
Analysis of the Regulatory Science Applied to a Single Portfolio of Eight Biosimilar Product Approvals by Four Key Regulatory Authorities


Abstract
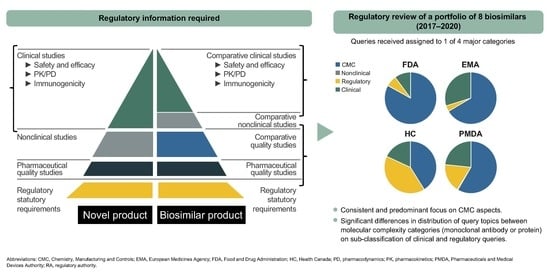
1. Introduction
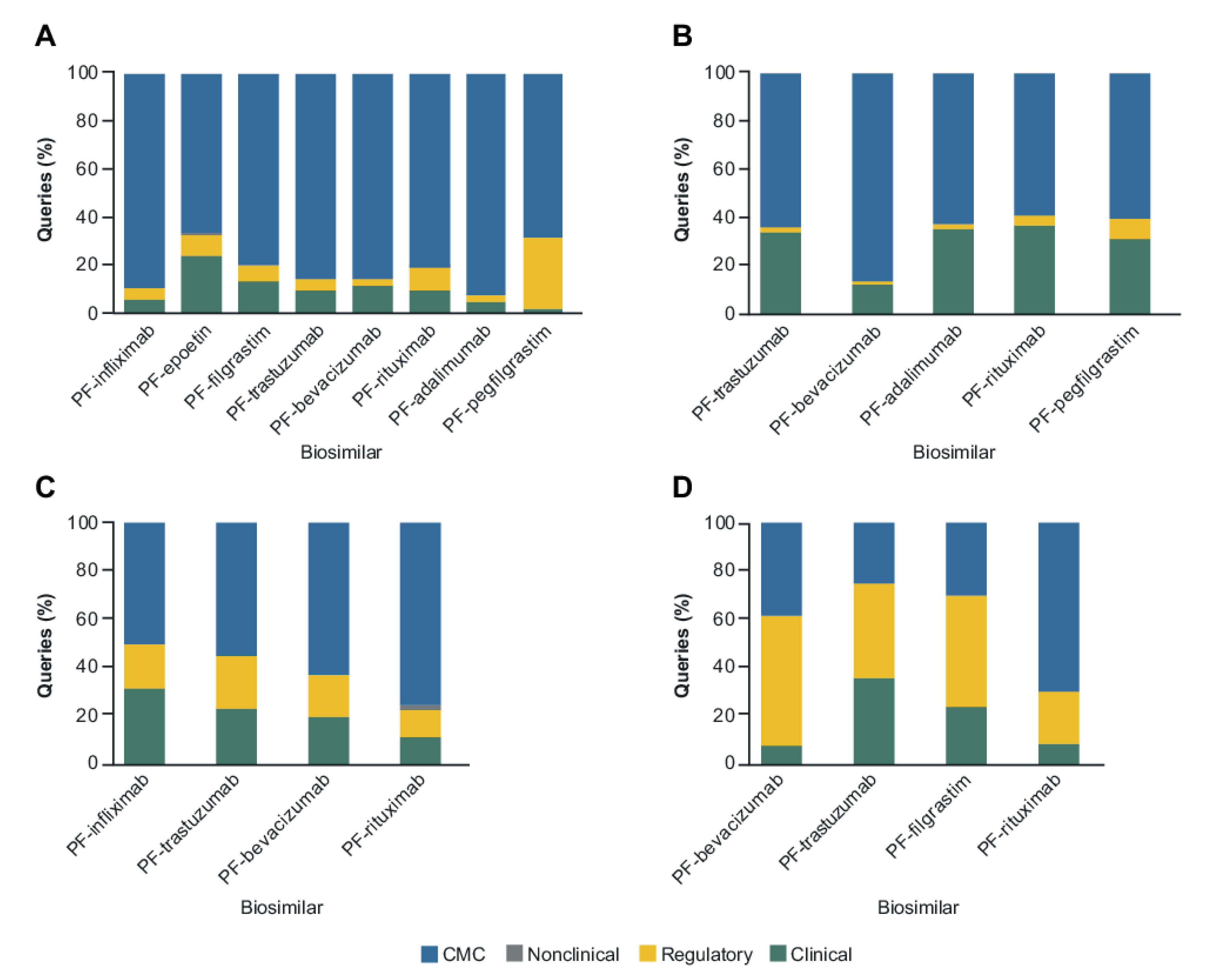
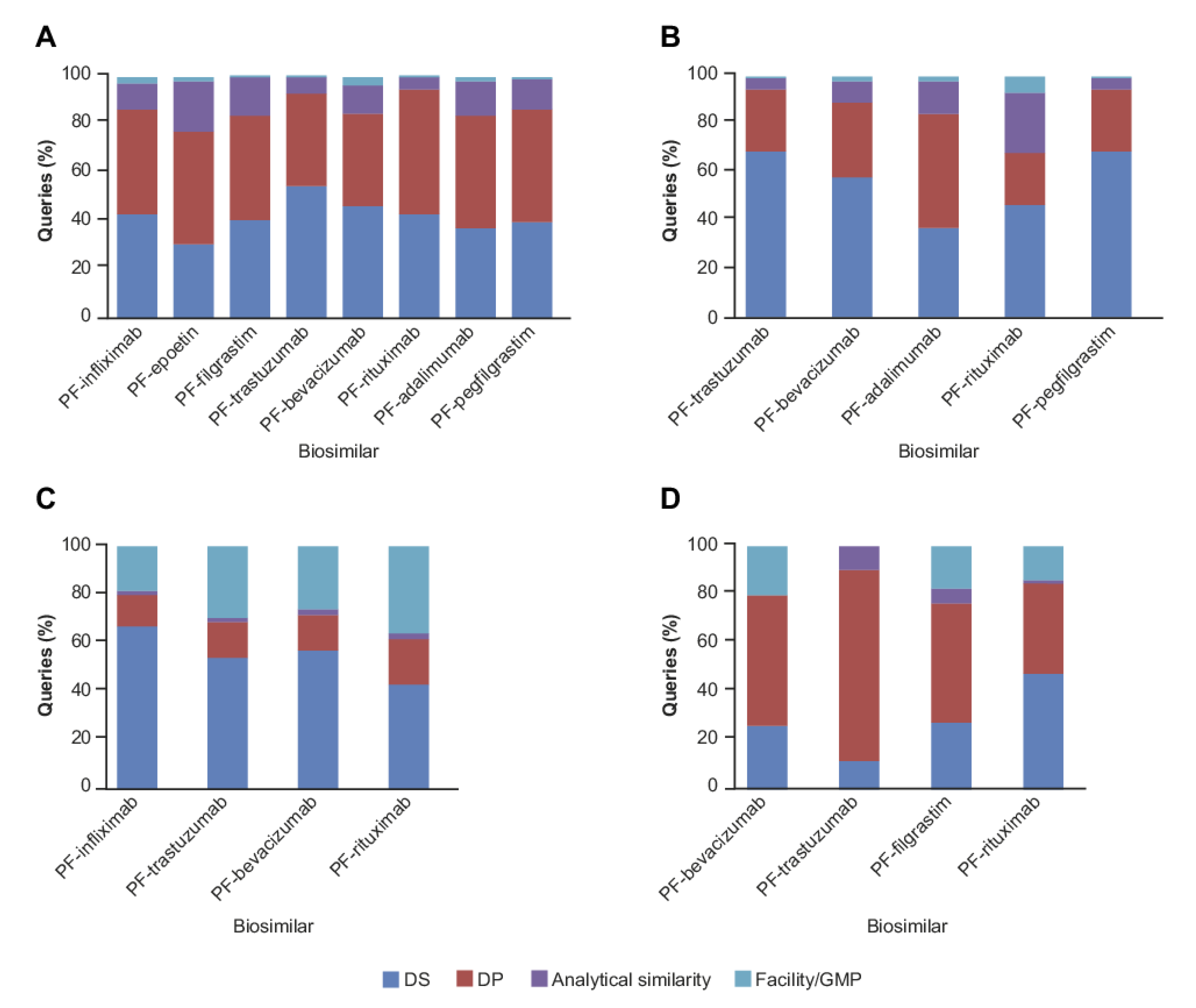
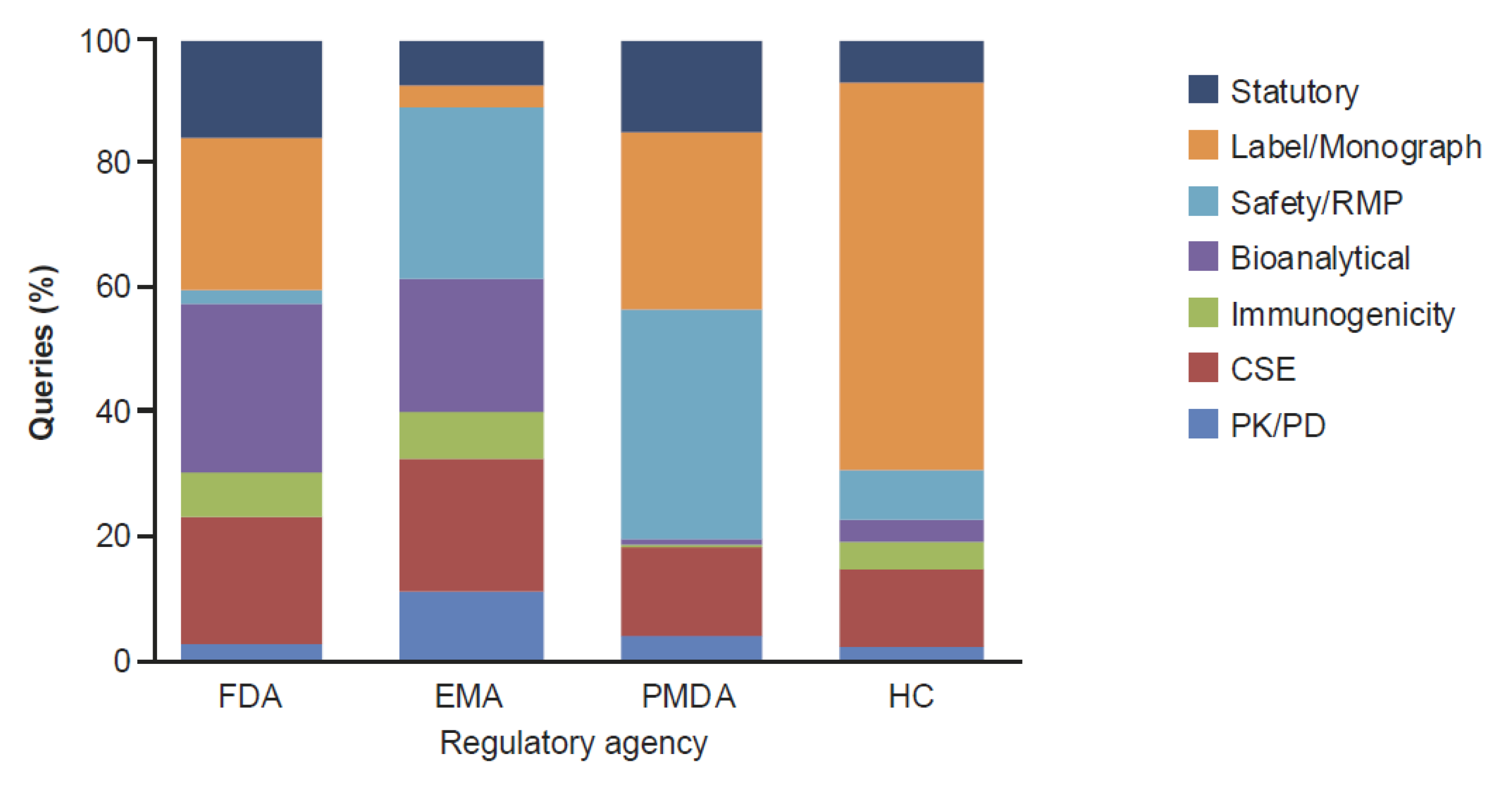
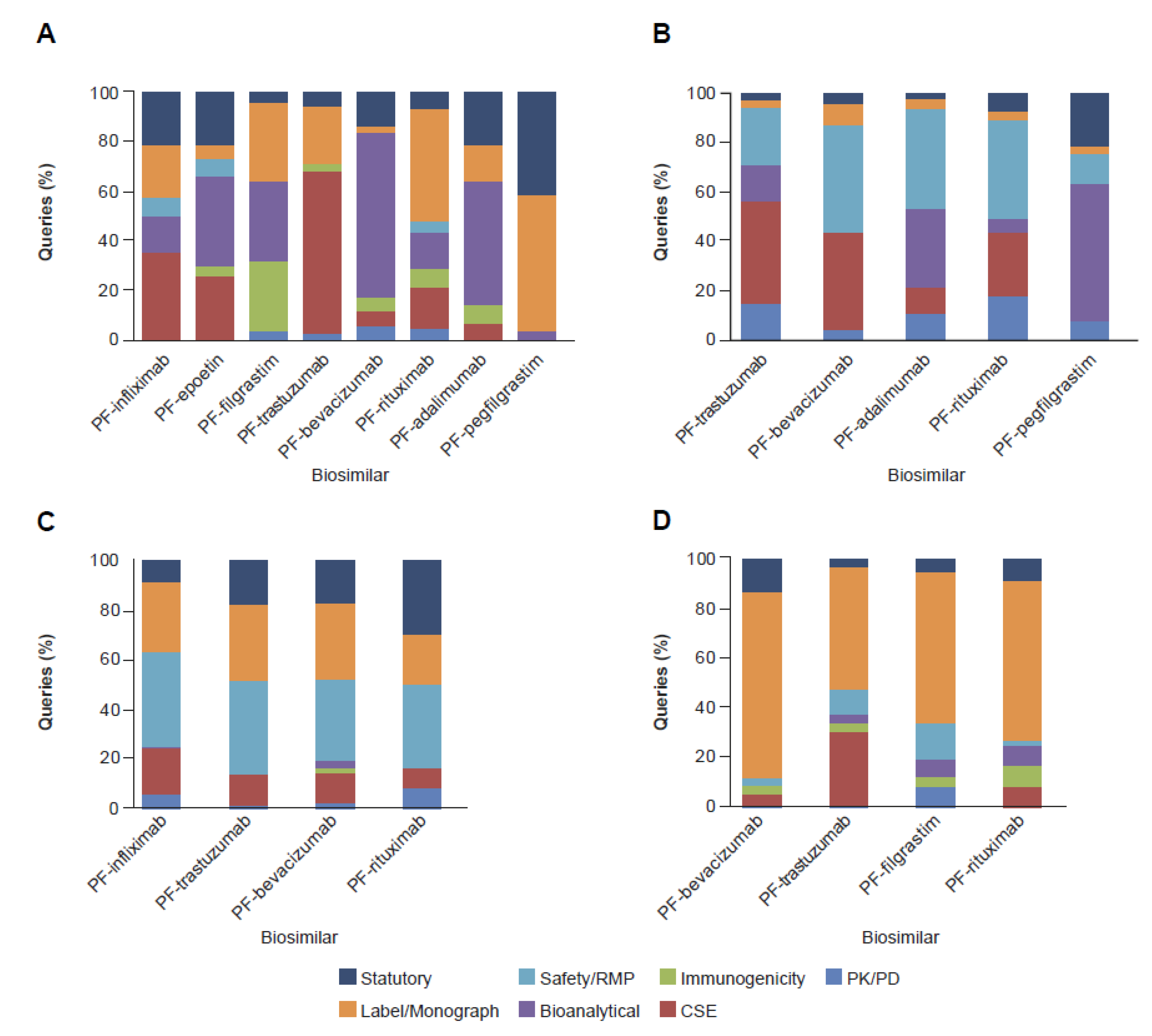
2. Results
2.1. CMC Category
2.2. Clinical and Regulatory Sub-Classification
3. Discussion
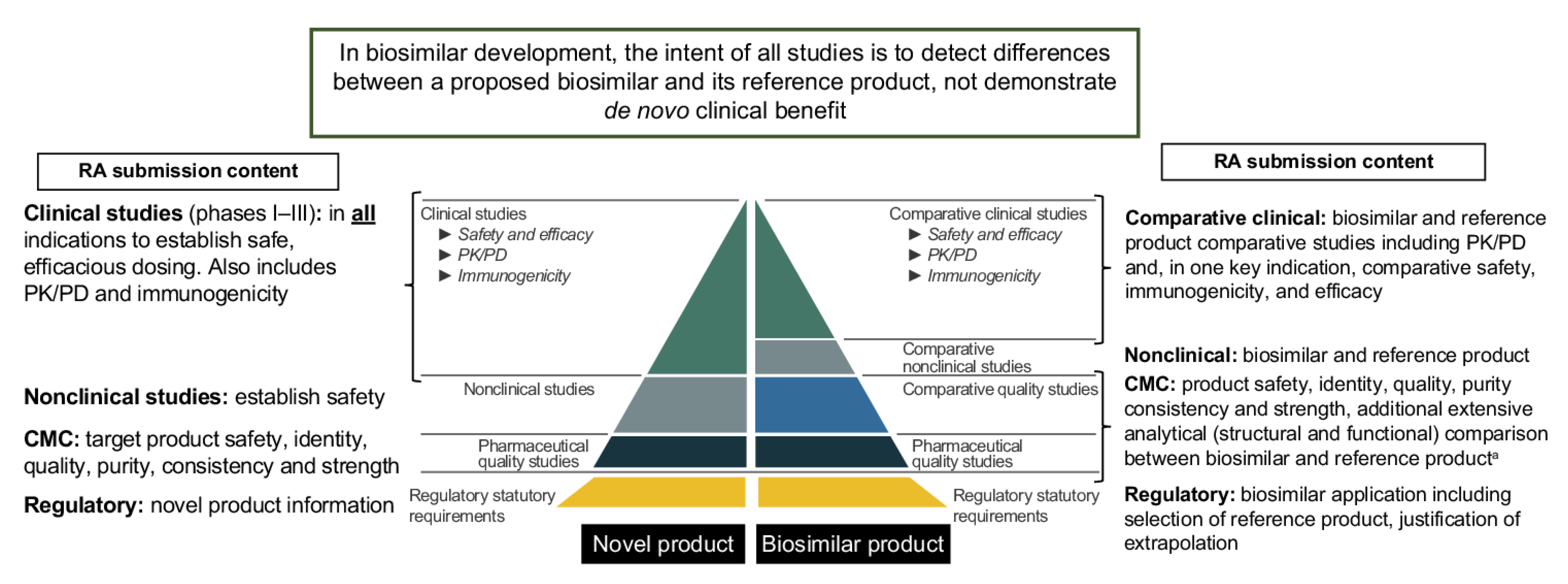
4. Materials and Methods
4.1. Major Classification
4.2. CMC Sub-Classification
4.3. Clinical and Regulatory Sub-Classification
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- US Food and Drug Administration. Biologics Price Competition and Innovation Act of 2009. Available online: https://www.fda.gov/media/78946/download (accessed on 13 August 2020).
- IQVIA. The Impact of Biosimilar Competition in Europe. Available online: https://ec.europa.eu/docsroom/documents/31642 (accessed on 25 November 2020).
- Ghosh, P.K. Similar Biologics: Global Opportunities and Issues. J. Pharm. Pharm. Sci. 2017, 19, 552–596. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.N.; Thorpe, R.; Knezevic, I. Survey Participants from 19 Countries. The Regulatory Landscape of Biosimilars: WHO Efforts and Progress Made from 2009 to 2019. Biologicals 2020, 65, 1–9. [Google Scholar] [CrossRef]
- Gamez-Belmonte, R.; Hernandez-Chirlaque, C.; Arredondo-Amador, M.; Aranda, C.J.; Gonzalez, R.; Martinez-Augustin, O.; Sanchez de Medina, F. Biosimilars: Concepts and Controversies. Pharmacol. Res. 2018, 133, 251–264. [Google Scholar] [CrossRef]
- Rugo, H.S.; Rifkin, R.M.; Declerck, P.; Bair, A.H.; Morgan, G. Demystifying Biosimilars: Development, Regulation and Clinical use. Future Oncol. 2019, 15, 777–790. [Google Scholar] [CrossRef]
- Wang, J.; Chow, S.C. On the Regulatory Approval Pathway of Biosimilar Products. Pharmaceuticals 2012, 5, 353–368. [Google Scholar] [CrossRef]
- ICH. Q5E Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q5e-comparability-biotechnologicalbiological-products-subject-changes-their-manufacturing-process (accessed on 13 August 2020).
- Schiestl, M.; Zabransky, M.; Sorgel, F. Ten Years of Biosimilars in Europe: Development and Evolution of the Regulatory Pathways. Drug Des. Devel. Ther. 2017, 11, 1509–1515. [Google Scholar] [CrossRef]
- Tariman, J.D. Biosimilars: Exploring the History, Science, and Progress. Clin. J. Oncol. Nurs. 2018, 22, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.V.; Wang, J.; Feagan, B.G.; Omoto, M. Biosimilars: State of Clinical and Regulatory Science. J. Pharm. Pharm. Sci. 2017, 20, 332–348. [Google Scholar] [CrossRef] [PubMed]
- Daller, J. Biosimilars: A Consideration of the Regulations in the United States and European Union. Regul. Toxicol. Pharmacol. 2016, 76, 199–208. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Biosimilar Development, Review, and Approval. Available online: https://www.fda.gov/drugs/biosimilars/biosimilar-development-review-and-approval (accessed on 13 August 2020).
- Isaacs, J.; Gonçalves, J.; Strohal, R.; Castañeda-Hernández, G.; Azevedo, V.; Dörner, T.; McInnes, I. The Biosimilar Approval Process: How Different is it? Consid. Med. 2017, 1. [Google Scholar] [CrossRef]
- European Medicines Agency. Biosimilar Medicines: Overview. Available online: https://www.ema.europa.eu/en/human-regulatory/overview/biosimilar-medicines-overview (accessed on 25 November 2020).
- European Medicines Agency. Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Non-Clinical and Clinical Issues. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-2.pdf (accessed on 13 August 2020).
- US Food and Drug Administration. Development of Therapeutic Protein Biosimilars: Comparative Analytical Assessment and Other Quality-Related Considerations Guidance for Industry DRAFT GUIDANCE. Available online: https://www.fda.gov/vaccines-blood-biologics/guidance-compliance-regulatory-information-biologics/biologics-guidances (accessed on 13 August 2020).
- Chapman, K.; Adjei, A.; Baldrick, P.; da Silva, A.; De Smet, K.; DiCicco, R.; Hong, S.S.; Jones, D.; Leach, M.W.; McBlane, J.; et al. Waiving In Vivo Studies for Monoclonal Antibody Biosimilar Development: National and Global Challenges. MAbs 2016, 8, 427–435. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Quality Issues (Revision 1). Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-0.pdf (accessed on 13 August 2020).
- Cilia, M.; Ruiz, S.; Richardson, P.; Salmonson, T.; Serracino-Inglott, A.; Wirth, F.; Borg, J.J. Quality Issues Identified During the Evaluation of Biosimilars by the European Medicines Agency’s Committee for Medicinal Products for Human Use. AAPS Pharm. Sci. Tech. 2018, 19, 489–511. [Google Scholar] [CrossRef] [PubMed]
- Christl, L. FDA’s Overview of the Regulatory Guidance for the Development and Approval of Biosimilar Products in the US. Available online: https://www.fda.gov/files/drugs/published/FDA%E2%80%99s-Overview-of-the-Regulatory-Guidance-for-the-Development-and-Approval-of-Biosimilar-Products-in-the-US.pdf (accessed on 13 August 2020).
- Tesser, J.R.; Furst, D.E.; Jacobs, I. Biosimilars and the Extrapolation of Indications for Inflammatory Conditions. Biologics 2017, 11, 5–11. [Google Scholar] [CrossRef]
- Weise, M.; Bielsky, M.C.; De Smet, K.; Ehmann, F.; Ekman, N.; Giezen, T.J.; Gravanis, I.; Heim, H.K.; Heinonen, E.; Ho, K.; et al. Biosimilars: What Clinicians Should Know. Blood 2012, 120, 5111–5117. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, H.; Lietzan, E.; Faccin, F.; Venema, J. Biosimilar Monoclonal Antibodies: The Scientific Basis for Extrapolation. Expert Opin. Biol. Ther. 2015, 15, 1633–1646. [Google Scholar] [CrossRef]
- European Medicines Agency. Medicines. Available online: https://www.ema.europa.eu/en/search/search/field_ema_web_topics%3Aname_field/Biosimilars (accessed on 13 August 2020).
- US Food and Drug Administration. Biosimilar Product Information: FDA-Approved Biosimilar Products. Available online: https://www.fda.gov/drugs/biosimilars/biosimilar-product-information (accessed on 13 August 2020).
- Greene, L.; Singh, R.M.; Carden, M.J.; Pardo, C.O.; Lichtenstein, G.R. Strategies for Overcoming Barriers to Adopting Biosimilars and Achieving Goals of the Biologics Price Competition and Innovation Act: A Survey of Managed Care and Specialty Pharmacy Professionals. J. Manag. Care. Spec. Pharm. 2019, 25, 904–912. [Google Scholar] [CrossRef]
- Halimi, V.; Daci, A.; Ancevska Netkovska, K.; Suturkova, L.; Babar, Z.U.; Grozdanova, A. Clinical and Regulatory Concerns of Biosimilars: A Review of Literature. Int. J. Environ. Res. Public Health 2020, 17, 5800. [Google Scholar] [CrossRef]
- Cazap, E.; Jacobs, I.; McBride, A.; Popovian, R.; Sikora, K. Global Acceptance of Biosimilars: Importance of Regulatory Consistency, Education, and Trust. Oncologist 2018, 23, 1188–1198. [Google Scholar] [CrossRef]
- World Health Organization. WHO Questions and Answers: Similar Biotherapeutic Products. Available online: http://www.who.int/biologicals/publications/trs/areas/biological_therapeutics/TRS_977_Annex_2.pdf (accessed on 13 August 2020).
- US Food and Drug Administration. Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product: Guidance for Industry. Available online: https://www.fda.gov/media/88622/download (accessed on 20 November 2020).
- Cohen, S.; Emery, P.; Greenwald, M.; Yin, D.; Becker, J.C.; Melia, L.A.; Li, R.; Gumbiner, B.; Thomas, D.; Spencer-Green, G.; et al. A Phase I Pharmacokinetics Trial Comparing PF-05280586 (a Potential Biosimilar) and Rituximab in Patients with Active Rheumatoid Arthritis. Br. J. Clin. Pharmacol. 2016, 82, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Palaparthy, R.; Udata, C.; Hua, S.Y.; Yin, D.; Cai, C.H.; Salts, S.; Rehman, M.I.; McClellan, J.; Meng, X. A Randomized Study Comparing the Pharmacokinetics of the Potential Biosimilar PF-06438179/GP1111 with Remicade® (Infliximab) in Healthy Subjects (REFLECTIONS B537-01). Expert Rev. Clin. Immunol. 2018, 14, 329–336. [Google Scholar] [CrossRef]
- Stalker, D.; Ramaiya, A.; Kumbhat, S.; Zhang, J.; Reid, S.; Martin, N. Pharmacodynamic and Pharmacokinetic Equivalences of Epoetin Hospira and Epogen® After Multiple Subcutaneous Doses to Healthy Male Subjects. Clin. Ther. 2016, 38, 1090–1101. [Google Scholar] [CrossRef] [PubMed]
- Stalker, D.; Reid, S.; Ramaiya, A.; Wisemandle, W.A.; Martin, N.E. Pharmacokinetic and Pharmacodynamic Equivalence of Epoetin Hospira and Epogen After Single Subcutaneous Doses to Healthy Male Subjects. Clin. Ther. 2016, 38, 1778–1788. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.M.; Ottery, F.D.; Borema, T.; Harris, S.; Levy, J.; May, T.B.; Moosavi, S.; Zhang, J.; Summers, M. PF-06881893 (Nivestym), a Filgrastim Biosimilar, Versus US-Licensed Filgrastim Reference Product (US-Neupogen®): Pharmacokinetics, Pharmacodynamics, Immunogenicity, and Safety of Single or Multiple Subcutaneous Doses in Healthy Volunteers. Biodrugs 2019, 33, 207–220. [Google Scholar] [CrossRef]
- Yin, D.; Barker, K.B.; Li, R.; Meng, X.; Reich, S.D.; Ricart, A.D.; Rudin, D.; Taylor, C.T.; Zacharchuk, C.M.; Hansson, A.G. A Randomized Phase 1 Pharmacokinetic Trial Comparing the Potential Biosimilar PF-05280014 with Trastuzumab in Healthy Volunteers (REFLECTIONS B327-01). Br. J. Clin. Pharmacol. 2014, 78, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Knight, B.; Rassam, D.; Liao, S.; Ewesuedo, R. A Phase I Pharmacokinetics Study Comparing PF-06439535 (a Potential Biosimilar) with Bevacizumab in Healthy Male Volunteers. Cancer Chemother. Pharmacol. 2016, 77, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Shirley, M. PF-06410293: An Adalimumab Biosimilar. Biodrugs 2020, 34, 695–698. [Google Scholar] [CrossRef]
- Moosavi, S.; Borema, T.; Ewesuedo, R.; Harris, S.; Levy, J.; May, T.B.; Summers, M.; Thomas, J.S.; Zhang, J.; Yao, H.M. PF-06881894, a Proposed Biosimilar to Pegfilgrastim, Versus US-Licensed and EU-Approved Pegfilgrastim Reference Products (Neulasta®): Pharmacodynamics, Pharmacokinetics, Immunogenicity, and Safety of Single or Multiple Subcutaneous Doses in Healthy Volunteers. Adv. Ther. 2020, 37, 3370–3391. [Google Scholar] [CrossRef]
- European Medicines Agency. Biosimilars in the EU: Information Guide for Healthcare Professionals. Available online: https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf (accessed on 10 December 2020).
- US Food and Drug Administration. Labeling for Biosimilar Products: Guidance for Industry. Available online: https://www.fda.gov/media/96894/download (accessed on 10 December 2020).
- Health Canada. Guidance Document: Information and Submission Requirements for Biosimilar Biologic Drugs. Available online: www.hc-sc.gc.ca/index-eng.php (accessed on 13 August 2020).
- Pharmaceutical and Food Safety Bureau Ministry of Health Labour and Welfare. Guideline for the Quality, Safety, and Efficacy Assurance of Follow-on Biologics. Available online: https://www.pmda.go.jp/files/000153851.pdf (accessed on 25 November 2020).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biosimilar | Regulatory Agency, Approval Date | Reference Product, Trade Name (INN) | Molecule Complexity | Therapy Area | ||||
|---|---|---|---|---|---|---|---|---|
| Product | INN (Trade Name), US/RoW | FDA | EMA | PMDA | HC | |||
| PF-filgrastim | filgrastim-aafi (Nivestym®)/ filgrastim (Nivestim) a | 20 July 2018 | 8 June 2010 e | − d | 20 April 2020 | Neupogen® (filgrastim) | Protein | Oncology |
| PF-epoetin | epoetin alfa-epbx (Retacrit®) b | 15 May 2018 | − d | − d | − d | Epogen®/Procrit® g (epoetin alfa) | Protein | Oncology |
| PF-epoetin | epoetin zeta (Retacrit®) b | − d | 18 December 2007 e | − d | − d | Epogen®/Procrit® g (epoetin alfa) | Protein | Oncology |
| PF-rituximab | rituximab-pvvr/ rituximab (Ruxience™) | 23 July 2019 | 1 April 2020 | 20 September 2019 | 4 May 2020 | Rituxan® (rituximab) | MAb | Oncology, inflammation h |
| PF-trastuzumab | trastuzumab-qyyp/trastuzumab (Trazimera™) | 11 March 2019 | 26 July 2018 | 21 September 2018 | 15 August 2019 | Herceptin® (trastuzumab) | MAb | Oncology |
| PF-bevacizumab | bevacizumab-bvzr/bevacizumab (Zirabev™) | 28 June 2019 | 14 February 2019 | 18 June 2019 | 14 June 2019 | Avastin® (bevacizumab) | MAb | Oncology |
| PF-pegfilgrastim | pegfilgrastim-apgf/pegfilgrastim (Nyvepria™) | 11 June 2020 | 20 November 2020 | − d | − d | Neulasta® (pegfilgrastim) | Protein | Oncology |
| PF-adalimumab | adalimumab-afzb (Abrilada™)/ adalimumab (Amsparity) c | 18 November 2019 | 13 February 2020 | − d | − d | Humira® (adalimumab) | MAb | Inflammation |
| PF-infliximab | infliximab-qbtx/infliximab (Ixifi™) | 13 December 2017 | Divested f | 2 July 2018 | − d | Remicade® (infliximab) | MAb | Inflammation |
| Classification Level | Category (Assignment Criteria) | |||
|---|---|---|---|---|
| Major | CMC (all aspects of drug substance [DS] and drug product [DP] manufacturing, testing, control and analytical similarity [including in vitro functional analysis]) | Nonclinical (in vivo animal studies and comparative toxicokinetics between biosimilar and reference product) | Clinical (all clinical studies including PK/PD studies in healthy volunteers or patients, comparative safety and efficacy, where appropriate, and immunogenicity) | Regulatory (statutory requirements of the submission including prescribing information, selection of reference product, justification of extrapolation, and regulatory procedural topics) |
| Sub-classification | DS (DS development, manufacture, control, storage, transportation and stability) DP (DP development, manufacture, control, storage, transportation and stability, including queries related to product testing activities [e.g., product sample testing for HC]) Analytical similarity (analytical similarity studies, data, and interpretation [including in vitro functional analysis]) GMP/Facility (GMP status of facilities involved in the development, manufacturing, and testing of the DS and DP, including queries issued following facility inspections conducted as part of an application (e.g., HC On-Site Evaluation and FDA pre-approval inspection) | Bioanalytical (assays used to generate and assess clinical data [all clinical studies]; this included the assay development, validation/qualification, and sample preparation across all clinical studies) PK/PD (data derived from the comparative PK/PD modeling) Immunogenicity (immunogenicity information derived from the clinical studies) Comparative Safety and Efficacy (CSE) (comparative study, generally conducted in only one clinical indication) Safety/RMP (safety, including the RMP where applicable [e.g., EMA, PMDA, HC] and pharmacovigilance) Label/Monograph (product information [e.g., US PI, EU SmPC, including the Canadian-specific product monograph]) a Statutory (justification of extrapolation, reference product selection, and general regulatory procedural topics [e.g., trade name approval]) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ingram, B.; Lumsden, R.S.; Radosavljevic, A.; Kobryn, C. Analysis of the Regulatory Science Applied to a Single Portfolio of Eight Biosimilar Product Approvals by Four Key Regulatory Authorities. Pharmaceuticals 2021, 14, 306. https://doi.org/10.3390/ph14040306
Ingram B, Lumsden RS, Radosavljevic A, Kobryn C. Analysis of the Regulatory Science Applied to a Single Portfolio of Eight Biosimilar Product Approvals by Four Key Regulatory Authorities. Pharmaceuticals. 2021; 14(4):306. https://doi.org/10.3390/ph14040306
Chicago/Turabian StyleIngram, Beverly, Rebecca S. Lumsden, Adriana Radosavljevic, and Christine Kobryn. 2021. "Analysis of the Regulatory Science Applied to a Single Portfolio of Eight Biosimilar Product Approvals by Four Key Regulatory Authorities" Pharmaceuticals 14, no. 4: 306. https://doi.org/10.3390/ph14040306
APA StyleIngram, B., Lumsden, R. S., Radosavljevic, A., & Kobryn, C. (2021). Analysis of the Regulatory Science Applied to a Single Portfolio of Eight Biosimilar Product Approvals by Four Key Regulatory Authorities. Pharmaceuticals, 14(4), 306. https://doi.org/10.3390/ph14040306