
Neuroinflammation in Sepsis: Molecular Pathways of Microglia Activation

 , , and
, , and
Abstract
1. Introduction
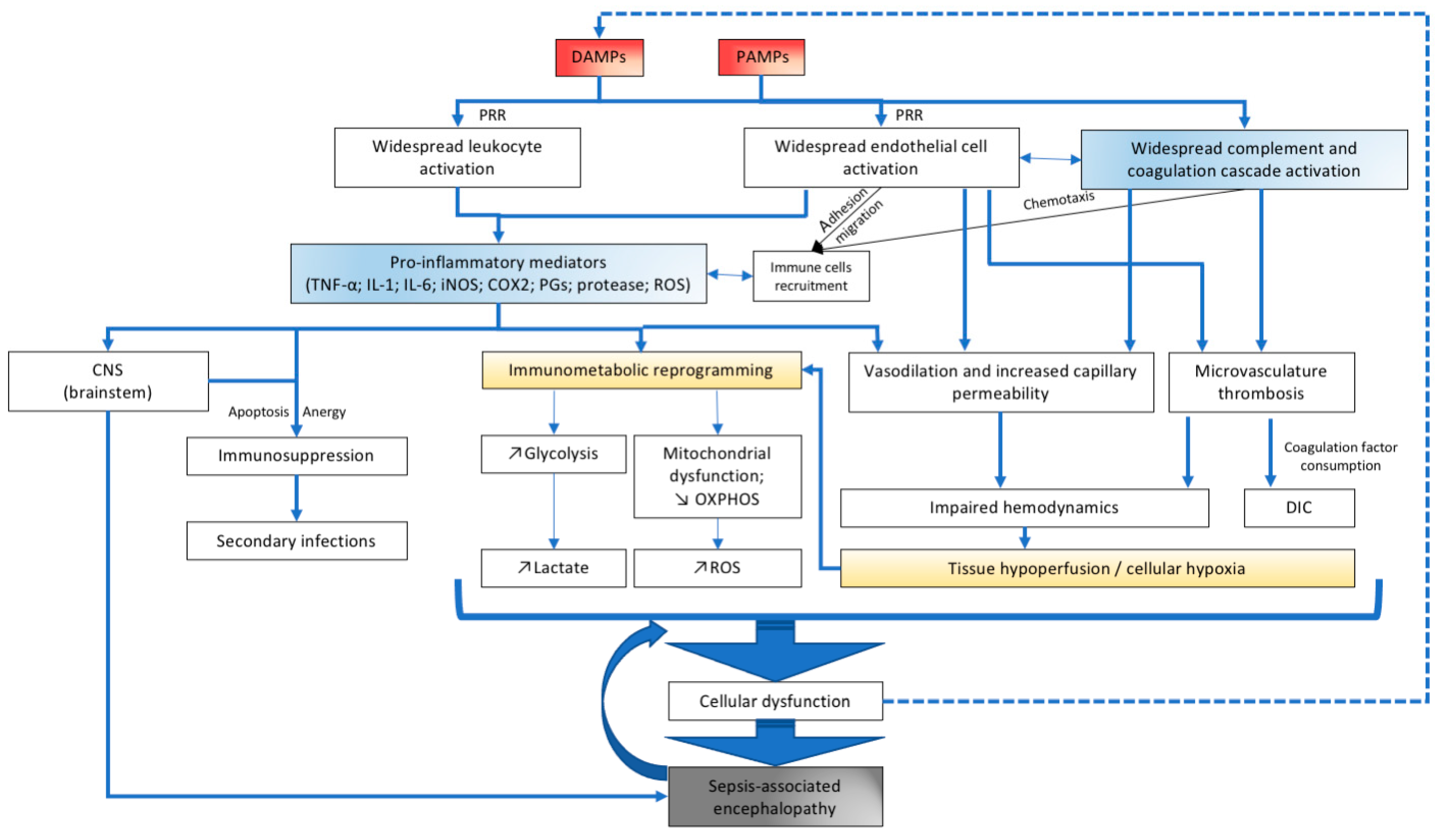
2. Neuroinflammation in Sepsis
3. The CNS Immune System
4. Microglia in Homeostasis
5. Microglia in Sepsis-Associated Encephalopathy
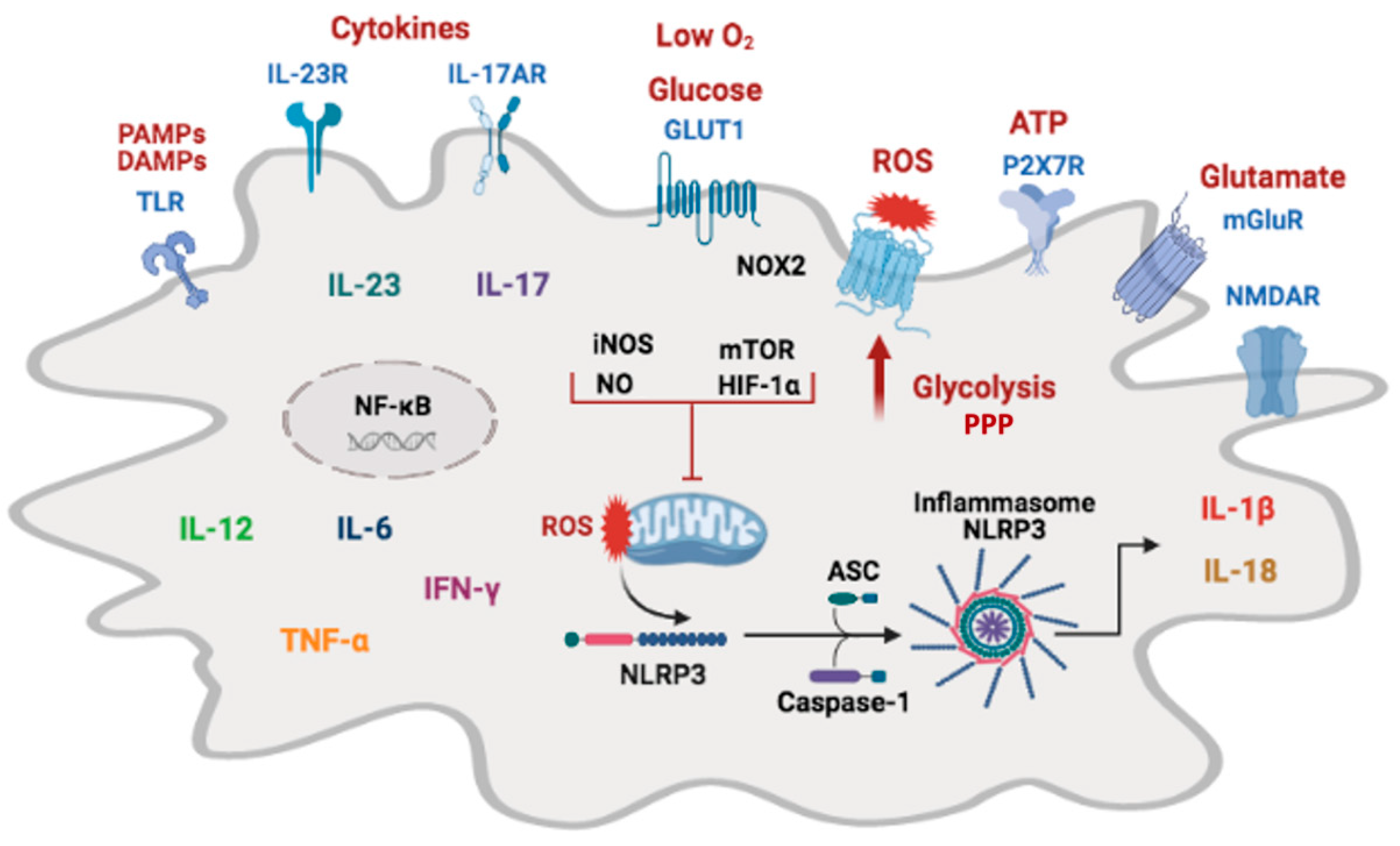
6. Microglia Metabolic Adaptations
7. Molecular Mechanisms That Regulate Microglial Immunometabolism
8. Microglia Immune Activation
8.1. Activation Phenotypes
8.2. Microglial Immune Receptors
8.3. Inflammasome Activation
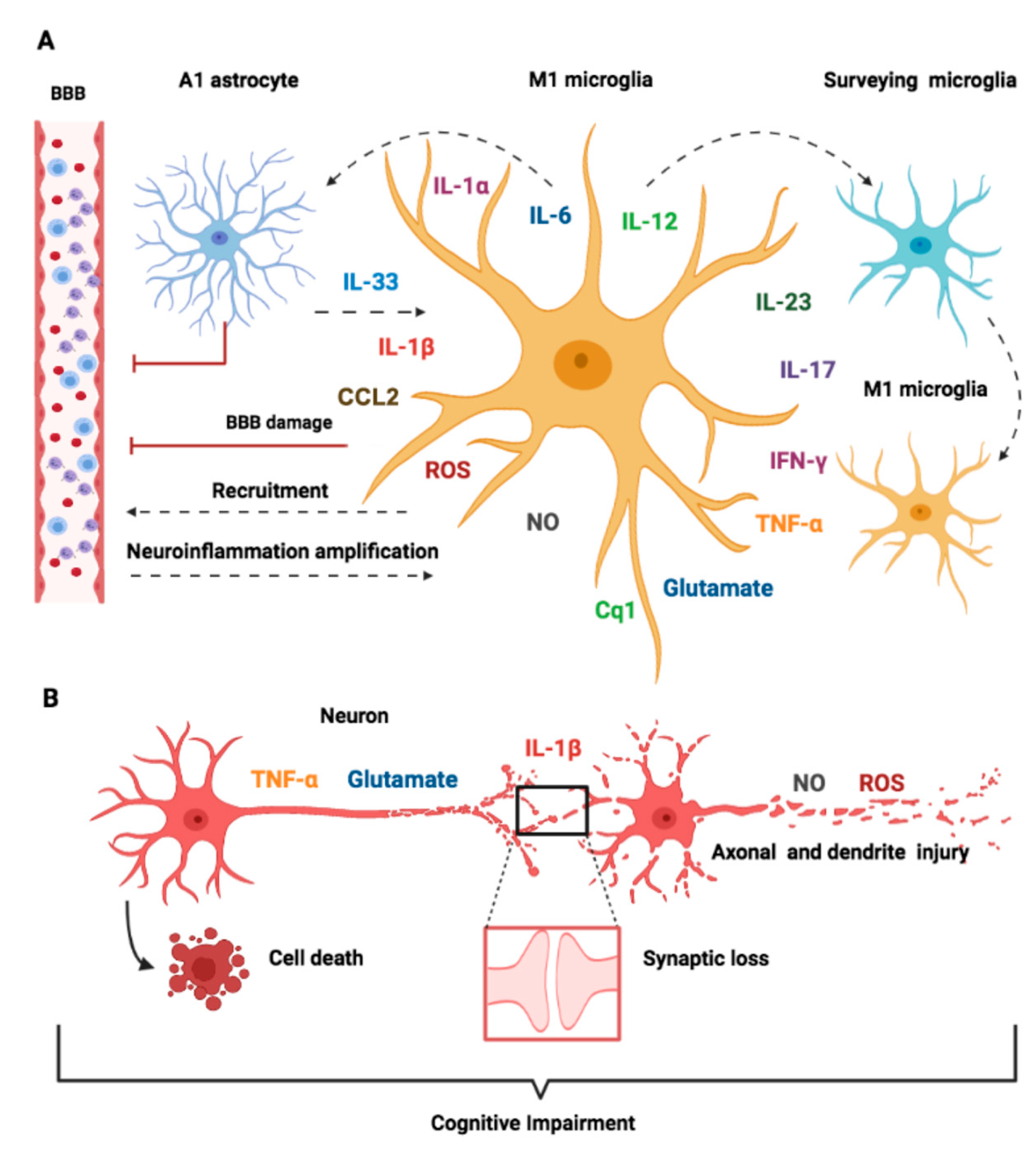
9. Crosstalk between Microglia and Other Cell Types
10. Molecular Mechanisms of Neuronal Toxicity in SAE
10.1. Cytokines
10.2. Inflammation-Mediated Glutamate Neurotoxicity
10.3. Oxidative Stress Pathways

{kind=link}
{kind=link}
{kind=link}
| Inflammatory Molecule | Functions | References |
|---|---|---|
| Cytokines | ||
| IL-1β | Pro-inflammatory cytokine secreted by microglia and infiltrating leukocytes. Initiates the host inflammatory response, induces synaptic dysfunction, suppresses hippocampal LTP; induces sickness behavior | [16,143,165,166] |
| IL-18 | Pro-inflammatory cytokine; induces the release of pro-inflammatory cytokines such as IL-1β, IL-6, IFN-γ, and IL-18 by glial cells. Induces sickness behavior, loss of appetite, sleep, and inhibition of LTP | [16,167,168] |
| IL-6 | Pleiotropic pro-inflammatory cytokine; stimulates migration of leukocytes, regulates the production of chemokines and expression of adhesion molecules, induces sickness behavior. High levels of IL-6 are strongly associated with mortality | [166,169,170] |
| IL-12 | Evokes neuroinflammation; expressed by microglia; involved in changes of the metal status; induces production of IFN-γ from NK and activated T cells | [170,171] |
| IL-17 | Induces the secretion of pro-inflammatory molecules (IL-1β, IL-23, IL-17, IL-6, MIP-2, NO), adhesion molecules, and neurotrophic factors by microglia; induces glial activation, microvascular pathology, and enhances neuroinflammation | [135,141,172] |
| IFN-γ | Upregulates cell surface molecules MHC class I and II, intercellular adhesion molecule I (ICAM-I), LPS receptor (CD14), Fc and complement receptors. Induces changes in the proteasome composition and release of cytokines (TNF-α, IL-1, and IL-6), NO, and complements (C1q, C2, C3, C4) | [104,173] |
| TNF-α | Pro-inflammatory cytokine; induces BBB disruption, infiltration of neutrophils, astrocytosis, and apoptosis of brain cells. Stimulates autocrine microglia activation and glutamate release by microglia and astrocytes and inhibits glutamate uptake. Suppresses hippocampal LTP | [148,165,170,174] |
| Chemokines | ||
| CCL2 (MCP-1) CXCL8 (IL-8) CXCL10 (IP-10), CXCL12 (SDF-1), CCL13 (MCP-4), CCL22 (MDC) CCL3 (MIP-1α) | Chemotatic cytokines (chemokines) that induce leukocyte migration, increase BBB permeability allowing infiltration of leukocytes; chemoattractants to neutrophils and microglia; produced in several brain regions, released by activated microglia | [66,136,175,176,177] |
| Reactive oxidant species | ||
| ROS, RNS, RSS | Mediators of oxidative stress; perform oxidation, nitrosylation, nitration, and sulfuration/polysulfidation reactions with endogenous molecules; change structure and function of proteins; promote lipid peroxidation altering membranes permeability, induce axonal damage and cytotoxicity; regulate gene transcription, ion transport, intermediary metabolism, and mitochondrial function; contribute to inflammasome activation | [41,64,65,178] |
| NO | Neurotoxic, vasodilator, mitochondrial inhibitor; gaseous signaling molecule; killing of pathogens | [32,130,132] |
| Neurotransmitters | ||
| Glutamate | Secreted by activated microglia and astrocytes; induces excitatory synapses; in high concentrations induces excitotoxic neuronal cell death; induces chemotaxis of microglia | [68,153,154,155,158,179] |
| ATP | Secreted by activated microglia, induces microglia chemotaxis, activation, and phagocytosis | [156,180] |
| Prostaglandins | ||
| PGE2 | Potent inflammatory mediator; induces cytokines secretion, vasodilation, endothelial permeability and BBB disruption | [181,182] |
| Matrix metalloproteinases | ||
| MMP2, MMP3, MMP8, MMP9, MMP12, MMP14 | Secreted by activated microglia, degrade the extracellular matrix contributing to tissue injury. MMP8 modulates TNF-α activation and stimulates the production of IL-6 and NO. MMP-3 and MMP-9 regulate IL-1β, IL-1Ra, iNOS, and IL-6 gene expression at the transcriptional level and that of TNF-α at the post-transcriptional level.MMP-2 and MMP-9 are associated with increased BBB permeability, and inhibition of MMP-9 and MMP-2 improves acute cognitive alterations associated with sepsis. | [183,184,185] |
11. Conclusions
Funding
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801. [Google Scholar] [CrossRef]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, Regional, and National Sepsis Incidence and Mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef]
- Kafa, I.M.; Bakirci, S.; Uysal, M.; Kurt, M.A. Alterations in the Brain Electrical Activity in a Rat Model of Sepsis-Associated Encephalopathy. Brain Res. 2010, 1354, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.M.; Urdanibia-Centelles, O.; Vedel-Larsen, E.; Thomsen, K.J.; Møller, K.; Olsen, K.S.; Lauritsen, A.Ø.; Eddelien, H.S.; Lauritzen, M.; Benedek, K. Continuous EEG Monitoring in a Consecutive Patient Cohort with Sepsis and Delirium. Neurocrit. Care 2020, 32, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Annane, D.; Sharshar, T. Cognitive Decline after Sepsis. Lancet Respir. Med. 2015, 3, 61–69. [Google Scholar] [CrossRef]
- Ely, E.W. Delirium as a Predictor of Mortality in Mechanically Ventilated Patients in the Intensive Care Unit. JAMA 2004, 291, 1753. [Google Scholar] [CrossRef]
- Eidelman, L.A. The Spectrum of Septic Encephalopathy. Definitions, Etiologies, and Mortalities. JAMA J. Am. Med. Assoc. 1996, 275, 470–473. [Google Scholar] [CrossRef]
- Iwashyna, T.J.; Ely, E.W.; Smith, D.M.; Langa, K.M. Long-Term Cognitive Impairment and Functional Disability among Survivors of Severe Sepsis. JAMA 2010, 304, 1787. [Google Scholar] [CrossRef]
- van Eijk, M.M.; Roes, K.C.B.; Honing, M.L.H.; Kuiper, M.A.; Karakus, A.; van der Jagt, M.; Spronk, P.E.; van Gool, W.A.; van der Mast, R.C.; Kesecioglu, J.; et al. Effect of Rivastigmine as an Adjunct to Usual Care with Haloperidol on Duration of Delirium and Mortality in Critically Ill Patients: A Multicentre, Double-Blind, Placebo-Controlled Randomised Trial. Lancet 2010, 376, 1829–1837. [Google Scholar] [CrossRef]
- Perry, V.H.; Cunningham, C.; Holmes, C. Systemic Infections and Inflammation Affect Chronic Neurodegeneration. Nat. Rev. Immunol. 2007, 7, 161–167. [Google Scholar] [CrossRef]
- Conde, J.R.; Streit, W.J. Effect of Aging on the Microglial Response to Peripheral Nerve Injury. Neurobiol. Aging 2006, 27, 1451–1461. [Google Scholar] [CrossRef]
- Bozza, F.A.; D’Avila, J.C.; Ritter, C.; Sonneville, R.; Sharshar, T.; Dal-Pizzol, F. Bioenergetics, Mitochondrial Dysfunction, and Oxidative Stress in the Pathophysiology of Septic Encephalopathy. Shock 2013, 39, 10–16. [Google Scholar] [CrossRef]
- Mazeraud, A.; Righy, C.; Bouchereau, E.; Benghanem, S.; Bozza, F.A.; Sharshar, T. Septic-Associated Encephalopathy: A Comprehensive Review. Neurotherapeutics 2020, 17, 392–403. [Google Scholar] [CrossRef]
- van Gool, W.A.; van de Beek, D.; Eikelenboom, P. Systemic Infection and Delirium: When Cytokines and Acetylcholine Collide. Lancet 2010, 375, 773–775. [Google Scholar] [CrossRef]
- Iacobone, E.; Bailly-Salin, J.; Polito, A.; Friedman, D.; Stevens, R.D.; Sharshar, T. Sepsis-Associated Encephalopathy and Its Differential Diagnosis. Crit. Care Med. 2009, 37, S331–S336. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From Inflammation to Sickness and Depression: When the Immune System Subjugates the Brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Kwon, A.; Kwak, B.O.; Kim, K.; Ha, J.; Kim, S.-J.; Bae, S.H.; Son, J.S.; Kim, S.-N.; Lee, R. Cytokine Levels in Febrile Seizure Patients: A Systematic Review and Meta-Analysis. Seizure 2018, 59, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.D.; Christensen, A.D.; Tewari, D.; McMahon, S.B.; Hamilton, J.A. Immune Cytokines and Their Receptors in Inflammatory Pain. Trends Immunol. 2018, 39, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, O.; Reid, M.B.; Van den Berghe, G.; Vanhorebeek, I.; Hermans, G.; Rich, M.M.; Larsson, L. The Sick and the Weak: Neuropathies/Myopathies in the Critically Ill. Physiol. Rev. 2015, 95, 1025–1109. [Google Scholar] [CrossRef]
- Aldana, B.I. Microglia-Specific Metabolic Changes in Neurodegeneration. J. Mol. Biol. 2019, 431, 1830–1842. [Google Scholar] [CrossRef]
- Labzin, L.I.; Heneka, M.T.; Latz, E. Innate Immunity and Neurodegeneration. Annu. Rev. Med. 2018, 69, 437–449. [Google Scholar] [CrossRef]
- González, H.; Elgueta, D.; Montoya, A.; Pacheco, R. Neuroimmune Regulation of Microglial Activity Involved in Neuroinflammation and Neurodegenerative Diseases. J. Neuroimmunol. 2014, 274, 1–13. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef]
- Balusu, S.; Van Wonterghem, E.; De Rycke, R.; Raemdonck, K.; Stremersch, S.; Gevaert, K.; Brkic, M.; Demeestere, D.; Vanhooren, V.; Hendrix, A.; et al. Identification of a Novel Mechanism of Blood–Brain Communication during Peripheral Inflammation via Choroid Plexus-derived Extracellular Vesicles. Embo Mol. Med. 2016, 8, 1162–1183. [Google Scholar] [CrossRef]
- Kealy, J.; Murray, C.; Griffin, E.W.; Lopez-Rodriguez, A.B.; Healy, D.; Tortorelli, L.S.; Lowry, J.P.; Watne, L.O.; Cunningham, C. Acute Inflammation Alters Brain Energy Metabolism in Mice and Humans: Role in Suppressed Spontaneous Activity, Impaired Cognition, and Delirium. J. Neurosci. 2020, 40, 5681–5696. [Google Scholar] [CrossRef]
- Sharshar, T.; Gray, F.; Poron, F.; Raphael, J.C.; Gajdos, P.; Annane, D. Multifocal Necrotizing Leukoencephalopathy in Septic Shock. Crit. Care Med. 2002, 30, 2371–2375. [Google Scholar] [CrossRef]
- Sharshar, T.; Annane, D.; de la Gradmaison, G.L.; Brouland, J.P.; Hopkinson, N.S.; Gray, F. The Neuropathology of Septic Shock. Brain Pathol. 2004, 14, 21–33. [Google Scholar] [CrossRef]
- Polito, A.; Eischwald, F.; Maho, A.-L.; Polito, A.; Azabou, E.; Annane, D.; Chrétien, F.; Stevens, R.D.; Carlier, R.; Sharshar, T. Pattern of Brain Injury in the Acute Setting of Human Septic Shock. Crit. Care 2013, 17, R204. [Google Scholar] [CrossRef]
- Ehler, J.; Barrett, L.K.; Taylor, V.; Groves, M.; Scaravilli, F.; Wittstock, M.; Kolbaske, S.; Grossmann, A.; Henschel, J.; Gloger, M.; et al. Translational Evidence for Two Distinct Patterns of Neuroaxonal Injury in Sepsis: A Longitudinal, Prospective Translational Study. Crit. Care 2017, 21, 262. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Cunningham, C. Microglia and Neurodegeneration: The Role of Systemic Inflammation. Glia 2013, 61, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Zrzavy, T.; Höftberger, R.; Berger, T.; Rauschka, H.; Butovsky, O.; Weiner, H.; Lassmann, H. Pro-Inflammatory Activation of Microglia in the Brain of Patients with Sepsis. Neuropathol. Appl. Neurobiol. 2019, 45, 278–290. [Google Scholar] [CrossRef] [PubMed]
- MEDAWAR, P.B. Immunity to Homologous Grafted Skin; the Fate of Skin Homografts Transplanted to the Brain, to Subcutaneous Tissue, and to the Anterior Chamber of the Eye. Br. J. Exp. Pathol. 1948, 29, 58–69. [Google Scholar] [PubMed]
- Galea, I.; Bechmann, I.; Perry, V.H. What Is Immune Privilege (Not)? Trends Immunol. 2007, 28, 12–18. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and Functional Features of Central Nervous System Lymphatic Vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A Dural Lymphatic Vascular System That Drains Brain Interstitial Fluid and Macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Brown, M.A. Innate Immunity in the Central Nervous System. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Pekna, M.; Messing, A.; Steinhäuser, C.; Lee, J.M.; Parpura, V.; Hol, E.M.; Sofroniew, M.V.; Verkhratsky, A. Astrocytes: A Central Element in Neurological Diseases. Acta Neuropathol. 2016, 131, 323–345. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Semmler, A.; Okulla, T.; Sastre, M.; Dumitrescu-Ozimek, L.; Heneka, M.T. Systemic Inflammation Induces Apoptosis with Variable Vulnerability of Different Brain Regions. J. Chem. Neuroanat. 2005, 30, 144–157. [Google Scholar] [CrossRef]
- Hernandes, M.S.; D’Avila, J.C.; Trevelin, S.C.; Reis, P.A.; Kinjo, E.R.; Lopes, L.R.; Castro-Faria-Neto, H.C.; Cunha, F.Q.; Britto, L.R.; Bozza, F.A. The Role of Nox2-Derived ROS in the Development of Cognitive Impairment after Sepsis. J. Neuroinflamm. 2014, 11, 36. [Google Scholar] [CrossRef]
- Pelvig, D.P.; Pakkenberg, H.; Stark, A.K.; Pakkenberg, B. Neocortical Glial Cell Numbers in Human Brains. Neurobiol. Aging 2008, 29, 1754–1762. [Google Scholar] [CrossRef]
- Mosser, C.-A.; Baptista, S.; Arnoux, I.; Audinat, E. Microglia in CNS Development: Shaping the Brain for the Future. Prog. Neurobiol. 2017, 149–150, 1–20. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Kierdorf, K.; Prinz, M. Microglia in Steady State. J. Clin. Investig. 2017, 127, 3201–3209. [Google Scholar] [CrossRef]
- Arnò, B.; Grassivaro, F.; Rossi, C.; Bergamaschi, A.; Castiglioni, V.; Furlan, R.; Greter, M.; Favaro, R.; Comi, G.; Becher, B.; et al. Neural Progenitor Cells Orchestrate Microglia Migration and Positioning into the Developing Cortex. Nat. Commun. 2014, 5, 5611. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Vinet, J.; van Weering, H.R.J.; Heinrich, A.; Kälin, R.E.; Wegner, A.; Brouwer, N.; Heppner, F.L.; van Rooijen, N.; Boddeke, H.W.G.M.; Biber, K. Neuroprotective Function for Ramified Microglia in Hippocampal Excitotoxicity. J. Neuroinflamm. 2012, 9, 515. [Google Scholar] [CrossRef]
- Masuch, A.; Shieh, C.-H.; van Rooijen, N.; van Calker, D.; Biber, K. Mechanism of Microglia Neuroprotection: Involvement of P2X7, TNFα, and Valproic Acid. Glia 2016, 64, 76–89. [Google Scholar] [CrossRef]
- Tremblay, M.-È.; Lowery, R.L.; Majewska, A.K. Microglial Interactions with Synapses Are Modulated by Visual Experience. PLoS Biol. 2010, 8, e1000527. [Google Scholar] [CrossRef]
- Milior, G.; Lecours, C.; Samson, L.; Bisht, K.; Poggini, S.; Pagani, F.; Deflorio, C.; Lauro, C.; Alboni, S.; Limatola, C.; et al. Fractalkine Receptor Deficiency Impairs Microglial and Neuronal Responsiveness to Chronic Stress. Brain Behav. Immun. 2016, 55, 114–125. [Google Scholar] [CrossRef]
- Hanisch, U.-K.; Kettenmann, H. Microglia: Active Sensor and Versatile Effector Cells in the Normal and Pathologic Brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- Liang, K.J.; Lee, J.E.; Wang, Y.D.; Ma, W.; Fontainhas, A.M.; Fariss, R.N.; Wong, W.T. Regulation of Dynamic Behavior of Retinal Microglia by CX3CR1 Signaling. Investig. Opthalmol. Vis. Sci. 2009, 50, 4444. [Google Scholar] [CrossRef] [PubMed]
- Pocock, J.M.; Kettenmann, H. Neurotransmitter Receptors on Microglia. Trends Neurosci. 2007, 30, 527–535. [Google Scholar] [CrossRef] [PubMed]
- de Haas, A.H.; Boddeke, H.W.G.M.; Biber, K. Region-Specific Expression of Immunoregulatory Proteins on Microglia in the Healthy CNS. Glia 2008, 56, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial Brain Region−dependent Diversity and Selective Regional Sensitivities to Aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef]
- Lauro, C.; Limatola, C. Metabolic Reprograming of Microglia in the Regulation of the Innate Inflammatory Response. Front. Immunol. 2020, 11, 493. [Google Scholar] [CrossRef] [PubMed]
- Hoogland, I.C.M.; Houbolt, C.; van Westerloo, D.J.; van Gool, W.A.; van de Beek, D. Systemic Inflammation and Microglial Activation: Systematic Review of Animal Experiments. J. Neuroinflamm. 2015, 12, 114. [Google Scholar] [CrossRef]
- Sandiego, C.M.; Gallezot, J.-D.; Pittman, B.; Nabulsi, N.; Lim, K.; Lin, S.-F.; Matuskey, D.; Lee, J.-Y.; O’Connor, K.C.; Huang, Y.; et al. Imaging Robust Microglial Activation after Lipopolysaccharide Administration in Humans with PET. Proc. Natl. Acad. Sci. USA 2015, 112, 12468–12473. [Google Scholar] [CrossRef]
- Lemstra, A.W.; Groen in’t Woud, J.C.M.; Hoozemans, J.J.M.; van Haastert, E.S.; Rozemuller, A.J.M.; Eikelenboom, P.; van Gool, W.A. Microglia Activation in Sepsis: A Case-Control Study. J. Neuroinflamm. 2007, 4, 4. [Google Scholar] [CrossRef]
- van Munster, B.C.; Aronica, E.; Zwinderman, A.H.; Eikelenboom, P.; Cunningham, C.; de Rooij, S.E.J.A. Neuroinflammation in Delirium: A Postmortem Case-Control Study. Rejuvenation Res. 2011, 14, 615–622. [Google Scholar] [CrossRef]
- Polito, A.; Brouland, J.P.; Porcher, R.; Sonneville, R.; Siami, S.; Stevens, R.D.; Guidoux, C.; Maxime, V.; de la Grandmaison, G.L.; Chrétien, F.C.; et al. Hyperglycaemia and Apoptosis of Microglial Cells in Human Septic Shock. Crit. Care 2011. [Google Scholar] [CrossRef]
- Kotas, M.E.; Medzhitov, R. Homeostasis, Inflammation, and Disease Susceptibility. Cell 2015, 160, 816–827. [Google Scholar] [CrossRef]
- Towner, R.A.; Garteiser, P.; Bozza, F.; Smith, N.; Saunders, D.; D′Avila, J.C.P.; Magno, F.; Oliveira, M.F.; Ehrenshaft, M.; Lupu, F.; et al. In Vivo Detection of Free Radicals in Mouse Septic Encephalopathy Using Molecular MRI and Immuno-Spin Trapping. Free Radic. Biol. Med. 2013, 65, 828–837. [Google Scholar] [CrossRef]
- Steckert, A.; Castro, A.; Quevedo, J.; Dal-Pizzol, F. Sepsis in the Central Nervous System and Antioxidant Strategies with Nacetylcysteine, Vitamins and Statins. Curr. Neurovasc. Res. 2014, 11, 83–90. [Google Scholar] [CrossRef]
- Reis, P.A.; Alexandre, P.C.B.; D’Avila, J.C.; Siqueira, L.D.; Antunes, B.; Estato, V.; Tibiriça, E.V.; Verdonk, F.; Sharshar, T.; Chrétien, F.; et al. Statins Prevent Cognitive Impairment after Sepsis by Reverting Neuroinflammation, and Microcirculatory/Endothelial Dysfunction. Brain Behav. Immun. 2017, 60, 293–303. [Google Scholar] [CrossRef]
- Bernier, L.-P.; York, E.M.; MacVicar, B.A. Immunometabolism in the Brain: How Metabolism Shapes Microglial Function. Trends Neurosci. 2020, 43, 854–869. [Google Scholar] [CrossRef]
- Kurtz, P.; D’Avila, J.C.; Prado, D.; Madeira, C.; Vargas-Lopes, C.; Panizzutti, R.; Azevedo, L.C.P.; Bozza, F.A. Cerebral Multimodal Monitoring in Sepsis: An Experimental Study. Shock 2019, 51, 228–234. [Google Scholar] [CrossRef]
- Szöllősi, D.; Hegedűs, N.; Veres, D.S.; Futó, I.; Horváth, I.; Kovács, N.; Martinecz, B.; Dénes, Á.; Seifert, D.; Bergmann, R.; et al. Evaluation of Brain Nuclear Medicine Imaging Tracers in a Murine Model of Sepsis-Associated Encephalopathy. Mol. Imaging Biol. 2018, 20, 952–962. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. We Need to Talk about the Warburg Effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.A. Can the Emerging Field of Immunometabolism Provide Insights into Neuroinflammation? Prog. Neurobiol. 2020, 184, 101719. [Google Scholar] [CrossRef]
- Williams, N.C.; O’Neill, L.A.J. A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front. Immunol. 2018, 9, 1. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate Is an Inflammatory Signal That Induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic Regulation of Microglia. Glia 2018, 66, 1200–1212. [Google Scholar] [CrossRef] [PubMed]
- Vilalta, A.; Brown, G.C. Deoxyglucose Prevents Neurodegeneration in Culture by Eliminating Microglia. J. Neuroinflamm. 2014, 11, 58. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhao, Y.; Sun, M.; Liu, S.; Li, B.; Zhang, L.; Yang, L. 2-Deoxy-d-Glucose Attenuates Sevoflurane-Induced Neuroinflammation through Nuclear Factor-Kappa B Pathway in Vitro. Toxicol. Vitr. 2014, 28, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Jiang, C.; Xue, B.; Zhu, S.; Wang, X. High Glucose Stimulates TNFα and MCP-1 Expression in Rat Microglia via ROS and NF-ΚB Pathways. Acta Pharmacol. Sin. 2011, 32, 188–193. [Google Scholar] [CrossRef]
- Yan, C.; Ma, Z.; Ma, H.; Li, Q.; Zhai, Q.; Jiang, T.; Zhang, Z.; Wang, Q. Mitochondrial Transplantation Attenuates Brain Dysfunction in Sepsis by Driving Microglial M2 Polarization. Mol. Neurobiol. 2020, 57, 3875–3890. [Google Scholar] [CrossRef]
- Gimeno-Bayón, J.; López-López, A.; Rodríguez, M.J.; Mahy, N. Glucose Pathways Adaptation Supports Acquisition of Activated Microglia Phenotype. J. Neurosci. Res. 2014, 92, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Moncada, S.; Bolaños, J.P. Nitric Oxide Switches on Glycolysis through the AMP Protein Kinase and 6-Phosphofructo-2-Kinase Pathway. Nat. Cell Biol. 2004, 6, 45–51. [Google Scholar] [CrossRef]
- Moncada, S.; Bolanos, J.P. Nitric Oxide, Cell Bioenergetics and Neurodegeneration. J. Neurochem. 2006, 97, 1676–1689. [Google Scholar] [CrossRef]
- Kakkar, P.; Singh, B.K. Mitochondria: A Hub of Redox Activities and Cellular Distress Control. Mol. Cell. Biochem. 2007, 305, 235–253. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 Receptor in Infection and Inflammation. Immunity 2017, 47, 15–31. [Google Scholar] [CrossRef]
- Ferrari, D.; Chiozzi, P.; Falzoni, S.; Dal Susino, M.; Melchiorri, L.; Baricordi, O.R.; Di Virgilio, F. Extracellular ATP Triggers IL-1 Beta Release by Activating the Purinergic P2Z Receptor of Human Macrophages. J. Immunol. 1997, 159, 1451–1458. [Google Scholar] [PubMed]
- Singer, M. The Role of Mitochondrial Dysfunction in Sepsis-Induced Multi-Organ Failure. Virulence 2014, 5, 66–72. [Google Scholar] [CrossRef] [PubMed]
- d’Avila, J.d.C.P.; Santiago, A.P.S.A.; Amâncio, R.T.; Galina, A.; Oliveira, M.F.; Bozza, F.A. Sepsis Induces Brain Mitochondrial Dysfunction. Crit. Care Med. 2008, 36, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, R.; Oda, Y. A Clinical Perspective of Sepsis-Associated Delirium. J. Intens. Care 2016, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, S.E.; O’Neill, L.A.J. HIF1α and Metabolic Reprogramming in Inflammation. J. Clin. Investig. 2016, 126, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.-I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507.e6. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.A.; Rao, N.A.; Aghajanirefah, A.; et al. MTOR- and HIF-1 -Mediated Aerobic Glycolysis as Metabolic Basis for Trained Immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef]
- Vanderhaeghen, T.; Vandewalle, J.; Libert, C. Hypoxia-inducible Factors in Metabolic Reprogramming during Sepsis. FEBS J. 2020, 287, 1478–1495. [Google Scholar] [CrossRef]
- Kumar, H.; Choi, D.-K. Hypoxia Inducible Factor Pathway and Physiological Adaptation: A Cell Survival Pathway? Mediat. Inflamm. 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Nair, S.; Sobotka, K.S.; Joshi, P.; Gressens, P.; Fleiss, B.; Thornton, C.; Mallard, C.; Hagberg, H. Lipopolysaccharide-induced Alteration of Mitochondrial Morphology Induces a Metabolic Shift in Microglia Modulating the Inflammatory Response in Vitro and in Vivo. Glia 2019, 67, 1047–1061. [Google Scholar] [CrossRef]
- Frey, D.; Jung, S.; Brackmann, F.; Richter-Kraus, M.; Trollmann, R. Hypoxia Potentiates LPS-Mediated Cytotoxicity of BV2 Microglial Cells In Vitro by Synergistic Effects on Glial Cytokine and Nitric Oxide System. Neuropediatrics 2015, 46, 321–328. [Google Scholar] [CrossRef]
- Bok, S.; Kim, Y.-E.; Woo, Y.; Kim, S.; Kang, S.-J.; Lee, Y.; Park, S.K.; Weissman, I.L.; Ahn, G.-O. Hypoxia-Inducible Factor-1α Regulates Microglial Functions Affecting Neuronal Survival in the Acute Phase of Ischemic Stroke in Mice. Oncotarget 2017, 8, 111508–111521. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Kapfhamer, D.; Minnella, A.M.; Kim, J.-E.; Won, S.J.; Chen, Y.; Huang, Y.; Low, L.H.; Massa, S.M.; Swanson, R.A. Bioenergetic State Regulates Innate Inflammatory Responses through the Transcriptional Co-Repressor CtBP. Nat. Commun. 2017, 8, 624. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-H.; Aid, S.; Kim, H.-W.; Jackson, S.H.; Bosetti, F. Inhibition of NADPH Oxidase Promotes Alternative and Anti-Inflammatory Microglial Activation during Neuroinflammation. J. Neurochem. 2012, 120, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 Polarization and Metabolic States. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M. Neuroinflammation and M2 Microglia: The Good, the Bad, and the Inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Lampron, A.; ElAli, A.; Rivest, S. Innate Immunity in the CNS: Redefining the Relationship between the CNS and Its Environment. Neuron 2013, 78, 214–232. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.G.; Muruve, D.A.; Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014, 15, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, H.; Ono, S.; Efron, P.A.; Scumpia, P.O.; Moldawer, L.L.; Mochizuki, H. ROLE OF TOLL-LIKE RECEPTORS IN THE DEVELOPMENT OF SEPSIS. Shock 2008, 29, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Spiller, S.; Elson, G.; Ferstl, R.; Dreher, S.; Mueller, T.; Freudenberg, M.; Daubeuf, B.; Wagner, H.; Kirschning, C.J. TLR4-Induced IFN-γ Production Increases TLR2 Sensitivity and Drives Gram-Negative Sepsis in Mice. J. Exp. Med. 2008, 205, 1747–1754. [Google Scholar] [CrossRef]
- Takeda, S.; Sato, N.; Morishita, R. Systemic Inflammation, Blood-Brain Barrier Vulnerability and Cognitive / Non-Cognitive Symptoms in Alzheimer Disease: Relevance to Pathogenesis and Therapy. Front. Aging Neurosci. 2014, 6, 1–8. [Google Scholar] [CrossRef]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 Forms an IL-1β-Processing Inflammasome with Increased Activity in Muckle-Wells Autoinflammatory Disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and Regulation of the Inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Vladimer, G.I.; Marty-Roix, R.; Ghosh, S.; Weng, D.; Lien, E. Inflammasomes and Host Defenses against Bacterial Infections. Curr. Opin. Microbiol. 2013, 16, 23–31. [Google Scholar] [CrossRef]
- Song, L.; Pei, L.; Yao, S.; Wu, Y.; Shang, Y. NLRP3 Inflammasome in Neurological Diseases, from Functions to Therapies. Front. Cell. Neurosci. 2017, 11, 63. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome Signalling in Brain Function and Neurodegenerative Disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin Activates the Inflammasome in Response to Toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of Inflammasome by Aggregated α–Synuclein, an Inflammatory Response in Synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Petrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate Immune Activation Through Nalp3 Inflammasome Sensing of Asbestos and Silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica Crystals and Aluminum Salts Activate the NALP3 Inflammasome through Phagosomal Destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host Cell Death and Inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wang, Y.; Zhou, R.; Li, Y.; Gao, Y.; Tu, D.; Wilson, B.; Song, S.; Feng, J.; Hong, J.-S.; et al. A Novel Role of NLRP3-Generated IL-1β in the Acute-Chronic Transition of Peripheral Lipopolysaccharide-Elicited Neuroinflammation: Implications for Sepsis-Associated Neurodegeneration. J. Neuroinflamm. 2020, 17, 64. [Google Scholar] [CrossRef]
- Liu, X.; Nemeth, D.P.; McKim, D.B.; Zhu, L.; DiSabato, D.J.; Berdysz, O.; Gorantla, G.; Oliver, B.; Witcher, K.G.; Wang, Y.; et al. Cell-Type-Specific Interleukin 1 Receptor 1 Signaling in the Brain Regulates Distinct Neuroimmune Activities. Immunity 2019, 50, 317–333.e6. [Google Scholar] [CrossRef]
- Cao, K.; Liao, X.; Lu, J.; Yao, S.; Wu, F.; Zhu, X.; Shi, D.; Wen, S.; Liu, L.; Zhou, H. IL-33/ST2 Plays a Critical Role in Endothelial Cell Activation and Microglia-Mediated Neuroinflammation Modulation. J. Neuroinflamm. 2018, 15, 136. [Google Scholar] [CrossRef]
- Nishioku, T.; Matsumoto, J.; Dohgu, S.; Sumi, N.; Miyao, K.; Takata, F.; Shuto, H.; Yamauchi, A.; Kataoka, Y. Tumor Necrosis Factor-α Mediates the Blood–Brain Barrier Dysfunction Induced by Activated Microglia in Mouse Brain Microvascular Endothelial Cells. J. Pharmacol. Sci. 2010, 112, 251–254. [Google Scholar] [CrossRef]
- Sumi, N.; Nishioku, T.; Takata, F.; Matsumoto, J.; Watanabe, T.; Shuto, H.; Yamauchi, A.; Dohgu, S.; Kataoka, Y. Lipopolysaccharide-Activated Microglia Induce Dysfunction of the Blood–Brain Barrier in Rat Microvascular Endothelial Cells Co-Cultured with Microglia. Cell. Mol. Neurobiol. 2010, 30, 247–253. [Google Scholar] [CrossRef]
- Wolf, Y.; Yona, S.; Kim, K.-W.; Jung, S. Microglia, Seen from the CX3CR1 Angle. Front. Cell. Neurosci. 2013, 7, 26. [Google Scholar] [CrossRef]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Microglia and Aging: The Role of the TREM2–DAP12 and CX3CL1-CX3CR1 Axes. Int. J. Mol. Sci. 2018, 19, 318. [Google Scholar] [CrossRef] [PubMed]
- Zujovic, V.; Benavides, J.; Vigé, X.; Carter, C.; Taupin, V. Fractalkine Modulates TNF-Secretion and Neurotoxicity Induced by Microglial Activation. Glia 2000, 29, 305–315. [Google Scholar] [CrossRef]
- Mizuno, T.; Kawanokuchi, J.; Numata, K.; Suzumura, A. Production and Neuroprotective Functions of Fractalkine in the Central Nervous System. Brain Res. 2003, 979, 65–70. [Google Scholar] [CrossRef]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.R.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of Microglial Neurotoxicity by the Fractalkine Receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. Neuroinflammation, Mast Cells, and Glia: Dangerous Liaisons. Neuroscientist 2017, 23, 478–498. [Google Scholar] [CrossRef]
- Zhang, X.; Dong, H.; Li, N.; Zhang, S.; Sun, J.; Zhang, S.; Qian, Y. Activated Brain Mast Cells Contribute to Postoperative Cognitive Dysfunction by Evoking Microglia Activation and Neuronal Apoptosis. J. Neuroinflamm. 2016, 13, 127. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, A.; Haruwaka, K.; Wake, H. Microglia: Lifelong Modulator of Neural Circuits. Neuropathol. Off. J. Jpn. Soc. Neuropathol. 2019, 39, 173–180. [Google Scholar] [CrossRef]
- Semmler, A.; Frisch, C.; Debeir, T.; Ramanathan, M.; Okulla, T.; Klockgether, T.; Heneka, M.T. Long-Term Cognitive Impairment, Neuronal Loss and Reduced Cortical Cholinergic Innervation after Recovery from Sepsis in a Rodent Model. Exp. Neurol. 2007, 204, 733–740. [Google Scholar] [CrossRef]
- Yokoo, H.; Chiba, S.; Tomita, K.; Takashina, M.; Sagara, H.; Yagisita, S.; Takano, Y.; Hattori, Y. Neurodegenerative Evidence in Mice Brains with Cecal Ligation and Puncture-Induced Sepsis: Preventive Effect of the Free Radical Scavenger Edaravone. PLoS ONE 2012, 7, e51539. [Google Scholar] [CrossRef] [PubMed]
- Weberpals, M.; Hermes, M.; Hermann, S.; Kummer, M.P.; Terwel, D.; Semmler, A.; Berger, M.; Schafers, M.; Heneka, M.T. NOS2 Gene Deficiency Protects from Sepsis-Induced Long-Term Cognitive Deficits. J. Neurosci. 2009, 29, 14177–14184. [Google Scholar] [CrossRef]
- Dal-Pizzol, F.; de Medeiros, G.F.; Michels, M.; Mazeraud, A.; Bozza, F.A.; Ritter, C.; Sharshar, T. What Animal Models Can Tell Us About Long-Term Psychiatric Symptoms in Sepsis Survivors: A Systematic Review. Neurotherapeutics 2021. [Google Scholar] [CrossRef] [PubMed]
- Klawonn, A.M.; Fritz, M.; Castany, S.; Pignatelli, M.; Canal, C.; Similä, F.; Tejeda, H.A.; Levinsson, J.; Jaarola, M.; Jakobsson, J.; et al. Microglial Activation Elicits a Negative Affective State through Prostaglandin-Mediated Modulation of Striatal Neurons. Immunity 2021, 54, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Tao, T.; Zhao, A.; Wen, L.; He, X.; Liu, Y.; Fu, Q.; Mi, W.; Lou, J. Blockade of IL-17A/IL-17R Pathway Protected Mice from Sepsis-Associated Encephalopathy by Inhibition of Microglia Activation. Mediat. Inflamm. 2019, 2019, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Warford, J.; Lamport, A.-C.; Kennedy, B.; Easton, A.S. Human Brain Chemokine and Cytokine Expression in Sepsis: A Report of Three Cases. Can. J. Neurol. Sci. J. Can. Des Sci. Neurol. 2017, 44, 96–104. [Google Scholar] [CrossRef]
- Michels, M.; Vieira, A.S.; Vuolo, F.; Zapelini, H.G.; Mendonça, B.; Mina, F.; Dominguini, D.; Steckert, A.; Schuck, P.F.; Quevedo, J.; et al. The Role of Microglia Activation in the Development of Sepsis-Induced Long-Term Cognitive Impairment. Brain Behav. Immun. 2015, 43, 54–59. [Google Scholar] [CrossRef]
- Michels, M.; Ávila, P.; Pescador, B.; Vieira, A.; Abatti, M.; Cucker, L.; Borges, H.; Goulart, A.I.; Junior, C.C.; Barichello, T.; et al. Microglial Cells Depletion Increases Inflammation and Modifies Microglial Phenotypes in an Animal Model of Severe Sepsis. Mol. Neurobiol. 2019. [Google Scholar] [CrossRef]
- Sonobe, Y.; Liang, J.; Jin, S.; Zhang, G.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Microglia Express a Functional Receptor for Interleukin-23. Biochem. Biophys. Res. Commun. 2008, 370, 129–133. [Google Scholar] [CrossRef]
- Kawanokuchi, J.; Shimizu, K.; Nitta, A.; Yamada, K.; Mizuno, T.; Takeuchi, H.; Suzumura, A. Production and Functions of IL-17 in Microglia. J. Neuroimmunol. 2008, 194, 54–61. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Lou, J.; Zhu, J.; He, M.; Deng, X.; Cai, Z. Neutralisation of Peritoneal IL-17A Markedly Improves the Prognosis of Severe Septic Mice by Decreasing Neutrophil Infiltration and Proinflammatory Cytokines. PLoS ONE 2012, 7, e46506. [Google Scholar] [CrossRef]
- Wong, M.-L.; Bongiorno, P.B.; Rettori, V.; McCann, S.M.; Licinio, J. Interleukin (IL) 1, IL-1 Receptor Antagonist, IL-10, and IL-13 Gene Expression in the Central Nervous System and Anterior Pituitary during Systemic Inflammation: Pathophysiological Implications. Proc. Natl. Acad. Sci. USA 1997, 94, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Moraes, C.A.; Santos, G.; Spohr, T.C.L.S.E.; D’Avila, J.C.; Lima, F.R.S.; Benjamim, C.F.; Bozza, F.A.; Gomes, F.C.A. Activated Microglia-Induced Deficits in Excitatory Synapses Through IL-1β: Implications for Cognitive Impairment in Sepsis. Mol. Neurobiol. 2015, 52, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Kim, H.J.; Shin, A.H.; Thayer, S.A. Synapse Loss Induced by Interleukin-1β Requires Pre-and Post-Synaptic Mechanisms. J. Neuroimmune Pharmacol. 2012, 7, 571–578. [Google Scholar] [CrossRef]
- Serantes, R.; Arnalich, F.; Figueroa, M.; Salinas, M.; Andrés-Mateos, E.; Codoceo, R.; Renart, J.; Matute, C.; Cavada, C.; Cuadrado, A.; et al. Interleukin-1β Enhances GABAA Receptor Cell-Surface Expression by a Phosphatidylinositol 3-Kinase/Akt Pathway. J. Biol. Chem. 2006, 281, 14632–14643. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Park, E.; You, B.; Jung, Y.; Park, A.R.; Park, S.G.; Lee, J.R. Neuronal Synapse Formation Induced by Microglia and Interleukin 10. PLoS ONE 2013, 8, e81218. [Google Scholar] [CrossRef]
- Raffaele, S.; Lombardi, M.; Verderio, C.; Fumagalli, M. TNF Production and Release from Microglia via Extracellular Vesicles: Impact on Brain Functions. Cells 2020, 9, 2145. [Google Scholar] [CrossRef]
- Welser-Alves, J.V.; Milner, R. Microglia Are the Major Source of TNF-α and TGF-Β1 in Postnatal Glial Cultures; Regulation by Cytokines, Lipopolysaccharide, and Vitronectin. Neurochem. Int. 2013, 63, 47–53. [Google Scholar] [CrossRef]
- Takeuchi, H.; Jin, S.; Wang, J.; Zhang, G.; Kawanokuchi, J.; Kuno, R.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Tumor Necrosis Factor-α Induces Neurotoxicity via Glutamate Release from Hemichannels of Activated Microglia in an Autocrine Manner. J. Biol. Chem. 2006, 281, 21362–21368. [Google Scholar] [CrossRef]
- Thomas, A.G.; O’Driscoll, C.M.; Bressler, J.; Kaufmann, W.; Rojas, C.J.; Slusher, B.S. Small Molecule Glutaminase Inhibitors Block Glutamate Release from Stimulated Microglia. Biochem. Biophys. Res. Commun. 2014, 443, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, P.; Domercq, M.; Brambilla, L.; Galli, R.; Schols, D.; De Clercq, E.; Vescovi, A.; Bagetta, G.; Kollias, G.; Meldolesi, J.; et al. CXCR4-Activated Astrocyte Glutamate Release via TNFα: Amplification by Microglia Triggers Neurotoxicity. Nat. Neurosci. 2001, 4, 702–710. [Google Scholar] [CrossRef]
- Barger, S.W.; Goodwin, M.E.; Porter, M.M.; Beggs, M.L. Glutamate Release from Activated Microglia Requires the Oxidative Burst and Lipid Peroxidation. J. Neurochem. 2007, 101, 1205–1213. [Google Scholar] [CrossRef]
- Reynolds, I.J.; Hastings, T.G. Glutamate Induces the Production of Reactive Oxygen Species in Cultured Forebrain Neurons Following NMDA Receptor Activation. J. Neurosci. 1995, 15, 3318–3327. [Google Scholar] [CrossRef]
- Freund, H.R.; Muggia-Sullam, M.; Peiser, J.; Melamed, E. Brain Neurotransmitter Profile Is Deranged during Sepsis and Septic Encephalopathy in the Rat. J. Surg. Res. 1985, 38, 267–271. [Google Scholar] [CrossRef]
- Toklu, H.Z.; Uysal, M.K.; Kabasakal, L.; Sirvanci, S.; Ercan, F.; Kaya, M. The Effects of Riluzole on Neurological, Brain Biochemical, and Histological Changes in Early and Late Term of Sepsis in Rats. J. Surg. Res. 2009, 152, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Domercq, M.; Vázquez-Villoldo, N.; Matute, C. Neurotransmitter Signaling in the Pathophysiology of Microglia. Front. Cell. Neurosci. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Christensen, R.N.; Ha, B.K.; Sun, F.; Bresnahan, J.C.; Beattie, M.S. Kainate Induces Rapid Redistribution of the Actin Cytoskeleton in Ameboid Microglia. J. Neurosci. Res. 2006, 84, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.J.; Nagarajah, R.; Banati, R.B.; Bennett, M.R. Glutamate Induces Directed Chemotaxis of Microglia. Eur. J. Neurosci. 2009, 29, 1108–1118. [Google Scholar] [CrossRef]
- Minghetti, L. Microglia as Effector Cells in Brain Damage and Repair: Focus on Prostanoids and Nitric Oxide. Prog. Neurobiol. 1998, 54, 99–125. [Google Scholar] [CrossRef]
- Taylor, D.L.; Diemel, L.T.; Cuzner, M.L.; Pocock, J.M. Activation of Group II Metabotropic Glutamate Receptors Underlies Microglial Reactivity and Neurotoxicity Following Stimulation with Chromogranin A, a Peptide up-Regulated in Alzheimer’s Disease. J. Neurochem. 2004, 82, 1179–1191. [Google Scholar] [CrossRef]
- Noda, M.; Beppu, K. Possible Contribution of Microglial Glutamate Receptors to Inflammatory Response upon Neurodegenerative Diseases. J. Neurol. Disord. 2013, 1, 131. [Google Scholar] [CrossRef]
- Beppu, K.; Kosai, Y.; Kido, M.A.; Akimoto, N.; Mori, Y.; Kojima, Y.; Fujita, K.; Okuno, Y.; Yamakawa, Y.; Ifuku, M.; et al. Expression, Subunit Composition, and Function of AMPA-Type Glutamate Receptors Are Changed in Activated Microglia; Possible Contribution of GluA2 (GluR-B)-Deficiency under Pathological Conditions. Glia 2013, 61, 881–891. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Huang, W.-Y.; Liu, K.-H.; Lin, S.; Chen, T.-Y.; Tseng, C.-Y.; Chen, H.-Y.; Wu, H.-M.; Hsu, K.-S. NADPH Oxidase 2 as a Potential Therapeutic Target for Protection against Cognitive Deficits Following Systemic Inflammation in Mice. Brain Behav. Immun. 2020, 84, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Prieto, G.A.; Tong, L.; Smith, E.D.; Cotman, C.W. TNFα and IL-1β but Not IL-18 Suppresses Hippocampal Long-Term Potentiation Directly at the Synapse. Neurochem. Res. 2019, 44, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Catalão, C.H.R.; Santos-Junior, N.N.; da Costa, L.H.A.; Souza, A.O.; Cárnio, E.C.; Sebollela, A.; Alberici, L.C.; Rocha, M.J.A. Simvastatin Prevents Long-Term Cognitive Deficits in Sepsis Survivor Rats by Reducing Neuroinflammation and Neurodegeneration. Neurotox. Res. 2020, 38, 871–886. [Google Scholar] [CrossRef]
- Wheeler, R.D.; Brough, D.; Le Feuvre, R.A.; Takeda, K.; Iwakura, Y.; Luheshi, G.N.; Rothwell, N.J. Interleukin-18 Induces Expression and Release of Cytokines from Murine Glial Cells: Interactions with Interleukin-1β. J. Neurochem. 2003, 85, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Alboni, S.; Cervia, D.; Sugama, S.; Conti, B. Interleukin 18 in the CNS. J. Neuroinflamm. 2010, 7, 9. [Google Scholar] [CrossRef]
- Remick, D.G.; Bolgos, G.; Copeland, S.; Siddiqui, J. Role of Interleukin-6 in Mortality from and Physiologic Response to Sepsis. Infect. Immun. 2005, 73, 2751–2757. [Google Scholar] [CrossRef]
- Orhun, G.; Tuzun, E.; Ergin Ozcan, P.; Ulusoy, C.; Yildirim, E.; Kucukerden, M.; Gurvit, H.; Ali, A.; Esen, F. Association between Inflammatory Markers and Cognitive Outcome in Patients with Acute Brain Dysfunction Due to Sepsis. Arch. Neuropsychiatry 2018, 56, 63–70. [Google Scholar] [CrossRef]
- Park, J.H.; Shin, S.H. Induction of IL-12 Gene Expression in the Brain in Septic Shock. Biochem. Biophys. Res. Commun. 1996, 224, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, J.; Krauthausen, M.; Hofer, M.J.; Heneka, M.T.; Campbell, I.L.; Müller, M. CNS-Targeted Production of IL-17A Induces Glial Activation, Microvascular Pathology and Enhances the Neuroinflammatory Response to Systemic Endotoxemia. PLoS ONE 2013, 8, e57307. [Google Scholar] [CrossRef]
- Calsavara, A.C.; Rodrigues, D.H.; Miranda, A.S.; Costa, P.A.; Lima, C.X.; Vilela, M.C.; Rachid, M.A.; Teixeira, A.L. Late Anxiety-Like Behavior and Neuroinflammation in Mice Subjected to Sublethal Polymicrobial Sepsis. Neurotox. Res. 2013, 24, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.J.; Jacob, A.; Cunningham, P.; Hensley, L.; Quigg, R.J. TNF Is a Key Mediator of Septic Encephalopathy Acting through Its Receptor, TNF Receptor-1. Neurochem. Int. 2008, 52, 447–456. [Google Scholar] [CrossRef]
- Thompson, W.L.; Karpus, W.J.; Van Eldik, L.J. MCP-1-Deficient Mice Show Reduced Neuroinflammatory Responses and Increased Peripheral Inflammatory Responses to Peripheral Endotoxin Insult. J. Neuroinflamm. 2008, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Kremlev, S. Differential Expression of Chemokines and Chemokine Receptors during Microglial Activation and Inhibition. J. Neuroimmunol. 2004, 149, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Herzig, D.S.; Luan, L.; Bohannon, J.K.; Toliver-Kinsky, T.E.; Guo, Y.; Sherwood, E.R. The Role of CXCL10 in the Pathogenesis of Experimental Septic Shock. Crit. Care 2014, 18, R113. [Google Scholar] [CrossRef] [PubMed]
- Cortese-Krott, M.M.; Koning, A.; Kuhnle, G.G.C.; Nagy, P.; Bianco, C.L.; Pasch, A.; Wink, D.A.; Fukuto, J.M.; Jackson, A.A.; Van Goor, H.; et al. The Reactive Species Interactome: Evolutionary Emergence, Biological Significance, and Opportunities for Redox Metabolomics and Personalized Medicine. Antioxid. Redox Signal. 2017, 27, 684–712. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, Y.; Nakaso, K.; Horikoshi, Y.; Morimoto, M.; Omotani, T.; Otsuki, A.; Inagaki, Y.; Sato, H.; Matsura, T. System Xc−in Microglia Is a Novel Therapeutic Target for Post-Septic Neurological and Psychiatric Illness. Sci. Rep. 2019, 9, 7562. [Google Scholar] [CrossRef] [PubMed]
- Gyoneva, S.; Davalos, D.; Biswas, D.; Swanger, S.A.; Garnier-Amblard, E.; Loth, F.; Akassoglou, K.; Traynelis, S.F. Systemic Inflammation Regulates Microglial Responses to Tissue Damage in Vivo. Glia 2014, 62, 1345–1360. [Google Scholar] [CrossRef]
- Kikuchi, D.S.; Campos, A.C.P.; Qu, H.; Forrester, S.J.; Pagano, R.L.; Lassègue, B.; Sadikot, R.T.; Griendling, K.K.; Hernandes, M.S. Poldip2 Mediates Blood-Brain Barrier Disruption in a Model of Sepsis-Associated Encephalopathy. J. Neuroinflamm. 2019, 16, 241. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Lan, X.; Zhang, J.; Xiao, S.; Cheng, X.; Wang, H.; Lu, D.; Zhu, L. USP8 Ameliorates Cognitive and Motor Impairments via Microglial Inhibition in a Mouse Model of Sepsis-Associated Encephalopathy. Brain Res. 2019, 1719, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.-J.; Han, J.E.; Woo, M.-S.; Shin, J.A.; Park, E.-M.; Kang, J.L.; Moon, P.G.; Baek, M.-C.; Son, W.-S.; Ko, Y.T.; et al. Matrix Metalloproteinase-8 Plays a Pivotal Role in Neuroinflammation by Modulating TNF-α Activation. J. Immunol. 2014, 193, 2384–2393. [Google Scholar] [CrossRef] [PubMed]
- Woo, M.S.; Park, J.S.; Choi, I.Y.; Kimf, W.K.; Kim, H.S. Inhibition of MMP-3 or -9 Suppresses Lipopolysaccharide-Induced Expression of Proinflammatory Cytokines and INOS in Microglia. J. Neurochem. 2008, 106, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Dal-Pizzol, F.; Rojas, H.A.; Dos Santos, E.M.; Vuolo, F.; Constantino, L.; Feier, G.; Pasquali, M.; Comim, C.M.; Petronilho, F.; Gelain, D.P.; et al. Matrix Metalloproteinase-2 and Metalloproteinase-9 Activities Are Associated with Blood-Brain Barrier Dysfunction in an Animal Model of Severe Sepsis. Mol. Neurobiol. 2013, 48, 62–70. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moraes, C.A.; Zaverucha-do-Valle, C.; Fleurance, R.; Sharshar, T.; Bozza, F.A.; d’Avila, J.C. Neuroinflammation in Sepsis: Molecular Pathways of Microglia Activation. Pharmaceuticals 2021, 14, 416. https://doi.org/10.3390/ph14050416
Moraes CA, Zaverucha-do-Valle C, Fleurance R, Sharshar T, Bozza FA, d’Avila JC. Neuroinflammation in Sepsis: Molecular Pathways of Microglia Activation. Pharmaceuticals. 2021; 14(5):416. https://doi.org/10.3390/ph14050416
Chicago/Turabian StyleMoraes, Carolina Araújo, Camila Zaverucha-do-Valle, Renaud Fleurance, Tarek Sharshar, Fernando Augusto Bozza, and Joana Costa d’Avila. 2021. "Neuroinflammation in Sepsis: Molecular Pathways of Microglia Activation" Pharmaceuticals 14, no. 5: 416. https://doi.org/10.3390/ph14050416
APA StyleMoraes, C. A., Zaverucha-do-Valle, C., Fleurance, R., Sharshar, T., Bozza, F. A., & d’Avila, J. C. (2021). Neuroinflammation in Sepsis: Molecular Pathways of Microglia Activation. Pharmaceuticals, 14(5), 416. https://doi.org/10.3390/ph14050416