis: A Review of Studies that Might be Contenders for Use as Antituberculosis Agents. ChemMedChem 2020, 15, 2207–2219. [Google Scholar] [CrossRef]
- Vischer, W.A. The experimental properties of G 30 320 (B 663)—A new anti-leprotic agent. Lepr. Rev. 1969, 40, 107–110. [Google Scholar] [CrossRef]
- Barry, V.C.; Conalty, M.L. Antituberculosis activity in the phenazine series: II. N3-substituted analinooposafranmes (rimino-compounds) and some derivatives. Int. J. Tuberc. Lung Dis. 1958, 78, 62–73. [Google Scholar]
- Browne, S.; Hogerzeil, L. “B 663” in the treatment of leprosy. Preliminary report of a pilot trial. Lepr. Rev. 1962, 33, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Barry, V.C.; Belton, J.G.; Conalty, M.L.; Denneny, J.M.; Edward, D.W.; O’sullivan, J.F.; Twomey, D.; Winder, F. A new series of phenazines (rimino-compounds) with high antituberculosis activity. Nature 1957, 179, 1013–1015. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Kassovska-Bratinova, S.; Teh, J.S.; Winkler, J.; Sullivan, K.; Isaacs, A.; Schechter, N.M.; Rubin, H. Reduction of clofazimine by mycobacterial type 2 NADH: Quinone oxidoreductase a pathway for the generation of bactericidal levels of reactive oxygen species. J. Biol. Chem. 2011, 286, 10276–10287. [Google Scholar] [CrossRef]
- Yawalkar, S.; Vischer, W. Lamprene (clofazimine) in leprosy. Lepr. Rev. 1979, 50, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, B.; Fu, L.; Zhu, H.; Guo, S.; Huang, H.; Yin, D.; Zhang, Y.; Lu, Y. In vitro and in vivo activities of the riminophenazine TBI-166 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2019, 63, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Sheng, L.; Liu, X.; Yang, S.; Liu, Z.; Li, Y. Determination of TBI-166, a novel antituberculotic, in Rat plasma by liquid chromatography–tandem mass spectrometry. Chromatographia 2014, 77, 1697–1703. [Google Scholar] [CrossRef]
- Lu, Y.; Zheng, M.; Wang, B.; Fu, L.; Zhao, W.; Li, P.; Xu, J.; Zhu, H.; Jin, H.; Yin, D. Clofazimine analogs with efficacy against experimental tuberculosis and reduced potential for accumulation. Antimicrob. Agents Chemother. 2011, 55, 5185–5193. [Google Scholar] [CrossRef]
- Li, Q.; Lu, X. New antituberculosis drugs targeting the respiratory chain. Chin. Chem. Lett. 2020, 31, 1357–1365. [Google Scholar] [CrossRef]
- Mohamed, M.; El-Hameed, A.; Sayed, A. Synthesis strategies and biological value of pyrrole and pyrrolopyrimidine. J. Adv. Pharm. Technol. Res. 2017, 1, 1–24. [Google Scholar] [CrossRef]
- Castro, A.J.; Gale, G.R.; Means, G.E.; Tertzakian, G. Antimicrobial properties of pyrrole derivatives. J. Med. Chem. 1967, 10, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.S.; Fathallah, S.S. Pyrroles and fused pyrroles: Synthesis and therapeutic activities. Mini. Rev. Org. Chem. 2014, 11, 477–507. [Google Scholar] [CrossRef]
- Deidda, D.; Lampis, G.; Fioravanti, R.; Biava, M.; Porretta, G.C.; Zanetti, S.; Pompei, R. Bactericidal activities of the pyrrole derivative BM212 against multidrug-resistant and intramacrophagic Mycobacterium tuberculosis strains. Antimicrob. Agents Chemother. 1998, 42, 3035–3037. [Google Scholar] [CrossRef]
- La Rosa, V.; Poce, G.; Canseco, J.O.; Buroni, S.; Pasca, M.R.; Biava, M.; Raju, R.M.; Porretta, G.C.; Alfonso, S.; Battilocchio, C.; et al. MmpL3 is the cellular target of the antitubercular pyrrole derivative BM212. Antimicrob. Agents Chemother. 2012, 56, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Meshcheryakov, V.A.; Poce, G.; Chng, S.-S. MmpL3 is the flippase for mycolic acids in mycobacteria. Proc. Natl. Acad. Sci. USA 2017, 114, 7993–7998. [Google Scholar] [CrossRef]
- Bhakta, S.; Scalacci, N.; Maitra, A.; Brown, A.K.; Dasugari, S.; Evangelopoulos, D.; McHugh, T.D.; Mortazavi, P.N.; Twist, A.; Petricci, E.; et al. Design and synthesis of 1-((1, 5-Bis (4-chlorophenyl)-2-methyl-1 H-pyrrol-3-yl) methyl)-4-methylpiperazine (BM212) and N-Adamantan-2-yl-N′-((E)-3, 7-dimethylocta-2, 6-dienyl) ethane-1, 2-diamine (SQ109) pyrrole hybrid derivatives: Discovery of potent antitubercular agents effective against multidrug-resistant mycobacteria. J. Med. Chem. 2016, 59, 2780–2793. [Google Scholar]
- Nikonenko, B.V.; Samala, R.; Einck, L.; Nacy, C.A. Rapid, simple in vivo screen for new drugs active against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2004, 48, 4550–4555. [Google Scholar] [CrossRef] [PubMed]
- Mahboub, B.; Vats, M. (Eds.) Tuberculosis: Current Issues in Diagnosis and Management.; BoD–Books on Demand: Rijeka, Croatia, 2013; pp. 1–480. [Google Scholar]
- Adhvaryu, M.; Vakharia, B. Drug-resistant tuberculosis: Emerging treatment options. Clin. Pharmacol. 2011, 3, 51–67. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Van den Boogaard, J.; Kibiki, G.S.; Kisanga, E.R.; Boeree, M.J.; Aarnoutse, R.E. New drugs against tuberculosis: Problems, progress, and evaluation of agents in clinical development. Antimicrob. Agents Chemother. 2009, 53, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Dessen, A.; Quemard, A.; Blanchard, J.S.; Jacobs, W.R.; Sacchettini, J.C. Crystal structure and function of the isoniazid target of Mycobacterium tuberculosis. Science 1995, 267, 1638–1641. [Google Scholar] [CrossRef] [PubMed]
- Rawat, R.; Whitty, A.; Tonge, P.J. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: Adduct affinity and drug resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 13881–13886. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Dubnau, E.; Quemard, A.; Balasubramanian, V.; Um, K.S.; Wilson, T.; Collins, D.; De Lisle, G.; Jacobs, W.R. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 1994, 263, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Heym, B.; Allen, B.; Young, D.; Cole, S. The catalase—Peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature 1992, 358, 591–593. [Google Scholar] [CrossRef]
- Shirude, P.S.; Madhavapeddi, P.; Naik, M.; Murugan, K.; Shinde, V.; Nandishaiah, R.; Bhat, J.; Kumar, A.; Hameed, S.; Holdgate, G.; et al. Methyl-thiazoles: A novel mode of inhibition with the potential to develop novel inhibitors targeting InhA in Mycobacterium tuberculosis. J. Med. Chem. 2013, 56, 8533–8542. [Google Scholar] [CrossRef]
- Kamsri, P.; Hanwarinroj, C.; Phusi, N.; Pornprom, T.; Chayajarus, K.; Punkvang, A.; Suttipanta, N.; Srimanote, P.; Suttisintong, K.; Songsiriritthigul, C.; et al. Discovery of new and potent InhA inhibitors as anti-tuberculosis agents: Structure based virtual screening validated by biological assays and x-ray crystallography. J. Chem. Inf. Model. 2019, 10, 226–234. [Google Scholar]
- Martínez-Hoyos, M.; Perez-Herran, E.; Gulten, G.; Encinas, L.; Álvarez-Gómez, D.; Alvarez, E.; Ferrer-Bazaga, S.; García-Pérez, A.; Ortega, F.; Angulo-Barturen, I.; et al. Antitubercular drugs for an old target: GSK693 as a promising InhA direct inhibitor. EBioMedicine 2016, 8, 291–301. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Alian, A.; Stroud, R.; Ortiz de Montellano, P.R. Pyrrolidine carboxamides as a novel class of inhibitors of enoyl acyl carrier protein reductase from Mycobacterium tuberculosis. J. Med. Chem. 2006, 49, 6308–6323. [Google Scholar] [CrossRef]
- Vasilyev, D.; Merrill, T.; Iwanow, A.; Dunlop, J.; Bowlby, M. A novel method for patch-clamp automation. Pflügers Arch. 2006, 452, 240–247. [Google Scholar] [CrossRef]
- Moulkrere, B.R.; Orena, B.S.; Mori, G.; Saffon-Merceron, N.; Rodriguez, F.; Lherbet, C.; Belkheiri, N.; Amari, M.; Hoffmann, P.; Fodili, M. Evaluation of heteroatom-rich derivatives as antitubercular agents with InhA inhibition properties. Med. Chem Res. 2018, 27, 308–320. [Google Scholar] [CrossRef]
- Barry, C.E., III; Lee, R.E.; Mdluli, K.; Sampson, A.E.; Schroeder, B.G.; Slayden, R.A.; Yuan, Y. Mycolic acids: Structure, biosynthesis and physiological functions. Prog. Lipid Res. 1998, 37, 143–179. [Google Scholar] [CrossRef]
- Vilchèze, C.; Morbidoni, H.R.; Weisbrod, T.R.; Iwamoto, H.; Kuo, M.; Sacchettini, J.C.; Jacobs, W.R. Inactivation of the inhA-encoded fatty acid synthase II (FASII) enoyl-acyl carrier protein reductase induces accumulation of the FASI end products and cell lysis of Mycobacterium smegmatis. J. Bacteriol. 2000, 182, 4059–4067. [Google Scholar] [CrossRef]
- Zhang, N.-N.; Liu, Z.-Y.; Liang, J.; Tang, Y.-X.; Qian, L.; Gao, Y.-M.; Zhang, T.Y.; Yan, M. Design, synthesis, and biological evaluation of m-amidophenol derivatives as a new class of antitubercular agents. MedChemComm 2018, 9, 1293–1304. [Google Scholar] [CrossRef]
- Flint, L.; Korkegian, A.; Parish, T. InhA inhibitors have activity against non-replicating Mycobacterium. PLoS ONE 2020, 15, e0239354. [Google Scholar] [CrossRef]
- Bonnett, S.A.; Dennison, D.; Files, M.; Bajpai, A.; Parish, T. A class of hydrazones are active against non-replicating Mycobacterium tuberculosis. PLoS ONE 2018, 13, e0198059. [Google Scholar] [CrossRef]
- Betts, J.C.; Lukey, P.T.; Robb, L.C.; McAdam, R.A.; Duncan, K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 2002, 43, 717–731. [Google Scholar] [CrossRef]
- McNeil, M.B.; Dennison, D.; Shelton, C.; Flint, L.; Korkegian, A.; Parish, T. Mechanisms of resistance against NITD-916, a direct inhibitor of Mycobacterium tuberculosis InhA. Tuberculosis 2017, 107, 133136. [Google Scholar] [CrossRef]
- Owens, R.C. An overview of harms associated with beta-lactam antimicrobials: Where do the carbapenems fit in? Crit. Care. 2008, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dhar, N.; Dubée, V.; Ballell, L.; Cuinet, G.; Hugonnet, J.-E.; Signorino-Gelo, F.; Barros, D.; Arthur, M.; McKinney, J.D. Rapid cytolysis of Mycobacterium tuberculosis by faropenem, an orally bioavailable β-lactam antibiotic. Antimicrob. Agents Chemother. 2015, 59, 1308–1319. [Google Scholar] [CrossRef] [PubMed]
- Milazzo, I.; Blandino, G.; Caccamo, F.; Musumeci, R.; Nicoletti, G.; Speciale, A. Faropenem, a new oral penem: Antibacterial activity against selected anaerobic and fastidious periodontal isolates. J. Antimicrob. Chemother. 2003, 51, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Rullas, J.; Dhar, N.; McKinney, J.D.; García-Pérez, A.; Lelievre, J.; Diacon, A.H.; Hugonnet, J.E.; Arthur, M.; Angulo-Barturen, I.; Barros-Aguirre, D.; et al. Combinations of β-lactam antibiotics currently in clinical trials are efficacious in a DHP-I-deficient mouse model of tuberculosis infection. Antimicrob. Agents Chemother. 2015, 59, 4997–4999. [Google Scholar] [CrossRef]
- Tanaka, E.; Kimoto, T.; Tsuyuguchi, K.; Suzuki, K.; Amitani, R. Successful treatment with faropenem and clarithromycin of pulmonary Mycobacterium abscessus infection. J. Infect. Chemother. 2002, 8, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Dubée, V.; Triboulet, S.; Mainardi, J.-L.; Ethève-Quelquejeu, M.; Gutmann, L.; Marie, A.; Dubost, L.; Hugonnet, J.E.; Arthur, M. Inactivation of Mycobacterium tuberculosis L, D-transpeptidase LdtMt1 by carbapenems and cephalosporins. Antimicrob. Agents Chemother. 2012, 56, 4189–4195. [Google Scholar] [CrossRef] [PubMed]
- Cordillot, M.; Dubée, V.; Triboulet, S.; Dubost, L.; Marie, A.; Hugonnet, J.-E.; Arthur, M.; Mainardi, J.L. In vitro cross-linking of Mycobacterium tuberculosis peptidoglycan by L, D-transpeptidases and inactivation of these enzymes by carbapenems. Antimicrob. Agents Chemother. 2013, 57, 5940–5945. [Google Scholar] [CrossRef]
- Van Rijn, S.P.; van Altena, R.; Akkerman, O.W.; van Soolingen, D.; van der Laan, T.; de Lange, W.C.; Kosterink, J.G.; van der Werf, T.S.; Alffenaar, J.W. Pharmacokinetics of ertapenem in patients with multidrug-resistant tuberculosis. Eur. Respir. J. 2016, 47, 1229–1234. [Google Scholar] [CrossRef]
- Musuka, S.; Srivastava, S.; Dona, C.W.S.; Meek, C.; Leff, R.; Pasipanodya, J.; Gumbo, T. Thioridazine pharmacokinetic-pharmacodynamic parameters “wobble” during treatment of tuberculosis: A theoretical basis for shorter-duration curative monotherapy with congeners. Antimicrob. Agents Chemother. 2013, 57, 5870–5877. [Google Scholar] [CrossRef]
- Teppler, H.; Gesser, R.M.; Friedland, I.R.; Woods, G.L.; Meibohm, A.; Herman, G.; Mistry, G.; Isaacs, R. Safety and tolerability of ertapenem. J. Antimicrob. Chemother. 2004, 53, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Zhanel, G.G.; Wiebe, R.; Dilay, L.; Thomson, K.; Rubinstein, E.; Hoban, D.J.; Noreddin, A.M.; Karlowsky, J.A. Comparative review of the carbapenems. Drugs 2007, 67, 1027–1052. [Google Scholar] [CrossRef]
- Van Rijn, S.P.; Srivastava, S.; Wessels, M.A.; van Soolingen, D.; Alffenaar, J.-W.C.; Gumbo, T. Sterilizing effect of ertapenem-clavulanate in a hollow-fiber model of tuberculosis and implications on clinical dosing. Antimicrob. Agents Chemother. 2017, 61, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tiberi, S.; D’Ambrosio, L.; De Lorenzo, S.; Viggiani, P.; Centis, R.; Sotgiu, G.; Alffenaar, J.W.; Migliori, G.B. Ertapenem in the treatment of multidrug-resistant tuberculosis: First clinical experience. Eur. Respir. J. 2016, 47, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Nix, D.E.; Majumdar, A.K.; Di Nubile, M.J. Pharmacokinetics and pharmacodynamics of ertapenem: An overview for clinicians. J. Antimicrob. Chemother. 2004, 53, 23–28. [Google Scholar] [CrossRef]
- Shah, P.M.; Isaacs, R.D. Ertapenem, the first of a new group of carbapenems. J. Antimicrob. Chemother. 2003, 52, 538–542. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Keating, G.M.; Perry, C.M. Ertapenem. Drugs 2005, 65, 2151–2178. [Google Scholar] [CrossRef]
- Lue, S.W.; Kelley, S.O. An aminoacyl-tRNA synthetase with a defunct editing site. Biochemistry 2005, 44, 3010–3016. [Google Scholar] [CrossRef]
- Palencia, A.; Li, X.; Bu, W.; Choi, W.; Ding, C.Z.; Easom, E.E.; Feng, L.; Hernandez, V.; Houston, P.; Liu, L.; et al. Discovery of novel oral protein synthesis inhibitors of Mycobacterium tuberculosis that target leucyl-tRNA synthetase. Antimicrob. Agents Chemother. 2016, 60, 6271–6280. [Google Scholar] [CrossRef]
- World Health Organization. Available online: https://apps.who.int/iris/handle/10665/275487 (accessed on 24 February 2021).
- Tenero, D.; Derimanov, G.; Carlton, A.; Tonkyn, J.; Davies, M.; Cozens, S.; Gresham, S.; Gaudion, A.; Puri, A.; Muliaditan, M.; et al. First-time-in-human study and prediction of early bactericidal activity for GSK3036656, a potent leucyl-tRNA synthetase inhibitor for tuberculosis treatment. Antimicrob. Agents Chemother. 2019, 63, 1–15. [Google Scholar] [CrossRef]
- Li, X.; Hernandez, V.; Rock, F.L.; Choi, W.; Mak, Y.S.; Mohan, M.; Mao, W.; Zhou, Y.; Easom, E.E.; Plattner, J.J.; et al. Discovery of a potent and specific M. tuberculosis Leucyl-tRNA synthetase inhibitor:(S)-3-(Aminomethyl)-4-chloro-7-(2-hydroxyethoxy) benzo [c][1, 2] oxaborol-1 (3 H)-ol (GSK656). J. Med. Chem. 2017, 60, 8011–8026. [Google Scholar] [CrossRef] [PubMed]
- Rullas, J.; García, J.I.; Beltrán, M.; Cardona, P.-J.; Cáceres, N.; García-Bustos, J.F.; Angulo-Barturen, I. Fast standardized therapeutic-efficacy assay for drug discovery against tuberculosis. Antimicrob. Agents Chemother. 2010, 54, 2262–2264. [Google Scholar] [CrossRef] [PubMed]
- Franzblau, S.G.; DeGroote, M.A.; Cho, S.H.; Andries, K.; Nuermberger, E.; Orme, I.M.; Mdluli, K.; Angulo-Barturen, I.; Dick, T.; Dartois, V.; et al. Comprehensive analysis of methods used for the evaluation of compounds against Mycobacterium tuberculosis. Tuberculosis 2012, 92, 453–488. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-Y.; Tasneen, R.; Tyagi, S.; Soni, H.; Converse, P.J.; Mdluli, K.; Nuermberger, E.L. Bactericidal and sterilizing activity of a novel regimen with bedaquiline, pretomanid, moxifloxacin, and pyrazinamide in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2017, 61, 1–8. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Mutant Gene | Mutation(s) | References |
|---|---|---|---|
| MXF | gyrA | D94G, D94N, and D94Y | Nosova et al. 2013; Groll et al. 2009 |
| GFX | gyrA gyrB | A90V, A94G, A94T, A94A, A94H and A89A Δ678, Δ679, and A533T | Nosova et al. 2013; Groll et al. 2009 |
| BDQ | atpE rv0678 | A63P, A63V, D28A, D28V, D28P, D28N, D28G, R124stop, L40F, T91P, and E21stop G66E, M1A, W42R, S53L, S53P, S63R, and S63G | Huitric et al. 2010; Segala et al. 2012; Zimenkov et al. 2017; Andries et al. 2014; Pang et al. 2017; Xu et al. 2018; Zhang et al. 2015 |
| LZD | rplC rrl | C154R G2299T, G2814T, G2270T, and G2746A | McNeil et al. 2017; Zhang et al. 2016; Pang 2017; Balasubramanian et al. 2014, Zhang et al. 2014; McNeil et al. 2017 |
| DM | ddn fgd1 | L107P and 59–101 (deletion) T960C | Schena et al. 2016, Fujiwara et al. 2017 |
| PM | ddn fgdi fbiC | V616, Y89H, and Y133D R212Q A2158A and C1114T | Haver et al. 2015 |
| SQ-109 | mmPL3 | A700T, L567P, Q40R, and T2055375C | Tahlan et al. 2012 |
| BTZ-043 | dprE1 | C387G, C387S, C387A, C387T, and C387N | Foo et al. 2016 |
| PBTZ-169 | dprE1 | C387G, C387S, C387A, C387T, and C387N | Foo et al. 2016; Chen et al. 2020 |
| OPC-167832 | dprE1 (rv3790) mmpS5-mmpL5 (rv0678) | C387G, C387S, C387A, C387T, C387N, and V388T G248C, A364S,T314H, and 84–85InsIS6110 | Hariguchi et al. 2020; Milano et al 2009 |
| TBA-7371 | dprE1 | Y314H | Gawad and Bonde 2018 |
| Q203 | qcrB | T313A | |
| NITD-916 | inhA fabG1InhA | S19W, I21M, I21V, F41L, F47L, S94A, M103T, D148E, M161L, R195G, I202F, G205S, G205A, G205R, A206E, G212D, G214P, I215S, L269R, Δ210, T162M, and R49H C-15T | McNeil 2017 |
| Chemical Class | Novel Drug | MW g/mol | LogP (ChemDraw) | Clinical Phase | Cellular Target | Tmax h | AUC mg.h/L a ng.h/mL b µg.h/mLc | T1/2 h | CYP450 Inhibition |
|---|---|---|---|---|---|---|---|---|---|
| Fluoroquinolones | Moxifloxacin | 401.43 | 1.60 | III | DNA gyrase and Topoisomerases | 1.5 | 26.9a | 11.5–15.6 | – |
| Gatifloxacin | 375.39 | 1.2 | III | DNA gyrase and Topoisomerases | 1–2 | 51.3 a | 7–14 | – | |
| DC-195a | 421.44 | 1.24 | Pre-clinical | DNA Replication | – | 15.8 a | 1.51–1.93 | Un-confirmed | |
| Diarylquinolones | Bedaquiline | 555.50 | 7.52 | IIb/III | ATP Synthase | 4–6 | 65 a | 21.7–24 | Moderate–high risk |
| TBAJ-876 | 657.56 | 6.08 | Pre-clinical | ATP Synthase | – | 4.61 c | – | – | |
| TBAJ-587 | 614.5 | 6.4 | Pre-clinical | ATP Synthase | – | 1.72 c | – | – | |
| Oxazolidinone | Linezolid | 337.35 | 0.58 | Iib | Protein synthesis | 1–2 | 210 a | 6–7.9 | – |
| Sutezolid | 353.41 | 1.3 | Iia | Protein synthesis | 0.5 | 31945 b | 2.8–4 | Substrate | |
| AZD5847 | 465.40 | 0.7 | Iia | Protein synthesis | 2–4 | 93.19 c | 7–11 | – | |
| Nitroimidazoles | Delamanid | 534.5 | — | III | Cell Wall Acids Synthesis | 4–5 | 2.9 a | 30–38 | Liver CYP3A |
| Pretomanid | 359.26 | — | III | Cell Wall, Lipids, and Protein synthesis | 4–5 | 53 c | 10–30 | Insignificant | |
| TBA 354 | 436.30 | — | II | Cell Wall, Lipids, and Protein synthesis | 2–6 | 22.7–242 c | 8–12 | Weak CYP3A4 | |
| Ethylenediamines | SQ109 | 330.50 | 4.44 | Iib/III | Cell Wall Acids Synthesis | 1 | 183.7–268.5 b | 19.6 | CYP2D6, CYP2C19 & weak CYP3A4 |
| Benzothiazinones | BTZ043 | 431.40 | — | Pre-clinical | DprE1 | 0.25 | 899 b | 1.22 | Low |
| PBTZ169 | 456.48 | 4.42 | II | DprE1 | 1.5–2.5 | 5478 b | 2.87 | – | |
| Carbostyrils | OPC-167832 | 456.85 | 2.82 | I | DprE1 | 0.5–1 | Dose dependent | 1.3–2.1 | – |
| TBA-7371 | 355.40 | 1.31 | I | DprE1 | – | 166–240 c | – | – | |
| Imidazopyridine amides | Q203 | 557.01 | 7.64 | Pre-clinical | QcrB | 2 | 44100 b | 23.4 | – |
| TB47 | 538.57 | 6.61 | QcrB | 3.2 | 33144 c | 35.6 | Insignificant | ||
| ND-11543 | 532.56 | 5.84 | QcrB | 2 | 11704 b | 24 | Substrate | ||
| Caprazamycins | CPZEN-45 | 688.70 | −2.59 | Pre-clinical | Cell wall Peptidoglycan biosynthesis (WecA) | – | – | – | – |
| Oxaboroles | GSK070 (3036656) | 257.48 | — | II | LeuRS | – | – | – | – |
| Riminophenazines | TBI-166 | 589.60 | 5.08 | Pre-clinical | DNA Synthesis (QcrB) | 2.7 | 2658.2 b | 20.4 | – |
| Hydrazides | LL3858 | 519.57 | 5.31 | I | Undefined | – | – | – | – |
| Pyrroles | BM212 | 414.40 | 5.30 | Lead Optimisation | MmpL3 Protein | – | – | – | – |
| Oxoborates | GSK-693 | 419.54 | 5.69 | Lead Optimisation | InhA | – | – | – | CYP3A4 |
| NITD-916 | 311.42 | 2.56 | Lead Optimisation | InhA | – | – | – | – | |
| Β-lactams | Faropenem | 285.31 | −1.62 | II | L,D-traspeptidase | 2 | 16.2 a | 1.2 | – |
| Ertapenem | 475.52 | −1.72 | II | L,D-traspeptidase | – | 544.9 a | 4 | – |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angula, K.T.; Legoabe, L.J.; Beteck, R.M. Chemical Classes Presenting Novel Antituberculosis Agents Currently in Different Phases of Drug Development: A 2010–2020 Review. Pharmaceuticals 2021, 14, 461. https://doi.org/10.3390/ph14050461
Angula KT, Legoabe LJ, Beteck RM. Chemical Classes Presenting Novel Antituberculosis Agents Currently in Different Phases of Drug Development: A 2010–2020 Review. Pharmaceuticals. 2021; 14(5):461. https://doi.org/10.3390/ph14050461
Chicago/Turabian StyleAngula, Klaudia T., Lesetja J. Legoabe, and Richard M. Beteck. 2021. "Chemical Classes Presenting Novel Antituberculosis Agents Currently in Different Phases of Drug Development: A 2010–2020 Review" Pharmaceuticals 14, no. 5: 461. https://doi.org/10.3390/ph14050461
APA StyleAngula, K. T., Legoabe, L. J., & Beteck, R. M. (2021). Chemical Classes Presenting Novel Antituberculosis Agents Currently in Different Phases of Drug Development: A 2010–2020 Review. Pharmaceuticals, 14(5), 461. https://doi.org/10.3390/ph14050461