
Therapeutic Strategies for Metastatic Triple-Negative Breast Cancers: From Negative to Positive


Abstract
1. Background
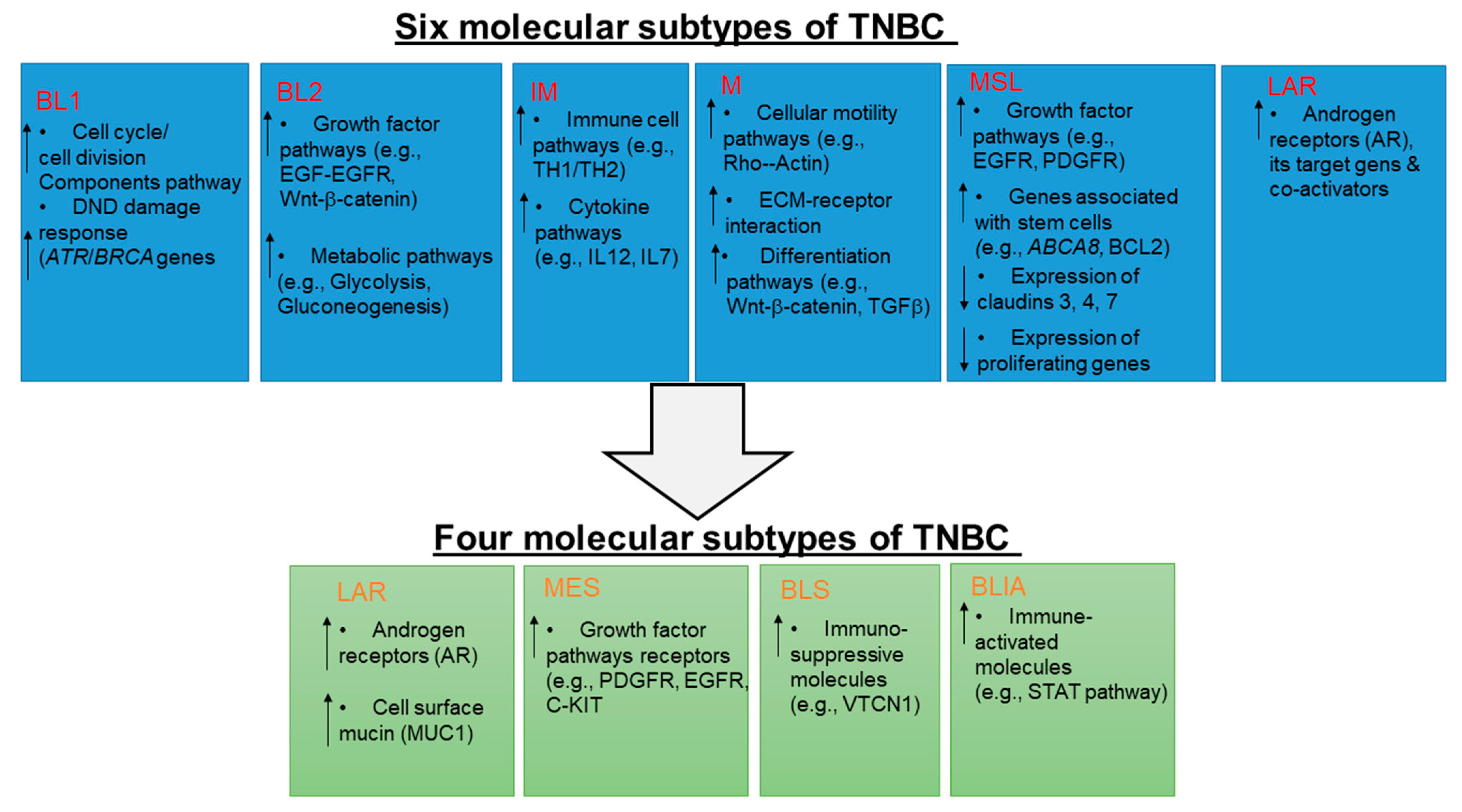
2. Molecular Subtypes of TNBC
3. Current FDA Approved Treatment Options
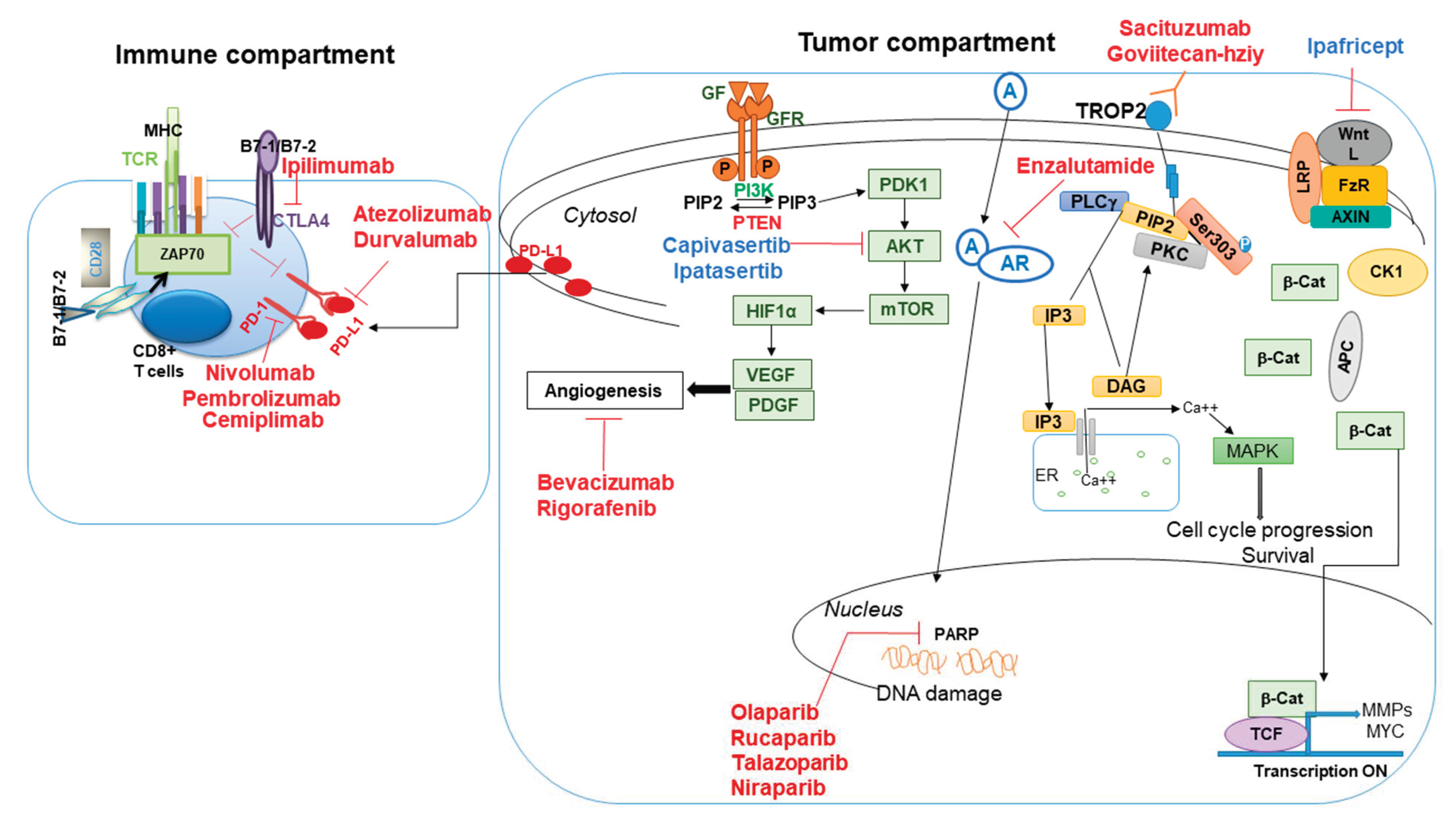
3.1. Alterations of DNA Damage Signaling Pathway and Targeted Therapeutics
3.2. Immune Therapies in TNBC Patients
3.3. Antibody–Drug Conjugates (ADC) as a Targeted Therapy
4. Promising Treatment Options with Drugs That Need FDA Approval
4.1. Antibody–Drug Conjugates (ADC)
4.2. Signaling Pathway-Targeted Therapies
5. Walking Forward
6. Limitations
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADC | antibody–drug conjugate |
| AR | androgen receptor |
| CBR | clinical benefit rate |
| CR | complete response |
| ctDNA | circulating tumor DNA |
| CPS | combines positive score |
| CTC | circulating tumor cells |
| CTLA4 | cytotoxic T-lymphocyte-associated protein 4 |
| DCR | disease control rate |
| DFS | disease-free survival |
| ER | estrogen receptor |
| HR | homologous recombination |
| HR | hazard ratio |
| HER2 | epidermal growth factor receptor 2 |
| ITT | intention to treat |
| IL | interleukin |
| NGS | next-generation sequencing |
| ORR | objective response rate |
| OS | overall survival |
| PARP | poly (ADP ribose) polymerase |
| PR | progesterone receptor |
| PR | partial response |
| PD-1 | programmed cell death protein 1 |
| PD-L1/2 | programmed death-ligand 1/2 |
| PFS | progression-free survival |
| TNBC | triple-negative breast cancer |
| VEGF | vascular endothelial growth factor |
References
- Malorni, L.; Shetty, P.B.; De Angelis, C.; Hilsenbeck, S.; Rimawi, M.F.; Elledge, R.; Osborne, C.K.; De Placido, S.; Arpino, G. Clinical and biologic features of triple-negative breast cancers in a large cohort of patients with long-term follow-up. Breast Cancer Res. Treat. 2012, 136, 795–804. [Google Scholar] [CrossRef]
- Billar, J.A.; Dueck, A.C.; Stucky, C.C.; Gray, R.J.; Wasif, N.; Northfelt, D.W.; McCullough, A.E.; Pockaj, B.A. Triple-negative breast cancers: Unique clinical presentations and outcomes. Ann. Surg. Oncol. 2010, 17 (Suppl. 3), 384–390. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.J.; Naidu, S.; Topham, A.K.; Guiles, F.; Xu, Y.; McCue, P.; Schwartz, G.F.; Park, P.K.; Rosenberg, A.L.; Brill, K.; et al. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: A single-institution compilation compared with the National Cancer Institute’s Surveillance, Epidemiology, and End Results database. Cancer 2007, 110, 876–884. [Google Scholar] [CrossRef]
- Stewart, P.A.; Luks, J.; Roycik, M.D.; Sang, Q.X.; Zhang, J. Differentially expressed transcripts and dysregulated signaling pathways and networks in African American breast cancer. PLoS ONE 2013, 8, e82460. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Toy, K.A.; Griffith, K.A.; Awuah, B.; Quayson, S.; Newman, L.A.; Kleer, C.G. Invasive breast carcinomas in Ghana: High frequency of high grade, basal-like histology and high EZH2 expression. Breast Cancer Res. Treat. 2012, 135, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Siddharth, S.; Sharma, D. Racial Disparity and Triple-Negative Breast Cancer in African-American Women: A Multifaceted Affair between Obesity, Biology, and Socioeconomic Determinants. Cancers 2018, 10, 514. [Google Scholar] [CrossRef]
- Parsons, H.A.; Burstein, H.J. Adjuvant Capecitabine in Triple-Negative Breast Cancer: New Strategies for Tailoring Treatment Recommendations. JAMA 2021, 325, 36–38. [Google Scholar] [CrossRef]
- Wang, X.; Wang, S.S.; Huang, H.; Cai, L.; Zhao, L.; Peng, R.J.; Lin, Y.; Tang, J.; Zeng, J.; Zhang, L.H.; et al. Effect of Capecitabine Maintenance Therapy Using Lower Dosage and Higher Frequency vs Observation on Disease-Free Survival Among Patients With Early-Stage Triple-Negative Breast Cancer Who Had Received Standard Treatment: The SYSUCC-001 Randomized Clinical Trial. JAMA 2021, 325, 50–58. [Google Scholar] [CrossRef]
- Cortes, J.; O’Shaughnessy, J.; Loesch, D.; Blum, J.L.; Vahdat, L.T.; Petrakova, K.; Chollet, P.; Manikas, A.; Dieras, V.; Delozier, T.; et al. Eribulin monotherapy versus treatment of physician’s choice in patients with metastatic breast cancer (EMBRACE): A phase 3 open-label randomised study. Lancet 2011, 377, 914–923. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Kandel, M.J.; Stadler, Z.; Masciari, S.; Collins, L.; Schnitt, S.; Harris, L.; Miron, A.; Richardson, A.; Garber, J.E. Prevalence of BRCA1 mutations in triple negative breast cancer (BC). J. Clin. Oncol. 2006, 24, 508. [Google Scholar] [CrossRef]
- Dey, N.; Smith, B.R.; Leyland-Jones, B. Targeting basal-like breast cancers. Curr. Drug Targets 2012, 13, 1510–1524. [Google Scholar] [CrossRef]
- Park, J.H.; Ahn, J.H.; Kim, S.B. How shall we treat early triple-negative breast cancer (TNBC): From the current standard to upcoming immuno-molecular strategies. ESMO Open 2018, 3, e000357. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Angulo, A.M.; Timms, K.M.; Liu, S.; Chen, H.; Litton, J.K.; Potter, J.; Lanchbury, J.S.; Stemke-Hale, K.; Hennessy, B.T.; Arun, B.K.; et al. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin. Cancer Res. 2011, 17, 1082–1089. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- McCann, K.E.; Hurvitz, S.A. Advances in the use of PARP inhibitor therapy for breast cancer. Drugs Context 2018, 7, 212540. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- FDA. Approves Olapraib for Germline BRCA-Mutated Metastatic Breast Cancer. Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm592357.htm (accessed on 15 March 2021).
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- FDA. Approves Talazoparib for gBRCAm HER-Negative Locally Advanced or Metastatic Breast Cancer. Available online: www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm623540.htm (accessed on 15 March 2021).
- Turner, N.C.; Telli, M.L.; Rugo, H.S.; Mailliez, A.; Ettl, J.; Grischke, E.M.; Mina, L.A.; Balmana Gelpi, J.; Fasching, P.A.; Hurvitz, S.A.; et al. Final results of aphase 2 study of talazoparib (TALA) following multiple cytotoxic regimens in advanded breast cancer patients (pts) with germline BRCA1/2 mutations (ABRAZO). J. Clin. Oncol. 2017, 35, 1007. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Goncalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Chopra, N.; Tovey, H.; Pearson, A.; Cutts, R.; Toms, C.; Proszek, P.; Hubank, M.; Dowsett, M.; Dodson, A.; Daley, F.; et al. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat. Commun. 2020, 11, 2662. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, S.K.; Schelman, W.R.; Wilding, G.; Moreno, V.; Baird, R.D.; Miranda, S.; Hylands, L.; Riisnaes, R.; Forster, M.; Omlin, A.; et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: A phase 1 dose-escalation trial. Lancet Oncol. 2013, 14, 882–892. [Google Scholar] [CrossRef]
- Dieras, V.; Han, H.S.; Kaufman, B.; Wildiers, H.; Friedlander, M.; Ayoub, J.P.; Puhalla, S.L.; Bondarenko, I.; Campone, M.; Jakobsen, E.H.; et al. Veliparib with carboplatin and paclitaxel in BRCA-mutated advanced breast cancer (BROCADE3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 1269–1282. [Google Scholar] [CrossRef]
- Stanton, S.E.; Adams, S.; Disis, M.L. Variation in the Incidence and Magnitude of Tumor-Infiltrating Lymphocytes in Breast Cancer Subtypes: A Systematic Review. JAMA Oncol. 2016, 2, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Safonov, A.; Jiang, T.; Bianchini, G.; Gyorffy, B.; Karn, T.; Hatzis, C.; Pusztai, L. Immune Gene Expression Is Associated with Genomic Aberrations in Breast Cancer. Cancer Res. 2017, 77, 3317–3324. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw, W.G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar] [CrossRef]
- Huang, J.; Wang, L.; Cong, Z.; Amoozgar, Z.; Kiner, E.; Xing, D.; Orsulic, S.; Matulonis, U.; Goldberg, M.S. The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1(-/-) murine model of ovarian cancer. Biochem. Biophys. Res. Commun. 2015, 463, 551–556. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.A.; Park, Y.H.; Delord, J.P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): An open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-Label Clinical Trial of Niraparib Combined With Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef]
- Teicher, B.A.; Chari, R.V. Antibody conjugate therapeutics: Challenges and potential. Clin. Cancer Res. 2011, 17, 6389–6397. [Google Scholar] [CrossRef]
- Won, K.A.; Spruck, C. Triplenegative breast cancer therapy: Current and future perspectives (Review). Int. J. Oncol. 2020, 57, 1245–1261. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Huang, J.F.; Qiu, J.R.; Zhang, H.L.; Tang, X.J.; Li, H.; Wang, C.J.; Wang, Z.C.; Feng, Z.Q.; Zhu, J. Significantly upregulated TACSTD2 and Cyclin D1 correlate with poor prognosis of invasive ductal breast cancer. Exp. Mol. Pathol. 2013, 94, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Mayer, I.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond, J.R.; O’Shaughnessy, J.; Moroose, R.L.; Santin, A.D.; Abramson, V.G.; et al. Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2019, 380, 741–751. [Google Scholar] [CrossRef]
- Modi, S.; Pusztai, L.; Forero, A.; Mita, M.; Miller, K.D.; Weise, A.; Krop, I.; Burris, H.; Kalinsky, K.; Tsai, M.; et al. Phase 1 study of the antibody-drug conjugate SGN-LIV1A in patients with heavily pretreated triple-negative metastatic breast cancer. Cancer Res. 2018, 78, 7445. [Google Scholar] [CrossRef]
- Cortés, J.; Diab, S.; Basho, R.K.; Oliveira, M.; Pluard, T.; Alemany, C.; Brown-Glaberman, U.; Meisel, J.; Boni, V.; Sinha, R.; et al. SGNLVA-002: Single-arm, open label phase Ib/II study of ladiratuzumab vedotin (LV) in combination with pembrolizumab for first-line treatment of patients with unresectable locally advanced or metastatic triple-negative breast cancer. J. Clin. Oncol. 2019, 37, 15. [Google Scholar] [CrossRef]
- Beerli, R.R.; Waldmeier, L.; Gebleux, R.; Pretto, F.; Grawunder, U. NBE-002, an anthracycline-based immune-stimulatory antibody drug conjugate (iADC) targeting ROR1 for the treatment of triple-negative breast cancer. Cancer Res. 2019, 79, 13. [Google Scholar] [CrossRef]
- Modi, S.; Ohtani, S.; Lee, C.C.; Wang, K.; Saxena, K.; Cameron, D.A. A phase III, multicenter, randomized, open label trial of [fam-] trastuzumab deruxtecan (DS-8201a) versus investigator’s choice in HER2-low breast cancer. J. Clin. Oncol. 2019, 37, 15. [Google Scholar] [CrossRef]
- Cancer, G.; Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef]
- Kim, S.B.; Dent, R.; Im, S.A.; Espie, M.; Blau, S.; Tan, A.R.; Isakoff, S.J.; Oliveira, M.; Saura, C.; Wongchenko, M.J.; et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017, 18, 1360–1372. [Google Scholar] [CrossRef]
- Dent, R.; Oliveira, M.; Isakoff, S.J.; Im, S.A.; Espié, M.; Blau, S.; Tan, A.R.; Saura, C.; Wongchenko, M.; Xu, N.; et al. Final Results fo the double blind placebo (PBO)-controlled randomised phase II LOTUS trial of first line ipatasertic (IPAT) + paclitaxel (PAC) for inoperable locally advancded/ metastatic triple-negative breast cancer (mTNBC). Ann. Oncol. 2020, 31, S62–S82. [Google Scholar] [CrossRef]
- Schmid, P.; Abraham, J.; Chan, S.; Wheatley, D.; Brunt, A.M.; Nemsadze, G.; Baird, R.D.; Park, Y.H.; Hall, P.S.; Perren, T.; et al. Capivasertib Plus Paclitaxel Versus Placebo Plus Paclitaxel As First-Line Therapy for Metastatic Triple-Negative Breast Cancer: The PAKT Trial. J. Clin. Oncol. 2020, 38, 423–433. [Google Scholar] [CrossRef]
- De, P.; Sun, Y.; Carlson, J.H.; Friedman, L.S.; Leyland-Jones, B.R.; Dey, N. Doubling down on the PI3K-AKT-mTOR pathway enhances the antitumor efficacy of PARP inhibitor in triple negative breast cancer model beyond BRCA-ness. Neoplasia 2014, 16, 43–72. [Google Scholar] [CrossRef]
- Juvekar, A.; Burga, L.N.; Hu, H.; Lunsford, E.P.; Ibrahim, Y.H.; Balmana, J.; Rajendran, A.; Papa, A.; Spencer, K.; Lyssiotis, C.A.; et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012, 2, 1048–1063. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.H.; Garcia-Garcia, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzman, M.; Grueso, J.; et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef]
- Yap, T.A.; Kristeleit, R.; Michalarea, V.; Pettitt, S.J.; Lim, J.S.J.; Carreira, S.; Roda, D.; Miller, R.; Riisnaes, R.; Miranda, S.; et al. Phase I Trial of the PARP Inhibitor Olaparib and AKT Inhibitor Capivasertib in Patients with BRCA1/2- and Non-BRCA1/2-Mutant Cancers. Cancer Discov. 2020, 10, 1528–1543. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Mao, Y.; Fei, X.C.; Shen, K.W. The impact of androgen receptor expression on breast cancer survival: A retrospective study and meta-analysis. PLoS ONE 2013, 8, e82650. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Pan, B.; Zhu, H.; Zhou, Y.; Mao, F.; Lin, Y.; Xu, Q.; Sun, Q. Prognostic value of androgen receptor in triple negative breast cancer: A meta-analysis. Oncotarget 2016, 7, 46482–46491. [Google Scholar] [CrossRef]
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A.; et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin. Cancer Res. 2013, 19, 5505–5512. [Google Scholar] [CrossRef] [PubMed]
- Traina, T.A.; Miller, K.; Yardley, D.A.; Eakle, J.; Schwartzberg, L.S.; O’Shaughnessy, J.; Gradishar, W.; Schmid, P.; Winer, E.; Kelly, C.; et al. Enzalutamide for the Treatment of Androgen Receptor-Expressing Triple-Negative Breast Cancer. J. Clin. Oncol. 2018, 36, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Schafer, J.M.; Pendleton, C.S.; Tang, L.; Johnson, K.C.; Chen, X.; Balko, J.M.; Gomez, H.; Arteaga, C.L.; et al. PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors. Breast Cancer Res. 2014, 16, 406. [Google Scholar] [CrossRef]
- Robles, A.J.; Cai, S.; Cichewicz, R.H.; Mooberry, S.L. Selective activity of deguelin identifies therapeutic targets for androgen receptor-positive breast cancer. Breast Cancer Res. Treat. 2016, 157, 475–488. [Google Scholar] [CrossRef]
- Wheler, J.J.; Janku, F.; Naing, A.; Li, Y.; Stephen, B.; Zinner, R.; Subbiah, V.; Fu, S.; Karp, D.; Falchook, G.S.; et al. TP53 Alterations Correlate with Response to VEGF/VEGFR Inhibitors: Implications for Targeted Therapeutics. Mol. Cancer Ther. 2016, 15, 2475–2485. [Google Scholar] [CrossRef]
- Leslie, K.K.; Filiaci, V.L.; Mallen, A.R.; Thiel, K.W.; Devor, E.J.; Moxley, K.; Richardson, D.; Mutch, D.; Secord, A.A.; Tewari, K.S.; et al. Mutated p53 portends improvement in outcomes when bevacizumab is combined with chemotherapy in advanced/recurrent endometrial cancer: An NRG Oncology study. Gynecol. Oncol. 2021, 161, 113–121. [Google Scholar] [CrossRef]
- Robert, N.J.; Dieras, V.; Glaspy, J.; Brufsky, A.M.; Bondarenko, I.; Lipatov, O.N.; Perez, E.A.; Yardley, D.A.; Chan, S.Y.; Zhou, X.; et al. RIBBON-1: Randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J. Clin. Oncol. 2011, 29, 1252–1260. [Google Scholar] [CrossRef]
- Brufsky, A.; Valero, V.; Tiangco, B.; Dakhil, S.R.; Brize, A.; Bousfoul, N.; Rugo, H.S.; Yardley, D.A. Impact of bevacuzumab (BEV) on efficacy of second-line chemotherapy (CT) for triple-negative breast cancer (TNBC): Analysis of RIBBON-2. J. Clin. Oncol. 2011, 29, 1010. [Google Scholar] [CrossRef]
- Burstein, H.J.; Elias, A.D.; Rugo, H.S.; Cobleigh, M.A.; Wolff, A.C.; Eisenberg, P.D.; Lehman, M.; Adams, B.J.; Bello, C.L.; DePrimo, S.E.; et al. Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J. Clin. Oncol. 2008, 26, 1810–1816. [Google Scholar] [CrossRef]
- De, P.; Carlson, J.H.; Jepperson, T.; Willis, S.; Leyland-Jones, B.; Dey, N. RAC1 GTP-ase signals Wnt-beta-catenin pathway mediated integrin-directed metastasis-associated tumor cell phenotypes in triple negative breast cancers. Oncotarget 2017, 8, 3072–3103. [Google Scholar] [CrossRef]
- Dey, N.; Barwick, B.G.; Moreno, C.S.; Ordanic-Kodani, M.; Chen, Z.; Oprea-Ilies, G.; Tang, W.; Catzavelos, C.; Kerstann, K.F.; Sledge, G.W., Jr.; et al. Wnt signaling in triple negative breast cancer is associated with metastasis. BMC Cancer 2013, 13, 537. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, J.; Rishi, A.K.; Reddy, K.B. Novel strategies to target chemoresistant triple-negative breast cancer. Genes Cancer 2020, 11, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Xie, W.; Zhao, H.; Wang, F.; Wang, Y.; He, Y.; Wang, T.; Zhang, K.; Yang, H.; Zhou, Z.; Shi, H.; et al. A novel humanized Frizzled-7-targeting antibody enhances antitumor effects of Bevacizumab against triple-negative breast cancer via blocking Wnt/beta-catenin signaling pathway. J. Exp. Clin. Cancer Res. 2021, 40, 30. [Google Scholar] [CrossRef]
- Le, P.N.; McDermott, J.D.; Jimeno, A. Targeting the Wnt pathway in human cancers: Therapeutic targeting with a focus on OMP-54F28. Pharmacol. Ther. 2015, 146, 1–11. [Google Scholar] [CrossRef]
- Jimeno, A.; Gordon, M.; Chugh, R.; Messersmith, W.; Mendelson, D.; Dupont, J.; Stagg, R.; Kapoun, A.M.; Xu, L.; Uttamsingh, S.; et al. A First-in-Human Phase I Study of the Anticancer Stem Cell Agent Ipafricept (OMP-54F28), a Decoy Receptor for Wnt Ligands, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 7490–7497. [Google Scholar] [CrossRef] [PubMed]
- Dotan, E.; Cardin, D.B.; Lenz, H.J.; Messersmith, W.; O’Neil, B.; Cohen, S.J.; Denlinger, C.S.; Shahda, S.; Astsaturov, I.; Kapoun, A.M.; et al. Phase Ib Study of Wnt Inhibitor Ipafricept with Gemcitabine and nab-paclitaxel in Patients with Previously Untreated Stage IV Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 5348–5357. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; LaBaff, A.; Wei, Y.; Nie, L.; Xia, W.; Huo, L.; Yamaguchi, H.; Hsu, Y.H.; Hsu, J.L.; Liu, D.; et al. Phosphorylation of EZH2 at T416 by CDK2 contributes to the malignancy of triple negative breast cancers. Am. J. Transl. Res. 2015, 7, 1009–1020. [Google Scholar]
- Puppe, J.; Opdam, M.; Schouten, P.C.; Jozwiak, K.; Lips, E.; Severson, T.; van de Ven, M.; Brambillasca, C.; Bouwman, P.; van Tellingen, O.; et al. EZH2 Is Overexpressed in BRCA1-like Breast Tumors and Predictive for Sensitivity to High-Dose Platinum-Based Chemotherapy. Clin. Cancer Res. 2019, 25, 4351–4362. [Google Scholar] [CrossRef]
- Yang, X.; Phillips, D.L.; Ferguson, A.T.; Nelson, W.G.; Herman, J.G.; Davidson, N.E. Synergistic activation of functional estrogen receptor (ER)-alpha by DNA methyltransferase and histone deacetylase inhibition in human ER-alpha-negative breast cancer cells. Cancer Res. 2001, 61, 7025–7029. [Google Scholar] [PubMed]
- Sharma, D.; Saxena, N.K.; Davidson, N.E.; Vertino, P.M. Restoration of tamoxifen sensitivity in estrogen receptor-negative breast cancer cells: Tamoxifen-bound reactivated ER recruits distinctive corepressor complexes. Cancer Res. 2006, 66, 6370–6378. [Google Scholar] [CrossRef]
- Yomtoubian, S.; Lee, S.B.; Verma, A.; Izzo, F.; Markowitz, G.; Choi, H.; Cerchietti, L.; Vahdat, L.; Brown, K.A.; Andreopoulou, E.; et al. Inhibition of EZH2 Catalytic Activity Selectively Targets a Metastatic Subpopulation in Triple-Negative Breast Cancer. Cell Rep. 2020, 30, 755–770.e756. [Google Scholar] [CrossRef]
- Cruz, C.; Llop-Guevara, A.; Garber, J.E.; Arun, B.K.; Perez, F.J.A.; Lluch, A.; Telli, M.L.; Fernandez, C.; Kahatt, C.; Galmarini, C.M.; et al. Multicenter Phase II Study of Lurbinectedin in BRCA-Mutated and Unselected Metastatic Advanced Breast Cancer and Biomarker Assessment Substudy. J. Clin. Oncol. 2018, 36, 3134–3143. [Google Scholar] [CrossRef]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Maxfield, K.; Zineh, I. Precision Dosing: A Clinical and Public Health Imperative. JAMA 2021, 325, 1505–1506. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Miao, D.; Demetri, G.D.; Adeegbe, D.; Rodig, S.J.; Shukla, S.; Lipschitz, M.; Amin-Mansour, A.; Raut, C.P.; Carter, S.L.; et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity 2017, 46, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Vidotto, T.; Melo, C.M.; Castelli, E.; Koti, M.; Dos Reis, R.B.; Squire, J.A. Emerging role of PTEN loss in evasion of the immune response to tumours. Br. J. Cancer 2020, 122, 1732–1743. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Name of the Drug(s) | Target | Patients Types | Clinical Trial Number |
|---|---|---|---|
| Ladiratuzumab vedotin (LV) | Zinc transporter LIV1 conjugated with MMAE | Advanced or metastatic TNBC | NCT01969643 |
| LV plus pembrolizumab | LIV1 and PD-1 | Front line therapy in TNBC patient | NCT03310957 |
| NBE-002 | ROR1 conjugated with anthracycline | Advanced solid tumors, including TNBC | NCT044410099 |
| Trastuzumab deruxtecan | HER2 conjugated with topoisomerase1 inhibitor | HER2-low un-resectable and/or metastatic breast cancer | NCT03734029 |
| Ipataserib (GDC-0068) (LOTUS trial) | AKT | PI3K-AKT activated TNBC | NCT02162719 |
| Ipataserib (GDC-0068) plus atezolizumab | AKT and PD-L1 | Locally advanced unresectable or metastatic triple-negative breast cancer | NCT04177108 |
| Capivasertib (AZD5363) (PAKT trial) | AKT | Advanced or metastatic TNBC | NCT02423603 |
| Alpelisib | P110 alpha (catalytic subunit of PI3K) | Advanced TNBC | NCT04251533 |
| Bicalutamide | Androgen receptor | Metastatic TNBC | NCT00468715 |
| Enzalutamide | Androgen receptor | Advanced, androgen receptor-positive, TNBC | NCT01889238 |
| Bicalutamide plus palbociclib | Androgen receptor and CDK4/6 | AR(+) metastatic breast cancer, including TNBC | NCT02605486 |
| Bicalutamide plus ribociclib | Androgen receptor and CDK4/6 | AR(+) TNBC | NCT03090165 |
| AZD8186 (single agent) and AZD8186 plus abiraterone acetate | PI3K beta/delta and androgen receptor | TNBC | NCT01884285 |
| Avastin plus everolimus | VEGF and mTORC1 | Locally advanced TNBC with tumors predicted insensitive to standard chemotherapy | NCT02456857 |
| Avastin | VEGF | Metastatic TNBC | NCT03577743 |
| Avastin plus atezolizumab | VEGF and PD-L1 | Metastatic TNBC | NCT04739670 |
| Azacitidine plus entinostat | DNA methyltransferase and HDAC | Advanced breast cancers including TNBC | NCT01349959 |
| Lurbinectedin | RNA polymerase II | Metastatic breast cancer, pancreatic cancer, and metastatic colorectal cancer | NCT02210364 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nandini, D.; Jennifer, A.; Pradip, D. Therapeutic Strategies for Metastatic Triple-Negative Breast Cancers: From Negative to Positive. Pharmaceuticals 2021, 14, 455. https://doi.org/10.3390/ph14050455
Nandini D, Jennifer A, Pradip D. Therapeutic Strategies for Metastatic Triple-Negative Breast Cancers: From Negative to Positive. Pharmaceuticals. 2021; 14(5):455. https://doi.org/10.3390/ph14050455
Chicago/Turabian StyleNandini, Dey, Aske Jennifer, and De Pradip. 2021. "Therapeutic Strategies for Metastatic Triple-Negative Breast Cancers: From Negative to Positive" Pharmaceuticals 14, no. 5: 455. https://doi.org/10.3390/ph14050455
APA StyleNandini, D., Jennifer, A., & Pradip, D. (2021). Therapeutic Strategies for Metastatic Triple-Negative Breast Cancers: From Negative to Positive. Pharmaceuticals, 14(5), 455. https://doi.org/10.3390/ph14050455