
Targeting Energy Expenditure—Drugs for Obesity Treatment



Abstract
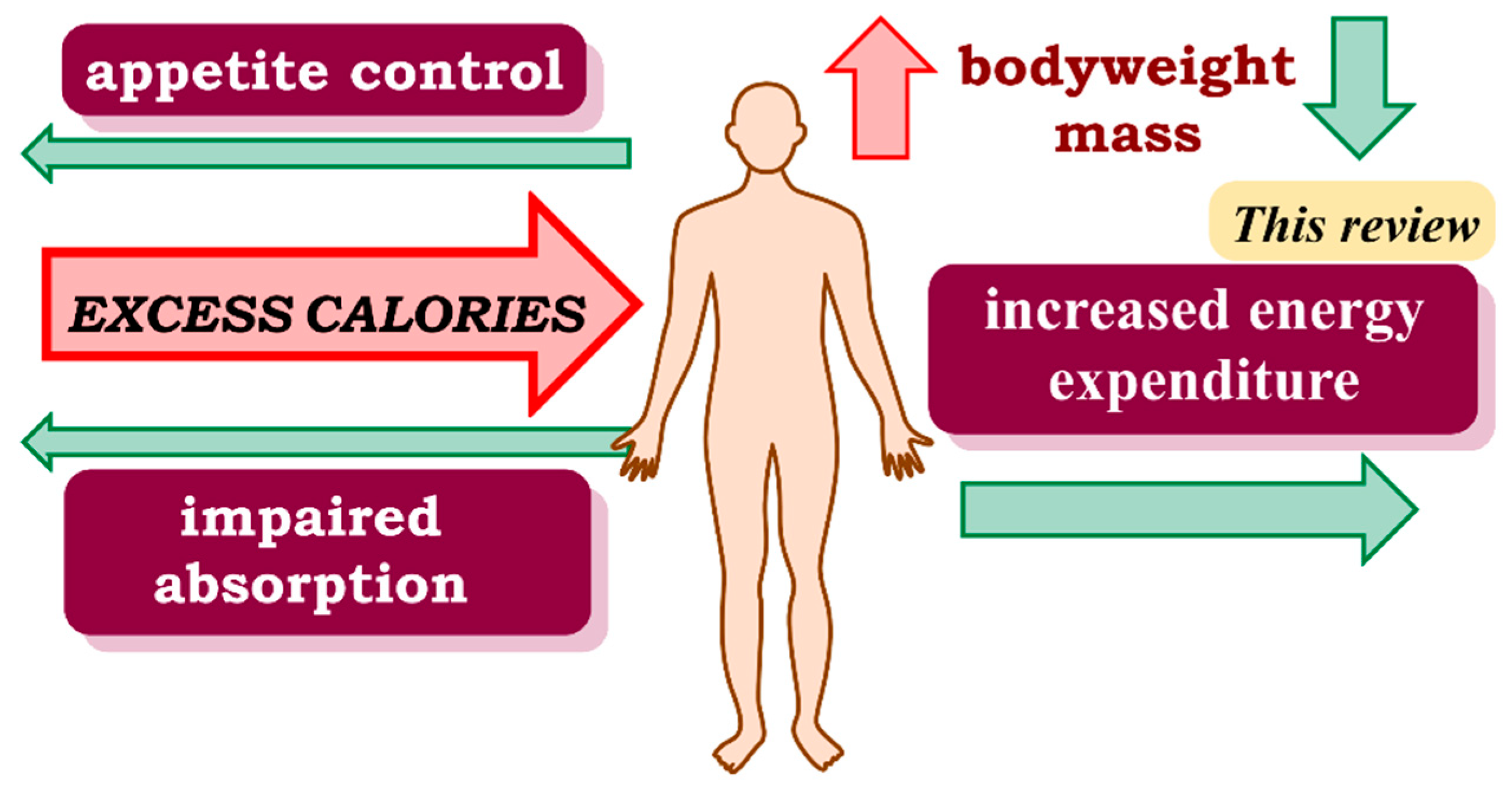
1. Introduction
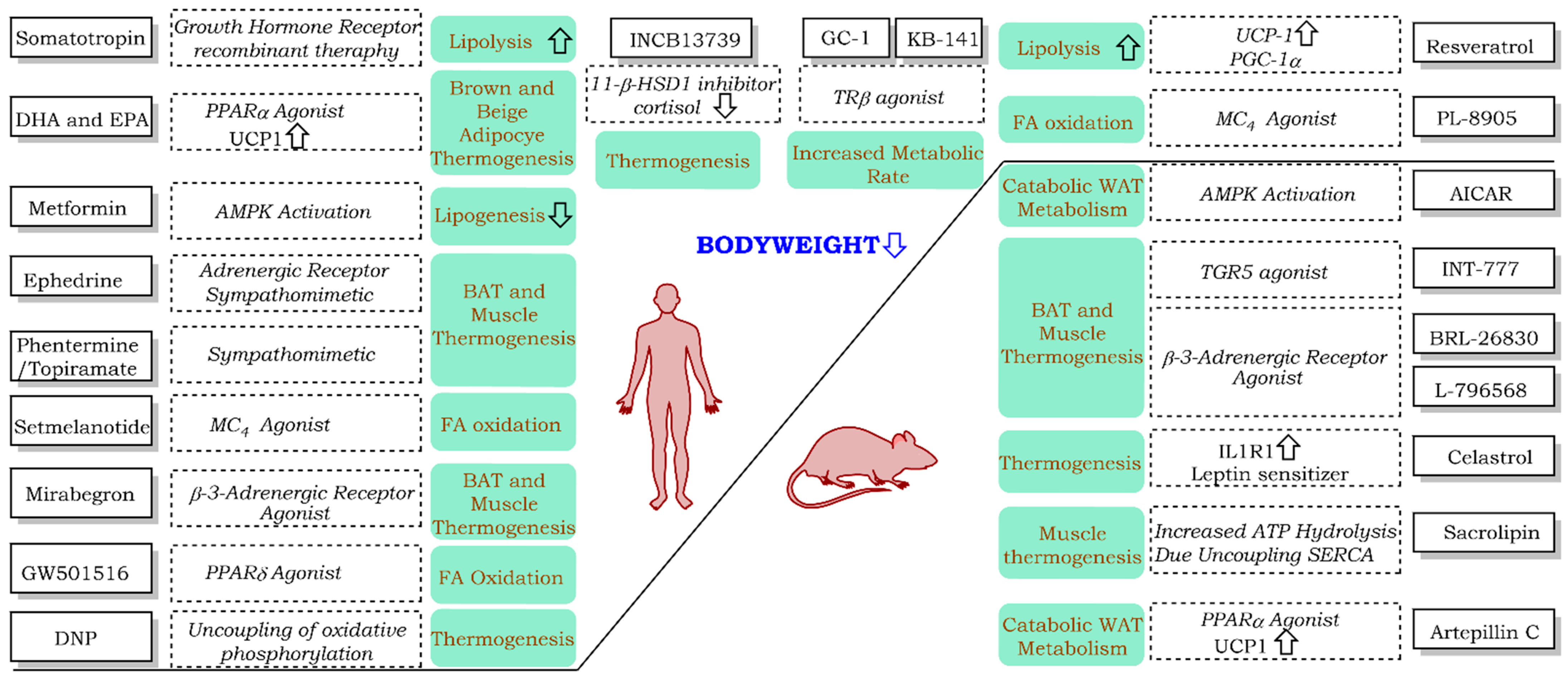
2. FDA -Approved and -Withdrawn Drugs
2.1. FDA-Approved
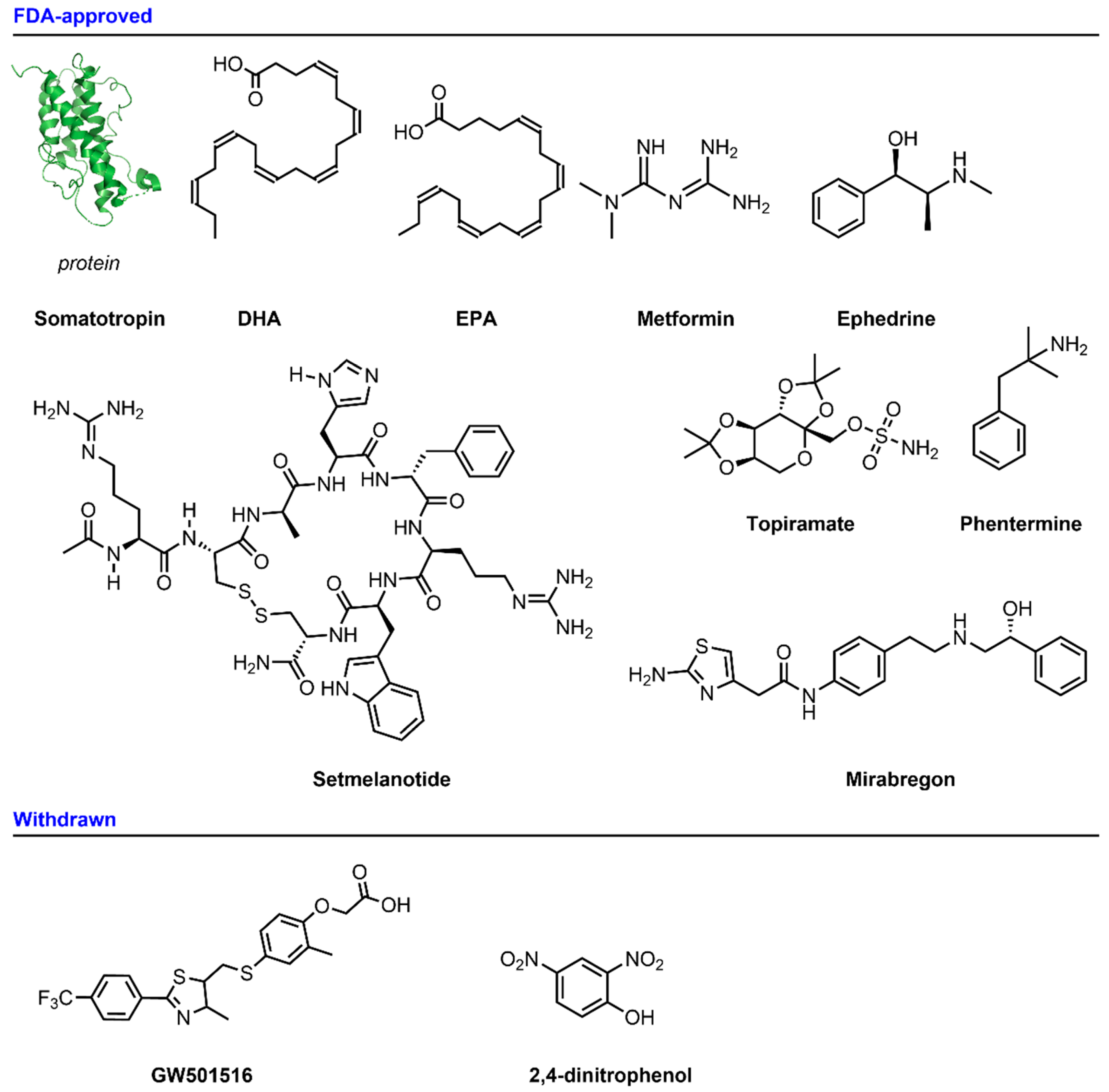
2.1.1. Somatotropin (HGH, Human Growth Hormone)
2.1.2. DHA and EPA
2.1.3. Setmelanotide
2.1.4. Metformin
2.1.5. Ephedrine
2.1.6. Phentermine/Topiramate
2.1.7. Mirabegron
2.2. Withdrawn
2.2.1. GW501516
2.2.2. 2.4-Dinitrophenol
3. Clinical Trials
3.1. INCB13739 (Oxophenylarsine)
3.2. GC-1 (Sobetirome or QRX-431)
3.3. Resveratrol
3.4. PL-8905
4. Treatments under Development
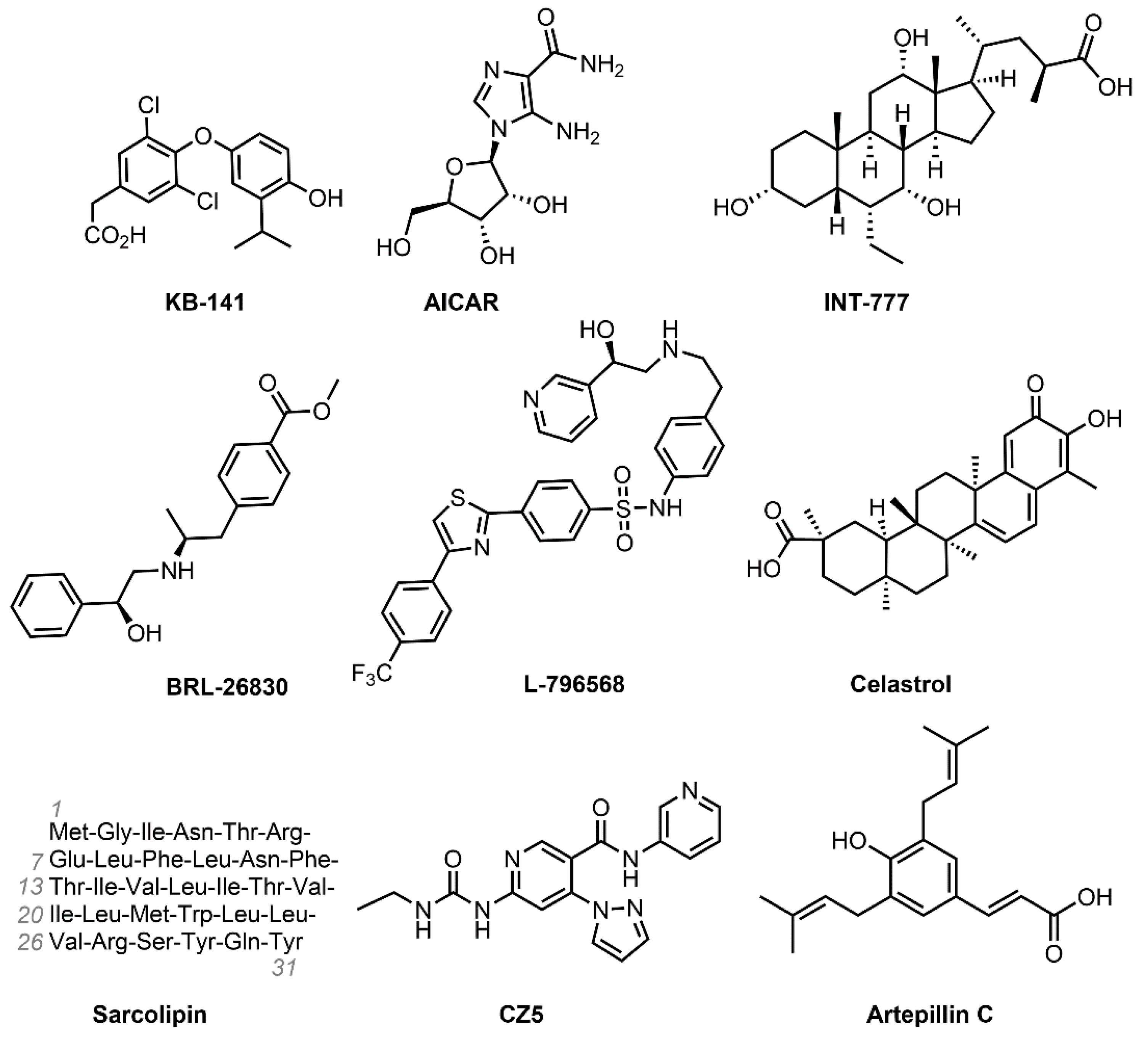
4.1. KB-141 (IH-5)
4.2. AICAR (Acadesine)
4.3. INT-777
4.4. BRL-26830 and L-796568
4.5. Celastrol
4.6. Sarcolipin
4.7. Artepillin C
5. Discussion
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- World Health Organisation. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 1 November 2019).
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef]
- Popkin, B.M.; Du, S.; Green, W.D.; Beck, M.A.; Algaith, T.; Herbst, C.H.; Alsukait, R.F.; Alluhidan, M.; Alazemi, N.; Shekar, M. Individuals with obesity and COVID-19: A global perspective on the epidemiology and biological relationships. Obes. Rev. 2020, 21, 17. [Google Scholar] [CrossRef]
- Wing, R.R.; Phelan, S. Long-term weight loss maintenance. Am. J. Clin. Nutr. 2005, 82, 222S–225S. [Google Scholar] [CrossRef]
- Tseng, Y.-H.; Cypess, A.M.; Kahn, C.R. Cellular bioenergetics as a target for obesity therapy. Nat. Rev. Drug Discov. 2010, 9, 465–482. [Google Scholar] [CrossRef]
- Betz, M.J.; Enerbäck, S. Targeting thermogenesis in brown fat and muscle to treat obesity and metabolic disease. Nat. Rev. Endocrinol. 2018, 14, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Kazak, L.; Chouchani, E.T.; Jedrychowski, M.P.; Erickson, B.K.; Shinoda, K.; Cohen, P.; Vetrivelan, R.; Lu, G.Z.; Laznik-Bogoslavski, D.; Hasenfuss, S.C.; et al. A Creatine-Driven Substrate Cycle Enhances Energy Expenditure and Thermogenesis in Beige Fat. Cell 2015, 163, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Harper, M.-E.; Green, K.; Brand, M.D. The Efficiency of Cellular Energy Transduction and Its Implications for Obesity. Annu. Rev. Nutr. 2008, 28, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Nakamura, K. Central regulation of brown adipose tissue thermogenesis and energy homeostasis dependent on food availability. Pflüger’s Arch. Gesammte Physiol. Menschen Tiere 2018, 470, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Enriori, P.J.; Sinnayah, P.; Simonds, S.E.; Rudaz, C.G.; Cowley, M.A. Leptin Action in the Dorsomedial Hypothalamus Increases Sympathetic Tone to Brown Adipose Tissue in Spite of Systemic Leptin Resistance. J. Neurosci. 2011, 31, 12189–12197. [Google Scholar] [CrossRef]
- Chellappa, K.; Perron, I.J.; Naidoo, N.; Baur, J.A. The leptin sensitizer celastrol reduces age-associated obesity and modulates behavioral rhythms. Aging Cell 2019, 18, e12874. [Google Scholar] [CrossRef] [PubMed]
- Geffner, M.; Reh, C.S. Somatotropin in the treatment of growth hormone deficiency and Turner syndrome in pediatric patients: A review. Clin. Pharmacol. Adv. Appl. 2010, 2, 111–122. [Google Scholar] [CrossRef]
- Scacchi, M.; Pincelli, A.; Cavagnini, F. Growth hormone in obesity. Int. J. Obes. 1999, 23, 260–271. [Google Scholar] [CrossRef]
- O’Sullivan, A.J.; Hoffman, D.M.; Kelly, J.J.; Freund, J.; Ho, K.K. Body composition and energy expenditure in acromegaly. J. Clin. Endocrinol. Metab. 1994, 78, 381–386. [Google Scholar] [CrossRef]
- Díez, J.J.; Sangiao-Alvarellos, S.; Cordido, F. Treatment with Growth Hormone for Adults with Growth Hormone Deficiency Syndrome: Benefits and Risks. Int. J. Mol. Sci. 2018, 19, 893. [Google Scholar] [CrossRef]
- Liang, S.; Xue, J.; Li, G. Effects of recombinant human growth hormone administration on cardiovascular risk factors in obese children with relative growth hormone deficiency. Lipids Heal. Dis. 2018, 17, 66. [Google Scholar] [CrossRef]
- ClinicalTrials.gov U.S. National Library of Medicine Growth Hormone in Obese Cases With Covid-19. Available online: https://clinicaltrials.gov/ct2/show/NCT04532554 (accessed on 3 March 2021).
- de Ronde, W.; Smit, D.L. Anabolic Androgenic Steroid Abuse in Young Males. Endocr. Connect. 2020, 9, R102–R111. [Google Scholar] [CrossRef] [PubMed]
- Kunz, H.E.; Dasari, S.; Lanza, I.R. EPA and DHA elicit distinct transcriptional responses to high-fat feeding in skeletal muscle and liver. Am. J. Physiol. Metab. 2019, 317, E460–E472. [Google Scholar] [CrossRef] [PubMed]
- Payahoo, L.; Ostadrahimi, A.; Farrin, N.; Khaje-Bishak, Y. Effects of n-3 Polyunsaturated Fatty Acid Supplementation on Serum Leptin Levels, Appetite Sensations, and Intake of Energy and Macronutrients in Obese People: A Randomized Clinical Trial. J. Diet. Suppl. 2017, 15, 596–605. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications‑A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef]
- Shang, T.; Liu, L.; Zhou, J.; Zhang, M.; Hu, Q.; Fang, M.; Wu, Y.; Yao, P.; Gong, Z. Protective effects of various ratios of DHA/EPA supplementation on high-fat diet-induced liver damage in mice. Lipids Health Dis. 2017, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, T.E.; Castro, É.; Belchior, T.; Andrade, M.L.; Chaves-Filho, A.B.; Peixoto, A.S.; Moreno, M.F.; Ortiz-Silva, M.; Moreira, R.J.; Inague, A.; et al. Fish Oil Protects Wild Type and Uncoupling Protein 1-Deficient Mice from Obesity and Glucose Intolerance by Increasing Energy Expenditure. Mol. Nutr. Food Res. 2019, 63, e1800813. [Google Scholar] [CrossRef]
- De Luis, D.; Domingo, J.C.; Izaola, O.; Casanueva, F.F.; Bellido, D.; Sajoux, I. Effect of DHA supplementation in a very low-calorie ketogenic diet in the treatment of obesity: A randomized clinical trial. Endocrinology 2016, 54, 111–122. [Google Scholar] [CrossRef]
- De la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2020. An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2021, 26, 627. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.Y.; Muniyappa, R.; Abel, B.S.; Mullins, K.P.; Staker, P.; Brychta, R.J.; Zhao, X.; Ring, M.; Psota, T.L.; Cone, R.D.; et al. RM-493, a Melanocortin-4 Receptor (MC4R) Agonist, Increases Resting Energy Expenditure in Obese Individuals. J. Clin. Endocrinol. Metab. 2015, 100, 1639–1645. [Google Scholar] [CrossRef]
- Bischof, J.M.; Van Der Ploeg, L.H.T.; Colmers, W.F.; Wevrick, R. Magel2-null mice are hyper-responsive to setmelanotide, a melanocortin 4 receptor agonist. Br. J. Pharmacol. 2016, 173, 2614–2621. [Google Scholar] [CrossRef] [PubMed]
- Clément, K.; Akker, E.V.D.; Argente, J.; Bahm, A.; Chung, W.K.; Connors, H.; De Waele, K.; Farooqi, I.S.; Gonneau-Lejeune, J.; Gordon, G.; et al. Efficacy and safety of setmelanotide, an MC4R agonist, in individuals with severe obesity due to LEPR or POMC deficiency: Single-arm, open-label, multicentre, phase 3 trials. Lancet Diabetes Endocrinol. 2020, 8, 960–970. [Google Scholar] [CrossRef]
- FDA Approves First Treatment for Weight Management for People with Certain Rare Genetic Conditions. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-approves-first-treatment-weight-management-people-certain-rare-genetic-conditions (accessed on 10 March 2020).
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetology 2017, 60, 1577–1585. [Google Scholar] [CrossRef]
- Viollet, B.; Guigas, B.; Garcia, N.S.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2011, 122, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Liu, N.-C.; Yu, I.-C.; Lin, H.-Y.; Lee, Y.-F.; Sparks, J.D.; Chen, L.-M.; Chang, C. Metformin Inhibits Nuclear Receptor TR4-Mediated Hepatic Stearoyl-CoA Desaturase 1 Gene Expression with Altered Insulin Sensitivity. Diabetes 2011, 60, 1493–1503. [Google Scholar] [CrossRef]
- Yerevanian, A.; Soukas, A.A. Metformin: Mechanisms in Human Obesity and Weight Loss. Curr. Obes. Rep. 2019, 8, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Hui, F.; Zhang, Y.; Ren, T.; Li, X.; Zhao, M.; Zhao, Q. Role of metformin in overweight and obese people without diabetes: A systematic review and network meta-analysis. Eur. J. Clin. Pharmacol. 2018, 75, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Tokubuchi, I.; Tajiri, Y.; Iwata, S.; Hara, K.; Wada, N.; Hashinaga, T.; Nakayama, H.; Mifune, H.; Yamada, K. Beneficial effects of metformin on energy metabolism and visceral fat volume through a possible mechanism of fatty acid oxidation in human subjects and rats. PLoS ONE 2017, 12, e0171293. [Google Scholar] [CrossRef] [PubMed]
- Carey, A.L.; Pajtak, R.; Formosa, M.F.; Van Every, B.; Bertovic, D.A.; Anderson, M.J.; Eikelis, N.; Lambert, G.W.; Kalff, V.; Duffy, S.J.; et al. Chronic ephedrine administration decreases brown adipose tissue activity in a randomised controlled human trial: Implications for obesity. Diabetology 2015, 58, 1045–1054. [Google Scholar] [CrossRef]
- Loh, R.; Kingwell, B.; Carey, A. Human brown adipose tissue as a target for obesity management; beyond cold-induced thermogenesis. Obes. Rev. 2017, 18, 1227–1242. [Google Scholar] [CrossRef]
- Abuzzahab, M.J.; Roth, C.L.; Shoemaker, A.H. Hypothalamic Obesity: Prologue and Promise. Horm. Res. Paediatr. 2019, 91, 128–136. [Google Scholar] [CrossRef]
- Wu, L.; Meng, J.; Shen, Q.; Zhang, Y.; Pan, S.; Chen, Z.; Zhu, L.-Q.; Lu, Y.; Huang, Y.; Zhang, G. Caffeine inhibits hypothalamic A1R to excite oxytocin neuron and ameliorate dietary obesity in mice. Nat. Commun. 2017, 8, 15904. [Google Scholar] [CrossRef]
- Halpern, B.; Mancini, M.C. Safety assessment of combination therapies in the treatment of obesity: Focus on naltrexone/bupropion extended release and phentermine-topiramate extended release. Expert Opin. Drug Saf. 2016, 16, 27–39. [Google Scholar] [CrossRef]
- Bray, G.A.; Hollander, P.; Klein, S.; Kushner, R.; Levy, B.; Fitchet, M.; Perry, B.H. A 6-Month Randomized, Placebo-Controlled, Dose-Ranging Trial of Topiramate for Weight Loss in Obesity. Obes. Res. 2003, 11, 722–733. [Google Scholar] [CrossRef]
- Ikegami, R.; Shimizu, I.; Sato, T.; Yoshida, Y.; Hayashi, Y.; Suda, M.; Katsuumi, G.; Li, J.; Wakasugi, T.; Minokoshi, Y.; et al. Gamma-Aminobutyric Acid Signaling in Brown Adipose Tissue Promotes Systemic Metabolic Derangement in Obesity. Cell Rep. 2018, 24, 2827–2837.e5. [Google Scholar] [CrossRef]
- Alfaris, N.; Minnick, A.M.; Hopkins, C.M.I.; Berkowitz, R.; Wadden, A.T. Combination phentermine and topiramate extended release in the management of obesity. Expert Opin. Pharmacother. 2015, 16, 1263–1274. [Google Scholar] [CrossRef] [PubMed]
- Dodgson, S.J.; Shank, R.P.; Maryanoff, B.E. Topiramate as an Inhibitor of Carbonic Anhydrase Isoenzymes. Epilepsia 2000, 41, S35–S39. [Google Scholar] [CrossRef] [PubMed]
- Hsia, D.S.; Gosselin, N.H.; Williams, J.; Farhat, N.; Marier, J.F.; Shih, W.; Peterson, C.; Siegel, R. A randomized, double-blind, placebo-controlled, pharmacokinetic and pharmacodynamic study of a fixed-dose combination of phentermine/topiramate in adolescents with obesity. Diabetes Obes. Metab. 2019, 22, 480–491. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; Weiner, L.S.; Roberts-Toler, C.; Elía, E.F.; Kessler, S.H.; Kahn, P.A.; English, J.; Chatman, K.; Trauger, S.A.; Doria, A.; et al. Activation of Human Brown Adipose Tissue by a β3-Adrenergic Receptor Agonist. Cell Metab. 2015, 21, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Hainer, V. Beta3-adrenoreceptor agonist mirabegron‑A potential antiobesity drug? Expert Opin. Pharmacother. 2016, 17, 2125–2127. [Google Scholar] [CrossRef]
- Loh, R.K.C.; Formosa, M.F.; La Gerche, A.; Reutens, A.T.; Kingwell, B.A.; Carey, A.L. Acute metabolic and cardiovascular effects of mirabegron in healthy individuals. Diabetes Obes. Metab. 2019, 21, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G. The Function of the Nuclear Receptor Peroxisome Proliferator–activated Receptor Delta in Energy Homeostasis. Nutr. Rev. 2003, 61, 387–390. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Doktorova, M.; Zwarts, I.; Van Zutphen, T.; Van Dijk, T.H.; Bloks, V.W.; Harkema, L.; De Bruin, A.; Downes, M.; Evans, R.M.; Verkade, H.J.; et al. Intestinal PPARδ protects against diet-induced obesity, insulin resistance and dyslipidemia. Sci. Rep. 2017, 7, 846. [Google Scholar] [CrossRef]
- Manickam, R.; Wahli, W. Roles of Peroxisome Proliferator-Activated Receptor β/δ in skeletal muscle physiology. Biochimie 2017, 136, 42–48. [Google Scholar] [CrossRef]
- Risérus, U.; Sprecher, D.; Johnson, T.; Olson, E.; Hirschberg, S.; Liu, A.; Fang, Z.; Hegde, P.; Richards, D.; Sarov-Blat, L.; et al. Activation of Peroxisome Proliferator-Activated Receptor (PPAR) Promotes Reversal of Multiple Metabolic Abnormalities, Reduces Oxidative Stress, and Increases Fatty Acid Oxidation in Moderately Obese Men. Diabetes 2008, 57, 332–339. [Google Scholar] [CrossRef]
- Olson, E.J.; Pearce, G.L.; Jones, N.P.; Sprecher, D.L. Lipid Effects of Peroxisome Proliferator-Activated Receptor-Δ Agonist GW501516 in Subjects with Low High-Density Lipoprotein Cholesterol. Arter. Thromb. Vasc. Biol. 2012, 32, 2289–2294. [Google Scholar] [CrossRef] [PubMed]
- Wall, C.E.; Yu, R.T.; Atkins, A.R.; Downes, M.; Evans, R.M. Nuclear receptors and AMPK: Can exercise mimetics cure diabetes? J. Mol. Endocrinol. 2016, 57, R49–R58. [Google Scholar] [CrossRef]
- Wang, X.; Sng, M.K.; Foo, S.; Chong, W.M.T.; Lee, W.L.; Tang, M.B.Y.; Xiaoling, W.; Luo, B.; Choong, C.; Wong, M.T.C.; et al. Early controlled release of peroxisome proliferator-activated receptor β/δ agonist GW501516 improves diabetic wound healing through redox modulation of wound microenvironment. J. Control Release 2015, 197, 138–147. [Google Scholar] [CrossRef]
- Colon-Gonzalez, F.; Kim, G.W.; Lin, J.E.; Valentino, M.A.; Waldman, S.A. Obesity pharmacotherapy: What is next? Mol. Asp. Med. 2013, 34, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Kintz, P.; Ameline, A.; Gheddar, L.; Raul, J. Testing for GW501516 (cardarine) in human hair using LC/MS–MS and confirmation by LC/HRMS. Drug Test. Anal. 2020, 12, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Grundlingh, J.; Dargan, P.I.; El-Zanfaly, M.; Wood, D.M. 2,4-Dinitrophenol (DNP): A Weight Loss Agent with Significant Acute Toxicity and Risk of Death. J. Med. Toxicol. 2011, 7, 205–212. [Google Scholar] [CrossRef]
- Petróczi, A.; Ocampo, J.A.V.; Shah, I.; Jenkinson, C.M.C.; New, R.; James, R.A.; Taylor, G.; Naughton, D.P. Russian roulette with unlicensed fat-burner drug 2,4-dinitrophenol (DNP): Evidence from a multidisciplinary study of the internet, bodybuilding supplements and DNP users. Subst. Abus. Treat. Prev. Policy 2015, 10, 39. [Google Scholar] [CrossRef]
- Sousa, D.; Carmo, H.; Bravo, R.R.; Carvalho, F.; Bastos, M.D.L.; De Pinho, P.G.; Da Silva, D.D. Diet aid or aid to die: An update on 2,4-dinitrophenol (2,4-DNP) use as a weight-loss product. Arch. Toxicol. 2020, 94, 1071–1083. [Google Scholar] [CrossRef]
- Nadler, J.E. Peripheral Neuritis Caused by Prolonged use of Dinitrophenol. J. Am. Med. Assoc. 1935, 105, 12. [Google Scholar] [CrossRef]
- Goldgof, M.; Xiao, C.; Chanturiya, T.; Jou, W.; Gavrilova, O.; Reitman, M.L. The Chemical Uncoupler 2,4-Dinitrophenol (DNP) Protects against Diet-induced Obesity and Improves Energy Homeostasis in Mice at Thermoneutrality. J. Biol. Chem. 2014, 289, 19341–19350. [Google Scholar] [CrossRef]
- Childress, E.S.; Alexopoulos, S.J.; Hoehn, K.L.; Santos, W.L. Small Molecule Mitochondrial Uncouplers and Their Therapeutic Potential. J. Med. Chem. 2018, 61, 4641–4655. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-Y.; Zhang, M.; Turner, N.; Zhang, L.-N.; Dong, T.-C.; Gu, M.; Leslie, S.J.; Li, J.-Y.; Nan, F.-J. A novel chemical uncoupler ameliorates obesity and related phenotypes in mice with diet-induced obesity by modulating energy expenditure and food intake. Diabetology 2013, 56, 2297–2307. [Google Scholar] [CrossRef]
- Murray, J.H.; Hargett, S.; Hoehn, K.L.; Santos, W.L. Anilinopyrazines as potential mitochondrial uncouplers. Bioorganic Med. Chem. Lett. 2020, 30, 127057. [Google Scholar] [CrossRef]
- Rosenstock, J.; Banarer, S.; Fonseca, V.A.; Inzucchi, S.E.; Sun, W.; Yao, W.; Hollis, G.; Flores, R.; Levy, R.; Williams, W.V.; et al. The 11- -Hydroxysteroid Dehydrogenase Type 1 Inhibitor INCB13739 Improves Hyperglycemia in Patients with Type 2 Diabetes Inadequately Controlled by Metformin Monotherapy. Diabetes Care 2010, 33, 1516–1522. [Google Scholar] [CrossRef]
- Morgan, S.; Tomlinson, J.W. 11β-Hydroxysteroid dehydrogenase type 1 inhibitors for the treatment of type 2 diabetes. Expert Opin. Investig. Drugs 2010, 19, 1067–1076. [Google Scholar] [CrossRef]
- Scott, J.S.; Bowker, S.S.; Deschoolmeester, J.; Gerhardt, S.; Hargreaves, D.; Kilgour, E.; Lloyd, A.; Mayers, R.M.; McCoull, W.; Newcombe, N.J.; et al. Discovery of a Potent, Selective, and Orally Bioavailable Acidic 11β-Hydroxysteroid Dehydrogenase Type 1 (11β-HSD1) Inhibitor: Discovery of 2-[(3S)-1-[5-(Cyclohexylcarbamoyl)-6-propylsulfanylpyridin-2-yl]-3-piperidyl]acetic Acid (AZD4017). J. Med. Chem. 2012, 55, 5951–5964. [Google Scholar] [CrossRef] [PubMed]
- Hewagalamulage, S.; Lee, T.; Clarke, I.; Henry, B. Stress, cortisol, and obesity: A role for cortisol responsiveness in identifying individuals prone to obesity. Domest. Anim. Endocrinol. 2016, 56, S112–S120. [Google Scholar] [CrossRef] [PubMed]
- Valsamakis, G.; Anwar, A.; Tomlinson, J.W.; Shackleton, C.H.L.; McTernan, P.G.; Chetty, R.; Wood, P.J.; Banerjee, A.K.; Holder, G.; Barnett, A.H.; et al. 11β-Hydroxysteroid Dehydrogenase Type 1 Activity in Lean and Obese Males with Type 2 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2004, 89, 4755–4761. [Google Scholar] [CrossRef]
- Villicev, C.M.; Freitas, F.R.S.; Aoki, M.S.; Taffarel, C.; Scanlan, T.S.; Moriscot, A.S.; Ribeiro, O.M.; Bianco, A.C.A.; Gouveia, C.H. Thyroid hormone receptor β-specific agonist GC-1 increases energy expenditure and prevents fat-mass accumulation in rats. J. Endocrinol. 2007, 193, 21–29. [Google Scholar] [CrossRef]
- Lindemann, J.L.; Webb, P. Sobetirome: The past, present and questions about the future. Expert Opin. Ther. Targets 2015, 20, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Grover, G.J.; Mellstrom, K.; Malm, J. Development of the Thyroid Hormone Receptor β-Subtype Agonist KB-141: A Strategy for Body Weight Reduction and Lipid Lowering with Minimal Cardiac Side Effects. Cardiovasc. Drug Rev. 2006, 23, 133–148. [Google Scholar] [CrossRef]
- Bryzgalova, G.; Effendic, S.; Khan, A.; Rehnmark, S.; Barbounis, P.; Boulet, J.; Dong, G.; Singh, R.; Shapses, S.; Malm, J.; et al. Anti-obesity, anti-diabetic, and lipid lowering effects of the thyroid receptor β subtype selective agonist KB-141. J. Steroid Biochem. Mol. Biol. 2008, 111, 262–267. [Google Scholar] [CrossRef]
- Grover, G.J.; Mellström, K.; Ye, L.; Malm, J.; Li, Y.-L.; Bladh, L.-G.; Sleph, P.G.; Smith, M.A.; George, R.; Vennström, B.; et al. Selective thyroid hormone receptor- activation: A strategy for reduction of weight, cholesterol, and lipoprotein (a) with reduced cardiovascular liability. Proc. Natl. Acad. Sci. USA 2003, 100, 10067–10072. [Google Scholar] [CrossRef]
- ClinicalTrials.gov U.S. National Library of Medicine Safety and Pharmacodynamic Study of Sobetirome in X-Linked Adrenoleukodystrophy (X-ALD). Available online: https://www.clinicaltrials.gov/ct2/show/NCT01787578?term=NCT01787578&draw=2&rank=1 (accessed on 25 February 2021).
- Fernández-Quintela, A.; Carpéné, C.; Fernández, M.; Aguirre, L.; Milton-Laskibar, I.; Contreras, J.; Portillo, M.P. Anti-obesity effects of resveratrol: Comparison between animal models and humans. J. Physiol. Biochem. 2016, 73, 417–429. [Google Scholar] [CrossRef]
- De Ligt, M.; Timmers, S.; Schrauwen, P. Resveratrol and obesity: Can resveratrol relieve metabolic disturbances? Biochim. et Biophys. ACTA (BBA)‑Mol. Basis Dis. 2015, 1852, 1137–1144. [Google Scholar] [CrossRef]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.-L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef] [PubMed]
- De Ligt, M.; Bergman, M.; Fuentes, R.M.; Essers, H.; Moonen-Kornips, E.; Havekes, B.; Schrauwen-Hinderling, V.B.; Schrauwen, P. No effect of resveratrol supplementation after 6 months on insulin sensitivity in overweight adults: A randomized trial. Am. J. Clin. Nutr. 2020, 112, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, J.P.L.; Palmer, D.; Meldal, M. MC4R Agonists: Structural Overview on Antiobesity Therapeutics. Trends Pharmacol. Sci. 2018, 39, 402–423. [Google Scholar] [CrossRef] [PubMed]
- Senese, R.; Cioffi, F.; Petito, G.; Goglia, F.; Lanni, A. Thyroid hormone metabolites and analogues. Endocrinology 2019, 66, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Erion, M.D.; Cable, E.E.; Ito, B.R.; Jiang, H.; Fujitaki, J.M.; Finn, P.D.; Zhang, B.-H.; Hou, J.; Boyer, S.H.; Van Poelje, P.D.; et al. Targeting thyroid hormone receptor-beta agonists to the liver reduces cholesterol and triglycerides and improves the therapeutic index. Proc. Natl. Acad. Sci. USA 2007, 104, 15490–15495. [Google Scholar] [CrossRef]
- Cheetham, S.C.; Jackson, H.C.; Vickers, S.P.; Dickinson, K.; Jones, R.B.; Heal, D.J. Novel targets for the treatment of obesity: A review of progress. Drug Discov. Today Ther. Strat. 2004, 1, 227–235. [Google Scholar] [CrossRef]
- Mirza, A.Z.; AlThagafi, I.I.; Shamshad, H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur. J. Med. Chem. 2019, 166, 502–513. [Google Scholar] [CrossRef]
- Suski, M.; Wiśniewska, A.; Stachowicz, A.; Olszanecki, R.; Kuś, K.; Białas, M.; Madej, J.; Korbut, R. The influence of AICAR‑direct activator of AMP-activated protein kinase (AMPK)‑on liver proteome in apoE-knockout mice. Eur. J. Pharm. Sci. 2017, 104, 406–416. [Google Scholar] [CrossRef]
- Gaidhu, M.P.; Frontini, A.; Hung, S.; Pistor, K.; Cinti, S.; Ceddia, R.B. Chronic AMP-kinase activation with AICAR reduces adiposity by remodeling adipocyte metabolism and increasing leptin sensitivity. J. Lipid Res. 2011, 52, 1702–1711. [Google Scholar] [CrossRef]
- Yoon, Y.; Lee, H.; Kang, R.; Bae, S. AICAR, an activator of AMPK, inhibits adipogenesis via the WNT/β-catenin pathway in 3T3-L1 adipocytes. Int. J. Mol. Med. 2011, 28, 65–71. [Google Scholar] [CrossRef]
- Comeglio, P.; Cellai, I.; Mello, T.; Filippi, S.; Maneschi, E.; Corcetto, F.; Corno, C.; Sarchielli, E.; Morelli, A.; Rapizzi, E.; et al. INT-767 prevents NASH and promotes visceral fat brown adipogenesis and mitochondrial function. J. Endocrinol. 2018, 238, 107–127. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, M.C.; Gilglioni, E.H.; de Boer, B.A.; Runge, J.H.; de Waart, D.R.; Salgueiro, C.L.; Ishii-Iwamoto, E.L.; Elferink, R.P.O.; Gaemers, I.C. Bile acid receptor agonists INT747 and INT777 decrease oestrogen deficiency-related postmenopausal obesity and hepatic steatosis in mice. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 2054–2062. [Google Scholar] [CrossRef]
- Wang, X.X.; Edelstein, M.H.; Gafter, U.; Qiu, L.; Luo, Y.; Dobrinskikh, E.; Lucia, S.; Adorini, L.; D’Agati, V.D.; Levi, J.; et al. G Protein-Coupled Bile Acid Receptor TGR5 Activation Inhibits Kidney Disease in Obesity and Diabetes. J. Am. Soc. Nephrol. 2015, 27, 1362–1378. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-Mediated Bile Acid Sensing Controls Glucose Homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of Bile Acids and Bile Acid Receptors in Metabolic Regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.P.; Asgharpour, A.; Mirshahi, F.; Park, S.H.; Liu, S.; Imai, Y.; Nadler, J.L.; Grider, J.R.; Murthy, K.S.; Sanyal, A.J. Activation of Transmembrane Bile Acid Receptor TGR5 Modulates Pancreatic Islet α Cells to Promote Glucose Homeostasis. J. Biol. Chem. 2016, 291, 6626–6640. [Google Scholar] [CrossRef]
- Finlin, B.S.; Memetimin, H.; Zhu, B.; Confides, A.L.; Vekaria, H.J.; El Khouli, R.H.; Johnson, Z.R.; Westgate, P.M.; Chen, J.; Morris, A.J.; et al. The β3-adrenergic receptor agonist mirabegron improves glucose homeostasis in obese humans. J. Clin. Investig. 2020, 130, 2319–2331. [Google Scholar] [CrossRef]
- Riis-Vestergaard, M.J.; Richelsen, B.; Bruun, J.M.; Li, W.; Hansen, J.B.; Pedersen, S.B. Beta-1 and Not Beta-3 Adrenergic Receptors May Be the Primary Regulator of Human Brown Adipocyte Metabolism. J. Clin. Endocrinol. Metab. 2020, 105, e994–e1005. [Google Scholar] [CrossRef]
- Larson, C.J. Translational Pharmacology and Physiology of Brown Adipose Tissue in Human Disease and Treatment. In Snake Venoms; J.B. Metzler: Berlin, Germany, 2018; pp. 381–424. [Google Scholar]
- VanBaak, M.; Hul, G.B.J.; Toubro, S.; Astrup, A.; Gottesdiener, K.M.; Saris, W.H.M.; Baak, M.A.; Desmet, M. Acute effect of L-796568, a novel?-adrenergic receptor agonist, on energy expenditure in obese men. Clin. Pharmacol. Ther. 2002, 71, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Larsen, T.M.; Toubro, S.A.; Van Baak, M.; Gottesdiener, K.M.; Larson, P.; Saris, W.H.M.; Astrup, A. Effect of a 28-d treatment with L-796568, a novel β3-adrenergic receptor agonist, on energy expenditure and body composition in obese men. Am. J. Clin. Nutr. 2002, 76, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lee, J.; Hernandez, M.A.S.; Mazitschek, R.; Ozcan, U. Treatment of Obesity with Celastrol. Cell 2015, 161, 999–1011. [Google Scholar] [CrossRef]
- Fang, P.; He, B.; Yu, M.; Shi, M.; Zhu, Y.; Zhang, Z.; Bo, P. Treatment with celastrol protects against obesity through suppression of galanin-induced fat intake and activation of PGC-1α/GLUT4 axis-mediated glucose consumption. Biochim. Biophys. ACTA (BBA)‑Mol. Basis Dis. 2019, 1865, 1341–1350. [Google Scholar] [CrossRef]
- Sáinz, N.; Barrenetxe, J.; Moreno-Aliaga, M.J.; Martínez, J.A. Leptin resistance and diet-induced obesity: Central and peripheral actions of leptin. Metabolism 2015, 64, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Quarta, C.; Sánchez-Garrido, M.A.; Tschöp, M.H.; Clemmensen, C. Renaissance of leptin for obesity therapy. Diabetology 2016, 59, 920–927. [Google Scholar] [CrossRef]
- Maurya, S.K.; Bal, N.C.; Sopariwala, D.H.; Pant, M.; Rowland, L.A.; Shaikh, S.A.; Periasamy, M. Sarcolipin Is a Key Determinant of the Basal Metabolic Rate, and Its Overexpression Enhances Energy Expenditure and Resistance against Diet-induced Obesity. J. Biol. Chem. 2015, 290, 10840–10849. [Google Scholar] [CrossRef] [PubMed]
- Bal, N.C.; Sahoo, S.K.; Maurya, S.K.; Periasamy, M. The Role of Sarcolipin in Muscle Non-shivering Thermogenesis. Front. Physiol. 2018, 9, 1217. [Google Scholar] [CrossRef]
- Maurya, S.K.; Herrera, J.L.; Sahoo, S.K.; Reis, F.C.; Vega, R.B.; Kelly, D.P.; Periasamy, M. Sarcolipin Signaling Promotes Mitochondrial Biogenesis and Oxidative Metabolism in Skeletal Muscle. Cell Rep. 2018, 24, 2919–2931. [Google Scholar] [CrossRef] [PubMed]
- Kohei, W.; Yoshinori, S.; Hiroshi, K. Brazilian propolis extract increases leptin expression in mouse adipocytes. Biomed. Res. 2015, 36, 343–346. [Google Scholar] [CrossRef]
- Nishikawa, S.; Aoyama, H.; Kamiya, M.; Higuchi, J.; Kato, A.; Soga, M.; Kawai, T.; Yoshimura, K.; Kumazawa, S.; Tsuda, T. Artepillin C, a Typical Brazilian Propolis-Derived Component, Induces Brown-Like Adipocyte Formation in C3H10T1/2 Cells, Primary Inguinal White Adipose Tissue-Derived Adipocytes, and Mice. PLoS ONE 2016, 11, e0162512. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, S.; Hyodo, T.; Aoyama, H.; Miyata, R.; Kumazawa, S.; Tsuda, T. Artepillin C, a Key Component of Brazilian Propolis, Induces Thermogenesis in Inguinal White Adipose Tissue of Mice through a Creatine-Metabolism-Related Thermogenic Pathway. J. Agric. Food Chem. 2019, 68, 1007–1014. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Target | Comment | Status | References |
|---|---|---|---|---|
| FDA-approved | ||||
| Somatotropin | Growth hormone receptor | Somatotropin reduces body mass in children without apparent side effects. | Phase IV (complete) for HGH deficiency, dwarfism, and obesity. | [12,13,14,15,16,17] |
| DHA and EPA | PPAR-α | n-3 PUFAs have shown weight loss mechanisms, such as increasing fat oxidation and energy expenditure, suppressing appetite and inducing apoptosis in adipocytes. | Phase IV (Completed) for obesity, insulin resistance, and as a dietary supplement. | [19,20,21,22,23] |
| Setmelanotide | Melanocortin 4 receptor (MC4r) | Setmelanotide (RM-493) is an MC4 receptor agonist approved by the FDA in 2020 to treat obesity. It does not have the adverse cardiovascular adverse effects shown by other MC4 receptor agonists. | FDA-approved and market distributed for leptin receptor deficiency, obesity, and POMC deficiency. | [25,26,27,28,81] |
| Metformin | AMP protein kinase | Metformin is currently the drug of first choice for the treatment of type 2 diabetes, following the guidelines of the American Diabetes Association and European Association of the Study of Diabetes. | FDA-approved for type 2 diabetes. | [30,31,32,33,34] |
| Ephedrine | Adrenergic receptors (ARs) | Ephedrine is defined as a sympathomimetic agent that replicates the adaptative thermogenic effects of chronic cold exposure in rodents; however, the treatment mechanism with sympathomimetic drugs in humans is still unknown. | FDA-approved for hypotension. | [36,37,38] |
| Phentermine/Topiramate | GABA receptor Carbonic anhydrase | Phentermine/topiramate: this combination of two drugs presents a very potent weight loss activity. After 52 weeks of treatment, an impressive median weight loss of 10.2 kg was produced at the maximum dose. | FDA-approved for chronic weight management. | [40,41,42,43,45] |
| Mirabegron | β-3-adrenergic receptor | The FDA approved mirabegron in 2012 for the treatment of overactive bladder syndrome (OAB). Approval at a daily dose of 50 mg was later given by other agencies from different regions, including Japan, the European Union, and Canada. | FDA-approved for overactive bladder. | [46,47,48] |
| Withdrawn | ||||
| GW501516 (Cardarine) | PPAR-δ (Peroxisome proliferator-activated receptor) | GW501516 is an anti-diabetic and anti-obesity treatment; however, because of the low specificity demonstrated in animal models, the oral administration of this compound is not considered a safe treatment. | Withdrawn from the market. | [49,50,51,52,54,55] |
| 2,4-Dinitrophenol | Mitochondrial oxidative phosphorylation | DNP was prohibited by the FDA in 1938. The modern chemical uncoupler CZ5 has shown no toxic effects in vivo. | Withdrawn from the market. | [58,60,62,63,64,65] |
| Clinical Trials | ||||
| INCB13739 | 11-β-hydroxysteroid dehydrogenase type 1 (11-β-HSD1) inhibitor | Improves positive metabolic response in obese males with 2 diabetes mellitus, with a 12-week treatment leading to a body weight reduction of between 0.6 and 1.1 kg. | Phase II for insulin resistance, obesity and type 2 diabetes. | [67,68,69,70] |
| GC1 | β-thyroid hormone receptor | GC-1 possesses all the beneficial metabolic properties of the active form of the thyroid hormone; however, preclinical animal studies and Phase I human clinical trials were withdrawn. | Phases I and II (for X-Linked adrenoleukodystrophy) but withdrawn. | [60,61,65,70] |
| Resveratrol | AMPK–SIRT1–PGC-1α axis | Resveratrol is a non-flavonoid polyphenol that has given promising results in animal models, producing energy expenditure and stimulating weight loss. However, a long-term study in humans showed that it does not affect body weight or body composition after 6 months of treatment. | Many different clinical trials concluded and ongoing, including Phase II. | [77,78,79,80] |
| PL-8905 | Melanocortin 4 receptor (MC4r) | PL-8905 is a macrocyclic peptide that shows a high selectivity for the MC4 receptor. In preclinical obesity, models have shown weight loss and glucose regulation. This compound has minimal side effects. | Clinical trials announced. | [26,81] |
| Under Development | ||||
| KB-141 | β-thyroid hormone receptor | KB-141 increases the metabolic rate and reduces the levels of plasma cholesterol without apparent cardiac side effects such as tachycardia. | Pre-clinical. | [73,74,83,84] |
| AICAR | AMP-activated protein kinase | AICAR can activate AMP-activated protein kinase (AMPK), which induces the inhibition of energy-consuming processes in numerous ways, switching on catabolic ATP-producing sites. | Pre-clinical. | [85,86,87,88] |
| INT777 | TGR5 (Takeda G-protein-coupled receptor 5) | INT-777 activity triggers thermogenesis in brown adipose tissue (BAT) and muscle, causing energy expenditure. | Pre-clinical. | [9,89,90,91,92,93,94] |
| BRL-26830 | β-3-adrenergic receptor | BRL-26830 causes a dose-dependent body weight loss in obese rats and mice without having this effect in lean counterparts. It increases energy expenditure with no effects on caloric intake, thereby preserving lean mass but reducing overweight. | Pre-clinical. | [95,96,97] |
| L-796568 | β-3-adrenergic receptor | L-796568 shows high potency and specificity characteristics that could be interesting for the development of new analogs for the treatment of obesity; however, the studies conducted 20 years ago were not conclusive. | Pre-clinical. | [96,98,99] |
| Celastrol | Leptin | Celastrol is a leptin sensitizer that induces adipogenesis inhibition, a metabolic increase in energy expenditure, and mitochondrial gene expression in mice fed a high-fat diet. | Pre-clinical. | [100,101,102,103] |
| Sarcolipin | SERCA uncoupling | Sarcolipin uncouples SERCA ATP hydrolysis from Ca2+ transport, thereby inducing muscle thermogenesis. | Pre-clinical. | [104,105,106] |
| Artepillin C | PPAR-γ is a ligand-activated transcription factor | Artepillin C (ArtC) promotes thermogenesis in vivo and acts as a peroxisome proliferator-activated receptor γ (PPAR-γ) agonist. | Pre-clinical. | [107,108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jimenez-Munoz, C.M.; López, M.; Albericio, F.; Makowski, K. Targeting Energy Expenditure—Drugs for Obesity Treatment. Pharmaceuticals 2021, 14, 435. https://doi.org/10.3390/ph14050435
Jimenez-Munoz CM, López M, Albericio F, Makowski K. Targeting Energy Expenditure—Drugs for Obesity Treatment. Pharmaceuticals. 2021; 14(5):435. https://doi.org/10.3390/ph14050435
Chicago/Turabian StyleJimenez-Munoz, Carlos M., Marta López, Fernando Albericio, and Kamil Makowski. 2021. "Targeting Energy Expenditure—Drugs for Obesity Treatment" Pharmaceuticals 14, no. 5: 435. https://doi.org/10.3390/ph14050435
APA StyleJimenez-Munoz, C. M., López, M., Albericio, F., & Makowski, K. (2021). Targeting Energy Expenditure—Drugs for Obesity Treatment. Pharmaceuticals, 14(5), 435. https://doi.org/10.3390/ph14050435