
Luminescent Phage-Based Detection of Klebsiella pneumoniae: From Engineering to Diagnostics

Abstract
1. Introduction
2. Results
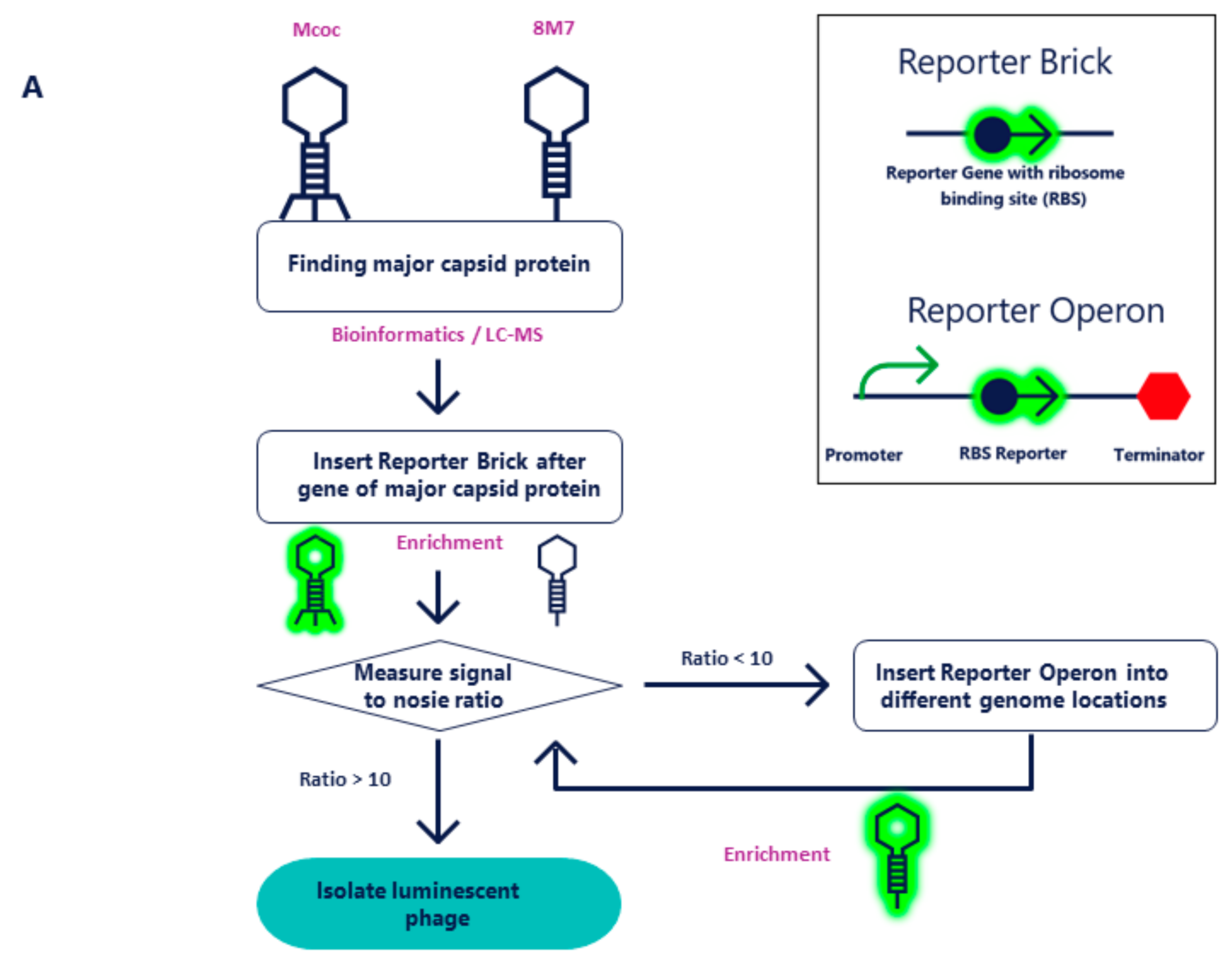
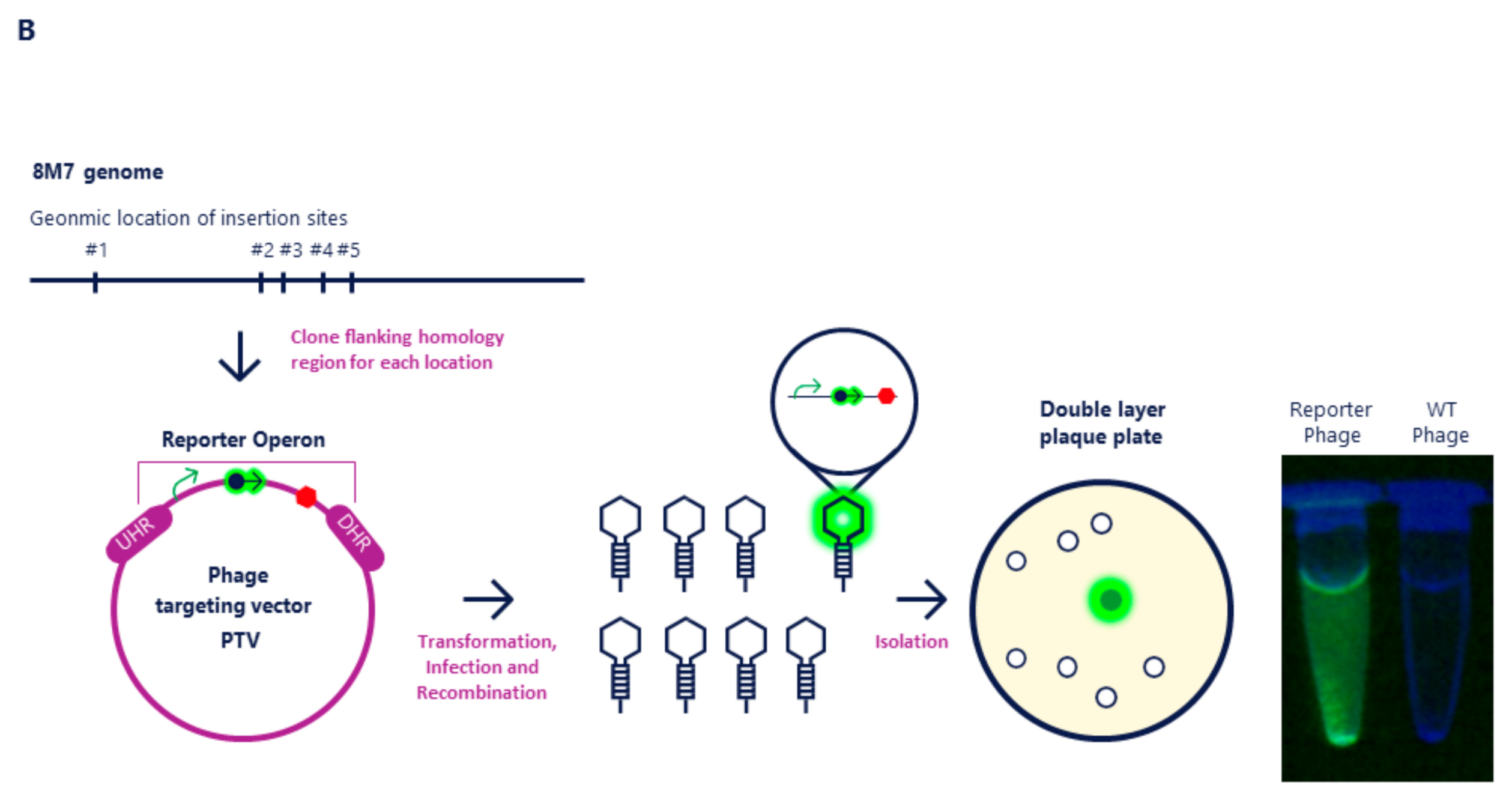
2.1. Molecular Cloning of NanoLuc into Mcoc and 8M7 Bacteriophages
2.2. Activity of Reporter Phages in Liquid Culture
2.3. Activity of Reporter Phages in Fecal Matrices
2.4. Stability of the Signal in Spiked Fecal Samples Stored under Different Conditions
3. Discussion
4. Materials and Methods
4.1. Stool and Sewage Samples
4.2. Klebsiella pneumonia Bacterial Strain
4.3. Polymerase Chain Reaction (PCR)
4.4. Recombinant Phage Mixture Generation
4.5. Recombinant Phage Isolation
4.6. Signal-To-Noise Ratio (SNR) Determination
4.7. Bacterial Preparation for Fecal Spiking
4.8. Phage Sensitivity Assay in Liquid
4.9. Sample Processing and Phage Sensitivity Assay in Fecal Matrices
4.10. Determination of Arbitrary Unit Cutoff for Positive Signal
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matasumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Paczosa, M.K.; Mecsas, J. Klebsiella pneumoniae: Going on the offense with a strong defense. Microbiol. Mol. Biol. Rev. 2016, 80, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Suda, W.; Luo, C.; Kawaguchi, T.; Motoo, I.; Narushima, S.; Kiguchi, Y.; Yasuma, K.; Watanabe, E.; Tanoue, T.; et al. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Science 2017, 358, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Francino, M.P. Antibiotics and the human gut microbiome: Dysbioses and accumulation of resistances. Front. Microbiol. 2016, 6. [Google Scholar] [CrossRef]
- Jernberg, C.; Löfmark, S.; Edlund, C.; Jansson, J.K. Long-term impacts of antibiotic exposure on the human intestinal microbiota. Microbiology 2010, 156, 3216–3223. [Google Scholar] [CrossRef]
- Combarros-Fuertes, P.; Fresno, J.M.; Estevinho, M.M.; Sousa-Pimenta, M.; Tornadijo, M.E.; Estevinho, L.M. Honey: Another alternative in the fight against antibiotic-resistant bacteria? Antibiotics 2020, 9, 774. [Google Scholar] [CrossRef]
- Rossiter, S.E.; Fletcher, M.H.; Wuest, W.M. Natural products as platforms to overcome antibiotic resistance. Chem. Rev. 2017, 117, 12415–12474. [Google Scholar] [CrossRef]
- Morehead, M.S.; Scarbrough, C. Emergence of global antibiotic resistance. Prim. Care Clin. Off. Pract. 2018, 45, 467–484. [Google Scholar] [CrossRef]
- Ndagi, U.; Falaki, A.A.; Abdullahi, M.; Lawal, M.M.; Soliman, M.E. Antibiotic resistance: Bioinformatics-based understanding as a functional strategy for drug design. RSC Adv. 2020, 10, 18451–18468. [Google Scholar] [CrossRef]
- Payne, R.J.; Phil, D.; Jansen, V.A. Phage therapy: The peculiar kinetics of self-replicating pharmaceuticals. Clin. Pharmacol. Ther. 2000, 68, 225–230. [Google Scholar] [CrossRef]
- Nobrega, F.L.; Vlot, M.; de Jonge, P.A.; Dreesens, L.L.; Beaumont, H.J.E.; Lavigne, R.; Dutilh, B.E.; Brouns, S.J.J. Targeting mechanisms of tailed bacteriophages. Nat. Rev. Microbiol. 2018, 16, 760–773. [Google Scholar] [CrossRef]
- Ács, N.; Gambino, M.; Brøndsted, L. Bacteriophage enumeration and detection methods. Front. Microbiol. 2020, 11, 4868. [Google Scholar] [CrossRef]
- Kropinski, A.M.; Mazzocco, A.; Waddell, T.E.; Lingohr, E.; Johnson, R.P. Enumeration of bacteriophages by double agar overlay plaque assay. In Bacteriophages: Methods and Protocols, Volume 1: Isolation, Characterization, and Interactions; Methods in Molecular, BiologyTM; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 69–76. ISBN 978-1-60327-164-6. [Google Scholar]
- Henry, M.; Biswas, B.; Vincent, L.; Mokashi, V.; Schuch, R.; Bishop-Lilly, K.A.; Sozhamannan, S. Development of a high throughput assay for indirectly measuring phage growth using the OmniLogTM system. Bacteriophage 2012, 2, 159–167. [Google Scholar] [CrossRef]
- Lammens, E.-M.; Nikel, P.I.; Lavigne, R. Exploring the synthetic biology potential of bacteriophages for engineering non-model bacteria. Nat. Commun. 2020, 11, 5294. [Google Scholar] [CrossRef]
- Lemire, S.; Yehl, K.M.; Lu, T.K. Phage-based applications in synthetic biology. Annu. Rev. Virol. 2018, 5, 453–476. [Google Scholar] [CrossRef]
- Loessner, M.J.; Rudolf, M.; Scherer, S. Evaluation of luciferase reporter bacteriophage A511::LuxAB for detection of Listeria monocytogenes in contaminated foods. Appl. Environ. Microbiol. 1997, 63, 2961–2965. [Google Scholar] [CrossRef]
- Loessner, M.J.; Rees, C.E.; Stewart, G.S.; Scherer, S. Construction of luciferase reporter bacteriophage A511::LuxAB for rapid and sensitive detection of viable Listeria cells. Appl. Environ. Microbiol. 1996, 62, 1133–1140. [Google Scholar] [CrossRef]
- Brownell, D.; King, J.; Caliando, B.; Sycheva, L.; Koeris, M. Engineering bacteriophage-based biosensors. In Bacteriophages; Methods in Molecular Biology; Clokie, M.R.J., Kropinski, A., Lavigne, R., Eds.; Springer New York: New York, NY, USA, 2019; Volume 1898, pp. 37–50. ISBN 978-1-4939-8939-3. [Google Scholar]
- Meile, S.; Sarbach, A.; Du, J.; Schuppler, M.; Saez, C.; Loessner, M.J.; Kilcher, S. Engineered reporter phages for rapid bioluminescence-based detection and differentiation of viable Listeria cells. Appl. Environ. Microbiol. 2020, 86. [Google Scholar] [CrossRef]
- Shin, J.; Jardine, P.; Noireaux, V. Genome replication, synthesis, and assembly of the bacteriophage T7 in a single cell-free reaction. ACS Synth. Biol. 2012, 1, 408–413. [Google Scholar] [CrossRef]
- Ando, H.; Lemire, S.; Pires, D.P.; Lu, T.K. Engineering modular viral scaffolds for targeted bacterial population editing. Cell Syst. 2015, 1, 187–196. [Google Scholar] [CrossRef]
- Kilcher, S.; Studer, P.; Muessner, C.; Klumpp, J.; Loessner, M.J. Cross-genus rebooting of custom-made, synthetic bacteriophage genomes in L-form bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, 567–572. [Google Scholar] [CrossRef]
- Davis, J.J.; Wattam, A.R.; Aziz, R.K.; Brettin, T.; Butler, R.; Butler, R.M.; Chlenski, P.; Conrad, N.; Dickerman, A.; Dietrich, E.M.; et al. The PATRIC Bioinformatics Resource Center: Expanding data and analysis capabilities. Nucleic Acids Res. 2020, 48, D606–D612. [Google Scholar] [CrossRef] [PubMed]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A Modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Tao, P.; Wu, X.; Tang, W.-C.; Zhu, J.; Rao, V. Engineering of bacteriophage T4 genome using CRISPR-Cas9. ACS Synth. Biol. 2017, 6, 1952–1961. [Google Scholar] [CrossRef]
- Hall, M.P.; Unch, J.; Binkowski, B.F.; Valley, M.P.; Butler, B.L.; Wood, M.G.; Otto, P.; Zimmerman, K.; Vidugiris, G.; Machleidt, T.; et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 2012, 7, 1848–1857. [Google Scholar] [CrossRef]
- Drulis-Kawa, Z.; Mackiewicz, P.; Kęsik-Szeloch, A.; Maciaszczyk-Dziubinska, E.; Weber-Dąbrowska, B.; Dorotkiewicz-Jach, A.; Augustyniak, D.; Majkowska-Skrobek, G.; Bocer, T.; Empel, J.; et al. Isolation and characterisation of KP34—A novel ΦKMV-like bacteriophage for Klebsiella pneumoniae. Appl. Microbiol. Biotechnol. 2011, 90, 1333–1345. [Google Scholar] [CrossRef] [PubMed]
- Pouillot, F.; Blois, H.; Iris, F. Genetically engineered virulent phage banks in the detection and control of emergent pathogenic bacteria. Biosecur. Bioterror. 2010, 8, 155–169. [Google Scholar] [CrossRef]
- Levin-Karp, A.; Barenholz, U.; Bareia, T.; Dayagi, M.; Zelcbuch, L.; Antonovsky, N.; Noor, E.; Milo, R. Quantifying translational coupling in E. coli synthetic operons using RBS modulation and fluorescent reporters. ACS Synth. Biol. 2013, 2, 327–336. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Coronel-Aguilera, C.P.; Romero, P.L.; Perry, L.; Minocha, U.; Rosenfield, C.; Gehring, A.G.; Paoli, G.C.; Bhunia, A.K.; Applegate, B. The use of a novel NanoLuc-based reporter phage for the detection of Escherichia coli O157:H7. Sci. Rep. 2016, 6, 33235. [Google Scholar] [CrossRef] [PubMed]
- Hinkley, T.C.; Garing, S.; Jain, P.; Williford, J.; Le Ny, A.-L.M.; Nichols, K.P.; Peters, J.E.; Talbert, J.N.; Nugen, S.R. A Syringe-based biosensor to rapidly detect low levels of Escherichia coli (ECOR13) in drinking water using engineered bacteriophages. Sensors 2020, 20, 1953. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Saxena, H.M. A new bacteriophage based luminescence assay for diagnosis of brucellosis. Indian J. Exp. Biol. 2017, 55, 7. [Google Scholar]
- Schofield, D.A.; Sharp, N.J.; Vandamm, J.; Molineux, I.J.; Spreng, K.A.; Rajanna, C.; Westwater, C.; Stewart, G.C. Bacillus anthracis diagnostic detection and rapid antibiotic susceptibility determination using ‘bioluminescent’ reporter phage. J. Microbiol. Methods 2013, 95, 156–161. [Google Scholar] [CrossRef]
- Schofield, D.A.; Molineux, I.J.; Westwater, C. Diagnostic bioluminescent phage for detection of yersinia pestis. J. Clin. Microbiol. 2009, 47, 3887–3894. [Google Scholar] [CrossRef]
- Willford, J.D.; Bisha, B.; Bolenbaugh, K.E.; Goodridge, L.D. Luminescence based enzyme-labeled phage (phazyme) assays for rapid detection of shiga toxin producing Escherichia coli serogroups. Bacteriophage 2011, 1, 101–110. [Google Scholar] [CrossRef][Green Version]
- Franche, N.; Vinay, M.; Ansaldi, M. Substrate-independent luminescent phage-based biosensor to specifically detect enteric bacteria such as E. coli. Environ. Sci. Pollut. Res. 2017, 24, 42–51. [Google Scholar] [CrossRef]
- Rose, C.; Parker, A.; Jefferson, B.; Cartmell, E. The characterization of feces and urine: A review of the literature to inform advanced treatment technology. Crit. Rev. Environ. Sci. Technol. 2015, 45, 1827–1879. [Google Scholar] [CrossRef]
- Nováková, J.; Izsáková, A.; Grivalský, T.; Ottmann, C.; Farkašovský, M. Improved method for high-efficiency electrotransformation of Escherichia coli with the large BAC plasmids. Folia Microbiol. 2014, 59, 53–61. [Google Scholar] [CrossRef]
- Abedon, S.T. Phage therapy dosing: The problem(s) with Multiplicity of Infection (MOI). Bacteriophage 2016, 6. [Google Scholar] [CrossRef]
- Baym, M.; Kryazhimskiy, S.; Lieberman, T.D.; Chung, H.; Desai, M.M.; Kishony, R. Inexpensive multiplexed library preparation for megabase-sized genomes. PLoS ONE 2015, 10, e0128036. [Google Scholar] [CrossRef]
- Antipov, D.; Hartwick, N.; Shen, M.; Raiko, M.; Lapidus, A.; Pevzner, P.A. PlasmidSPAdes: Assembling plasmids from whole genome sequencing data. Bioinformatics 2016, 32, 3380–3387. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Publication | Target Bacteria | Matrix | Reporter Phage and Detection Method | Sensitivity | Stability of Signal |
|---|---|---|---|---|---|
| Present | Klebsiella pneumonia strain (KP2H7) | Feces (requires dilution of 1:10 to overcome presence of inhibitors) which allows diagnostics of gut microbiome in chronic diseases | Mcoc and 8M7 NanoLuc | 100 cells per 100 mg feces | Possibility of both on-site POC diagnostics and diagnostics by a central laboratory. Useful diagnostic assay with the potential for field application |
| Zhang et al. [34] | E. coli O157:H7 | Food matrix incl. ground beef | ɸV10 NanoLuc | Detection of a very low quantity of contaminating E. coli O157:H7 (5–6 cells) in 7–9 h | N/A |
| Hinkley et al. [35] | E. coli ECOR13 | Drinking water | T7 NanoLuc | Identification of less than 20 colony forming units (CFU) E. coli in 100 mL drinking water within 5 h (0.2CFU/mL) | N/A |
| Gupta et al. [36] | Brucella abortus; Brucella melitensis | Clinical samples—aborted cattle fetus stomach contents | Brucella phage Luciferase | Average increase of luminescence was 10.03 fold | Useful diagnostic assay with the potential for field application |
| Schofield et al. [37] | Bacillus anthracis | Blood samples | Wβ luxAB | 105 CFU/mL | N/A |
| Schofield et al. [38] | Yersenia pestis | Rapid diagnostic detection of cultivated Y. pestis isolates or infected clinical serum specimens | φA1122 luxAB | 103 CFU/mL within 60min | |
| Willford et al. [39] | Shiga toxin producing E. coli | Food; drinking water | Phazyme Enzyme-labeled phage | 105–106 CFU/mL in pure culture | In a simple and rapid manner, with minimal need for instrumentation to interpret the test result |
| Franche et al. [40] | Enterobacteriaceae | Water | HK620; HK97; GFP | 104 bacteria/mL in 1.5 h | Neither concentration nor enrichment step required |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zelcbuch, L.; Yitzhaki, E.; Nissan, O.; Gidron, E.; Buchshtab, N.; Kario, E.; Kredo-Russo, S.; Zak, N.B.; Bassan, M. Luminescent Phage-Based Detection of Klebsiella pneumoniae: From Engineering to Diagnostics. Pharmaceuticals 2021, 14, 347. https://doi.org/10.3390/ph14040347
Zelcbuch L, Yitzhaki E, Nissan O, Gidron E, Buchshtab N, Kario E, Kredo-Russo S, Zak NB, Bassan M. Luminescent Phage-Based Detection of Klebsiella pneumoniae: From Engineering to Diagnostics. Pharmaceuticals. 2021; 14(4):347. https://doi.org/10.3390/ph14040347
Chicago/Turabian StyleZelcbuch, Lior, Elad Yitzhaki, Olga Nissan, Eliya Gidron, Nufar Buchshtab, Edith Kario, Sharon Kredo-Russo, Naomi B. Zak, and Merav Bassan. 2021. "Luminescent Phage-Based Detection of Klebsiella pneumoniae: From Engineering to Diagnostics" Pharmaceuticals 14, no. 4: 347. https://doi.org/10.3390/ph14040347
APA StyleZelcbuch, L., Yitzhaki, E., Nissan, O., Gidron, E., Buchshtab, N., Kario, E., Kredo-Russo, S., Zak, N. B., & Bassan, M. (2021). Luminescent Phage-Based Detection of Klebsiella pneumoniae: From Engineering to Diagnostics. Pharmaceuticals, 14(4), 347. https://doi.org/10.3390/ph14040347