
Targeting Bacterial Sortases in Search of Anti-virulence Therapies with Low Risk of Resistance Development

 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Sortases’ Role in Virulence Mechanisms
3. Screening Protocols for Identification of Sortases Inhibitors
4. Sortase A Inhibitors
4.1. Plant Extracts
4.2. Thiol Reactive Reagents
4.3. Vinyl Sulfones
4.4. 3-Aryl Acrylic Acids and Derivatives
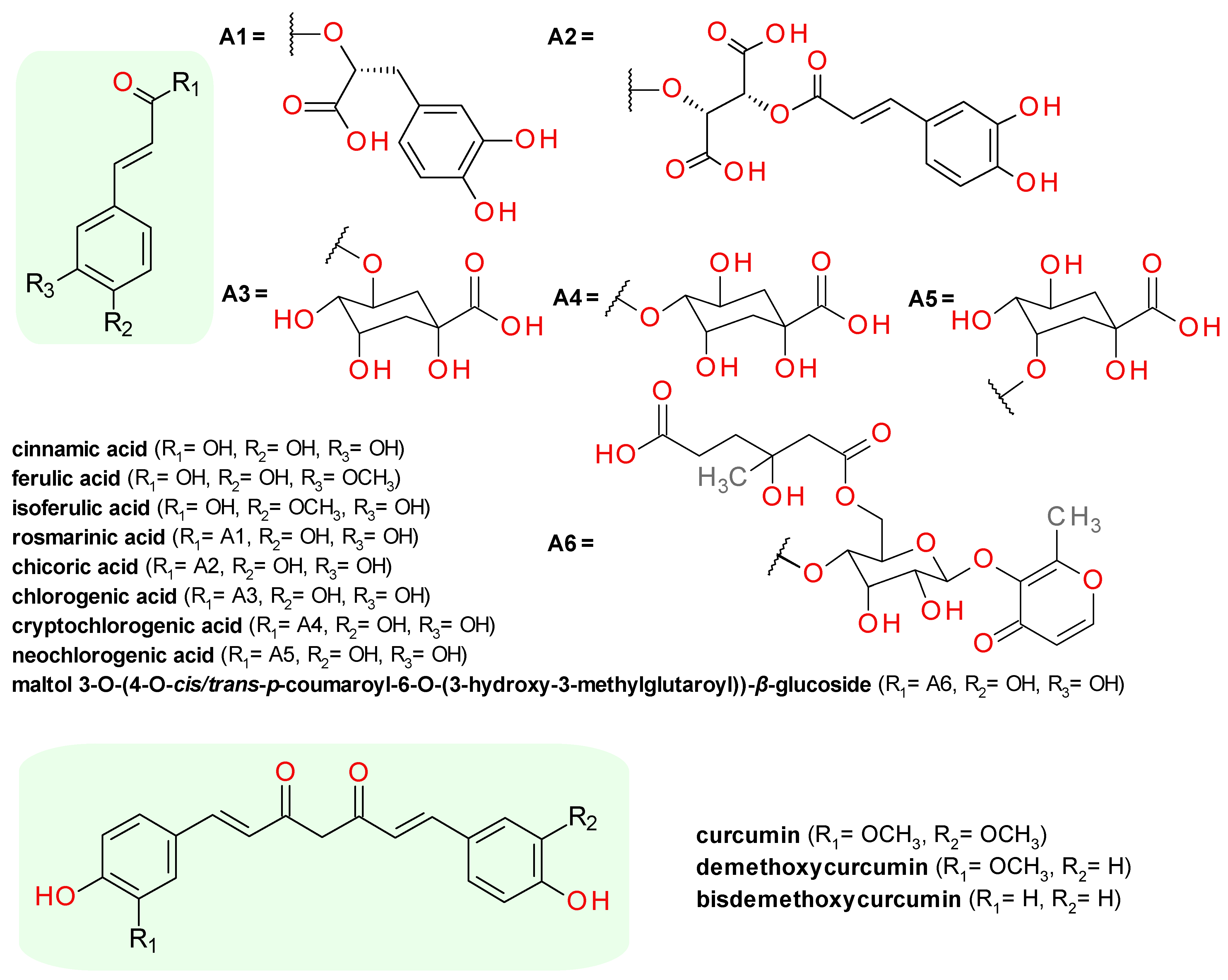
4.4.1. Cinnamic Acid Derivatives
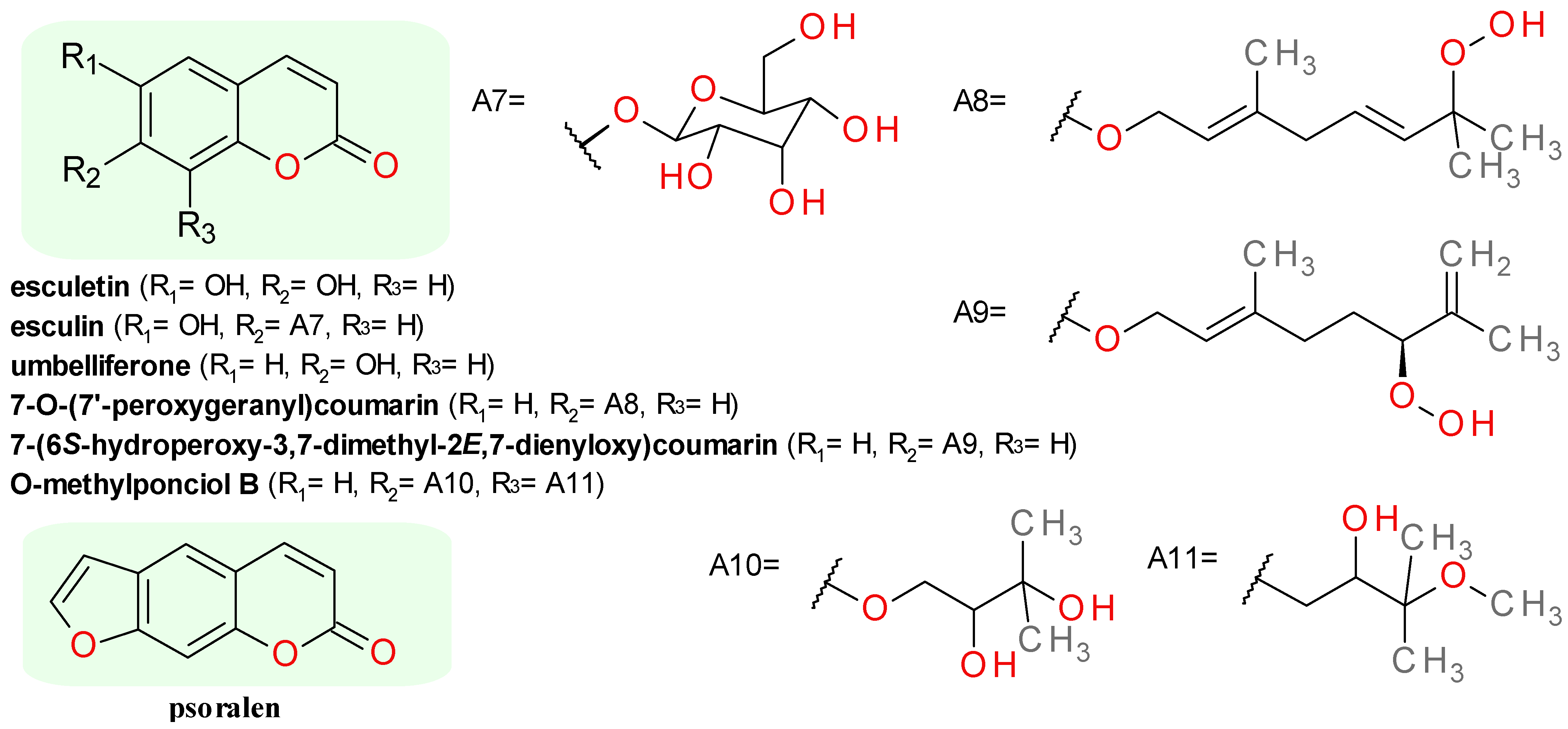
4.4.2. Coumarin Derivatives
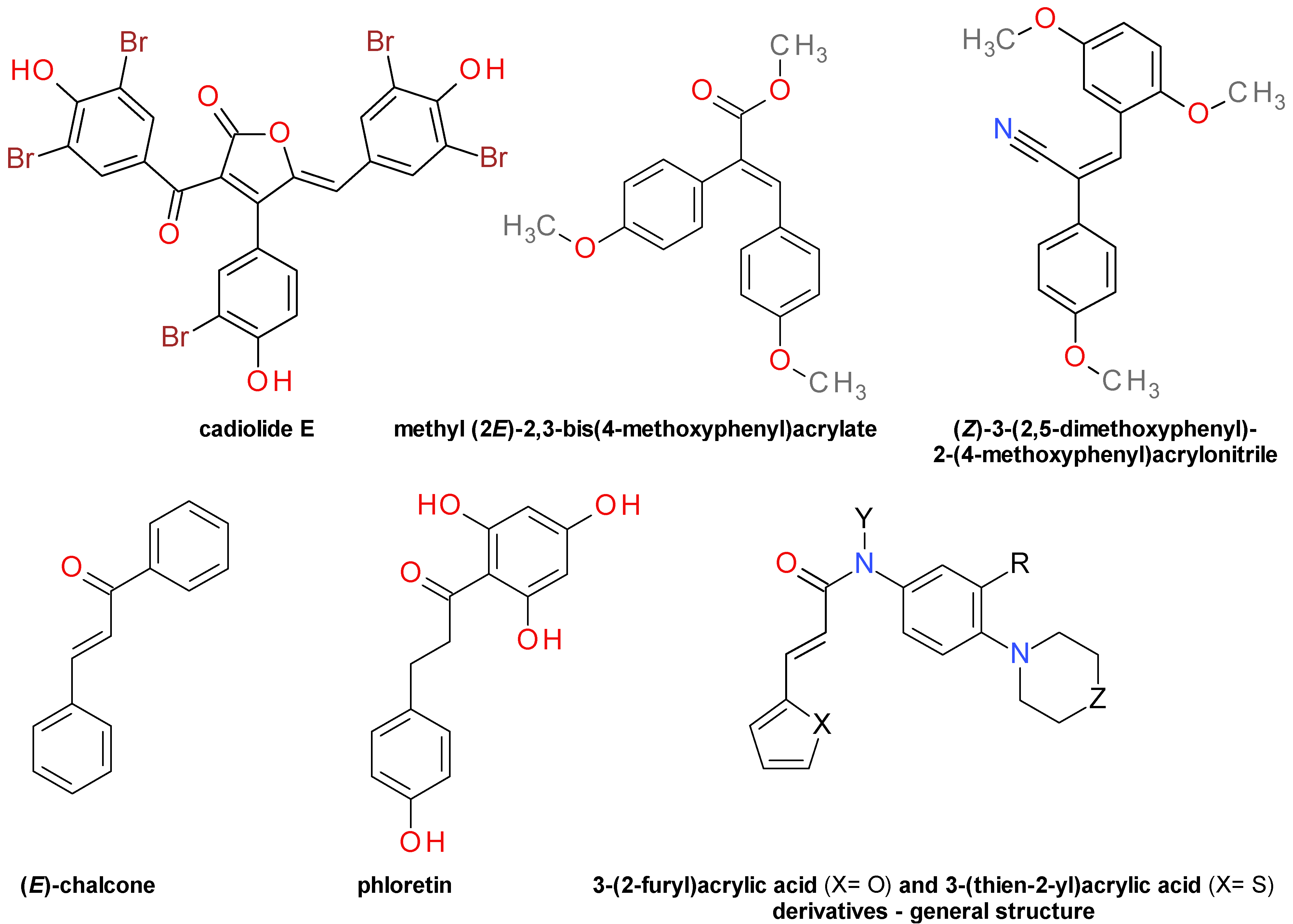
4.4.3. Aromatic Furanone Derivatives
4.4.4. Diarylacrylonitriles
4.4.5. Chalcones Derivatives
4.4.6. 3-(2-Furyl)Acrylic Acid and 3-(Thien-2-yl)Acrylic Acid Derivatives
4.5. Flavonoids
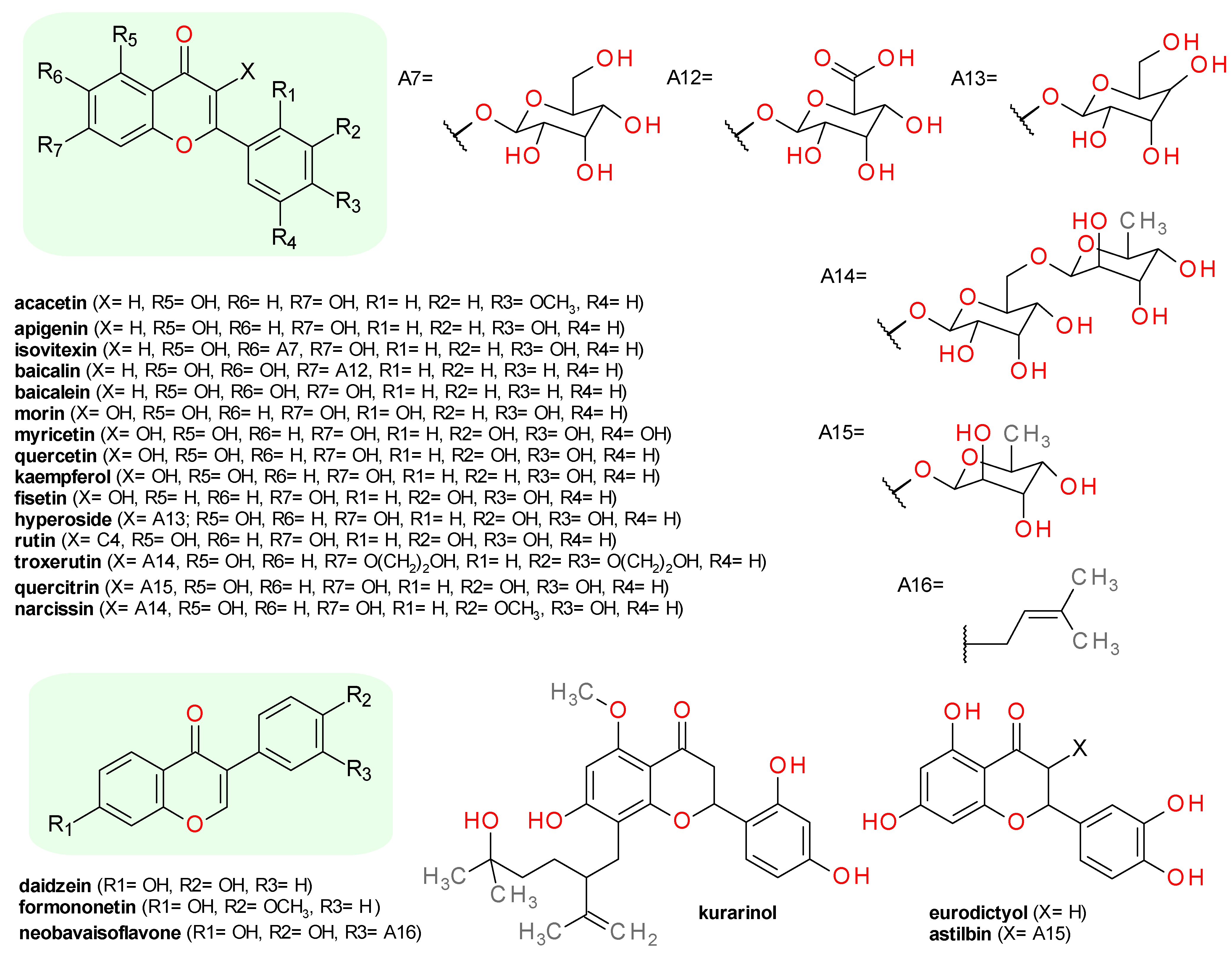
4.5.1. Flavones
4.5.2. Flavonols
4.5.3. Isoflavones
4.5.4. Flavanones
4.5.5. Flavanonols
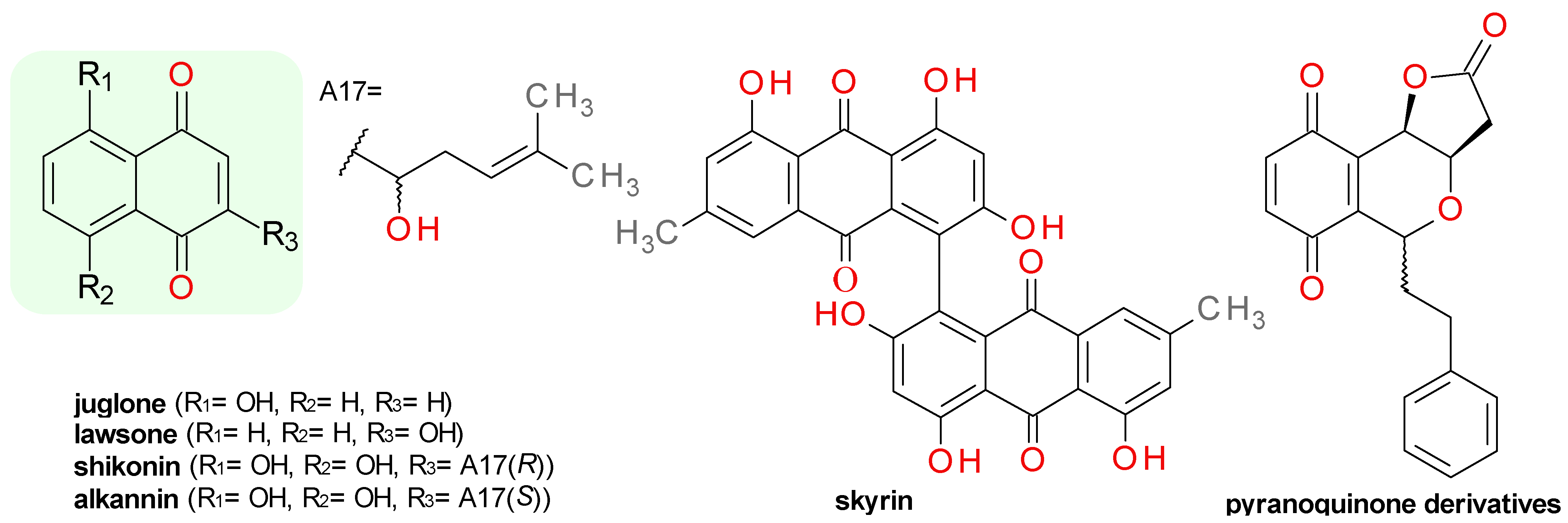
4.6. Quinone Derivatives
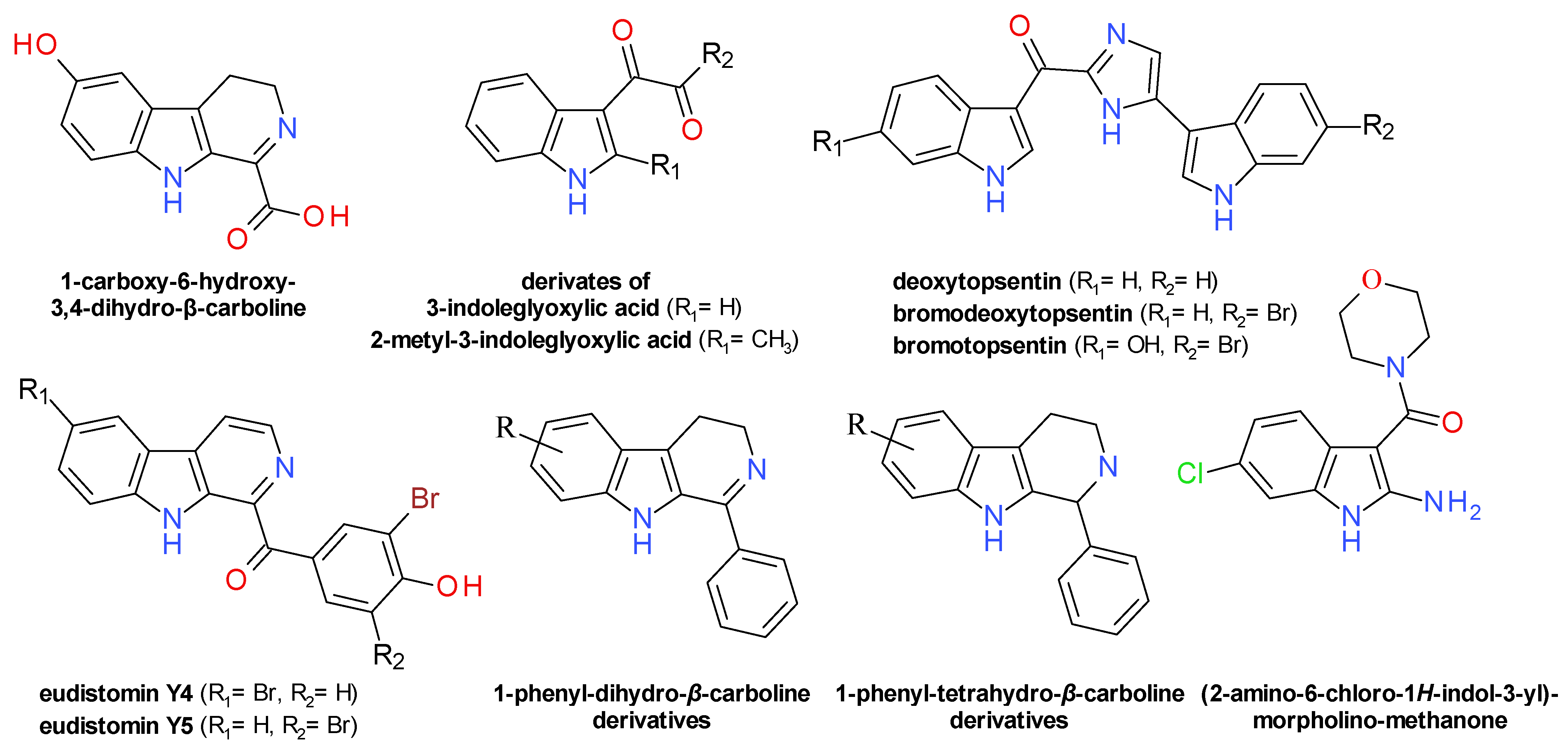
4.7. Indoles Derivatives
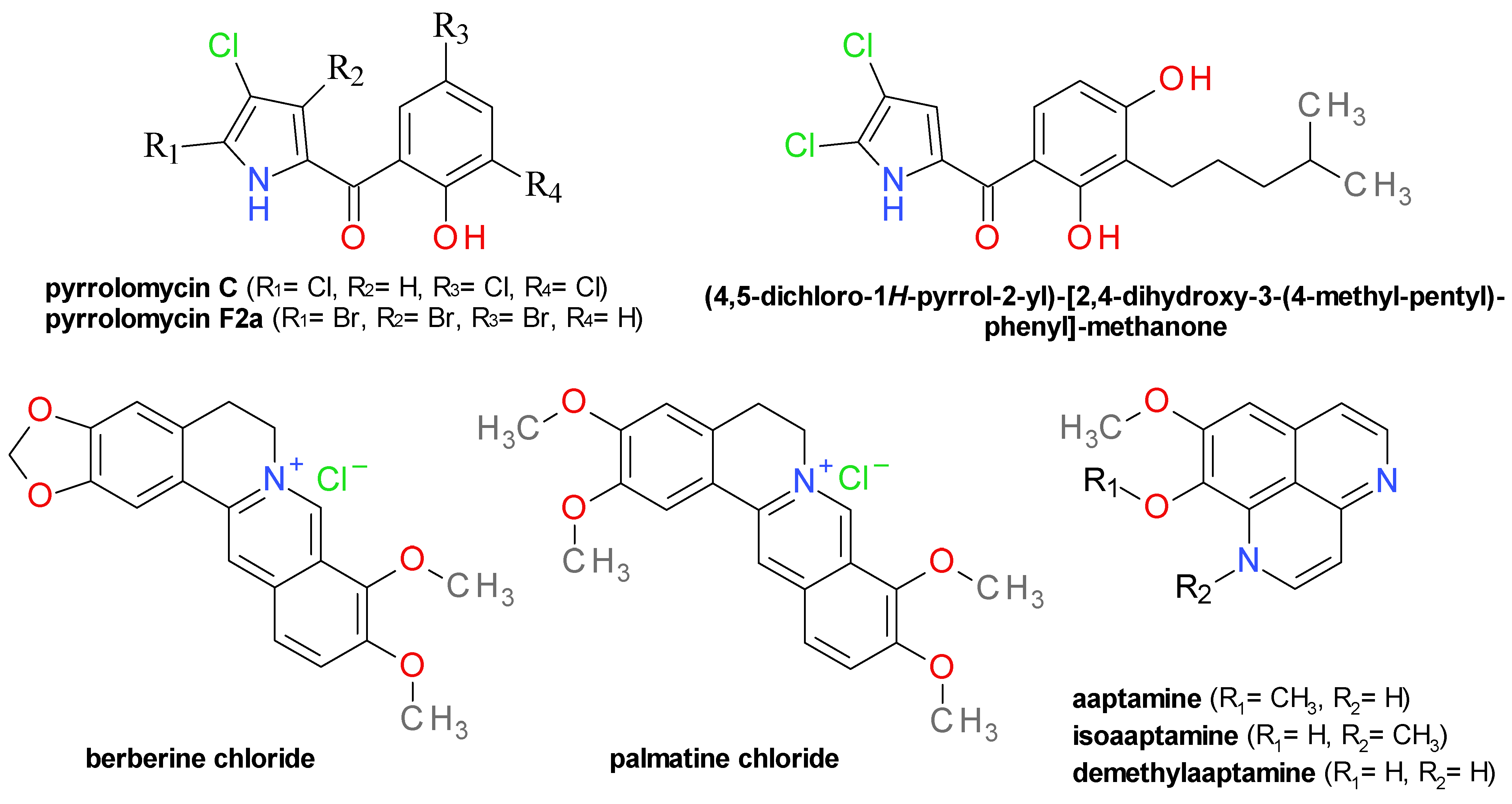
4.8. Pyrrolomycins and Analogues
4.9. Isoquinoline Derivatives
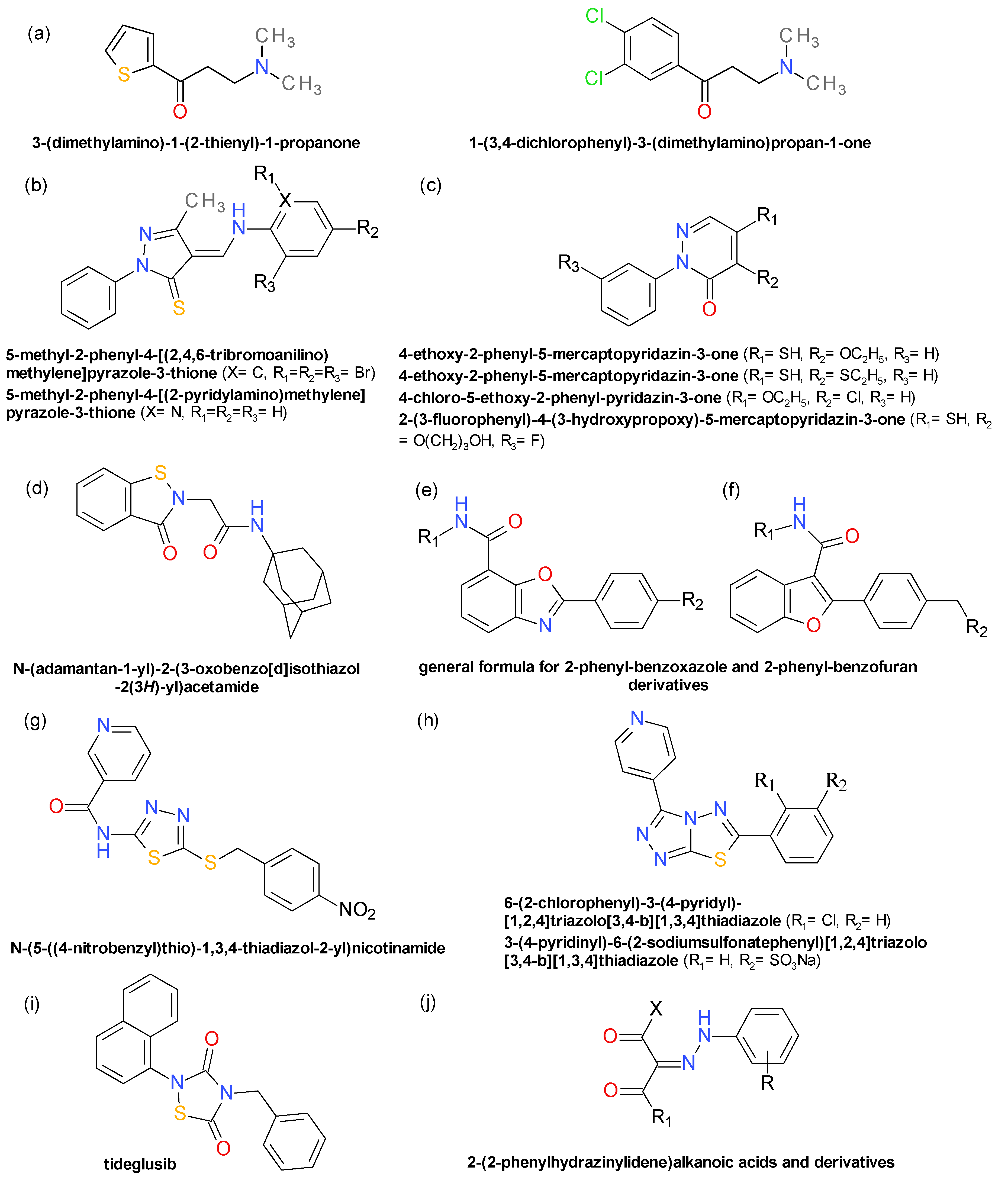
4.10. Aryl β-Aminoethyl Ketones
4.11. Pyrazolethiones and Pyridazinones
4.12. Benzisothiazolinones
4.13. Derivatives of 2-Phenyl-Benzoxazole and 2-Phenyl-Benzofuran
4.14. Thiadiazoles Derivatives. Triazolothiadiazoles Derivatives
4.15. 1,2,4-Thiadiazolidine-3,5-Dione Derivatives
4.16. 2-(2-Phenylhydrazinylidene)Alkanoic Acids and Derivatives
4.17. Various Structures
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Calvert, M.B.; Jumde, V.R.; Titz, A. Pathoblockers or Antivirulence Drugs as a New Option for the Treatment of Bacterial Infections. Beilstein J. Org. Chem. 2018, 14, 2607–2617. [Google Scholar] [CrossRef] [PubMed]
- Imperi, F.; Fiscarelli, E.V.; Visaggio, D.; Leoni, L.; Visca, P. Activity and Impact on Resistance Development of Two Antivirulence Fluoropyrimidine Drugs in Pseudomonas aeruginosa. Front. Cell. Infect. Microbiol. 2019, 9, 49. [Google Scholar] [CrossRef]
- Belete, T.M. Novel Targets to Develop New Antibacterial Agents and Novel Alternatives to Antibacterial Agents. Hum. Microbiome J. 2019, 11, 100052. [Google Scholar] [CrossRef]
- van Duin, D.; Paterson, D.L. Multidrug-Resistant Bacteria in the Community: Trends and Lessons Learned. Infect. Dis. Clin. N. Am. 2016, 30, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Zrelovs, N.; Kurbatska, V.; Rudevica, Z.; Leonchiks, A.; Fridmanis, D. Sorting out the Superbugs: Potential of Sortase A Inhibitors among Other Antimicrobial Strategies to Tackle the Problem of Antibiotic Resistance. Antibiotics 2021, 10, 164. [Google Scholar] [CrossRef] [PubMed]
- Martínez, O.F.; Cardoso, M.H.; Ribeiro, S.M.; Franco, O.L. Recent Advances in Anti-Virulence Therapeutic Strategies with a Focus on Dismantling Bacterial Membrane Microdomains, Toxin Neutralization, Quorum-Sensing Interference and Biofilm Inhibition. Front. Cell. Infect. Microbiol. 2019, 9, 74. [Google Scholar] [CrossRef] [PubMed]
- Navarre, W.W.; Schneewind, O. Surface Proteins of Gram-Positive Bacteria and Mechanisms of Their Targeting to the Cell Wall Envelope. Microbiol. Mol. Biol. Rev. 1999, 63, 174–229. [Google Scholar] [CrossRef]
- Fischetti, V.A. Surface Proteins on Gram-Positive Bacteria. Microbiol. Spectr. 2019, 7, 7. [Google Scholar] [CrossRef]
- Mazmanian, S.K. Staphylococcus Aureus Sortase, an Enzyme that Anchors Surface Proteins to the Cell Wall. Science 1999, 285, 760–763. [Google Scholar] [CrossRef]
- Kappel, K.; Wereszczynski, J.; Clubb, R.T.; McCammon, J.A. The Binding Mechanism, Multiple Binding Modes, and Allosteric Regulation of Staphylococcus Aureus Sortase A Probed by Molecular Dynamics Simulations. Protein Sci. 2012, 12, 1858–1871. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Liu, G.; Jensen, E.R.; Lenoy, E.; Schneewind, O. Staphylococcus Aureus Sortase Mutants Defective in the Display of Surface Proteins and in the Pathogenesis of Animal Infections. Proc. Natl. Acad. Sci. USA 2000, 97, 5510–5515. [Google Scholar] [CrossRef] [PubMed]
- Ton-That, H.; Schneewind, O. Anchor Structure of Staphylococcal Surface Proteins: IV. Inhibitors of the Cell Wall Sorting Reaction. J. Biol. Chem. 1999, 274, 24316–24320. [Google Scholar] [CrossRef] [PubMed]
- Ton-That, H.; Liu, G.; Mazmanian, S.K.; Faull, K.F.; Schneewind, O. Purification and characterization of Sortase, the Transpeptidase that Cleaves Surface Proteins of Staphylococcus Aureus at the LPXTG motif. Proc. Natl. Acad. Sci. USA 1999, 96, 12424–12429. [Google Scholar] [CrossRef] [PubMed]
- Kattke, M.D.; Chan, A.H.; Duong, A.; Sexton, D.L.; Sawaya, M.R.; Cascio, D.; Elliot, M.A.; Clubb, R.T. Crystal Structure of the Streptomyces Coelicolor Sortase E1 Transpeptidase Provides Insight into the Binding Mode of the Novel class e Sorting Signal. PLoS ONE 2016, 11, e0167763. [Google Scholar] [CrossRef]
- Spirig, T.; Weiner, E.M.; Clubb, R.T. Sortase Enzymes in Gram-Positive Bacteria. Mol. Microbiol. 2011, 82, 1044–1059. [Google Scholar] [CrossRef] [PubMed]
- Jacobitz, A.W.; Kattke, M.D.; Wereszczynski, J.; Clubb, R.T. Sortase Transpeptidases: Structural Biology and Catalytic Mechanism. Adv. Protein Chem. Struct. Biol. 2017, 109, 223–264. [Google Scholar]
- Mazmanian, S.K.; Ton-That, H.; Su, K.; Schneewind, O. An Iron-Regulated Sortase Anchors a Class of Surface Protein During Staphylococcus Aureus Pathogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 2293–2298. [Google Scholar] [CrossRef]
- Oh, K.-B.; Kim, S.-H.; Lee, J.; Cho, W.-J.; Lee, T.; Kim, S. Discovery of Diarylacrylonitriles as a Novel Series of Small Molecule Sortase A Inhibitors. J. Med. Chem. 2004, 47, 2418–2421. [Google Scholar] [CrossRef]
- Wang, L.; Bi, C.; Cai, H.; Liu, B.; Zhong, X.; Deng, X.; Wang, T.; Xiang, H.; Niu, X.; Wang, D. The Therapeutic Effect of Chlorogenic Acid against Staphylococcus Aureus Infection through Sortase A Inhibition. Front. Microbiol. 2015, 6, 1031. [Google Scholar] [CrossRef]
- Wallock-Richards, D.J.; Marles-Wright, J.; Clarke, D.J.; Maitra, A.; Dodds, M.; Hanley, B.; Campopiano, D.J. Molecular basis of Streptococcus Mutans Sortase A Inhibition by the Flavonoid Natural Product Trans-Chalcone. Chem. Commun. 2015, 51, 10483–10485. [Google Scholar] [CrossRef]
- Hu, P.; Huang, P.; Chen, M.W. Curcumin Reduces Streptococcus Mutans Biofilm Formation by Inhibiting Sortase A Activity. Arch. Oral Biol. 2013, 58, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Song, M.; Pan, J.; Shen, X.; Liu, W.; Zhang, X.; Li, H.; Deng, X. Quercetin Impairs Streptococcus Pneumoniae Biofilm Formation by Inhibiting Sortase a Activity. J. Cell. Mol. Med. 2018, 22, 6228–6237. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, Y.; Zhang, B.; Niu, X.; Song, M.; Luo, Z.; Lu, G.; Liu, B.; Zhao, X.; Wang, J.; et al. Inhibition of Sortase A by Chalcone Prevents Listeria Monocytogenes Infection. Biochem. Pharmacol. 2016, 106, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Gosschalk, J.E.; Chang, C.; Sue, C.K.; Siegel, S.D.; Wu, C.; Kattke, M.D.; Yi, S.W.; Damoiseaux, R.; Jung, M.E.; Ton-That, H.; et al. A Cell-Based Screen in Actinomyces oris to Identify Sortase Inhibitors. Sci. Rep. 2020, 10, 8520. [Google Scholar] [CrossRef] [PubMed]
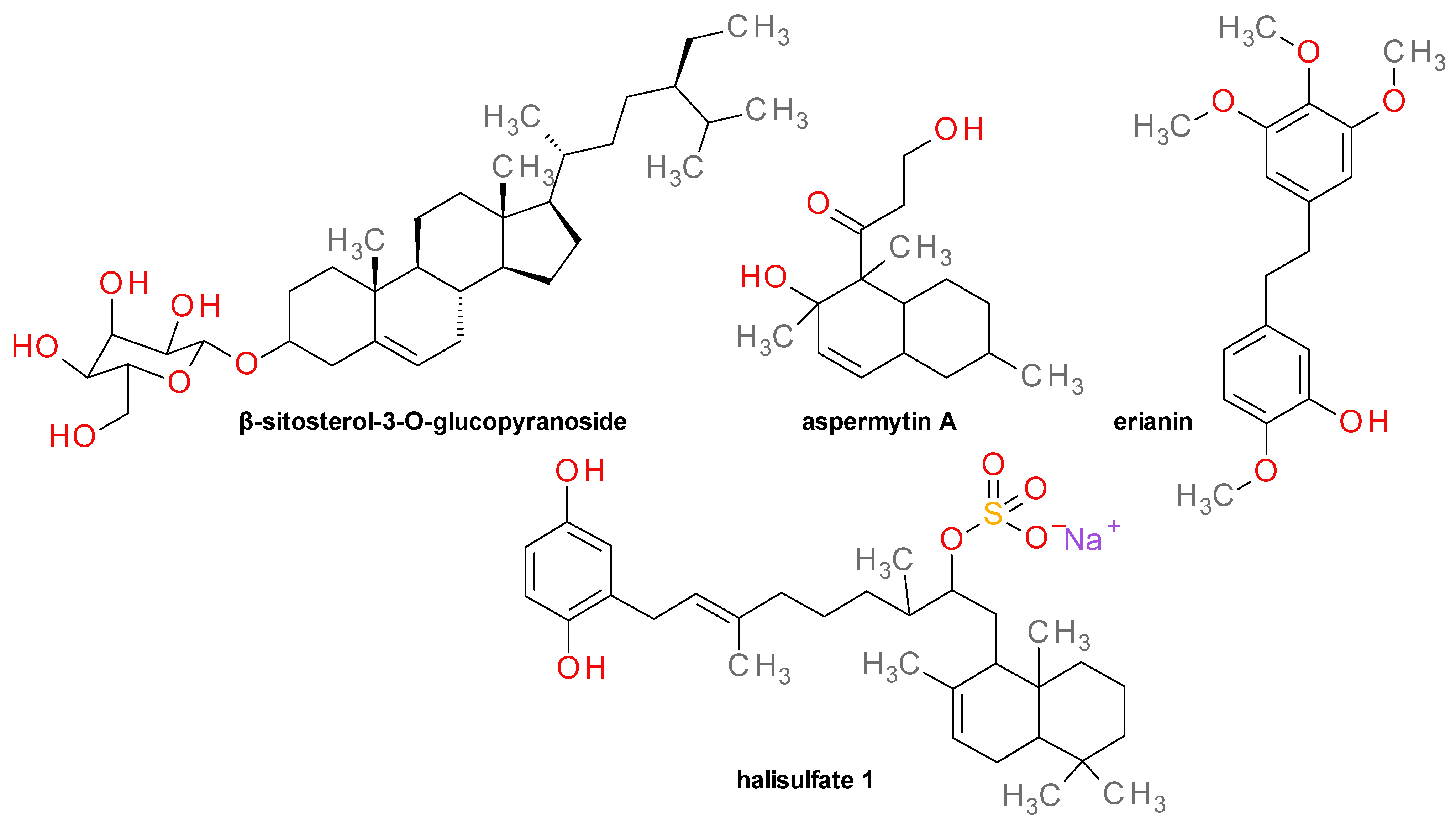
- Kim, S.-H.; Shin, D.-S.; Oh, M.-N.; Chung, S.-C.; Lee, J.-S.; Chang, I.-M.; Oh, K.-B. Inhibition of Sortase, a Bacterial Surface Protein Anchoring Transpeptidase, by Beta-Sitosterol-3-O-Glucopyranoside from Fritillaria Verticillata. Biosci. Biotechnol. Biochem. 2003, 67, 2477–2479. [Google Scholar] [CrossRef]
- Kruger, R.G.; Dostal, P.; McCafferty, D.G. Development of a High-Performance Liquid Chromatography Assay and Revision of Kinetic Parameters for the Staphylococcus Aureus Sortase Transpeptidase SrtA. Anal. Biochem. 2004, 326, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Parrino, B.; Cusimano, M.G.; Spanò, V.; Montalbano, A.; Barraja, P.; Schillaci, D.; Cirrincione, G.; Diana, P.; Cascioferro, S. New Thiazole Nortopsentin Analogues Inhibit Bacterial Biofilm Formation. Mar. Drugs 2018, 16, 274. [Google Scholar] [CrossRef]
- Nitulescu, G.; Nicorescu, I.M.; Olaru, O.T.; Ungurianu, A.; Mihai, D.P.; Zanfirescu, A.; Nitulescu, G.M.; Margina, D. Molecular Docking and Screening Studies of New Natural Sortase A Inhibitors. Int. J. Mol. Sci. 2017, 18, 2217. [Google Scholar] [CrossRef]
- Guo, Y.; Cai, S.; Gu, G.; Guo, Z.; Long, Z. Recent Progress in the Development of Sortase A Inhibitors as Novel Anti-Bacterial Virulence Agents. RSC Adv. 2015, 5, 49880–49889. [Google Scholar] [CrossRef]
- Cascioferro, S.; Totsika, M.; Schillaci, D. Sortase A: An Ideal Target for Anti-Virulence Drug Development. Microb. Pathog. 2014, 77, 105–112. [Google Scholar] [CrossRef]
- Maresso, A.W.; Schneewind, O. Sortase as a Target of Anti-Infective Therapy. Pharmacol. Rev. 2008, 60, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-W.; Chang, I.-M.; Oh, K.-B. Inhibition of the Bacterial Surface Protein Anchoring Transpeptidase Sortase by Medicinal Plants. Biosci. Biotechnol. Biochem. 2002, 66, 2751–2754. [Google Scholar] [CrossRef] [PubMed]
- Park, B.-S.; Kim, J.-G.; Kim, M.-R.; Lee, S.-E.; Takeoka, G.R.; Oh, K.-B.; Kim, J.-H. Curcuma longa L. Constituents Inhibit Sortase A and Staphylococcus aureus Cell Adhesion to Fibronectin. J. Agric. Food Chem. 2005, 53, 9005–9009. [Google Scholar] [CrossRef]
- Won, T.H.; Song, I.H.; Kim, K.H.; Yang, W.Y.; Lee, S.K.; Oh, D.C.; Oh, W.K.; Oh, K.B.; Shin, J. Bioactive Metabolites from the Fruits of Psoralea Corylifolia. J. Nat. Prod. 2015, 78, 666–673. [Google Scholar] [CrossRef]
- Marraffini, L.A.; Dedent, A.C.; Schneewind, O. Sortases and the Art of Anchoring Proteins to the Envelopes of Gram-Positive Bacteria. Microbiol. Mol. Biol. Rev. 2006, 70, 192–221. [Google Scholar] [CrossRef]
- Frankel, B.A.; Bentley, M.; Kruger, R.G.; McCafferty, D.G. Vinyl Sulfones: Inhibitors of SrtA, a Transpeptidase Required for Cell Wall Protein Anchoring and Virulence in Staphylococcus Aureus. J. Am. Chem. Soc. 2004, 126, 3404–3405. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtsev, K.V.; Bentley, M.L.; McCafferty, D.G. Probing of the cis-5-Phenyl Proline Scaffold as a Platform for the Synthesis of Mechanism-Based Inhibitors of the Staphylococcus Aureus Sortase SrtA Isoform. Bioorg. Med. Chem. 2009, 17, 2886–2893. [Google Scholar] [CrossRef]
- Guzman, J.D. Natural Cinnamic Acids, Synthetic Derivatives and Hybrids with Antimicrobial Activity. Molecules 2014, 19, 19292–19349. [Google Scholar] [CrossRef]
- Yang, W.Y.; Won, T.H.; Ahn, C.H.; Lee, S.H.; Yang, H.C.; Shin, J.; Oh, K.B. Streptococcus Mutans Sortase A Inhibitory Metabolites from the Flowers of Sophora Japonica. Bioorganic Med. Chem. Lett. 2015, 25, 1394–1397. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Song, I.-H.; Lee, J.-H.; Yang, W.-Y.; Oh, K.-B.; Shin, J. Sortase A Inhibitory Metabolites from the Roots of Pulsatilla Koreana. Bioorg. Med. Chem. Lett. 2014, 24, 44–48. [Google Scholar] [CrossRef]
- Bi, C.; Wang, L.; Niu, X.; Cai, H.; Zhong, X.; Deng, X.; Wang, T.; Wang, D. The Use of Chlorogenic Acid and its Analogues as Inhibitors: An Investigation of the Inhibition of sortase A of Staphylococcus Aureus Using Molecular Docking and Dynamic Simulation. Biotechnol. Lett. 2016, 38, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Magesh, S.; Chen, Y.; Hu, L. Small Molecule Modulators of Keap1-Nrf2-ARE Pathway as Potential Preventive and Therapeutic Agents. Med. Res. Rev. 2012, 32, 687–726. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Chung, B.; Lee, W.-H.; Lee, J.; Suh, Y.; Oh, D.-C.; Oh, K.-B.; Shin, J. Sortase A-Inhibitory Coumarins from the Folk Medicinal Plant Poncirus Trifoliata. J. Nat. Prod. 2020, 83, 3004–3011. [Google Scholar] [CrossRef] [PubMed]
- Won, T.H.; Jeon, J.E.; Kim, S.H.; Lee, S.H.; Rho, B.J.; Oh, D.C.; Oh, K.B.; Shin, J. Brominated Aromatic Furanones and Related Esters from the Ascidian Synoicum sp. J. Nat. Prod. 2012, 75, 2055–2061. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Cho, E.; Park, J.S.; Won, T.H.; Seo, S.-Y.; Oh, D.-C.; Oh, K.-B.; Shin, J. Isocadiolides A-H: Polybrominated Aromatics from a Synoicum sp. Ascidian. J. Nat. Prod. 2020, 83, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.B.; Oh, M.N.; Kim, J.G.; Shin, D.S.; Shin, J. Inhibition of Sortase-Mediated Staphylococcus Aureus Adhesion to Fibronectin via Fibronectin-Binding Protein by Sortase Inhibitors. Appl. Microbiol. Biotechnol. 2006, 70, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.-B.; Nam, K.-W.; Ahn, H.; Shin, J.; Kim, S.; Mar, W. Therapeutic Effect of (Z)-3-(2,5-Dimethoxyphenyl)-2-(4-Methoxyphenyl) Acrylonitrile (DMMA) Against Staphylococcus Aureus Infection in a Murine Model. Biochem. Biophys. Res. Commun. 2010, 396, 440–444. [Google Scholar] [CrossRef]
- Zhang, B.; Teng, Z.; Li, X.; Lu, G.; Deng, X.; Niu, X.; Wang, J. Chalcone Attenuates Staphylococcus aureus Virulence by Targeting Sortase A and Alpha-Hemolysin. Front. Microbiol. 2017, 8, 1715. [Google Scholar] [CrossRef]
- Cho, H.; Chung, B.; Kim, C.K.; Oh, D.C.; Oh, K.B.; Shin, J. Spatholobus suberectus Dunn. Constituents Inhibit Sortase A and Staphylococcus Aureus Cell Clumping to Fibrinogen. Arch. Pharm. Res. 2017, 40, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, B.; Teng, Z.; Zhou, X.; Wang, X.; Zhang, B.; Lu, G.; Niu, X.; Yang, Y.; Deng, X. Phloretin Attenuates Listeria Monocytogenes Virulence Both in Vitro and in Vivo by Simultaneously Targeting Listeriolysin O and sortase A. Front. Cell. Infect. Microbiol. 2017, 7. [Google Scholar] [CrossRef]
- Chenna, B.C.; King, J.R.; Shinkre, B.A.; Glover, A.L.; Lucius, A.L.; Velu, S.E. Synthesis and Structure Activity Relationship Studies of Novel Staphylococcus Aureus Sortase A Inhibitors. Eur. J. Med. Chem. 2010, 45, 3752–3761. [Google Scholar] [CrossRef] [PubMed]
- Uivarosi, V.; Munteanu, A.-C.; Nițulescu, G.M. Chapter 2—An Overview of Synthetic and Semisynthetic Flavonoid Derivatives and Analogues: Perspectives in Drug Discovery. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 60, pp. 29–84. [Google Scholar]
- Bi, C.; Dong, X.; Zhong, X.; Cai, H.; Wang, D.; Wang, L. Acacetin Protects Mice from Staphylococcus Aureus Bloodstream Infection by Inhibiting the Activity of Sortase A. Molecules 2016, 21, 1285. [Google Scholar] [CrossRef] [PubMed]
- Mu, D.; Xiang, H.; Dong, H.; Wang, D.; Wang, T. Isovitexin, a Potential Candidate Inhibitor of Sortase a of Staphylococcus Aureus USA300. J. Microbiol. Biotechnol. 2018, 28, 1426–1432. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Gao, Y.; Wang, H.; Niu, X.; Wang, J. Baicalin Weakens Staphylococcus Aureus Pathogenicity by Targeting Sortase B. Front. Cell. Infect. Microbiol. 2018, 8, 418. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Xu, L.; Zhang, T.; Deng, X.; Wang, J. A Potential Bio-Control Agent from Baical Skullcap Root against Listeriosis via the Inhibition of Sortase A and Listeriolysin O. J. Cell. Mol. Med. 2019, 23, 2042–2051. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Kim, J.-G.; Lee, T.-H.; Oh, K.-B. Flavonols Inhibit Sortases and Sortase-Mediated Staphylococcus aureus Clumping to Fibrinogen. Biol. Pharm. Bull. 2006, 29, 1751–1755. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.; Zanfirescu, A.; Olaru, O.T.; Nicorescu, I.M.; Nitulescu, G.M.; Margina, D. Structural Analysis of Sortase A Inhibitors. Molecules 2016, 21, 1591. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, F.; Bi, C.; Wang, L.; Zhong, X.; Cai, H.; Deng, X.; Niu, X.; Wang, D. Quercitrin, an Inhibitor of Sortase A, Interferes with the Adhesion of Staphylococcal Aureus. Molecules 2015, 20, 6533–6543. [Google Scholar] [CrossRef]
- Huang, P.; Hu, P.; Zhou, S.; Li, Q.; Chen, W. Morin Inhibits Sortase A and Subsequent Biofilm Formation in Streptococcus mutans. Curr. Microbiol. 2014, 68, 47–52. [Google Scholar] [CrossRef]
- Yang, W.-Y.; Kim, C.-K.; Ahn, C.-H.; Kim, H.; Shin, J.; Oh, K.-B. Flavonoid Glycosides Inhibit Sortase A and Sortase A-Mediated Aggregation of Streptococcus Mutans, an Oral Bacterium Responsible for Human Dental Caries. J. Microbiol. Biotechnol. 2016, 26, 1557–1565. [Google Scholar] [CrossRef]
- Park, W.; Ahn, C.H.; Cho, H.; Kim, C.K.; Shin, J.; Oh, K.B. Inhibitory Effects of Flavonoids from Spatholobus Suberectus on Sortase a and Sortase a-Mediated Aggregation of Streptococcus Mutans. J. Microbiol. Biotechnol. 2017, 27, 1457–1460. [Google Scholar] [CrossRef] [PubMed]
- Oh, I.; Yang, W.-Y.; Chung, S.-C.; Kim, T.-Y.; Oh, K.-B.; Shin, J. In Vitro Sortase a Inhibitory and Antimicrobial Activity of Flavonoids Isolated from the Roots of Sophora Flavescens. Arch. Pharm. Res. 2011, 34, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, Q.; Li, J.; Jing, S.; Jin, Y.; Yang, L.; Yu, H.; Wang, D.; Wang, T.; Wang, L. Eriodictyol as a Potential Candidate Inhibitor of Sortase A Protects Mice From Methicillin-Resistant Staphylococcus Aureus-Induced Pneumonia. Front. Microbiol. 2021, 12, 268. [Google Scholar]
- Wang, J.; Shi, Y.; Jing, S.; Dong, H.; Wang, D.; Wang, T. Astilbin Inhibits the Activity of Sortase A from Streptococcus Mutans. Molecules 2019, 24, 465. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Ruan, Y.T.; Yin, Z.P.; Zhang, Q.F. A Comparison of Solubility, Stability, and Bioavailability Between Astilbin and Neoastilbin Isolated from Smilax Glabra Rhizoma. Molecules 2020, 25, 4728. [Google Scholar] [CrossRef]
- Hou, X.; Wang, M.; Wen, Y.; Ni, T.; Guan, X.; Lan, L.; Zhang, N.; Zhang, A.; Yang, C.-G. Quinone Skeleton as a New Class of Irreversible Inhibitors against Staphylococcus Aureus Sortase A. Bioorg. Med. Chem. Lett. 2018, 28, 1864–1869. [Google Scholar] [CrossRef]
- Nitulescu, G.; Mihai, D.P.; Nicorescu, I.M.; Olaru, O.T.; Ungurianu, A.; Zanfirescu, A.; Nitulescu, G.M.; Margina, D. Discovery of Natural Naphthoquinones as Sortase A Inhibitors and Potential Anti-Infective Solutions Against Staphylococcus Aureus. Drug Dev. Res. 2019, 80, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Thappeta, K.R.V.; Zhao, L.N.; Nge, C.E.; Crasta, S.; Leong, C.Y.; Ng, V.; Kanagasundaram, Y.; Fan, H.; Ng, S.B. In-Silico Identified new Natural Sortase a Inhibitors Disrupt S. Aureus Biofilm Formation. Int. J. Mol. Sci. 2020, 21, 8601. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-S.; Yoon, K.-M.; Han, Y.-R.; Lee, K.J.; Chung, S.-C.; Kim, T.-I.; Lee, S.-H.; Shin, J.; Oh, K.-B. 5-Hydroxyindole-Type Alkaloids, as Candida Albicans Isocitrate Lyase Inhibitors, from the Tropical Sponge Hyrtios sp. Bioorg. Med. Chem. Lett. 2009, 19, 1051–1053. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Han, Y.-R.; Park, W.; Nam, S.-H.; Oh, K.-B.; Lee, H.-S. Synthetic Analogs of Indole-Containing Natural Products as Inhibitors of Sortase A and Isocitrate Lyase. Bioorg. Med. Chem. Lett. 2010, 20, 6882–6885. [Google Scholar] [CrossRef]
- Wójcik, M.; Eleftheriadis, N.; Zwinderman, M.R.H.; Dömling, A.S.S.; Dekker, F.J.; Boersma, Y.L. Identification of Potential Antivirulence Agents by Substitution-Oriented Screening for Inhibitors of Streptococcus Pyogenes Sortase A. Eur. J. Med. Chem. 2019, 161, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.-B.; Mar, W.; Kim, S.; Kim, J.-Y.; Oh, M.-N.; Kim, J.-G.; Shin, D.; Sim, C.J.; Shin, J. Bis(indole) Alkaloids as Sortase A Inhibitors from the Sponge Spongosorites sp. Bioorg. Med. Chem. Lett. 2005, 15, 4927–4931. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Cho, E.; Hwang, J.-Y.; Park, S.C.; Chung, B.; Kwon, O.-S.; Sim, C.J.; Oh, D.-C.; Oh, K.-B.; Shin, J. Bioactive Bis(indole) Alkaloids from a Spongosorites sp. Sponge. Mar. Drugs 2021, 19, 3. [Google Scholar] [CrossRef] [PubMed]
- Parrino, B.; Carbone, D.; Cascioferro, S.; Pecoraro, C.; Giovannetti, E.; Deng, D.; Di Sarno, V.; Musella, S.; Auriemma, G.; Cusimano, M.G.; et al. 1,2,4-Oxadiazole Topsentin Analogs as Staphylococcal Biofilm Inhibitors Targeting the Bacterial Transpeptidase Sortase A. Eur. J. Med. Chem. 2021, 209, 112892. [Google Scholar] [CrossRef]
- Won, T.H.; Jeon, J.-E.; Lee, S.-H.; Rho, B.J.; Oh, K.-B.; Shin, J. Beta-Carboline Alkaloids Derived from the Ascidian Synoicum sp. Bioorg. Med. Chem. 2012, 20, 4082–4087. [Google Scholar] [CrossRef] [PubMed]
- Szabó, T.; Volk, B.; Milen, M. Recent Advances in the Synthesis of β-Carboline Alkaloids. Molecules 2021, 26, 663. [Google Scholar] [CrossRef]
- Wang, W.; Nam, S.-J.; Lee, B.-C.; Kang, H. β-Carboline Alkaloids from a Korean Tunicate Eudistoma sp. J. Nat. Prod. 2008, 71, 163–166. [Google Scholar] [CrossRef]
- Cascioferro, S.; Raimondi, M.V.; Cusimano, M.G.; Raffa, D.; Maggio, B.; Daidone, G.; Schillaci, D. Pharmaceutical Potential of Synthetic and Natural Pyrrolomycins. Molecules 2015, 20, 21658–21671. [Google Scholar] [CrossRef]
- Kim, S.H.; Shin, D.S.; Oh, M.N.; Chung, S.C.; Lee, J.S.; Oh, K.B. Inhibition of the Bacterial Surface Protein Anchoring Transpeptidase Sortase by Isoquinoline Alkaloids. Biosci. Biotechnol. Biochem. 2004, 68, 421–424. [Google Scholar] [CrossRef]
- Park, S.C.; Chung, B.; Lee, J.; Cho, E.; Hwang, J.-Y.; Oh, D.-C.; Shin, J.; Oh, K.-B. Sortase A-Inhibitory Metabolites from a Marine-Derived Fungus Aspergillus sp. Mar. Drugs 2020, 18, 359. [Google Scholar] [CrossRef]
- Raimondi, M.V.; Listro, R.; Cusimano, M.G.; La Franca, M.; Faddetta, T.; Gallo, G.; Schillaci, D.; Collina, S.; Leonchiks, A.; Barone, G. Pyrrolomycins as Antimicrobial Agents. Microwave-Assisted Organic Synthesis and Insights into their Antimicrobial Mechanism of Action. Bioorg. Med. Chem. 2019, 27, 721–728. [Google Scholar] [CrossRef]
- Wang, G.; Wang, X.; Sun, L.; Gao, Y.; Niu, X.; Wang, H. Novel Inhibitor Discovery of Staphylococcus Aureus Sortase B and the Mechanism Confirmation via Molecular Modeling. Molecules 2018, 23, 977. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.H.; Chung, S.C.; Shin, J.; Lee, S.H.; Kim, T.I.; Lee, H.S.; Oh, K.B. Aaptamines as Sortase A Inhibitors from the Tropical Sponge Aaptos Aaptos. Bioorganic Med. Chem. Lett. 2007, 17, 5366–5369. [Google Scholar] [CrossRef] [PubMed]
- Maresso, A.W.; Wu, R.; Kern, J.W.; Zhang, R.; Janik, D.; Missiakas, D.M.; Duban, M.E.; Joachimiak, A.; Schneewind, O. Activation of Inhibitors by Sortase Triggers Irreversible Modification of the Active Site. J. Biol. Chem. 2007, 282, 23129–23139. [Google Scholar] [CrossRef]
- Suree, N.; Yi, S.W.; Thieu, W.; Marohn, M.; Damoiseaux, R.; Chan, A.; Jung, M.E.; Clubb, R.T. Discovery and Structure–Activity Relationship Analysis of Staphylococcus Aureus Sortase a Inhibitors. Bioorg. Med. Chem. 2009, 17, 7174–7185. [Google Scholar] [CrossRef]
- Chan, A.H.; Yi, S.W.; Weiner, E.M.; Amer, B.R.; Sue, C.K.; Wereszczynski, J.; Dillen, C.A.; Senese, S.; Torres, J.Z.; McCammon, J.A.; et al. NMR Structure-Based Optimization of Staphylococcus Aureus Sortase A Pyridazinone Inhibitors. Chem. Biol. Drug Des. 2017, 90, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Zhulenkovs, D.; Rudevica, Z.; Jaudzems, K.; Turks, M.; Leonchiks, A. Discovery and Structure–Activity Relationship Studies of Irreversible Benzisothiazolinone-Based Inhibitors against Staphylococcus Aureus Sortase A Transpeptidase. Bioorg. Med. Chem. 2014, 22, 5988–6003. [Google Scholar] [CrossRef]
- Zhang, Y.; Bao, J.; Deng, X.-X.; He, W.; Fan, J.-J.; Jiang, F.-Q.; Fu, L. Synthesis, Biological Evaluation and Molecular Docking of 2-Phenyl-Benzo[d]Oxazole-7-Carboxamide Derivatives as Potential Staphylococcus Aureus Sortase A Inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 4081–4085. [Google Scholar] [CrossRef]
- He, W.; Zhang, Y.; Bao, J.; Deng, X.; Batara, J.; Casey, S.; Guo, Q.; Jiang, F.; Fu, L. Synthesis, Biological Evaluation and Molecular Docking Analysis of 2-Phenyl-Benzofuran-3-Carboxamide Derivatives as Potential Inhibitors of Staphylococcus Aureus Sortase A. Bioorganic Med. Chem. 2017, 25, 1341–1351. [Google Scholar] [CrossRef]
- Wehrli, P.M.; Uzelac, I.; Olsson, T.; Jacso, T.; Tietze, D.; Gottfries, J. Discovery and Development of Substituted Thiadiazoles as Inhibitors of Staphylococcus Aureus Sortase A. Bioorganic Med. Chem. 2019, 27, 115043. [Google Scholar] [CrossRef]
- Yang, T.; Zhang, T.; Guan, X.N.; Dong, Z.; Lan, L.; Yang, S.; Yang, C.G. Tideglusib and Its Analogues as Inhibitors of Staphylococcus aureus SrtA. J. Med. Chem. 2020, 63, 8442–8457. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, J.M.; Fuertes, A.; Orozco, L.; Del Monte-Millán, M.; Delgado, E.; Medina, M. Evidence for Irreversible Inhibition of Glycogen Synthase Kinase-3β by Tideglusib. J. Biol. Chem. 2012, 287, 893–904. [Google Scholar] [CrossRef]
- Maggio, B.; Raffa, D.; Raimondi, M.V.; Cascioferro, S.; Plescia, F.; Schillaci, D.; Cusimano, M.G.; Leonchiks, A.; Zhulenkovs, D.; Basile, L.; et al. Discovery of a New Class of Sortase a Transpeptidase Inhibitors to Tackle Gram-Positive Pathogens: 2-(2-Phenylhydrazinylidene)Alkanoic Acids and Related Derivatives. Molecules 2016, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, P.; He, X.; Yuan, Z.-W.; Yin, Z.-Q.; Fu, H.; Lin, J.; He, C.; Liang, X.; Lv, C.; Shu, G.; et al. Erianin Against Staphylococcus aureus Infection via Inhibiting Sortase A. Toxins 2018, 10, 385. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Jeon, J.E.; Lee, Y.J.; Lee, H.S.; Sim, C.J.; Oh, K.B.; Shin, J. Sesterterpenes from the Tropical Sponge Coscinoderma sp. J. Nat. Prod. 2011, 74, 1805–1811. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nitulescu, G.; Margina, D.; Zanfirescu, A.; Olaru, O.T.; Nitulescu, G.M. Targeting Bacterial Sortases in Search of Anti-virulence Therapies with Low Risk of Resistance Development. Pharmaceuticals 2021, 14, 415. https://doi.org/10.3390/ph14050415
Nitulescu G, Margina D, Zanfirescu A, Olaru OT, Nitulescu GM. Targeting Bacterial Sortases in Search of Anti-virulence Therapies with Low Risk of Resistance Development. Pharmaceuticals. 2021; 14(5):415. https://doi.org/10.3390/ph14050415
Chicago/Turabian StyleNitulescu, Georgiana, Denisa Margina, Anca Zanfirescu, Octavian Tudorel Olaru, and George Mihai Nitulescu. 2021. "Targeting Bacterial Sortases in Search of Anti-virulence Therapies with Low Risk of Resistance Development" Pharmaceuticals 14, no. 5: 415. https://doi.org/10.3390/ph14050415
APA StyleNitulescu, G., Margina, D., Zanfirescu, A., Olaru, O. T., & Nitulescu, G. M. (2021). Targeting Bacterial Sortases in Search of Anti-virulence Therapies with Low Risk of Resistance Development. Pharmaceuticals, 14(5), 415. https://doi.org/10.3390/ph14050415