3.2. General Procedure for the Synthesis
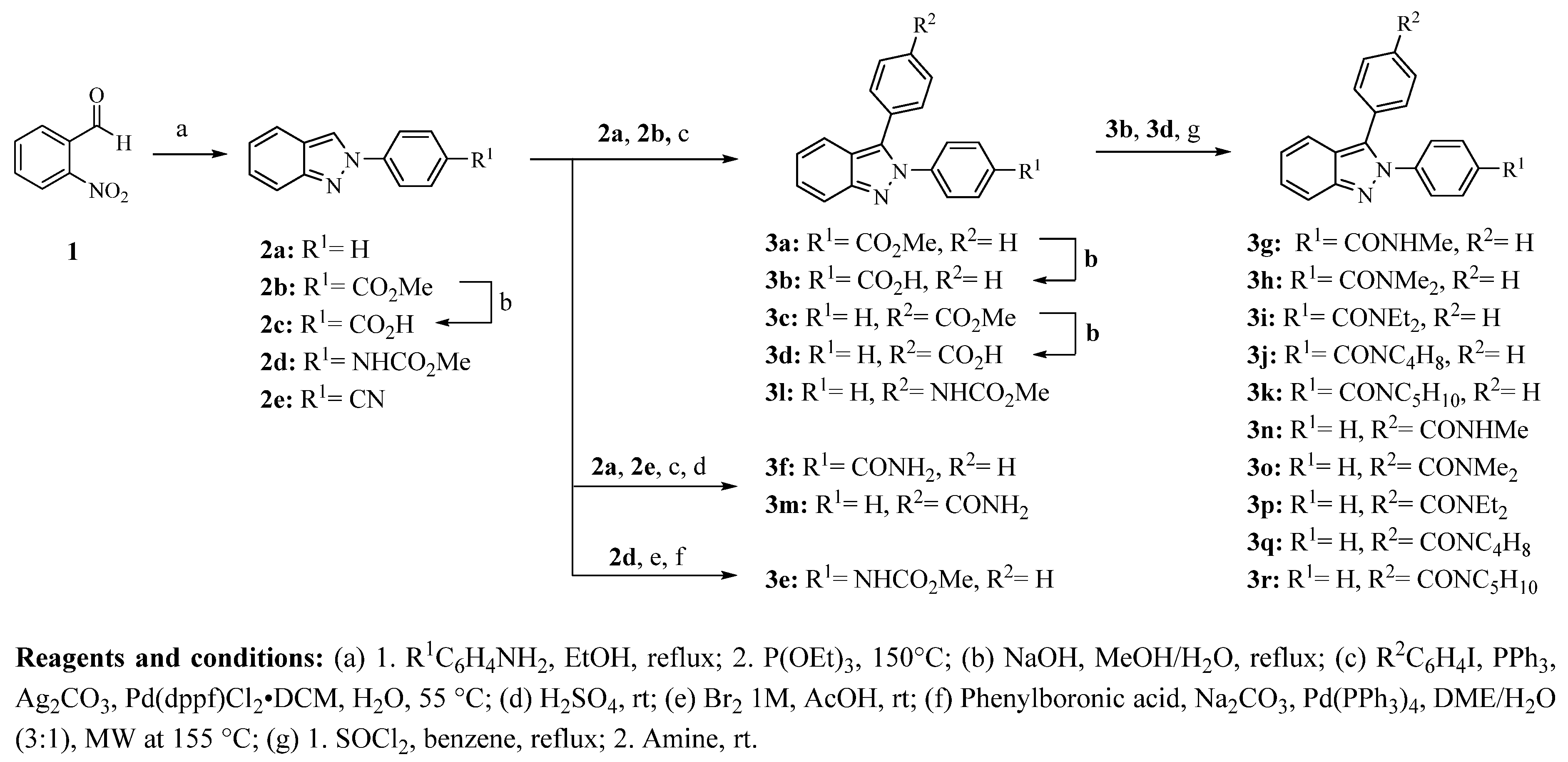
3.2.1. 2-Phenyl-2H-Indazole Derivatives (2a, 2b, 2d, and 2e)
2-Phenyl-2
H-indazole derivatives were synthesized employing a slight modification of the Cadogan method [
17,
18]. A mixture of 2-nitrobenzaldehyde (200 mg, 1.32 mmol) and aniline or the corresponding substituted aniline (1.32 mmol) was dissolved in ethanol (10 mL) and heated at reflux for 2 h. Then, the solvent was removed under vacuum to give the appropriate 1-(2-nitrophenyl)-
N-phenylmethanimine. This same reaction was carried out at room temperature to obtain the methyl 4-((2-nitrobenzylidene)amino)benzoate. Later, triethyl phosphite (3.96 mmol) was added and heated at 150 °C employing an oil bath for 2 h under N
2 atmosphere, until the starting material was consumed. After cooling, the mixture was treated with 20 mL of 5% hydrogen peroxide solution, and the product was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were dried over Na
2SO
4 and concentrated in vacuo. The crude product was purified by column chromatography using hexane/ethyl acetate (80:20). Only compound
2b was isolated by vacuum filtration and washed with cold ethanol to give a pure compound.
2-Phenyl-2
H-indazole (
2a). White solid (60% yield); m.p.: 81.2–81.6 °C (lit. [
18]: 81–82 °C); the spectroscopic data matched previously reported data [
17,
24]:
1H NMR (600 MHz, CDCl
3) δ 8.40 (d,
J = 0.9 Hz, 1H), 7.91–7.88 (m, 2H), 7.79 (dd,
J = 8.8, 0.9 Hz, 1H), 7.70 (dt,
J = 8.5, 1.0 Hz, 1H), 7.54–7.50 (m, 2H), 7.41–7.37 (m, 1H), 7.32 (ddd,
J = 8.8, 6.6, 1.0 Hz, 1H), 7.11 (ddd,
J = 8.4, 6.6, 0.7 Hz, 1H);
13C NMR (151 MHz, CDCl
3): δ 149.78, 140.52, 129.54, 127.88, 126.81, 122.76, 122.44, 120.99, 120.39, 120.37, 117.94.
Methyl 4-(2
H-indazol-2-yl) benzoate (
2b). After completion of the reaction, the product was filtrated and washed with cold MeOH. White solid (38% yield); m.p.: 185.8–186.2 °C (lit. [
25]: 186–187 °C); the spectroscopic data matched previously reported data [
17]:
1H NMR (600 MHz, CDCl
3) δ 8.47 (d,
J = 0.7 Hz, 1H), 8.22–8.18 (m, 2H), 8.02–7.99 (m, 2H), 7.77 (dd,
J = 8.8, 0.8 Hz, 1H), 7.69 (d,
J = 8.5 Hz, 1H), 7.33 (ddd,
J = 8.8, 6.6, 1.0 Hz, 1H), 7.14–7.10 (m, 1H), 3.95 (s, 3H);
13C NMR (151 MHz, CDCl
3) δ 166.19, 150.19, 143.64, 131.16, 129.27, 127.45, 123.01, 122.98, 120.47, 120.26, 118.06, 52.33.
Methyl (4-(2H-indazol-2-yl)phenyl)carbamate (2d). Pale orange solid (17% yield); m.p.: 163.7–165.0 °C; 1H NMR (600 MHz, CDCl3) δ 8.46 (bs, 1H), 8.38 (d, J = 0.9 Hz, 1H), 7.83–7.79 (m, 2H), 7.76 (dd, J = 8.8, 0.9 Hz, 1H), 7.70 (d, J = 8.4 Hz, 1H), 7.64 (d, J = 7.6 Hz, 2H), 7.31 (ddd, J = 8.7, 6.6, 1.1 Hz, 1H), 7.10 (ddd, J = 8.4, 6.6, 0.8 Hz, 1H), 3.80 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 154.27, 149.48, 138.52, 135.46, 126.59, 122.62, 122.20, 121.44, 120.30, 120.25, 119.20, 117.62, 52.17; MS [M+H]+ m/z 268.12.
4-(2
H-indazol-2-yl)benzonitrile (
2e). Yellow solid (19% yield); m.p.; 162.0–163.5 °C; (lit. [
24]: 163.4–164.6 °C); the spectroscopic data matched previously reported data [
24]:
1H NMR (600 MHz, CDCl
3) δ 8.47 (d,
J = 0.9 Hz, 1H), 8.10–8.05 (m, 2H), 7.85–7.80 (m, 2H), 7.76 (dd,
J = 8.8, 0.9 Hz, 1H), 7.70 (dt,
J = 8.5, 1.0 Hz, 1H), 7.35 (ddd,
J = 8.8, 6.6, 1.1 Hz, 1H), 7.14 (ddd,
J = 8.5, 6.6, 0.8 Hz, 1H);
13C NMR (151 MHz, CDCl
3) δ 150.37, 143.33, 133.69, 127.87, 123.36, 123.14, 120.85, 120.49, 120.37, 118.13, 118.08, 111.18; MS [M + H]
+ m/
z 220.11.
3.2.2. 4-(2H-Indazol-2-yl) Benzoic Acid (2c)
Methyl 4-(2H-indazol-2-yl) benzoate (300 mg, 1.26 mmol) was dissolved in methanol (10 mL) and an aqueous solution of NaOH (3.77 mmol in 5 mL of water) was added. The reaction mixture was heated under reflux for 5 h. After completion, the reaction mixture was cooled on an ice bath and acidified to pH 1 with HCl to induce precipitation. The solid was filtered under vacuum and dried.
4-(2
H-Indazol-2-yl) benzoic acid (
2c). White solid (96% yield); m.p.: 288.3–288.5 °C (lit. [
25]: 286–288 °C); the spectroscopic data matched previously reported data [
17]:
1H NMR (600 MHz, DMSO-
d6) δ 13.14 (bs, 1H), 9.22 (s, 1H), 8.26 (d,
J = 8.6 Hz, 2H), 8.15 (d,
J = 8.6 Hz, 2H), 7.79 (d,
J = 8.4 Hz, 1H), 7.74 (d,
J = 8.7 Hz, 1H), 7.41–7.30 (m, 1H), 7.19–7.08 (m, 1H);
13C NMR (151 MHz, DMSO-
d6) δ 166.47, 149.23, 142.84, 130.83, 129.65, 127.28, 122.55, 122.44, 122.03, 120.99, 119.86, 117.49.
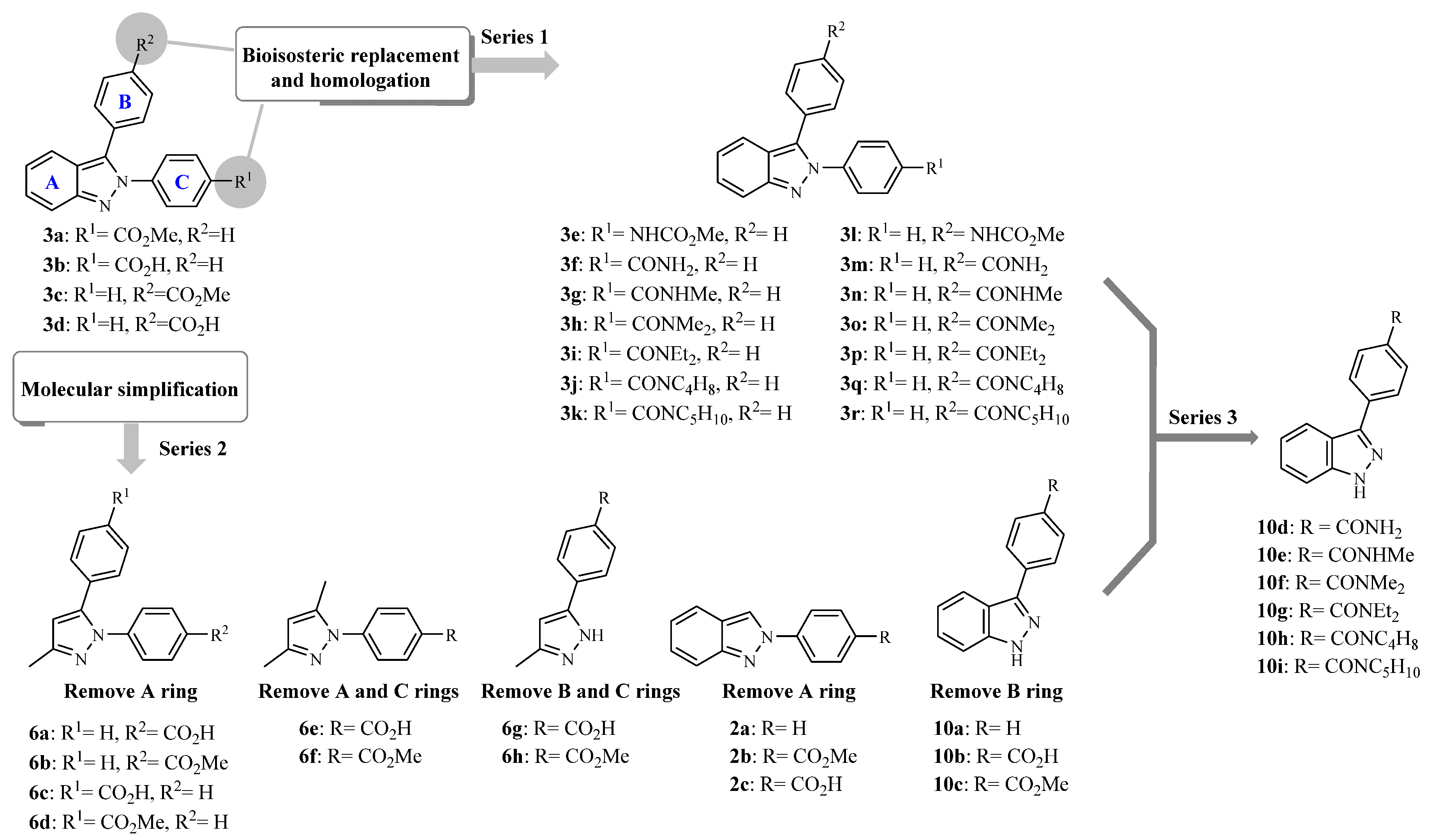
3.2.3. 2,3-Diphenyl-2H-Indazole Derivatives (3a, 3c, and 3l)
2,3-Diphenyl-2
H-indazole derivatives
3a,
3c, and
3l were synthesized by a palladium-catalyzed arylation previously described by Ohnmacht et al. [
19]. Compounds
3a and
3c were synthesized using the appropriate 2-phenyl-2
H-indazole and the substituted 4-iodobenzene, whereas compound
3l was synthesized from 2-phenyl-2
H-indazole and methyl(4-bromophenyl) carbamate.
Methyl 4-(3-phenyl-2
H-indazol-2-yl) benzoate (
3a). Pale yellow solid (40% yield); m.p.: 152.4
–154.9 °C (lit. [
17]: 152.4
–154.9 °C); the spectroscopic data matched previously reported data [
17]:
1H NMR (600 MHz, CDCl
3) δ 8.09–8.03 (m, 2H), 7.80 (d,
J = 8.8 Hz, 1H), 7.69 (d,
J = 8.5 Hz, 1H), 7.57–7.49 (m, 2H), 7.41 (dt,
J = 3.7, 1.2 Hz, 3H), 7.35 (dd,
J = 7.5, 2.0 Hz, 3H), 7.15 (ddd,
J = 8.5, 6.6, 0.8 Hz, 1H), 3.93 (s, 3H);
13C NMR (151 MHz, CDCl
3) δ 166.19, 149.33, 143.75, 135.68, 130.40, 129.68, 129.61, 128.96, 128.64, 127.44, 125.68, 122.88, 122.06, 120.52, 117.79, 52.32.
Methyl 4-(2-phenyl-2
H-indazol-3-yl) benzoate (
3c). Pale yellow solid (76% yield): m.p.: 164.5–166.3 °C (lit. [
17]: 164.5–166.3 °C) the spectroscopic data matched previously reported data [
17,
26]:
1H NMR (600 MHz, CDCl
3) δ 8.09–8.03 (m, 2H), 7.82 (d,
J = 8.8 Hz, 1H), 7.72 (dt,
J = 8.5, 0.9 Hz, 1H), 7.46–7.36 (m, 8H), 7.19 (ddd,
J = 8.5, 6.5, 0.6 Hz, 1H), 3.93 (s, 3H);
13C NMR (151 MHz, CDCl
3) δ 166.55, 149.08, 139.99, 134.37, 134.13, 129.97, 129.66, 129.49, 129.18, 128.59, 127.14, 126.04, 123.18, 121.90, 120.09, 118.02, 52.29.
Methyl (4-(2-phenyl-2H-indazol-3-yl)phenyl)carbamate (3l). Pale brown solid (26% yield): m.p.: 197.9–199.7 °C; 1H NMR (600 MHz, DMSO-d6) δ 9.85 (s, 1H), 7.70 (ddt, J = 24.0, 8.5, 0.9 Hz, 2H), 7.53 (d, J = 8.6 Hz, 2H), 7.50–7.41 (m, 5H), 7.37 (ddd, J = 8.7, 6.6, 1.1 Hz, 1H), 7.30–7.26 (m, 2H), 7.14 (ddd, J = 8.5, 6.5, 0.8 Hz, 1H), 3.68 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 154.36, 148.61, 140.36, 139.82, 135.37, 130.46, 129.52, 128.89, 127.25, 126.45, 123.41, 122.67, 121.46, 120.95, 118.66, 117.78, 52.20; MS [M+H]+ m/z 344.1381.
3.2.4. 2,3-Diphenyl-2H-Indazole Derivatives (3b and 3d)
Employing the hydrolysis method 3.2.2 described above, compounds 3b and 3d were prepared from their esters 3a or 3c, respectively.
4-(3-Phenyl-2
H-indazol-2-yl) benzoic acid (
3b). White solid (70% yield); m.p.: 129.2–130.1 °C (lit. [
17]: 129.2–130.1 °C); the spectroscopic data matched previously reported data [
17]:
1H NMR (600 MHz, DMSO-
d6) δ 8.01 (d,
J = 8.6 Hz, 2H), 7.77 (d,
J = 8.8 Hz, 1H), 7.69 (d,
J = 8.5 Hz, 1H), 7.57 (d,
J = 8.6 Hz, 2H), 7.44 (dddd,
J = 11.9, 7.6, 5.4, 3.9 Hz, 6H), 7.21–7.15 (m, 1H);
13C NMR (151 MHz, DMSO-
d6) δ 166.49, 148.52, 143.08, 135.26, 130.53, 130.15, 129.52, 129.03, 128.95, 128.71, 127.26, 125.99, 122.81, 121.38, 120.40, 117.49.
4-(2-Phenyl-2
H-indazol-3-yl) benzoic acid (
3d). White solid (88% yield); mp: 296.2–298.2 °C (lit. [
17]: 296.2–298.2 °C); the spectroscopic data matched previously reported data [
17]:
1H NMR (600 MHz, DMSO-
d6) δ δ 7.92 (d,
J = 8.2 Hz, 2H), 7.75 (d,
J = 8.7 Hz, 1H), 7.71 (d,
J = 8.5 Hz, 1H), 7.50–7.41 (m, 5H), 7.38 (ddd,
J = 8.7, 6.6, 0.9 Hz, 1H), 7.28 (d,
J = 8.3 Hz, 2H), 7.19–7.13 (m, 1H);
13C NMR (151 MHz, DMSO-
d6) δ 169.13, 148.11, 140.15, 139.79, 135.10, 129.32, 129.17, 128.98, 128.37, 128.26, 126.77, 125.88, 122.34, 121.02, 120.38, 117.29.
3.2.5. 2,3-Diphenyl-2H-Indazole Derivatives (3f and 3m)
The proper 2-phenyl-2
H-indazole (
2a or
2e) and 4-bromobenzonitrile or 4-iodobenzene were reacted employing the previously described method by Ohnmacht et al. [
19] to give the benzonitrile derivative. Then, the intermediate was dissolved and stirred in H
2SO
4 (1 mL) at room temperature overnight. After completion, the mixture was poured into ice-water (15 mL) to induce precipitation. The solid was filtered under vacuum and dried.
4-(3-Phenyl-2H-indazol-2-yl)benzamide (3f). White solid (57% yield): m.p.: 221.6–222.8 °C; 1H NMR (600 MHz, DMSO-d6) δ 8.09 (s, 1H), 7.97–7.92 (m, 2H), 7.76 (dt, J = 8.8, 0.9 Hz, 1H), 7.68 (dt, J = 8.5, 1.0 Hz, 1H), 7.55–7.37 (m, 9H), 7.18 (ddd, J = 8.5, 6.6, 0.8 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 167.30, 148.85, 142.33, 135.65, 134.40, 129.97, 129.50, 129.45, 129.09, 128.77, 127.59, 126.14, 123.16, 121.78, 120.81, 117.91; MS [M+H]+ m/z 314.1294.
4-(2-Phenyl-2H-indazol-3-yl)benzamide (3m). Pale yellow solid (34% yield): m.p.: 230.6–232.7 °C; 1H NMR (600 MHz, DMSO-d6) δ 8.05 (s, 1H), 7.93 (d, J = 8.3 Hz, 2H), 7.77 (d, J = 8.7 Hz, 1H), 7.72 (d, J = 8.5 Hz, 1H), 7.47 (ddd, J = 15.3, 11.3, 7.9 Hz, 8H), 7.40 (dd, J = 11.7, 3.7 Hz, 1H), 7.20 (dd, J = 8.1, 6.9 Hz, 1H); 13C NMR (151 MHz, DMSO-d6) δ 167.63, 148.69, 140.16, 134.64, 134.28, 132.31, 129.70, 129.64, 129.16, 128.40, 127.41, 126.57, 123.30, 121.71, 120.68, 117.96; MS [M+H]+ m/z 314.1281.
3.2.6. Methyl (4-(3-Phenyl-2H-indazol-2-yl)phenyl) Carbamate (3e)
Methyl (4-(phenyl-2
H-indazole-2-yl)phenyl) carbamate
2d (500 mg, 1.88 mmol) was dissolved in acetic acid (5 mL). Bromine (1.8 mL of 1 M solution in acetic acid) was slowly added at 0–5 °C and then led to warm at room temperature and stirred overnight. After completion of the reaction, ice-water was added and the solid formed was filtered and dried under vacuum. The crude intermediate (0.5 mmol) was treated with phenylboronic acid (0.55 mmol), Na
2CO
3 (1.5 mmol), Pd(PPh
3)
4 (0.01 mmol) and 3 mL of DME/water (3:1). The mixture was heated under microwave irradiation at 155 °C for 30 min [
20]. After cooling, the solvent was removed under vacuum and the obtained product was purified by column chromatography using hexane/ethyl acetate (60:40).
Methyl (4-(3-phenyl-2H-indazol-2-yl)phenyl)carbamate (3e). White solid (58% yield): m.p.: 201.5–202.5 °C; 1H NMR (600 MHz, DMSO-d6) δ 9.91 (s, 1H), 7.73 (dt, J = 8.8, 0.9 Hz, 1H), 7.67 (dt, J = 8.5, 1.0 Hz, 1H), 7.53 (d, J = 8.9 Hz, 2H), 7.48–7.44 (m, 2H), 7.43–7.39 (m, 1H), 7.39–7.31 (m, 5H), 7.15 (ddd, J = 8.5, 6.6, 0.8 Hz, 1H), 3.69 (s, 3H); 13C NRM (151 MHz, DMSO-d6) δ 154.39, 148.48, 139.76, 135.26, 134.59, 129.87, 129.71, 129.32, 128.83, 127.16, 127.02, 122.81, 121.45, 120.71, 118.52, 117.77, 52.25; MS [M+H]+ m/z 344.1391.
3.2.7. 2,3-Diphenyl-2H-Indazole Carboxamides (3g–k and 3n–r)
Method A: To a solution of carboxylic acids 3b or 3d (250 mg, 0.8 mmol) in benzene (5 mL), SOCl2 (0.35 mL, 4.8 mmol) was added. The mixture was heated at 70 °C for 4 h. After completion, the excess of SOCl2 was distilled-off at reduced pressure (3 × 5 mL of benzene) to give the acyl chloride intermediate and then stirred with an excess of the adequate amine (16 mmol) at room temperature for 30 min. The mixture was poured in methanol and heated until solids were dissolved. Water was added to induces the precipitation of the compound. The formed solid was separated by vacuum filtration. Finally, the crude product was purified by column chromatography using hexane/ethyl acetate (80:20).
N,N-Diethyl-4-(3-phenyl-2H-indazol-2-yl)benzamide (3i). Pale yellow solid (80% yield); m.p.: 136.5–138.0 °C. 1H NMR (600 MHz, CDCl3) δ 7.79 (d, J = 8.8 Hz, 1H), 7.71 (d, J = 8.5 Hz, 1H), 7.52–7.48 (m, 2H), 7.48–7.35 (m, 8H), 7.15 (ddd, J = 8.3, 6.5, 0.5 Hz, 1H), 3.55 (s, 2H), 3.24 (s, 2H), 1.25 (s, 3H), 1.09 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 170.25, 149.18, 140.78, 137.00, 135.61, 129.71, 129.57, 128.99, 128.57, 127.29, 127.21, 125.93, 122.73, 121.91, 120.59, 117.72, 43.35, 39.47, 14.21, 12.90; MS [M+H]+ m/z 370.23.
(4-(3-Phenyl-2H-indazol-2-yl)phenyl)(pyrrolidin-1-yl)methanone (3j). Pale brown solid (40% yield); m.p.: 186.1–186.2 °C; 1H NMR (600 MHz, CDCl3) δ 7.79 (d, J = 8.8 Hz, 1H), 7.71 (d, J = 8.5 Hz, 1H), 7.61–7.52 (m, 2H), 7.52–7.45 (m, 2H), 7.45–7.34 (m, 6H), 7.18–7.12 (m, 1H), 3.65 (t, J = 7.0 Hz, 2H), 3.40 (t, J = 6.6 Hz, 2H), 2.00–1.93 (m, 2H), 1.92–1.85 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 168.60, 149.15, 141.22, 136.84, 135.56, 129.66, 129.56, 128.95, 128.54, 127.97, 127.26, 125.70, 122.71, 121.90, 120.54, 117.70, 49.56, 46.29, 26.40, 24.41; MS [M+H]+ m/z 368.24.
(4-(3-Phenyl-2H-indazol-2-yl)phenyl)(piperidin-1-yl)methanone (3k). White solid (73% yield); m.p.: 136.0–138.0 °C; 1H NMR (600 MHz, CDCl3) δ 7.81 (d, J = 8.8 Hz, 1H), 7.73 (d, J = 8.5 Hz, 1H), 7.48–7.34 (m, 10H), 7.20–7.14 (m, 1H), 3.71 (bs, 2H), 3.38 (bs, 2H), 1.85–1.57 (bs, 6H); 13C NMR (151 MHz, CDCl3) 169.56, 149.00, 140.02, 136.12, 134.41, 131.01, 129.56, 129.13, 128.45, 127.41, 127.04, 126.02, 122.85, 121.81, 120.22, 117.89, 48.77, 43.20, 26.59, 25.54, 24.54; MS [M+H]+ m/z 382.26.
N,N-Diethyl-4-(2-phenyl-2H-indazol-3-yl)benzamide (3p). White solid (88 % yield); m.p.: 148.4–149.6 °C;1H NMR (600 MHz, CDCl3) δ 7.81 (d, J = 8.8 Hz, 1H), 7.73 (d, J = 8.5 Hz, 1H), 7.49–7.36 (m, 10H), 7.19–7.14 (m, 1H), 3.56 (bs, 2H), 3.29 (bs, 2H), 1.26 (bs, 3H), 1.14 (bs, 3H); 13C NMR (151 MHz, CDCl3) δ 170.57, 149.03, 140.06, 136.93, 134.47, 130.79, 129.59, 129.13, 128.47, 127.06, 126.92, 126.05, 122.86, 121.80, 120.24, 117.92, 43.39, 39.41, 14.25, 12.89; MS [M+H]+ m/z 370.26.
(4-(2-Phenyl-2H-indazol-3-yl)phenyl)(pyrrolidin-1-yl)methanone (3q). White solid (37% yield); m.p.: 167.0–168.8 °C; 1H NMR (600 MHz, CDCl3) δ 7.81 (d, J = 8.8 Hz, 1H), 7.72 (dt, J = 8.5, 0.8 Hz, 1H), 7.56 (d, J = 8.3 Hz, 2H), 7.46–7.36 (m, 8H), 7.17 (ddd, J = 8.4, 6.5, 0.7 Hz, 1H), 3.66 (t, J = 7.0 Hz, 2H), 3.46 (t, J = 6.6 Hz, 2H), 2.01–1.95 (m, 2H), 1.94–1.88 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 168.95, 149.03, 140.04, 136.82, 134.46, 131.39, 129.44, 129.15, 128.49, 127.68, 127.09, 126.04, 122.90, 121.84, 120.23, 117.91, 49.60, 46.33, 26.46, 24.43; MS [M+H]+ m/z 368.24.
(4-(2-Phenyl-2H-indazol-3-yl)phenyl)(piperidin-1-yl)methanone (3r). White solid (90% yiled); m.p.: 198.2–199.2 °C; 1H NMR (600 MHz, CDCl3) δ 7.81 (d, J = 8.8 Hz, 1H), 7.73 (d, J = 8.5 Hz, 1H), 7.55–7.28 (m, 10H), 7.20–7.14 (m, 1H), 3.72 (bs, 2H), 3.38 (bs, 2H), 1.69 (bs, 6H).13C NMR (151 MHz, CDCl3) δ 169.56, 149.00, 140.02, 136.12, 134.41, 131.01, 129.56, 129.13, 128.45, 127.41, 127.04, 126.02, 122.85, 121.81, 120.22, 117.89, 48.77, 43.22, 26.57, 25.59, 24.54; MS [M+H]+ m/z 382.26.
Method B: The acyl chlorides were synthesized employing the same procedure as described in method A. Next, the crude acyl chloride was mixed with methylamine hydrochloride or N,N-dimethylamine hydrochloride (1 mmol) and dissolved in anhydrous CH2Cl2 (3 mL). Then, triethylamine (1.0 mmol) was slowly added to the mixture and stirred at room temperature for 2 h. After completion, the mixture was concentrated in vacuo. The product was purified by column chromatography using hexane/ethyl acetate (70:30).
N-Methyl-4-(3-phenyl-2H-indazol-2-yl)benzamide (3g). Pale brown solid (43% yield); m.p.:194.3–195.4 °C. 1H NMR (600 MHz, CDCl3) δ 7.78 (dt, J = 8.9, 0.9 Hz, 1H), 7.77–7.74 (m, 2H), 7.70 (dt, J = 8.5, 0.9 Hz, 1H), 7.53–7.43 (m, 2H), 7.43–7.30 (m, 6H), 7.15 (ddd, J = 8.5, 6.5, 0.8 Hz, 1H), 6.38 (d, J = 4.3 Hz, 1H), 3.01 (d, J = 4.8 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ 167.20, 149.22, 142.42, 135.66, 134.18, 129.65, 129.53, 128.94, 128.60, 127.68, 127.40, 125.88, 122.81, 121.95, 120.53, 117.66, 26.89; MS [M+H]+ m/z 328.1439.
N,N-Dimethyl-4-(3-phenyl-2H-indazol-2-yl)benzamide (3h). Pale brown solid (70% yield); m.p.:153.0–154.5 °C. 1H NMR (600 MHz, DMSO-d6) δ 7.75 (d, J = 8.7 Hz, 1H), 7.68 (d, J = 8.5 Hz, 1H), 7.53–7.35 (m, 10H), 7.20–7.14 (m, 1H), 2.99 (s, 3H), 2.91 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 169.52, 148.78, 140.70, 136.78, 135.56, 129.95, 129.48, 129.43, 129.08, 128.23, 127.53, 126.32, 123.12, 121.67, 120.82, 117.89, 35.18; MS [M+H]+ m/z 342.24.
N-Methyl-4-(2-phenyl-2H-indazol-3-yl)benzamide (3n). Pale brown solid (62% yield); m.p.: 235.7–236.6 °C; 1H NMR (600 MHz, DMSO-d6) δ 8.52 (d, J = 4.5 Hz, 1H), 7.89–7.86 (m, 2H), 7.79–7.75 (m, 1H), 7.72 (dd, J = 8.5, 1.0 Hz, 1H), 7.51–7.44 (m, 7H), 7.40 (ddd, J = 8.6, 6.6, 0.9 Hz, 1H), 7.23–7.17 (m, 1H), 2.79 (d, J = 4.6 Hz, 3H); 13C NMR (151 MHz, DMSO-d6) 166.40, 148.69, 140.16, 134.62. 134.54, 132.10, 129.76, 129.63, 129.14, 127.98, 127.41, 126.56, 123.29, 121.69, 120.67, 117.97, 26.72; MS [M+H]+ m/z 328.1442.
N,N-Dimethyl-4-(2-phenyl-2H-indazol-3-yl)benzamide (3o). White solid (82% yield); m.p.: 166.2–168.0 °C; 1H NMR (600 MHz, DMSO-d6) 7.76 (dt, J = 8.7, 0.7 Hz, 1H), 7.72 (dt, J = 8.5, 0.8 Hz, 1H), 7.51–7.44 (m, 7H), 7.43–7.38 (m, 3H), 7.19 (ddd, J = 8.5, 6.6, 0.7 Hz, 1H), 2.99 (s, 3H), 2.92 (s, 3H); 13C NMR (151 MHz, DMSO-d6) 174.63, 153.43, 144.91, 141.38, 139.46, 135.24, 134.48, 134.37, 133.89, 132.67, 132.15, 131.28, 127.94, 126.42, 125.46, 122.70, 39.94; MS [M+H]+ m/z 342.21.
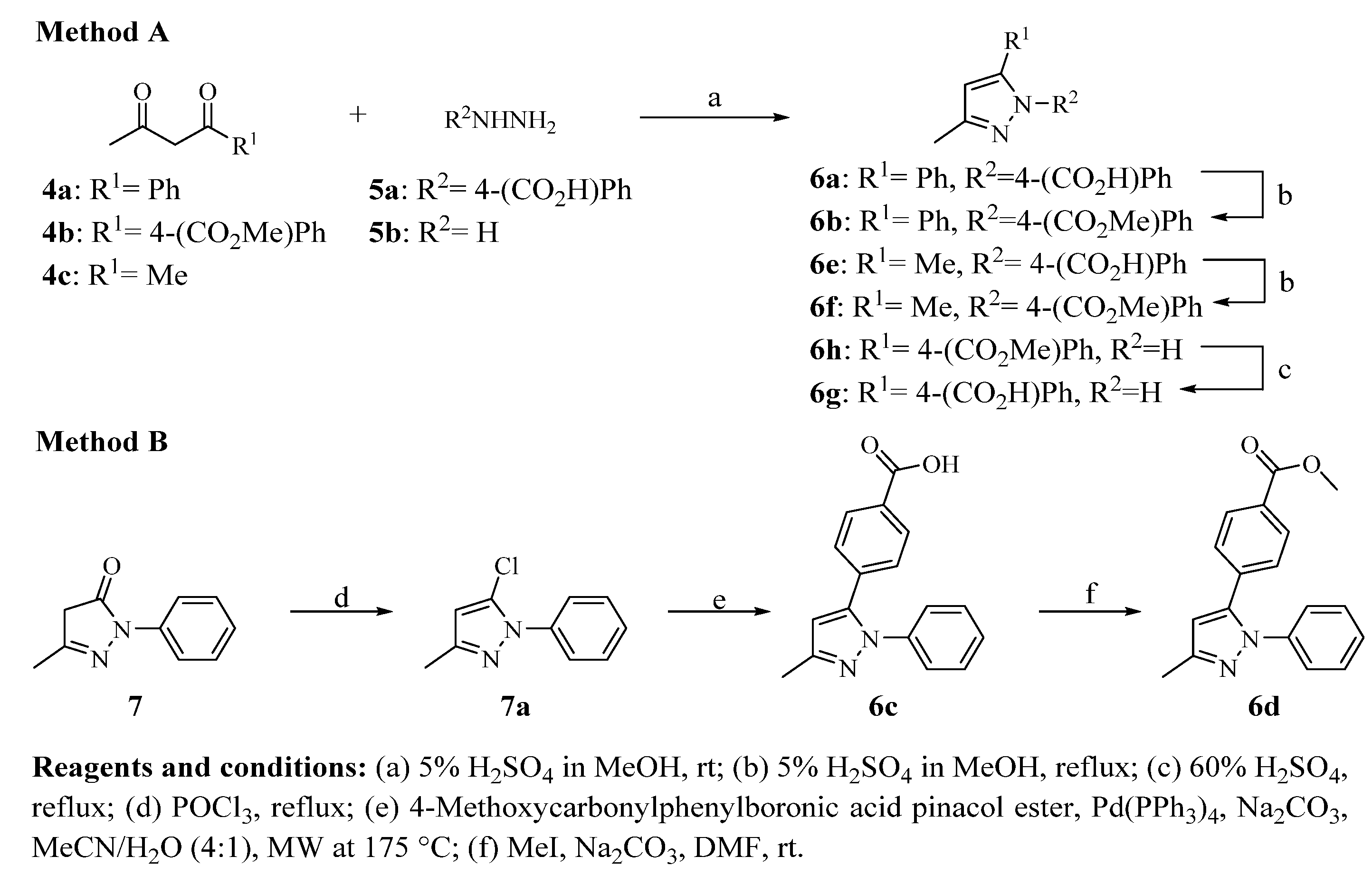
3.2.8. Synthesis of Pyrazole Derivatives (6a, 6e, and 6h)
To a solution of 1,3-dicarbonyl compound (2 mmol) in 5 mL of a 5% methanolic solution of H
2SO
4, hydrazine hydrate or 4-hydrazinylbenzoic acid (2 mmol) was added. The reaction mixture was stirred at room temperature for 2–5 h and then cooled on an ice bath. To induce a complete precipitation, water (2–5 mL) was added. The resulting solid was separated in vacuo and dried [
21].
4-(3-Methyl-5-phenyl-1
H-pyrazol-1-yl)benzoic acid (
6a). White solid (91% yield); m.p.: 126.3–128.3 °C; the spectroscopic data matched previously reported data [
27]
1H NMR (600 MHz, CDCl
3) δ 8.06–8.01 (m, 2H), 7.40–7.36 (m, 2H), 7.36–7.31 (m, 3H), 7.25–7.20 (m, 2H), 6.34 (s, 1H), 3.49 (s, 1H), 2.41 (s, 3H);
13C NMR (151 MHz CDCl
3) δ 170.56, 150.50, 144.12, 144.10, 130.96, 130.43, 128.73, 128.66, 128.56, 127.58, 124.29, 109.03, 13.53; MS [M+H]
+ m/
z 279.17.
4-(3,5-Dimethyl-1
H-pyrazol-1-yl)benzoic acid (
6e). White solid (90% yield); m.p.: 160.7–161.8 °C; the spectroscopic data matched previously reported data [
28]:
1H NMR (600 MHz, CDCl
3) 8.19 (d,
J = 8.7 Hz, 2H), 7.59 (d,
J = 8.7 Hz, 2H), 6.05 (s, 1H), 2.39 (d,
J = 0.6 Hz, 3H), 2.33 (s, 3H); RMN
13C (151 MHz, CDCl
3) δ 170.33, 150.06, 143.94, 139.73, 131.15, 127.74; MS [M+H]
+ m/
z 217.10.
Methyl 4-(3-methyl-1H-pyrazol-5-yl)benzoate (6h). Yellow solid (82% yield); m.p.: 188.0–191 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.80 (bs, 1H), 7.97 (d, J = 8.5 Hz, 2H), 7.90 (d, J = 8.4 Hz, 2H), 6.56 (s, 1H), 3.86 (s, 3H), 2.27 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 166.48, 130.06, 128.42, 125.38, 125.09, 102.41, 52.47, 11.22; MS [M+H]+ m/z 217.10.
3.2.9. Pyrazole Derivatives (6b and 6f)
A solution of carboxylic acid (6a or 6e, 1.1 mmol) in 5 mL of a 5% methanolic solution of H2SO4 was heated under reflux for 2 h. When the reaction was completed, the mixture was neutralized with a 10% aqueous solution of NaHCO3. The resulting solid was separated under vacuum filtration and dried.
Methyl 4-(3-methyl-5-phenyl-1H-pyrazol-1-yl)benzoate (6b). White solid (84% yield); m.p.: 100.2–101.6 °C; 1H NMR (600 MHz, CDCl3) δ 7.97 (d, J = 8.8 Hz, 2H), 7.37–7.33 (m, 2H), 7.33–7.29 (m, 3H), 7.24–7.19 (m, 2H), 6.33 (s, 1H), 3.90 (s, 3H), 2.39 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 166.38, 150.28, 143.95, 143.63, 130.51, 130.29, 128.67, 128.56, 128.41, 128.18, 124.19, 108.84, 52.14, 13.56; MS [M+H]+ m/z 293.19.
Methyl 4-(3,5-dimethyl-1H-pyrazol-1-yl)benzoate (6f). White solid (73% yield); m.p.: 60.2–61.2 °C; 1H NMR (600 MHz, CDCl3) 8.12 (d, J = 8.8 Hz, 2H), 7.55 (d, J = 8.8 Hz, 2H), 6.03 (s, 1H), 3.94 (s, 3H), 2.37 (d, J = 0.6 Hz, 3H), 2.30 (s, 3H); 13C NMR (151 MHz, CDCl3) 166.38, 149.87, 143.61, 139.53, 130.52, 128.25, 123.54, 108.16, 52.19, 13.48, 12.78; MS [M+H]+ m/z 231.13.
3.2.10. 4-(3-Methyl-1H-pyrazol-5-yl) Benzoic Acid (6g)
Methyl 4-(3-methyl-1H-pyrazol-5-yl) benzoate 6h (250 mg, 1.1 mmol) was dissolved in a 5 mL of a 60% aqueous solution of H2SO4 and heated under reflux for 2 h. After completion, the reaction was poured into 10 mL of iced water. The resulting solid was filtered in vacuo, washed with cold water and dried.
4-(3-Methyl-1H-pyrazol-5-yl)benzoic acid (6g). Yellow solid (95% yield); m.p.: 285–286 °C; 1H NMR (600 MHz, DMSO-d6) δ 7.96 (d, J = 8.5 Hz, 2H), 7.88 (d, J = 8.5 Hz, 2H), 6.55 (d, J = 0.7 Hz, 1H), 2.29–2.26 (m, 3H); 13C NMR (151 MHz, DMSO-d6) δ 167.55, 148.45, 141.87, 137.76, 130.20, 129.65, 125.27, 102.40, 11.40; MS [M+H]+ m/z 203.13.
3.2.11. 4-(3-Methyl-1-phenyl-1H-pyrazol-5-yl) Benzoic Acid (6c)
5-Methyl-2-phenyl-2,4-dihydro-3H-pyrazol-3-one (1.0 g, 5.74 mmol) was dissolved in POCl3 (1.5 mL) and heated under reflux for 5 h. The mixture was cooled on an ice bath and quenched with 10 mL of water. The product was extracted with ethyl acetate (3 × 10 mL), the combined organic layers were dried over Na2SO4 and concentrated in vacuo. The resulting oily product (200 mg, 1.0 mmol) was mixed with 4-carbomethoxyphenylboronic acid pinacol ester (300 mg, 1.1 mmol), Pd(PPh3)4 (116 mg, 0.1 mmol), Na2CO3 (424 mg, 4 mmol) and 5 mL of MeCN/H2O (4:1). The reaction was heated under microwave irradiation at 175 °C for 20 min twice. Then, the solvent was removed in vacuo. The resulting crude product was purified by column chromatography using hexane/ethyl acetate (80:20).
4-(3-Methyl-1-phenyl-1H-pyrazol-5-yl)benzoic acid (6c). Pale brown solid (55% yield); m.p.: 221.0–223.3 °C; 1H NMR (600 MHz, CDCl3) δ 8.05–7.97 (m, 2H), 7.37–7.32 (m, 2H), 7.32–7.28 (m, 3H), 7.27 (dd, J = 8.4, 1.4 Hz, 2H), 6.41 (s, 1H), 2.41 (s, 3H).; 13C NMR (151 MHz, CDCl3) δ 170.85, 149.72, 142.48, 139.73, 135.69, 130.23, 129.04, 128.77, 128.47, 127.56, 125.24, 108.41, 13.46; MS [M+H]+ m/z 279.17.
3.2.12. Methyl 4-(3-Methyl-1-phenyl-1H-pyrazol-5-yl) Benzoate (6d)
A mixture of 4-(3-methyl-1-phenyl-1H-pyrazol-5-yl) benzoic acid 6c (200 mg, 0.7 mmol) and Na2CO3 (75 mg, 0.7 mmol), DMF (2 mL) and water (0.5 mL) was stirred for 15 min at room temperature. Afterward, methyl iodide (0.7 mmol) was added to the mixture and stirred at room temperature for 1 h. Then, the reaction was poured into water (10 mL) and extracted with ethyl acetate (3 × 5 mL). The combined organic layers were dried over Na2SO4 and concentrated under vacuum. The crude product was purified by recrystallization from ethanol.
Methyl 4-(3-methyl-1-phenyl-1
H-pyrazol-5-yl)benzoate (
6d). White solid (82% yield); 92.0–94.3 °C; the spectroscopic data matched previously reported data [
29]
1H NMR (600 MHz, CDCl
3) 7.95 (d,
J = 8.6 Hz, 2H), 7.36–7.31 (m, 2H), 7.31–7.23 (m, 5H), 6.39 (s, 1H), 3.91 (s, 3H), 2.39 (s, 3H);
13C NMR (151 MHz, CDCl
3) 166.57, 149.64, 142.54, 139.87, 135.02, 129.63, 129.47, 128.99, 128.40, 127.42, 125.17, 108.28, 52.18, 13.52; MS [M+H]
+ m/
z 293.15.
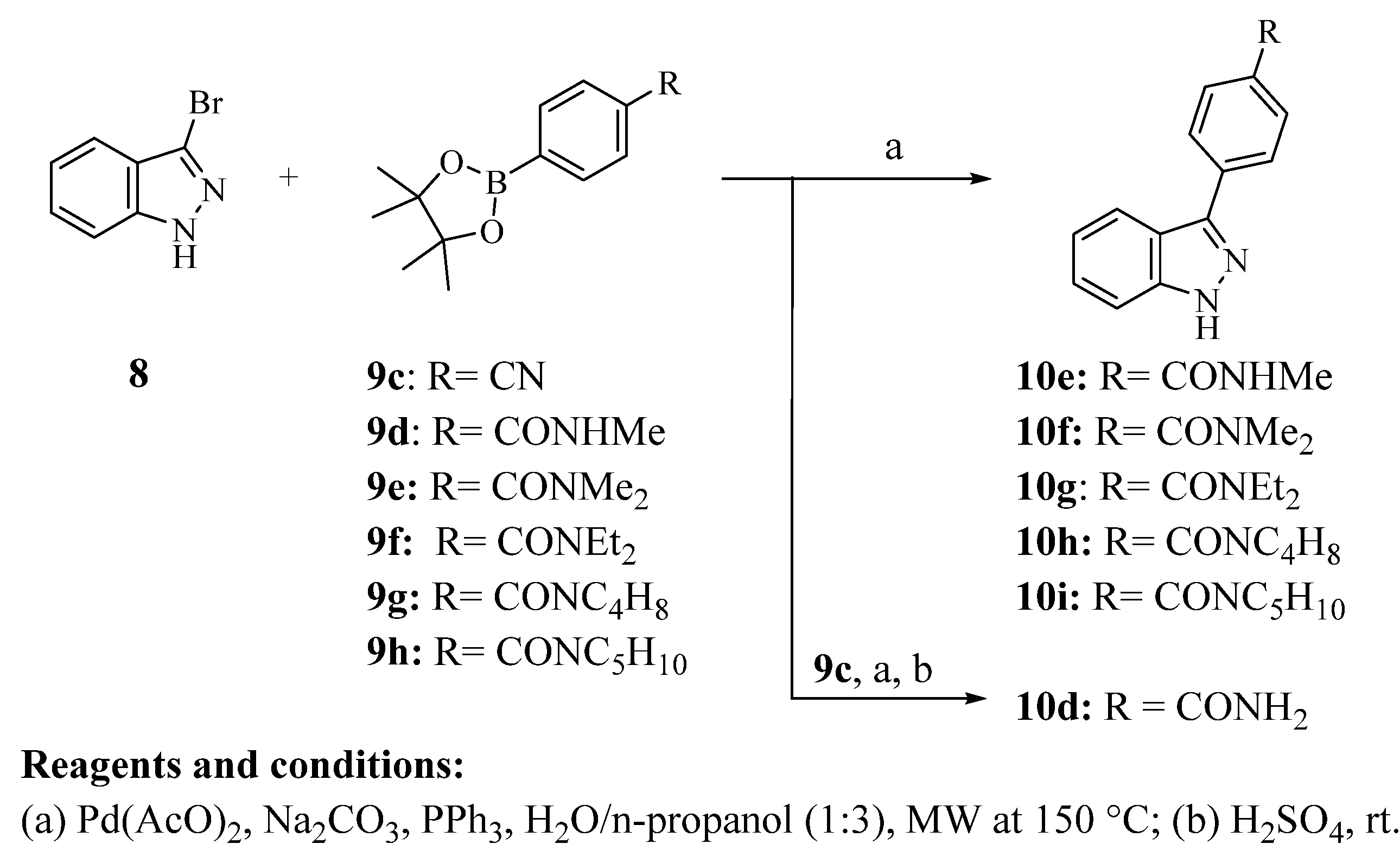
3.2.13. Synthesis of 3-Phenyl-1H-Indazoles (10a, 10b, and 10d–i)
A mixture of 3-bromo-1H-indazole (200 mg, 1.01 mmol), phenylboronic acid or appropriate aryl boronic acid pinacol ester (1.11 mmol), Na2CO3 (128 mg, 1.21 mmol), PPh3 (8 mg, 0.03 mmol), Pd(OAc)2 (2.2 mg, 0.01 mmol) and 5 mL of n-propanol/H2O (3:1) was heated under microwave irradiation at 150 °C for 20 min. The solvent was removed in vacuo and the resulting residue was purified by column chromatography using hexane/ethyl acetate (50:50).
3-Phenyl-1
H-indazole (
10a). The product was purified using column chromatography and hexane/ethyl acetate (80:20) as mobile phase. White solid (80% yield). m.p.: 105.2–106.8 °C [lit. [
30]: 106–107 °C]; the spectroscopic data matched previously reported data [
31,
32]:
1H NMR (600 MHz, CDCl
3) δ 11.44 (bs, 1H), 8.05–7.99 (m, 3H), 7.56–7.51 (m, 2H), 7.47–7.42 (m, 1H), 7.37–7.33 (m, 1H), 7.29–7.25 (m, 1H), 7.23–7.19 (m, 1H);
13C NMR (151 MHz, CDCl
3) δ 145.74, 141.68, 133.56, 128.92, 128.16, 127.71, 126.77, 121.33, 121.08, 120.95, 110.21; Purity (qNMR, %
w/
w): 98.89 ± 2.29.
4-(1
H-Indazol-3-yl)benzoic acid (
10b). White solid (82% yield); m.p.: 283.5–286 °C; the spectroscopic data matched previously reported data [
32]:
1H NMR (600 MHz, DMSO-
d6) δ 13.52 (bs, 1H), 13.05 (bs, 1H), 8.19–8.14 (m, 3H), 8.12–8.08 (m, 2H), 7.64 (d,
J = 8.4 Hz, 1H), 7.48–7.42 (m, 1H), 7.26 (ddd,
J = 7.8, 6.8, 0.8 Hz, 1H);
13C NMR (151 MHz, DMSO-
d6) δ 172.33, 148.26, 147.20, 146.82, 143.12, 135.17, 134.68, 131.70, 131.48, 126.67, 125.81, 125.76, 125.28, 116.01; Purity (qNMR, %
w/
w): 96.48 ± 0.42.
4-(1H-Indazol-3-yl)benzamide (10d). Pale brown solid (45% yield); m.p.: 253.0–255.5 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.43 (s, 1H), 8.14 (d, J = 8.3 Hz, 1H), 8.12–8.09 (m, 3H), 8.04 (d, J = 8.5 Hz, 2H), 7.63 (d, J = 8.4 Hz, 1H), 7.49–7.41 (m, 2H), 7.25 (ddd, J = 7.9, 6.9, 0.7 Hz, 1H); 13C NMR (151 MHz, DMSO-d6) δ 168.04, 142.74, 142.03, 136.86, 133.54, 128.59, 126.71, 126.68, 121.80, 121.08, 120.52, 111.16; MS [M+H]+ m/z 238.14; Purity (qNMR, % w/w): 95.23±0.71.
4-(1H-Indazol-3-yl)-N-methylbenzamide (10e). White solid (34% yield); m.p.: 239.5–241.0 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.41 (s, 1H), 8.55 (d, J = 4.5 Hz, 1H), 8.14 (d, J = 8.2 Hz, 1H), 8.10 (d, J = 8.4 Hz, 2H), 7.99 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 8.4 Hz, 1H), 7.47–7.41 (m, 1H), 7.28–7.22 (m, 1H), 3.38 (s, 3H); 13C NMR (151 MHz DMSO-d6) δ 166.17, 142.17, 141.47, 136.12, 133.22, 127.59, 126.22, 126.13, 121.23, 120.54, 119.96, 110.60, 26.19; MS [M+H]+ m/z 252.17; Purity (qNMR, % w/w): 96.59 ± 0.80.
4-(1
H-Indazol-3-yl)-
N,N-dimethylbenzamide (
10f). White solid (48% yield); m.p.: 160.5–163.0 °C; the spectroscopic data matched previously reported data [
12]:
1H NMR (600 MHz, DMSO-
d6) δ 13.38 (s, 1H), 8.12 (d,
J = 8.3 Hz, 1H), 8.07 (d,
J = 8.3 Hz, 2H), 7.62 (d,
J = 8.4 Hz, 1H), 7.56 (d,
J = 8.3 Hz, 2H), 7.46–7.40 (m, 1H), 7.24 (ddd,
J = 7.9, 6.8, 0.8 Hz, 1H), 3.01 (d,
J = 16.2 Hz, 6H);
13C NMR (151 MHz, DMSO-
d6) δ 169.76, 142.30, 141.47, 135.30, 134.58, 127.60, 126.27, 126.12, 121.16, 120.47, 119.92, 110.60, 34.71; MS [M+H]
+ m/
z: 266.20; Purity (qNMR, %
w/
w): 97.56 ± 1.07.
N,N-Diethyl-4-(1H-indazol-3-yl)benzamide (10g). White solid (53% yield); m.p.: 259.3–261.5 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.37 (s, 1H), 8.12 (d, J = 8.2 Hz, 1H), 8.07 (d, J = 8.3 Hz, 2H), 7.62 (d, J = 8.4 Hz, 1H), 7.50 (d, J = 8.3 Hz, 2H), 7.46–7.40 (m, 1H), 7.27–7.21 (m, 1H), 3.46 (s, 2H), 3.27 (s, 2H), 1.16 (bs,3H), 1.10 (bs, 3H); 13C NMR (151 MHz, DMSO-d6) δ 169.62, 142.31, 141.47, 136.17, 134.25, 126.71, 126.42, 126.11, 121.14, 120.47, 119.90, 110.59, 42.78, 13.98, 12.74; MS [M+H]+ m/z 294.23; Purity (qNMR, % w/w): 96.68 ± 0.33.
(4-(1H-Indazol-3-yl)phenyl)(pyrrolidin-1-yl)methanone (10h). White solid (21% yield); m.p.: 201.0–202.3 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.39 (s, 1H), 8.12 (d, J = 8.3 Hz, 1H), 8.07 (dd, J = 6.6, 1.8 Hz, 2H), 7.70–7.66 (m, 2H), 7.62 (d, J = 8.4 Hz, 1H), 7.46–7.40 (m, 1H), 7.24 (ddd, J = 7.9, 6.9, 0.8 Hz, 1H), 3.52–3.46 (m, 4H), 1.91–1.81 (m, 4H); 13C NMR (151 MHz DMSO-d6) δ 167.79, 142.28, 141.47, 135.97, 135.04, 134.91, 127.70, 126.17, 126.11, 121.18, 120.46, 119.93, 110.60, 48.88, 45.91, 25.93, 23.82; MS [M+H]+ m/z 292.20; Purity (qNMR, % w/w): 99.12 ± 1.25.
(4-(1H-indazol-3-yl)phenyl)(piperidin-1-yl)methanone (10i). White solid (42% yield); m.p.: 193.5–195.5 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.38 (s, 1H), 8.12 (d, J = 8.2 Hz, 1H), 8.07 (d, J = 8.2 Hz, 2H), 7.62 (d, J = 8.6 Hz, 1H), 7.52 (d, J = 8.2 Hz, 2H), 7.46–7.39 (m, 1H), 7.27–7.20 (m, 1H), 3.61 (bs, 2H), 3.36 (bs, 2H), 1.65–14.5 (m, 6H); 13C NMR (151 MHz, DMSO-d6) δ 169.08, 142.84, 142.00, 135.86, 135.06, 127.83, 126.94, 126.66, 121.69, 121.01, 120.45, 111.13, 48.56, 42.84, 26.47, 25.74, 24.51; MS [M+H]+ m/z 306.23; Purity (qNMR, % w/w): 97.32 ± 0.62.
3.2.14. Methyl 4-(1H-Indazol-3-yl) Benzoate (10c)
Employing the esterification method 3.2.9. described above, compound 10c was synthesized from acid derivative 10b.
Methyl 4-(1
H-indazol-3-yl)benzoate (
10c). Yellow solid (94% yield); m.p.: 205.5–208.0 °C; the spectroscopic data matched previously reported data [
16]:
1H NMR (600 MHz, DMSO-
d6) δ 13.52 (s, 1H), 8.19 (d,
J = 8.4 Hz, 2H), 8.16 (d,
J = 8.1 Hz, 1H), 8.11 (d,
J = 8.4 Hz, 2H), 7.64 (d,
J = 8.4 Hz, 1H), 7.45 (dd,
J = 11.3, 3.8 Hz, 1H), 7.27 (dd,
J = 11.2, 3.9 Hz, 1H), 3.90 (s, 3H);
13C NMR (151 MHz DMSO-
d6) δ 165.95, 141.78, 141.53, 138.24, 129.73, 128.20, 126.54, 126.24, 121.46, 120.47, 120.00, 110.74, 52.07; Purity (qNMR, %
w/
w): 96.76 ± 1.06.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}