
ε2-Phages Are Naturally Bred and Have a Vastly Improved Host Range in Staphylococcus aureus over Wild Type Phages

 ,
,  , , , , and
, , , , and
Abstract
1. Introduction
2. Results
2.1. Study Conducted on a Panel of S. aureus Strains Which Reflects the Clonal Complex Prevalance in Human Infections
2.2. The Plaquing Host Range of Wild Type Phages Is Much Higher Than Their Kinetic Host Range
2.3. Phage Breeding Strongly Increased the Kinetic and Plaquing Host Range
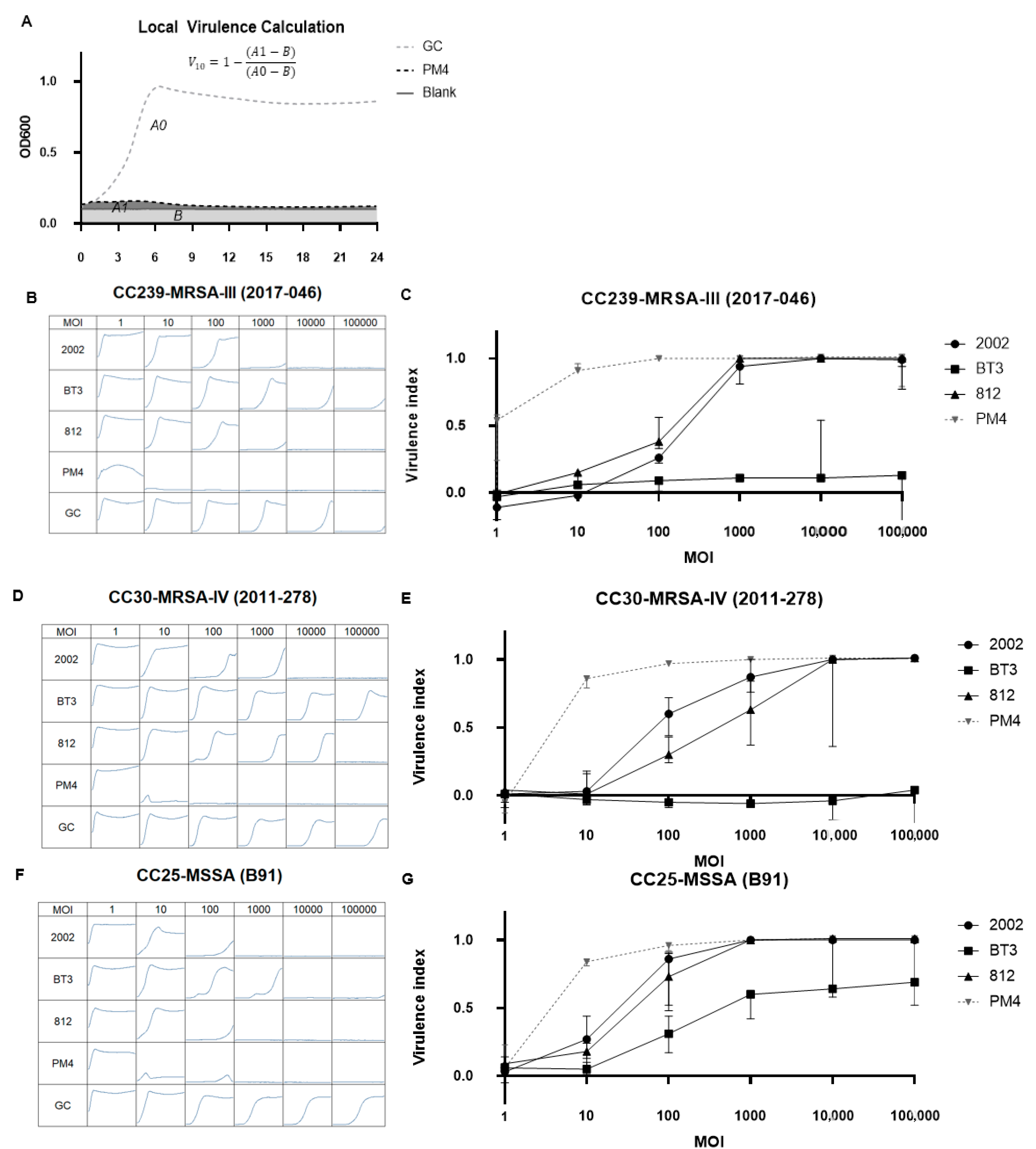
2.4. ε2 Phages Have Increased Virulence Compared to Wild Types in Bacterial Suspensions
2.5. The Cocktail PM-399 Has a KHR of 92% and Does Not Form Resistance at a Lab Scale
2.6. Wild-Type and Bred ε2-Phages Phages Belong to the Herelleviridae Family and Are Deemed Safe for Therapeutic Use
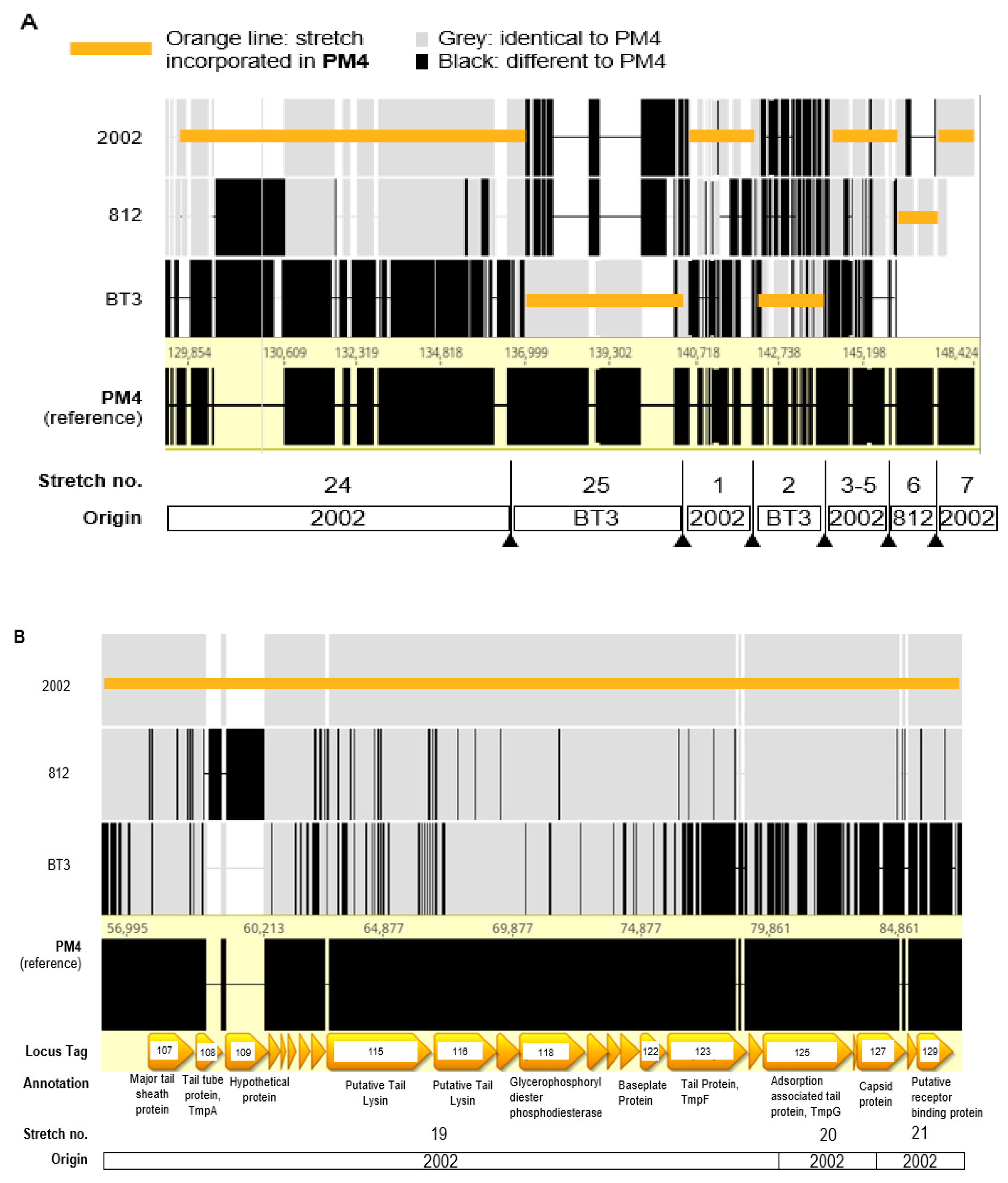
2.7. ε2-Phages with the Best Host Ranges Have Genomes Intercrossed from up to Three Ancestors
2.8. Single Point Mutations on ε2-Phages Bred from Romulus and Remus Increased the Host Range
3. Discussion
4. Materials and Methods
4.1. Collection of S. aureus Strains and Growth Conditions
4.2. Collection of Phages
4.3. Propagation of Phages and Cocktail Preparation
4.4. Plaquing Host Range and Efficiency of Plaquing
4.5. Kinetic Host Range
4.6. Breeding
4.7. Resistance Formation
4.8. Virulence Index
4.9. Genomic Analysis
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gordillo Altamirano, F.L.; Barr, J.J. Phage therapy in the postantibiotic era. Clin. Microbiol. Rev. 2019, 32, 2. [Google Scholar] [CrossRef]
- Górski, A.; Borysowski, J.; Międzybrodzki, R. Phage Therapy: Towards a Successful Clinical Trial. Antibiotics 2020, 9, 827. [Google Scholar] [CrossRef]
- Pires, D.P.; Melo, L.D.R.; Vilas Boas, D.; Sillankorva, S.; Azeredo, J. Phage therapy as an alternative or complementary strategy to prevent and control biofilm-related infections. Curr. Opin. Microbiol. 2017, 39, 48–56. [Google Scholar] [CrossRef]
- Lin, D.M.; Koskella, B.; Lin, H.C. Phage therapy: An alternative to antibiotics in the age of multi-drug resistance. World J. Gastrointest. Pharmacol. Ther. 2017, 8, 162. [Google Scholar] [CrossRef] [PubMed]
- Storms, Z.J.; Teel, M.R.; Mercurio, K.; Sauvageau, D. The Virulence Index: A Metric for Quantitative Analysis of Phage Virulence. Phage 2020, 1, 27–36. [Google Scholar] [CrossRef]
- Kortright, K.E.; Chan, B.K.; Koff, J.L.; Turner, P.E. Phage Therapy: A Renewed Approach to Combat Antibiotic-Resistant Bacteria. Cell Host Microbe 2019, 25, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Levin, B.R.; Bull, J.J. Population and evolutionary dynamics of phage therapy. Nat. Rev. Microbiol. 2004, 2, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Roach, D.R.; Leung, C.Y.; Henry, M.; Morello, E.; Singh, D.; Di Santo, J.P.; Weitz, J.S.; Debarbieux, L. Synergy between the Host Immune System and Bacteriophage Is Essential for Successful Phage Therapy against an Acute Respiratory Pathogen. Cell Host Microbe 2017, 22, 38.e4–47.e4. [Google Scholar] [CrossRef] [PubMed]
- Oechslin, F. Resistance Development to Bacteriophages Occurring during Bacteriophage Therapy. Viruses 2018, 10, 351. [Google Scholar] [CrossRef] [PubMed]
- Mutti, M.; Corsini, L. Robust Approaches for the Production of Active Ingredient and Drug Product for Human Phage Therapy. Front. Microbiol. 2019, 10, 2289. [Google Scholar] [CrossRef]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.-A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): A randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 2018, 19, 2–3. [Google Scholar] [CrossRef]
- Pirnay, J.P.; De Vos, D.; Verbeken, G.; Merabishvili, M.; Chanishvili, N.; Vaneechoutte, M.; Zizi, M.; Laire, G.; Lavigne, R.; Huys, I.; et al. The phage therapy paradigm: Prêt-à-porter or sur-mesure? Pharm. Res. 2011, 28, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Abedon, S.T.; Loc-Carrillo, C. Phage cocktails and the future of phage therapy. Future Microbiol. 2013, 8, 769–783. [Google Scholar] [CrossRef]
- Rohde, C.; Resch, G.; Pirnay, J.P.; Blasdel, B.G.; Debarbieux, L.; Gelman, D.; Górski, A.; Hazan, R.; Huys, I.; Kakabadze, E.; et al. Expert opinion on three phage therapy related topics: Bacterial phage resistance, phage training and prophages in bacterial production strains. Viruses 2018, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- Lenneman, B.R.; Fernbach, J.; Loessner, M.J.; Lu, T.K.; Kilcher, S. Enhancing phage therapy through synthetic biology and genome engineering. Curr. Opin. Biotechnol. 2021, 68, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Cleto, S.; Sillankorva, S.; Azeredo, J.; Lu, T.K. Genetically Engineered Phages: A Review of Advances over the Last Decade. Microbiol. Mol. Biol. Rev. 2016. [Google Scholar] [CrossRef]
- Burrowes, B.; Molineux, I.; Fralick, J. Directed in Vitro Evolution of Therapeutic Bacteriophages: The Appelmans Protocol. Viruses 2019, 11, 241. [Google Scholar] [CrossRef] [PubMed]
- Mapes, A.C.; Trautner, B.W.; Liao, K.S.; Ramig, R.F. Development of expanded host range phage active on biofilms of multi-drug resistant Pseudomonas aeruginosa. Bacteriophage 2016, 6, e1096995. [Google Scholar] [CrossRef] [PubMed]
- Yehl, K.; Lemire, S.; Yang, A.C.; Ando, H.; Mimee, M.; Torres, M.D.T.; de la Fuente-Nunez, C.; Lu, T.K. Engineering Phage Host-Range and Suppressing Bacterial Resistance Through Phage Tail Fiber Mutagenesis. Cell 2019, 179, 459–469.e9. [Google Scholar] [CrossRef] [PubMed]
- Selle, K.; Fletcher, J.R.; Tuson, H.; Schmitt, D.S.; McMillan, L.; Vridhambal, G.S.; Rivera, A.J.; Montgomery, S.A.; Fortier, L.-C.; Barrangou, R.; et al. In Vivo Targeting of Clostridioides difficile Using Phage-Delivered CRISPR-Cas3 Antimicrobials. mBio 2020. [Google Scholar] [CrossRef] [PubMed]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Kwan, T.; Liu, J.; DuBow, M.; Gros, P.; Pelletier, J. The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proc. Natl. Acad. Sci. USA 2005, 102, 5174–5179. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Wolz, C. Phages of Staphylococcus aureus and their impact on host evolution. Infect. Genet. Evol. 2014, 21, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Favor, A.H.; Llanos, C.D.; Youngblut, M.D.; Bardales, J.A. Optimizing bacteriophage engineering through an accelerated evolution platform. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Botka, T.; Pantůček, R.; Mašlaňová, I.; Benešík, M.; Petráš, P.; Růžičková, V.; Havlíčková, P.; Varga, M.; Žemličková, H.; Koláčková, I.; et al. Lytic and genomic properties of spontaneous host-range Kayvirus mutants prove their suitability for upgrading phage therapeutics against staphylococci. Sci. Rep. 2019, 9, 5475. [Google Scholar] [CrossRef] [PubMed]
- Hershey, A.D.; Rotman, R. Genetic Recombination between Host-Range and Plaque-Type Mutants of Bacteriophage in Single Bacterial Cells. Genetics 1949, 34, 44–71. [Google Scholar] [PubMed]
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Kattula, D.; Burkert, F. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Zimmerli, W.; Trampuz, A.; Ochsner, P.E. Current concepts: Prosthetic-joint infections. N. Engl. J. Med. 2004, 351, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Kourtis, A.P.; Hatfield, K.; Baggs, J.; Mu, Y.; See, I.; Epson, E.; Nadle, J.; Kainer, M.A.; Dumyati, G.; Petit, S.; et al. Vital Signs: Epidemiology and Recent Trends in Methicillin-Resistant and in Methicillin-Susceptible Staphylococcus aureus Bloodstream Infections—United States. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Hepburn, L.; Hijnen, D.J.; Sellman, B.R.; Mustelin, T.; Sleeman, M.A.; May, R.D.; Strickland, I. The complex biology and contribution of Staphylococcus aureus in atopic dermatitis, current and future therapies. Br. J. Dermatol. 2017, 177, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Dunyach-Remy, C.; Ngba Essebe, C.; Sotto, A.; Lavigne, J.-P. Staphylococcus aureus Toxins and Diabetic Foot Ulcers: Role in Pathogenesis and Interest in Diagnosis. Toxins 2016, 8, 209. [Google Scholar] [CrossRef] [PubMed]
- Spratt, B.G. Exploring the concept of clonality in bacteria. Methods Mol. Biol. 2004, 266, 323–352. [Google Scholar] [PubMed]
- Feil, E.J.; Cooper, J.E.; Grundmann, H.; Robinson, D.A.; Enright, M.C.; Berendt, T.; Peacock, S.J.; Smith, J.M.; Murphy, M.; Spratt, B.G.; et al. How clonal is Staphylococcus aureus? J. Bacteriol. 2003, 185, 3307–3316. [Google Scholar] [CrossRef] [PubMed]
- Stefani, S.; Chung, D.R.; Lindsay, J.A.; Friedrich, A.W.; Kearns, A.M.; Westh, H.; MacKenzie, F.M. Meticillin-resistant Staphylococcus aureus (MRSA): Global epidemiology and harmonisation of typing methods. Int. J. Antimicrob. Agents 2012, 39, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Monecke, S.; Luedicke, C.; Slickers, P.; Ehricht, R. Molecular epidemiology of Staphylococcus aureus in asymptomatic carriers. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 1159–1165. [Google Scholar] [CrossRef]
- Rasmussen, G.; Monecke, S.; Ehricht, R.; Söderquist, B. Prevalence of Clonal Complexes and Virulence Genes among Commensal and Invasive Staphylococcus aureus Isolates in Sweden. PLoS ONE 2013, 8, e77477. [Google Scholar] [CrossRef]
- Pantůček, R.; Rosypalová, A.; Doškař, J.; Kailerová, J.; Růžičková, V.; Borecká, P.; Snopková, Š.; Horváth, R.; Götz, F.; Rosypal, S. The polyvalent staphylococcal phage φ812: Its host-range mutants and related phages. Virology 1998, 246, 241–252. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vandersteegen, K.; Mattheus, W.; Ceyssens, P.-J.; Bilocq, F.; De Vos, D.; Pirnay, J.-P.; Noben, J.-P.; Merabishvili, M.; Lipinska, U.; Hermans, K.; et al. Microbiological and Molecular Assessment of Bacteriophage ISP for the Control of Staphylococcus aureus. PLoS ONE 2011, 6, e24418. [Google Scholar] [CrossRef]
- Gutiérrez, D.; Vandenheuvel, D.; Martínez, B.; Rodríguez, A.; Lavigne, R.; García, P. Two phages, phiIPLA-RODI and phiIPLA-C1C, lyse mono-and dual-species staphylococcal biofilms. Appl. Environ. Microbiol. 2015, 81, 3336–3348. [Google Scholar] [CrossRef]
- Vandersteegen, K.; Kropinski, A.M.; Nash, J.H.E.; Noben, J.-P.; Hermans, K.; Lavigne, R. Romulus and Remus, Two Phage Isolates Representing a Distinct Clade within the Twortlikevirus Genus, Display Suitable Properties for Phage Therapy Applications. J. Virol. 2013, 87, 3237–3247. [Google Scholar] [CrossRef]
- Lehman, S.M.; Mearns, G.; Rankin, D.; Cole, R.A.; Smrekar, F.; Branston, S.D.; Morales, S. Design and preclinical development of a phage product for the treatment of antibiotic-resistant staphylococcus aureus infections. Viruses 2019, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Kvachadze, L.; Balarjishvili, N.; Meskhi, T.; Tevdoradze, E.; Skhirtladze, N.; Pataridze, T.; Adamia, R.; Topuria, T.; Kutter, E.; Rohde, C.; et al. Evaluation of lytic activity of staphylococcal bacteriophage Sb-1 against freshly isolated clinical pathogens. Microb. Biotechnol. 2011, 4, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Kaźmierczak, Z.; Górski, A.; Dąbrowska, K. Facing Antibiotic Resistance: Staphylococcus aureus Phages as a Medical Tool. Viruses 2014, 6, 2551–2570. [Google Scholar] [CrossRef] [PubMed]
- Kanjilal, S.; Abdul Sater, M.R.; Thayer, M.; Lagoudas, G.K.; Kim, S.; Blainey, P.C.; Gradc, Y.H. Trends in antibiotic susceptibility in staphylococcus aureus in Boston, Massachusetts, from 2000 to 2014. J. Clin. Microbiol. 2018, 56. [Google Scholar] [CrossRef] [PubMed]
- Luedicke, C.; Slickers, P.; Ehricht, R.; Monecke, S. Molecular fingerprinting of Staphylococcus aureus from bone and joint infections. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.A.; Reyes, J.; Carvajal, L.P.; Rincon, S.; Diaz, L.; Panesso, D.; Ibarra, G.; Rios, R.; Munita, J.M.; Salles, M.J.; et al. A prospective cohort multicenter study of molecular epidemiology and phylogenomics of Staphylococcus aureus bacteremia in nine Latin American countries. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Vybiral, D.; Takáč, M.; Loessner, M.; Witte, A.; Von Ahsen, U.; Bläsi, U. Complete nucleotide sequence and molecular characterization of two lytic Staphylococcus aureus phages: 44AHJD and P68. FEMS Microbiol. Lett. 2003, 219, 275–283. [Google Scholar] [CrossRef]
- Tzipilevich, E.; Pollak-fiyaksel, O.; Ben-yehuda, S.; Genetics, M.; Carolina, N. Bacteria elicit a phage tolerance response subsequent to infection of their neighbors. bioRxiv 2021. [Google Scholar] [CrossRef]
- Pirnay, J.-P.; Blasdel, B.G.; Bretaudeau, L.; Buckling, A.; Chanishvili, N.; Clark, J.R.; Corte-Real, S.; Debarbieux, L.; Dublanchet, A.; De Vos, D.; et al. Quality and Safety Requirements for Sustainable Phage Therapy Products. Pharm. Res. 2015, 32, 2173–2179. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Guo, X.; Dong, K.; Zhang, Y.; Li, Q.; Zhu, Y.; Zeng, L.; Tang, R.; Li, L. Safety assessment of Staphylococcus phages of the family Myoviridae based on complete genome sequences. Sci. Rep. 2017, 7, 41259. [Google Scholar] [CrossRef] [PubMed]
- O’Flaherty, S.; Coffey, A.; Edwards, R.; Meaney, W.; Fitzgerald, G.F.; Ross, R.P. Genome of Staphylococcal Phage K: A New Lineage of Myoviridae Infecting Gram-Positive Bacteria with a Low G+C Content. J. Bacteriol. 2004, 186, 2862–2871. [Google Scholar] [CrossRef]
- Grundmann, H.; Aanensen, D.M.; van den Wijngaard, C.C.; Spratt, B.G.; Harmsen, D.; Friedrich, A.W. Geographic Distribution of Staphylococcus aureus Causing Invasive Infections in Europe: A Molecular-Epidemiological Analysis. PLoS Med. 2010, 7, e1000215. [Google Scholar] [CrossRef] [PubMed]
- Onsea, J.; Soentjen, P.; Djebara, S.; Merabishvili, M.; Depypere, M.; Spriet, I.; De Munter, P.; Debaveye, Y.; Nijs, S.; Vanderschot, P.; et al. Bacteriophage Application for Difficult-to-treat Musculoskeletal Infections: Development of a Standardized Multidisciplinary Treatment Protocol. Viruses 2019, 11, 891. [Google Scholar] [CrossRef] [PubMed]
- Mulzer, J.; Trampuz, A.; Potapov, E. V Treatment of chronic left ventricular assist device infection with local application of bacteriophages. Eur. J. Cardio-Thoracic Surg. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ferry, T.; Kolenda, C.; Batailler, C.; Gustave, C.A.; Lustig, S.; Malatray, M.; Fevre, C.; Josse, J.; Petitjean, C.; Chidiac, C.; et al. Phage Therapy as Adjuvant to Conservative Surgery and Antibiotics to Salvage Patients With Relapsing S. aureus Prosthetic Knee Infection. Front. Med. 2020, 7, 570572. [Google Scholar] [CrossRef] [PubMed]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii infection. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Djebara, S.; Maussen, C.; De Vos, D.; Merabishvili, M.; Damanet, B.; Pang, K.; De Leenheer, P.; Strachinaru, I.; Soentjens, P.; Pirnay, J.-P. Processing Phage Therapy Requests in a Brussels Military Hospital: Lessons Identified. Viruses 2019, 11, 265. [Google Scholar] [CrossRef]
- Bull, J.J.; Gill, J.J. The habits of highly effective phages: Population dynamics as a framework for identifying therapeutic phages. Front. Microbiol. 2014, 5, 618. [Google Scholar] [CrossRef] [PubMed]
- Cano, E.J.; Caflisch, K.M.; Bollyky, P.L.; Van Belleghem, J.D.; Patel, R.; Fackler, J.; Brownstein, M.J.; Horne, B.; Biswas, B.; Henry, M.; et al. Phage Therapy for Limb-threatening Prosthetic Knee Klebsiella pneumoniae Infection: Case Report and In Vitro Characterization of Anti-biofilm Activity. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gerlach, D.; Du, X.; Larsen, J.; Stegger, M.; Kuhner, P.; Peschel, A.; Xia, G.; Winstel, V. An accessory wall teichoic acid glycosyltransferase protects Staphylococcus aureus from the lytic activity of Podoviridae. Sci. Rep. 2015, 5, 17219. [Google Scholar] [CrossRef] [PubMed]
- Peters, R.P.H.; Van Agtmael, M.A.; Gierveld, S.; Danner, S.A.; Groeneveld, A.B.J.; Vandenbroucke-Grauls, C.M.J.E.; Savelkoul, P.H.M. Quantitative detection of Staphylococcus aureus and Enterococcus faecalis DNA in blood to diagnose bacteremia in patients in the intensive care unit. J. Clin. Microbiol. 2007, 45, 3641–3646. [Google Scholar] [CrossRef]
- Hesse, S.; Rajaure, M.; Wall, E.; Johnson, J.; Bliskovsky, V.; Gottesman, S.; Adhya, S. Phage resistance in multidrug-resistant klebsiella pneumoniae st258 evolves via diverse mutations that culminate in impaired adsorption. mBio 2020, 11. [Google Scholar] [CrossRef]
- Gibson, S.B.; Green, S.I.; Liu, C.G.; Salazar, K.C.; Clark, J.R.; Terwilliger, A.L.; Kaplan, H.B.; Maresso, A.W.; Trautner, B.W.; Ramig, R.F. Constructing and Characterizing Bacteriophage Libraries for Phage Therapy of Human Infections. Front. Microbiol. 2019, 10, 2537. [Google Scholar] [CrossRef]
- Azam, A.H.; Hoshiga, F.; Takeuchi, I.; Miyanaga, K.; Tanji, Y. Analysis of phage resistance in Staphylococcus aureus SA003 reveals different binding mechanisms for the closely related Twort-like phages ɸSA012 and ɸSA039. Appl. Microbiol. Biotechnol. 2018, 102, 8963–8977. [Google Scholar] [CrossRef]
- Bertozzi Silva, J.; Storms, Z.; Sauvageau, D. Host receptors for bacteriophage adsorption. FEMS Microbiol. Lett. 2016, 363, fnw002. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.R.; Yip, T.K.S.; Myles, B.; Laughlin, L. Roles of genes 38, 39, and 40 in shutoff of host biosyntheses during infection of Bacillus subtilis by bacteriophage SPO1. Virology 2009, 392, 271–274. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sergueev, K.V.; Filippov, A.A.; Farlow, J.; Su, W.; Kvachadze, L.; Balarjishvili, N.; Kutateladze, M.; Nikolich, M.P. Correlation of host range expansion of therapeutic bacteriophage sb-1 with allele state at a hypervariable repeat locus. Appl. Environ. Microbiol. 2019, 85. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.; McAuliffe, O.; Paul Ross, R.; O’Mahony, J.; Coffey, A. Development of a broad-host-range phage cocktail for biocontrol. Bioeng. Bugs 2011, 2, 31–37. [Google Scholar] [CrossRef]
- Mirzaei, M.K.; Nilsson, A.S. Isolation of phages for phage therapy: A comparison of spot tests and efficiency of plating analyses for determination of host range and efficacy. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Clinical and Laboratory Standards Institute M100 Performance Standards for Antimicrobial Susceptibility Testing, 27 ed.; Clinical and Laboratory Standards: Wayne, PA, USA, 2017.
- Dziuginta, J.; Moodley, A. A Rapid Bacteriophage DNA Extraction Method. Methods Protoc. 2018, 1, 27. [Google Scholar] [CrossRef]
- Yamamoto, K.R.; Alberts, B.M.; Benzinger, R.; Lawhorne, L.; Treiber, G. Rapid bacteriophage sedimentation in the presence of polyethylene glycol and its application to large-scale virus purification. Virology 1970, 40, 734–744. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Stewart, C.R.; Casjens, S.R.; Cresawn, S.G.; Houtz, J.M.; Smith, A.L.; Ford, M.E.; Peebles, C.L.; Hatfull, G.F.; Roger, W.; Huang, W.M.; et al. The Genome of Bacillus subtilis Bacteriophage SPO1. J. Mol. Biol. 2010, 388, 48–70. [Google Scholar] [CrossRef] [PubMed]
- McNair, K.; Bailey, B.A.; Edwards, R.A. PHACTS, a computational approach to classifying the lifestyle of phages. Bioinformatics 2012, 28, 614–618. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clonal Complex | # of Isolates Used in This Study | % of Total Strains in This Study | % MRSA | Literature Range % of Strains a |
|---|---|---|---|---|
| CC45 | 19 | 17% | 16% | 4–26% |
| CC30 | 16 | 15% | 31% | 4–23% |
| CC8 | 15 | 14% | 40% | 7–27% |
| CC22 | 9 | 8% | 89% | 2–3% |
| CC5 | 8 | 7% | 88% | 8–35% |
| CC398 | 7 | 6% | 100% | 1% |
| CC25 | 6 | 5% | 0% | 7–9% |
| CC101 | 3 | 3% | 0% | 8% |
| CC12 | 3 | 3% | 0% | 5% |
| CC15 | 3 | 3% | 0% | 6–14% |
| CC80 | 2 | 2% | 100% | 1% |
| CC9 | 2 | 2% | 0% | 2% |
| CC88 | 2 | 2% | 0% | 2% |
| CC6 | 2 | 2% | 100% | N/A |
| CC239 | 2 | 2% | 100% | 4% |
| CC1 | 2 | 2% | 100% | 2–8% |
| CC96 | 1 | 1% | 0% | 1% |
| CC59 | 1 | 1% | 100% | 1% |
| CC121 | 1 | 1% | 0% | 3% |
| CC7 | 1 | 1% | 0% | 3% |
| CC97 | 1 | 1% | 0% | 1% |
| CC772 | 1 | 1% | 100% | N/A |
| CC60 | 1 | 1% | 100% | N/A |
| CC395 | 1 | 1% | 0% | N/A |
| CC49 | 1 | 1% | 0% | 2% |
| Total no. | 110 | 100% | 43% | - |
| Phage Name | Reference | Genome Length (bp) | Accession Number |
|---|---|---|---|
| 2002 | This study | 145,076 | MW528836 |
| BT3 | This study | 146,878 | MW546073 |
| 812 | Pantůček et al., 1998 This study | 150,390 148,660 | MH844528.1 MW546072 |
| Remus | Vandersteegen et al., 2013 This study | 134,641 141,985 | JX846612 MW546076 |
| Romulus | Vandersteegen et al., 2013 This study | 131,332 136,651 | JX846613.1 MW546077 |
| PM4 | This study | 148,627 | MW546074 |
| PM9 | This study | 148,495 | MW546064 |
| PM22 | This study | 140,558 | MW546065 |
| PM25 | This study | 142,467 | MW546066 |
| PM28 | This study | 142,406 | MW546067 |
| PM32 | This study | 148,303 | MW546070 |
| PM34 | This study | 143,062 | MW546068 |
| PM36 | This study | 143,035 | MW546069 |
| PM56 | This study | 136,653 | MW546071 |
| PM93 | This study | 144,038 | MW546075 |
| P66 | Kwan et al., 2005 | 18,199 | NC_007046.1 |
| P68 | Vybiral et al., 2003 | 18,227 | NC_004679.1 |
| From (nt) | To (nt) | Module | Stretch | PM4 | PM25 | PM22 | PM32 | PM28 | PM34 | PM36 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2299 | LTR (left) | 1 | 2002 | BT3/812 | BT3/2002 | 2002 | 2002 | 812 | 812 |
| 2300 | 2885 | LTR (left) | 2 | BT3 | 812 | BT3 | BT3 | BT3 | BT3 | 812 |
| 2886 | 4248 | LTR (left) | 3 | BT3/2002 | 812 | 2002/BT3 | 2002/BT3 | BT3/2002 | BT3/2002 | 812 |
| 4249 | 5604 | LTR (left) | 4 | 2002 | 812 | 2002 | 2002 | 2002 | 2002 | 812 |
| 5605 | 6298 | LTR (left) | 5 | 2002/812 | 2002/812/BT3 | 2002/812 | 2002/812 | 2002/812 | 812 | 812 |
| 6299 | 7508 | LTR (left) | 6 | 812 | 812 | 812 | 812 | 812 | 812 | 812 |
| 7509 | 8343 | LTR (left) | 7 | 812/2002 | 812 | 812/2002 | 812/2002 | 812/2002 | 812 | 812 |
| 8344 | 10,181 | Replication | 8 | 2002 | 2002 | 2002 | 2002 | 2002 | 2002 | 2002 |
| 10,182 | 10,849 | Replication | 9 | 2002 | 2002 | 2002 | 2002 | 2002 | 812 | 2002 |
| 10,850 | 11,837 | Replication | 10 | 2002 | 2002 | 2002 | 2002 | 2002 | 2002 | 2002 |
| 11,838 | 21,147 | Replication | 11 | 2002 | 2002 | 2002 | 2002 | 2002 | 2002 | 812/2002 |
| 21,148 | 34,935 | Replication Lysis | 12 | 2002 | 2002 | 2002 | 2002 | 2002 | 2002 | 812 |
| 34,936 | 35,732 | Lysis | 13 | BT3 | BT3 | BT3 | BT3 | BT3 | BT3 | BT3 |
| 35,733 | 43,441 | Lysis Structural | 14 | 2002 | 2002 | 2002 | 2002 | 2002 | 2002 | 2002 |
| 43,442 | 45,265 | Structural | 15 | 2002 | 2002 | 2002 | 2002 | 812 | 2002 | 2002 |
| 45,266 | 46,723 | Structural | 16 | 2002 | 2002 | 2002 | 2002 | 2002 | 2002 | 2002 |
| 46,724 | 48,054 | Structural | 17 | 2002 | 2002 | 2002 | 2002 | 2002 | 2002 | BT3/2002 |
| 48,055 | 57,822 | Structural | 18 | 2002 | 2002 | 2002 | 2002 | 2002 | 2002 | 812 |
| 57,823 | 79,061 | Structural | 19 | 2002 | 2002/812 | 2002 | 812/2002/BT3 | 2002/812 | 2002/812 | 812 |
| 79,062 | 83,269 | Structural | 20 | 2002 | 2002 | 2002 | 2002 | 2002 | 2002 | 2002 |
| 83,270 | 95,880 | Structural Replication | 21 | 2002 | 2002 | 2002 | 2002 | 2002 | 2002 | 812 |
| 95,881 | 104,924 | Replication Unclear | 22 | 2002/BT3 | 2002/BT3 | 2002 | 2002/812/BT3 | 2002/BT3 | 2002/812/BT3 | 812 |
| 104,925 | 106,889 | Unclear | 23 | 2002 | 2002 | 2002 | 2002 | 2002 | 2002 | 812 |
| 106,890 | 137,479 | Unclear | 24 | 2002 | 2002/812 | 2002 | 2002/812/BT3 | 2002 | 2002/812 | 812 |
| 137,480 | 140,284 | Unclear | 25 | BT3 | BT3 | BT3 | BT3 | BT3 | 812 | 812 |
| 140,285 | 142,583 | LTR (right) | 1 | 2002 | BT3/812 | BT3/2002 | 2002 | 2002 | 812 | 812 |
| 142,584 | 143,169 | LTR (right) | 2 | BT3 | 812 | BT3 | BT3 | BT3 | BT3 | 812 |
| 143,170 | 144,532 | LTR (right) | 3 | BT3/2002 | 812 | 2002/BT3 | 2002/BT3 | BT3/2002 | BT3/2002 | 812 |
| 144,533 | 145,888 | LTR (right) | 4 | 2002 | 812 | 2002 | 2002 | 2002 | 2002 | 812 |
| 145,889 | 146,582 | LTR (right) | 5 | 2002/812 | 2002/812/BT3 | 2002/812 | 2002/812 | 2002/812 | 812 | 812 |
| 146,583 | 147,792 | LTR (right) | 6 | 812 | 812 | 812 | 812 | 812 | 812 | 812 |
| 147,793 | 148,627 | LTR (right) | 7 | 812/2002 | 812 | 812/2002 | 812/2002 | 812/2002 | 812 | 812 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sáez Moreno, D.; Visram, Z.; Mutti, M.; Restrepo-Córdoba, M.; Hartmann, S.; Kremers, A.I.; Tišáková, L.; Schertler, S.; Wittmann, J.; Kalali, B.; et al. ε2-Phages Are Naturally Bred and Have a Vastly Improved Host Range in Staphylococcus aureus over Wild Type Phages. Pharmaceuticals 2021, 14, 325. https://doi.org/10.3390/ph14040325
Sáez Moreno D, Visram Z, Mutti M, Restrepo-Córdoba M, Hartmann S, Kremers AI, Tišáková L, Schertler S, Wittmann J, Kalali B, et al. ε2-Phages Are Naturally Bred and Have a Vastly Improved Host Range in Staphylococcus aureus over Wild Type Phages. Pharmaceuticals. 2021; 14(4):325. https://doi.org/10.3390/ph14040325
Chicago/Turabian StyleSáez Moreno, David, Zehra Visram, Michele Mutti, Marcela Restrepo-Córdoba, Susana Hartmann, Ana Isabel Kremers, Lenka Tišáková, Susanne Schertler, Johannes Wittmann, Benham Kalali, and et al. 2021. "ε2-Phages Are Naturally Bred and Have a Vastly Improved Host Range in Staphylococcus aureus over Wild Type Phages" Pharmaceuticals 14, no. 4: 325. https://doi.org/10.3390/ph14040325
APA StyleSáez Moreno, D., Visram, Z., Mutti, M., Restrepo-Córdoba, M., Hartmann, S., Kremers, A. I., Tišáková, L., Schertler, S., Wittmann, J., Kalali, B., Monecke, S., Ehricht, R., Resch, G., & Corsini, L. (2021). ε2-Phages Are Naturally Bred and Have a Vastly Improved Host Range in Staphylococcus aureus over Wild Type Phages. Pharmaceuticals, 14(4), 325. https://doi.org/10.3390/ph14040325