
A Robust Bioassay of the Human Bradykinin B2 Receptor That Extends Molecular and Cellular Studies: The Isolated Umbilical Vein



Abstract
1. The Kallikrein-Kinin System
2. Hereditary Angioedema as the Therapeutic Showcase of the KKS

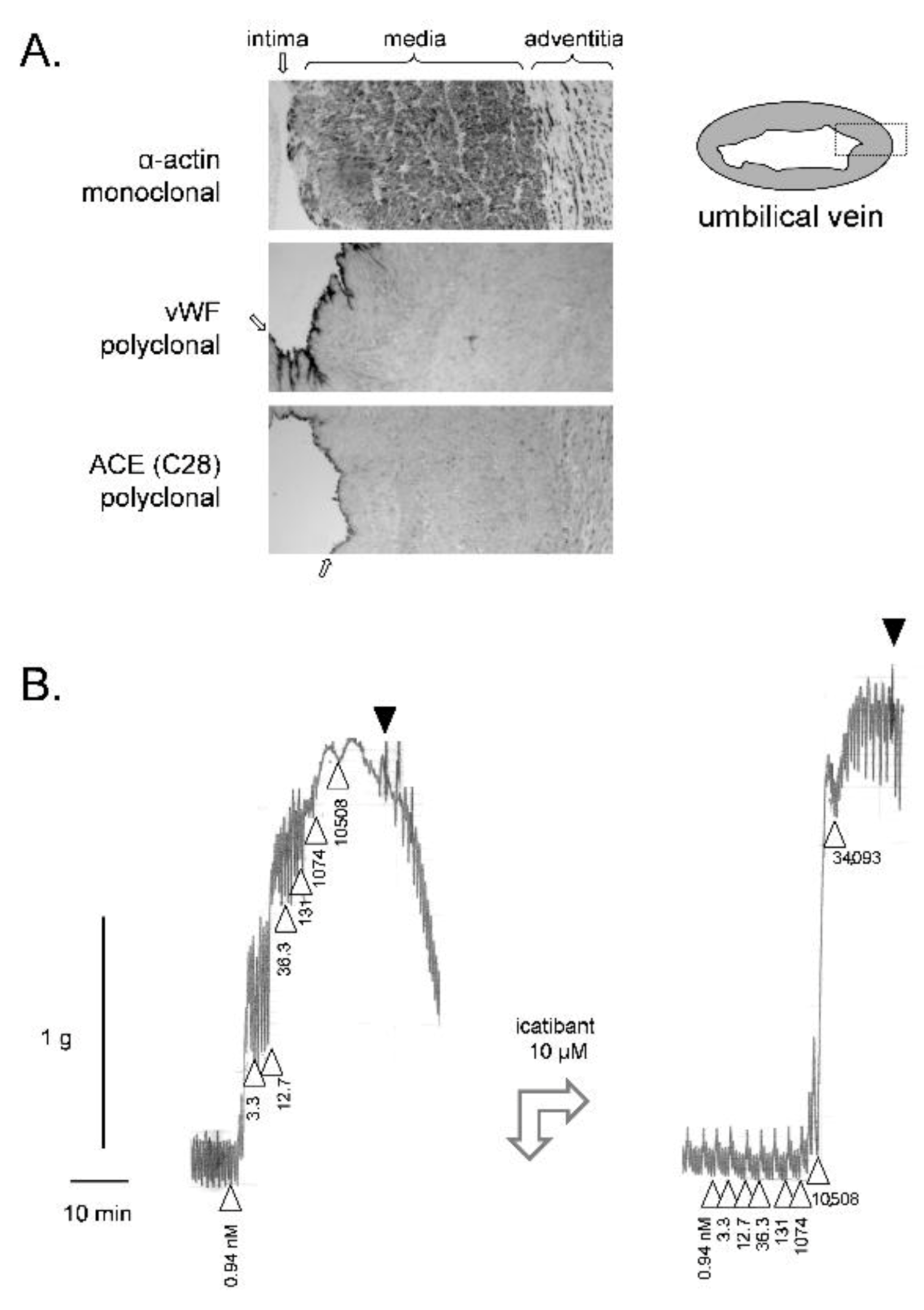{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
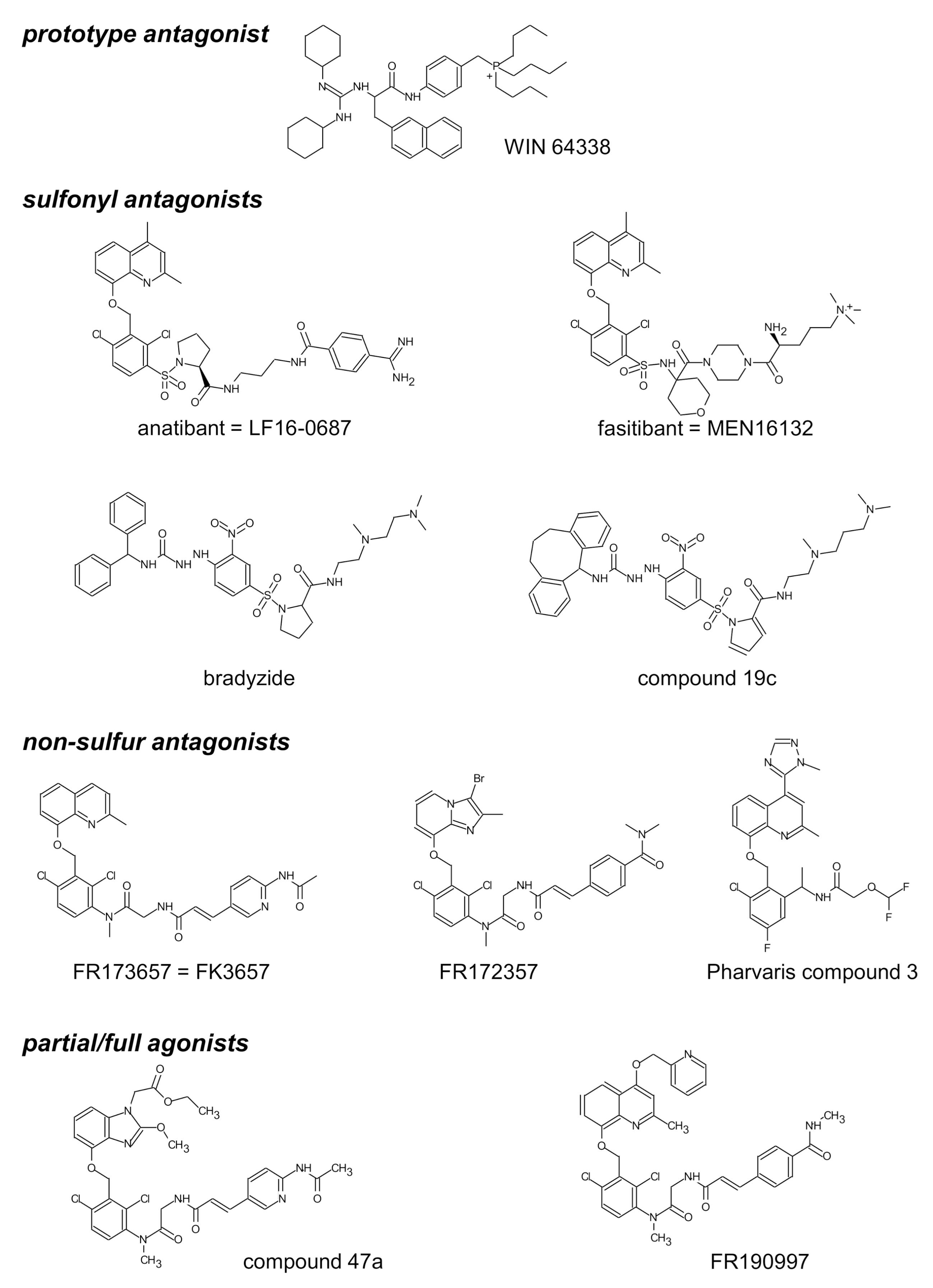
| Antagonist | pA2 ± s.e.m. | Slope ± s.e.m. | Specificity (sp): Inactive against Listed Agonists; Other Remarks | Refs. |
|---|---|---|---|---|
| selected peptide antagonists | ||||
| icatibant (Hoe 140) | 8.2 ± 0.26 | −0.83 ± 0.26 | = d-Arg[Hyp3, Thi5, D-Tic7, Oic8]-BK; sole B2R ligand in clinical use | [37] |
| 8.00 ± 0.11 | −1.06 ± 0.14 | sp: 5-HT, histamine | [38] | |
| 8.42 ± 0.07 | −0.99 ± 0.06 | sp: weak at B1R (pA2 5.48) | [39] | |
| 8.18 ± 0.28 | ≈−1 | [40] | ||
| 8.06 ± 0.37 | −0.85 ± 0.16 | sp: B1R agonist | [36] | |
| MEN 11270 | 8.14 ± 0.22 | −0.95 ± 0.11 | = conformationally constrained derivative of icatibant; sp: 5-HT, noradrenaline | [41] |
| B-9430 | 7.70 | −1.10 | = d-Arg-[Hyp3, Igl5, D-Igl7, Oic8]-BK | [42] |
| B-10380 | 6.83 ± 0.04 | −1.00 | B-9430 N-terminally extended with the green-emitting fluorophore carboxyfluorescein-ε-aminocaproyl | [43] |
| B-10665 | 6.83 | B-9430 N-terminally extended with the infrared-emitting fluorophore Cy7 | [44] | |
| selected non-peptide antagonists | ||||
| WIN 64338 | 5.99 ± 0.08 | −1.15 ± 0.08 | sp: histamine, f-Met-Leu-Phe, 5-HT, U46619 | [37] |
| 6.06 ±0.12 | [40] | |||
| bradyzide | ≈ 5.42 | [45] | ||
| Compound 19c | 7.53 ± 0.24 | −1.14 ± 0.18 | [45] | |
| FR 173657 | 7.80 ± 0.30 | ≈−1 | [40] | |
| 8.22 | −1.00 | [46] | ||
| anatibant (LF16-0687) | 9.1 ± 0.2 | ≈−1 | [47] | |
| 8.3 | sp: 5-HT | [48] | ||
| 8.46 ± 0.10 | −1.11 ± 0.07 | pA2 against the partial agonist activity of Compound 47a | [49] | |
| FR 172357 | 8.65 | −0.99 | sp: 5-HT, noradrenaline, endothelin-1, B1R agonist | [50] |
| Pharvaris Compound 3 | 9.67 ± 0.42 | −0.76 ± 0.13 | sp: 5-HT, U46619, B1R agonist; reversible at 1–10 nM | [36] |
3. The Need for a Human Bioassay for the B2R Antagonists
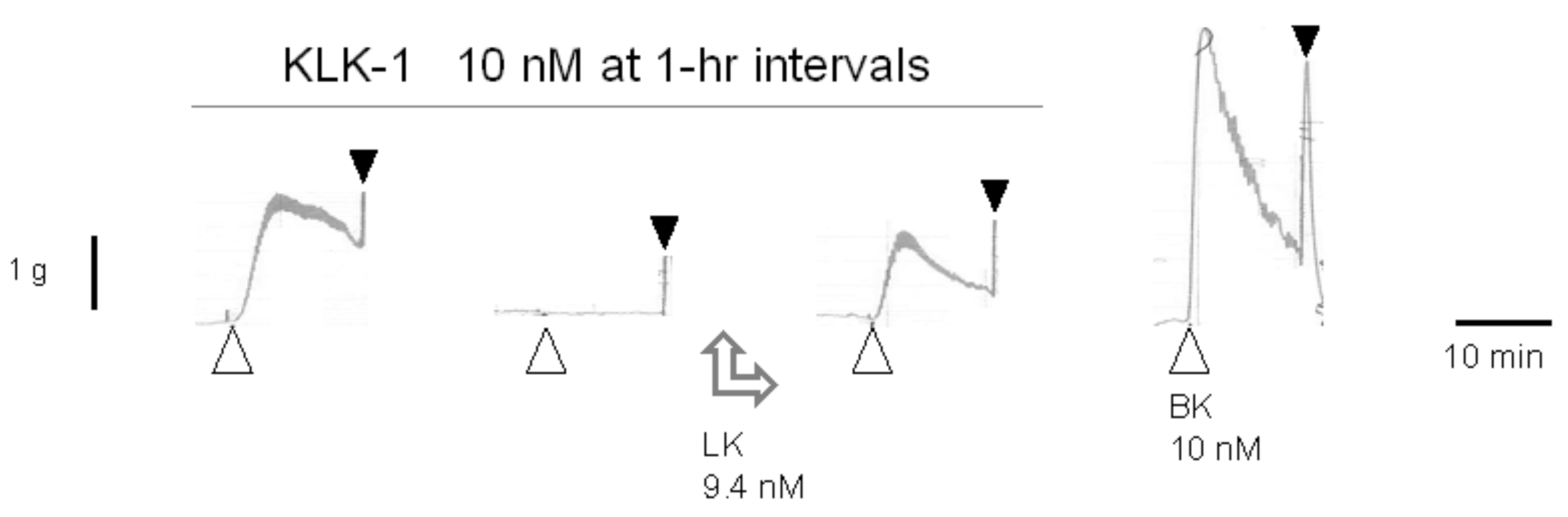
4. Nonconventional Ligands of the B2R Assessed Using the Umbilical Vein
5. Prodrug B2R Agonists Activated by Vascular Peptidases
6. The B1R in the Umbilical Vein
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACE | angiotensin-I converting enzyme |
| Arg-CP | arginine carboxypeptidase |
| B1R | bradykinin B1 receptor |
| B2R | bradykinin B2 receptor |
| BK | bradykinin |
| C1-INH | C1-esterase inhibitor |
| EC50 | half maximal effective concentration |
| Emax | maximal effect |
| F12 | gene encoding factor XII |
| FXII | coagulation factor XII |
| GPCR | G protein coupled receptor |
| HAE | hereditary angioedema |
| HK | high molecular weight kininogen |
| IL-1 | interleukin-1 |
| KKS | kallikrein-kinin system |
| KLK-1 | tissue kallikrein |
| KLK1 | gene encoding tissue kallikrein |
| KLKB-1 | gene encoding plasma prekallikrein |
| KNG1 | gene encoding kininogens |
| LC-MS | liquid chromatography–mass spectrometry |
| LK | low molecular weight kininogen |
| Lys-BK | lysyl-bradykinin, kallidin |
| pA2 | a measure of affinity of the antagonist for its receptor |
| PLG | gene encoding plasminogen |
| s.e.m. | standard error of the mean |
| SERPINA4 | gene encoding kallistatin |
| SERPING1 | gene encoding C1-INH |
| TNF-α | tumor necrosis factor-α |
| vWF | Von Willebrand factor |
References
- Bhoola, K.D.; Figueroa, C.D.; Worthy, K. Bioregulation of kinins: Kallikreins, kininogens, and kininases. Pharmacol. Rev. 1992, 44, 1–80. [Google Scholar] [PubMed]
- Blais, C.; Marceau, F.; Rouleau, J.L.; Adam, A. The kallikrein-kininogen-kinin systems: Lessons from the quantification of endogenous kinins. Peptides 2000, 21, 1903–1940. [Google Scholar] [CrossRef]
- Alhenc-Gelas, F.; Bouby, N.; Girolami, J.P. Kallikrein/K1, Kinins, and ACE/Kininase II in homeostasis and in disease insight from human and experimental genetic studies, therapeutic implication. Front. Med. (Lausanne) 2019, 6, 136. [Google Scholar] [CrossRef] [PubMed]
- Kakoki, M.; Smithies, O. The kallikrein-kinin system in health and in diseases of the kidney. Kidney Int. 2009, 75, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.P. The bradykinin-forming cascade: A historical perspective. Chem. Immunol. Allergy 2014, 100, 205–213. [Google Scholar]
- Manolis, A.J.; Marketou, M.E.; Gavras, I.; Gavras, H. Cardioprotective properties of bradykinin: Role of the B2 receptor. Hypertens. Res. 2010, 33, 772–777. [Google Scholar] [CrossRef]
- Leeb-Lundberg, L.M.; Marceau, F.; Müller-Esterl, W.; Pettibone, D.J.; Zuraw, B.L. International union of pharmacology. XLV. Classification of the kinin receptor family: From molecular mechanisms to pathophysiological consequences. Pharmacol. Rev. 2005, 57, 27–77. [Google Scholar] [CrossRef] [PubMed]
- Moreau, M.E.; Garbacki, N.; Molinaro, G.; Brown, N.J.; Marceau, F.; Adam, A. The kallikrein–kinin system: Current and future pharmacological targets. J. Pharmacol. Sci. 2005, 99, 6–38. [Google Scholar] [CrossRef]
- Bergaya, S.; Meneton, P.; Bloch-Faure, M.; Mathieu, E.; Alhenc-Gelas, F.; Lévy, B.I.; Boulanger, C.M. Decreased flow-dependent dilation in carotid arteries of tissue kallikrein-knockout mice. Circ. Res. 2001, 88, 593–599. [Google Scholar] [CrossRef]
- Meneton, P.; Bloch-Faure, M.; Hagege, A.A.; Ruetten, H.; Huang, W.; Bergaya, S.; Ceiler, D.; Gehring, D.; Martins, I.; Salmon, G.; et al. Cardiovascular abnormalities with normal blood pressure in tissue kallikrein-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 2634–2639. [Google Scholar] [CrossRef]
- Björkqvist, J.; Jämsä, A.; Renné, T. Plasma kallikrein: The bradykinin-producing enzyme. Thromb. Haemost. 2013, 110, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Cyr, M.; Lepage, Y.; Blais, C.; Gervais, N.; Cugno, M.; Rouleau, J.L.; Adam, A. Bradykinin and des-Arg9-bradykinin metabolic pathways and kinetics of activation of human plasma. Am. J. Physiol. Heart Circul. Physiol. 2001, 281, H275–H283. [Google Scholar] [CrossRef]
- Regoli, D.; Barabé, J. Pharmacology of bradykinin and related kinins. Pharmacol. Rev. 1980, 32, 1–46. [Google Scholar]
- Veeravalli, K.K.; Akula, A. Involvement of nitric oxide and prostaglandin pathways in the cardioprotective actions of bradykinin in rats with experimental myocardial infarction. Pharmacol. Res. 2004, 49, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Marketou, M.; Kintsurashvili, E.; Papanicolaou, K.N.; Lucero, H.A.; Gavras, I.; Gavras, H. Cardioprotective effects of a selective B2 receptor agonist of bradykinin post-acute myocardial infarct. Am. J. Hypertens. 2010, 23, 562–568. [Google Scholar] [CrossRef]
- Potier, L.; Waeckel, L.; Vincent, M.P.; Chollet, C.; Gobeil, F., Jr.; Marre, M.; Bruneval, P.; Richer, C.; Roussel, R.; Alhenc-Gelas, F.; et al. Selective kinin receptor agonists as cardioprotective agents in myocardial ischemia and diabetes. J. Pharmacol. Exp. Ther. 2013, 346, 23–30. [Google Scholar] [CrossRef]
- Bossi, F.; Fischetti, F.; Regoli, D.; Durigutto, P.; Frossi, B.; Gobeil, F., Jr.; Ghebrehiwet, B.; Peerschke, E.I.; Cicardi, M.; Tedesco, F. Novel pathogenic mechanism and therapeutic approaches to angioedema associated with C1 inhibitor deficiency. J. Allergy Clin. Immunol. 2009, 124, 1304–1310. [Google Scholar] [CrossRef] [PubMed]
- Marceau, F.; Hess, J.F.; Bachvarov, D.R. The B1 receptors for kinins. Pharmacol. Rev. 1998, 50, 357–386. [Google Scholar]
- Marceau, F.; Regoli, D. Bradykinin receptor ligands: Therapeutic perspectives. Nat. Rev. Drug Disc. 2004, 3, 845–852. [Google Scholar] [CrossRef]
- Ratnoff, O.D.; Lepow, I.H. Some properties of an esterase derived from preparations of the first component of complement. J. Exp. Med. 1957, 106, 327–343. [Google Scholar] [CrossRef]
- Schapira, M.; Scott, C.F.; Colman, R.W. Contribution of plasma protease inhibitors to the inactivation of kallikrein in plasma. J. Clin. Investig. 1982, 69, 462–468. [Google Scholar] [CrossRef]
- de Agostini, A.; Ujnen, H.R.; Pixley, R.A.; Colman, R.W.; Schapira, M. Inactivation of factor XII active fragment in normal plasma. J. Clin. Investig. 1984, 73, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Harpel, P.C.; Lewin, M.F.; Kaplan, A.P. Distribution of plasma kallikrein between C1 inactivator and alpha2-macroglobulin in plasma utilizing a new assay for alpha2-macroglobulin–kallikrein complexes. J. Biol. Chem. 1985, 260, 4257–4263. [Google Scholar] [CrossRef]
- Chao, J.; Chao, L. Biochemistry, regulation and potential function of kallistatin. Biol. Chem. Hoppe Seyler 1995, 376, 705–713. [Google Scholar]
- Caballero, T.; Baeza, M.; Cabañas, R.; Campos, A.; Cimbollek, S.; Gómez-Traseira, C.; González-Quevedo, T.; Guilarte, M.; Jurado-Palomo, J.; Larco, J.I.; et al. Consensus statement on the diagnosis, management, and treatment of angioedema mediated by bradykinin. Part II. Treatment, follow-up, and special situations. J. Investig. Allergol. Clin. Immunol. 2011, 21, 422–441. [Google Scholar]
- Kaplan, A.P.; Joseph, K. Pathogenesis of hereditary angioedema: The role of the bradykinin-forming cascade. Immunol Allergy Clin. N. Am. 2017, 37, 513–525. [Google Scholar] [CrossRef]
- Marcelino-Rodriguez, I.; Callero, A.; Mendoza-Alvarez, A.; Perez-Rodriguez, E.; Barrios-Recio, J.; Garcia-Robaina, J.C.; Flores, C. Bradykinin-mediated angioedema: An update of the genetic causes and the impact of genomics. Front. Genet. 2019, 10, 900. [Google Scholar] [CrossRef]
- Bork, K.; Wulff, K.; Meinke, P.; Wagner, N.; Hardt, J.; Witzke, G. A novel mutation in the coagulation factor 12 gene in subjects with hereditary angioedema and normal C1-inhibitor. Clin. Immunol. 2011, 141, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Dewald, G. A missense mutation in the plasminogen gene, within the plasminogen kringle 3 domain, in hereditary angioedema with normal C1 inhibitor. Biochem. Biophys. Res. Commun. 2018, 498, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Bork, K.; Wulff, K.; Steinmüller-Magin, L.; Brænne, I.; Staubach-Renz, P.; Witzke, G.; Hardt, J. Hereditary angioedema with a mutation in the plasminogen gene. Allergy 2018, 73, 442–450. [Google Scholar] [CrossRef]
- Bork, K.; Wulff, K.; Rossmann, H.; Steinmüller-Magin, L.; Braenne, I.; Witzke, G.; Hardt, J. Hereditary angioedema cosegregating with a novel kininogen 1 gene mutation changing the N-terminal cleavage site of bradykinin. Allergy 2019, 74, 2479–2481. [Google Scholar] [CrossRef]
- Nicola, S.; Rolla, G.; Brussino, L. Breakthroughs in hereditary angioedema management: A systematic review of approved drugs and those under research. Drugs Context. 2019, 8, 212605. [Google Scholar]
- Cicardi, M.; Levy, R.J.; McNeil, D.L.; Li, H.H.; Sheffer, A.L.; Campion, M.; Horn, P.T.; Pullman, W.E. Ecallantide for the treatment of acute attacks in hereditary angio- edema. N. Engl. J. Med. 2010, 363, 523–531. [Google Scholar] [CrossRef]
- Perego, F.; Wu, M.A.; Valerieva, A.; Caccia, S.; Suffritti, C.; Zanichelli, A.; Bergamaschini, L.; Cicardi, M. Current and emerging biologics for the treatment of hereditary angioedema. Exp. Opin. Biol. Ther. 2019, 19, 517–526. [Google Scholar] [CrossRef]
- Zuraw, B.; Lumry, W.R.; Johnston, D.T.; Aygören-Pürsün, E.; Banerji, A.; Bernstein, J.A.; Christiansen, S.C.; Jacobs, J.S.; Sitz, K.V.; Gower, R.G.; et al. Oral once-daily berotralstat for the prevention of hereditary angioedema attacks: A randomized, double-blind, placebo-controlled phase 3 trial. J. Allergy Clin. Immunol. 2020. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Lesage, A.; Gibson, C.; Marceau, F.; Ambrosi, H.-D.; Saupe, J.; Katzer, W.; Loenders, B.; Charest-Morin, X.; Knolle, J. In vitro pharmacological profile of new small molecule bradykinin B2 receptor antagonists. Front. Pharmacol. 2020, 11, 916. [Google Scholar] [CrossRef]
- Marceau, F.; Levesque, L.; Drapeau, G.; Rioux, F.; Salvino, J.M.; Wolfe, H.; Seoane, P.R.; Sawutz, D.G. Effects of peptide and nonpeptide antagonists of bradykinin B2 receptors on the venoconstrictor action of bradykinin. J. Pharmacol. Exp. Ther. 1994, 269, 1136–1143. [Google Scholar]
- Félétou, M.; Martin, C.A.; Molimard, M.; Naline, E.; Germain, M.; Thurieau, C.; Fauchère, J.L.; Canet, E.; Advenier, C. In vitro effects of HOE 140 in human bronchial and vascular tissue. Eur. J. Pharmacol. 1995, 274, 57–64. [Google Scholar] [CrossRef]
- Gobeil, F.; Pheng, L.H.; Badini, I.; Nguyen-Le, X.K.; Pizard, A.; Rizzi, A.; Blouin, D.; Regoli, D. Receptors for kinins in the human isolated umbilical vein. Br. J. Pharmacol. 1996, 118, 289–294. [Google Scholar] [CrossRef]
- Paquet, J.L.; Luccarini, J.M.; Fouchet, C.; Defrêne, E.; Loillier, B.; Robert, C.; Bélichard, P.; Cremers, B.; Pruneau, D. Pharmacological characterization of the bradykinin B2 receptor: Inter-species variability and dissociation between binding and functional responses. Br. J. Pharmacol. 1999, 126, 1083–1090. [Google Scholar] [CrossRef]
- Meini, S.; Quartara, L.; Rizzi, A.; Patacchini, R.; Cucchi, P.; Giolitti, A.; Calò, G.; Regoli, D.; Criscuoli, M.; Maggi, C.A. MEN 11270, a novel selective constrained peptide antagonist with high affinity at the human B2 kinin receptor. J. Pharmacol. Exp. Ther. 1999, 289, 1250–1256. [Google Scholar] [PubMed]
- Bawolak, M.-T.; Gera, L.; Stewart, J.M.; Marceau, F. B-9972 (D-Arg-[Hyp3, Igl5, Oic7, Igl8]-bradykinin) is an inactivation-resistant agonist of the bradykinin B2 receptor derived from the peptide antagonist B-9430 (D-Arg-[Hyp3, Igl5, D-Igl7, Oic8]-bradykinin): Pharmacologic profile and effective induction of receptor degradation. J. Pharmacol. Exp. Ther. 2007, 323, 534–546. [Google Scholar] [PubMed]
- Bawolak, M.-T.; Gera, L.; Bouthillier, J.; Stewart, J.M.; Adam, A.; Marceau, F. A fluorescent version of the bradykinin B2 receptor antagonist B-9430: Pharmacological characterization and use in live cell imaging. Peptides 2008, 29, 1626–1630. [Google Scholar] [CrossRef]
- Gera, L.; Charest-Morin, X.; Jean, M.; Bachelard, H.; Marceau, F. Infrared-emitting, peptidase-resistant fluorescent ligands of the bradykinin B2 receptor: Application to cytofluorometry and imaging. BMC Res. Notes 2016, 9, 452. [Google Scholar] [CrossRef] [PubMed]
- Marceau, F.; Fortin, J.-P.; Morissette, G.; Dziadulewicz, E.K. A non-peptide antagonist unusually selective for the human form of the bradykinin B2 receptor. Int. Immunopharmacol. 2003, 3, 1529–1536. [Google Scholar] [CrossRef]
- Rizzi, A.; Gobeil, F.; Bogoni, G.; Calò, G.; Campobasso, C.; Inamura, N.; Regoli, D. Antagonistic effects of FR 173657 on human, pig, rabbit, and guinea pig kinin receptors: An in vitro study. Can. J. Physiol. Pharmacol. 1997, 75, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Pruneau, D.; Paquet, J.L.; Luccarini, J.M.; Defrêne, E.; Fouchet, C.; Franck, R.M.; Loillier, B.; Robert, C.; Bélichard, P.; Duclos, H.; et al. Pharmacological profile of LF 16-0687, a new potent non-peptide bradykinin B2 receptor antagonist. Immunopharmacology 1999, 43, 187–194. [Google Scholar] [CrossRef]
- Bawolak, M.-T.; Marceau, F. Does zaltoprofen antagonize the bradykinin receptors? Regul. Peptides 2007, 140, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Bawolak, M.-T.; Fortin, S.; Bouthillier, J.; Adam, A.; Gera, L.; C.-Gaudreault, R.; Marceau, F. Effects of inactivation-resistant agonists on the signalling, desensitization and down-regulation of bradykinin B2 receptors. Br. J. Pharmacol. 2009, 158, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, A.; Rizzi, C.; Amadesi, S.; Calò, G.; Varani, K.; Inamura, N.; Regoli, D. Pharmacological characterisation of the first non-peptide bradykinin B2 receptor agonist FR 190997: An in vitro study on human, rabbit and pig vascular B2 receptors. Naunyn Schmiedebergs Arch. Pharmacol. 1999, 360, 361–367. [Google Scholar] [CrossRef]
- Rhaleb, N.E.; Rouissi, N.; Jukic, D.; Regoli, D.; Henke, S.; Breipohl, G.; Knolle, J. Pharmacological characterization of a new highly potent B2 receptor antagonist (HOE 140: D-Arg-[Hyp3,Thi5,D-Tic7,Oic8]bradykinin). Eur. J. Pharmacol. 1992, 210, 115–120. [Google Scholar] [CrossRef]
- Houle, S.; Larrivée, J.-F.; Bachvarova, M.; Bouthillier, J.; Bachvarov, D.R.; Marceau, F. Antagonist-induced intracellular sequestration of rabbit bradykinin B2 receptor. Hypertension 2000, 35, 1319–1325. [Google Scholar] [CrossRef]
- Félétou, M.; Germain, M.; Thurieau, C.; Fauchère, J.L.; Canet, E. Agonistic and antagonistic properties of the bradykinin B2 receptor antagonist, Hoe 140, in isolated blood vessels from different species. Br. J. Pharmacol. 1994, 112, 683–689. [Google Scholar] [CrossRef]
- Griesbacher, T.; Lembeck, F. Analysis of the antagonistic actions of HOE 140 and other novel bradykinin analogues on the guinea-pig ileum. Eur. J. Pharmacol. 1992, 211, 393–398. [Google Scholar] [CrossRef]
- Altura, B.M.; Malaviya, D.; Reich, C.F.; Orkin, L.R. Effects of vasoactive agents on isolated human umbilical arteries and veins. Am. J. Physiol. 1972, 222, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Tuvemo, T. Role of prostaglandins, prostacyclin, and thromboxanes in the control of the umbilical-placental circulation. Semin. Perinatol. 1980, 4, 91–95. [Google Scholar] [PubMed]
- Marceau, F.; deBlois, D.; Laplante, C.; Petitclerc, E.; Pelletier, G.; Grose, J.H.; Hugli, T.E. Contractile effect of the chemotactic factors f-Met-Leu-Phe and C5a on the human isolated umbilical artery. Role of cyclooxygenase products and tissue macrophages. Circ. Res. 1990, 67, 1059–1070. [Google Scholar] [CrossRef]
- Paczkowski, N.J.; Finch, A.M.; Whitmore, J.B.; Short, A.J.; Wong, A.K.; Monk, P.N.; Cain, S.A.; Fairlie, D.P.; Taylor, S.M. Pharmacological characterization of antagonists of the C5a receptor. Br. J. Pharmacol. 1999, 128, 1461–1466. [Google Scholar] [CrossRef]
- Lorigo, M.; Mariana, M.; Feiteiro, J.; Cairrao, E. How is the human umbilical artery regulated? J. Obstet. Gynaecol. Res. 2018, 55, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Fryer, R.M.; Segreti, J.; Banfor, P.N.; Widomski, D.L.; Backes, B.J.; Lin, C.W.; Ballaron, S.J.; Cox, B.F.; Trevillyan, J.M.; Reinhart, G.A.; et al. Effect of bradykinin metabolism inhibitors on evoked hypotension in rats: Rank efficacy of enzymes associated with bradykinin-mediated angioedema. Br. J. Pharmacol. 2008, 153, 947–955. [Google Scholar] [CrossRef]
- Gaudreau, P.; Barabé, J.; St-Pierre, S.; Regoli, D. Pharmacological studies of kinins in venous smooth muscle. Can. J. Physiol. Pharmacol. 1981, 59, 371–379. [Google Scholar] [CrossRef]
- Gera, L.; Roy, C.; Bawolak, M.-T.; Bouthillier, J.; Adam, A.; Marceau, F. Met-Lys-bradykinin-Ser-Ser, a peptide produced by the neutrophil from kininogen, is metabolically activated by angiotensin converting enzyme in vascular tissue. Pharmacol. Res. 2011, 64, 528–534. [Google Scholar] [CrossRef]
- Koumbadinga, G.A.; Bawolak, M.T.; Marceau, E.; Adam, A.; Gera, L.; Marceau, F. A ligand-based approach to investigate the expression and function of angiotensin converting enzyme in intact human umbilical vein endothelial cells. Peptides 2010, 31, 1546–1554. [Google Scholar] [CrossRef]
- Arunlakshana, O.; Schild, H.O. Some quantitative uses of drug antagonists. Br. J. Pharmacol. Chemother. 1959, 14, 48–58. [Google Scholar] [CrossRef]
- Marceau, F.; Bawolak, M.-T.; Fortin, J.-P.; Morissette, G.; Roy, C.; Bachelard, H.; Gera, L.; Charest-Morin, X. Bifunctional ligands of the bradykinin B2 and B1 receptors: An exercise in peptide hormone plasticity. Peptides 2018, 105, 37–50. [Google Scholar] [CrossRef]
- Sawutz, D.G.; Salvino, J.M.; Dolle, R.E.; Casiano, F.; Ward, S.J.; Houck, W.T.; Faunce, D.M.; Douty, B.D.; Baizman, E.; Awad, M.M.; et al. The nonpeptide WIN 64338 is a bradykinin B2 receptor antagonist. Proc. Natl. Acad. Sci. USA 1994, 91, 4693–4697. [Google Scholar] [CrossRef]
- Lupala, C.S.; Gomez-Gutierrez, P.; Perez, J.J. New insights into the stereochemical requirements of the bradykinin B2 receptor antagonists binding. J. Comput. Aided Mol. Des. 2016, 30, 85–101. [Google Scholar] [CrossRef]
- Burgess, G.M.; Perkins, M.N.; Rang, H.P.; Campbell, E.A.; Brown, M.C.; McIntyre, P.; Urban, L.; Dziadulewicz, E.K.; Ritchie, T.J.; Hallett, A.; et al. Bradyzide, a potent non-peptide B2 bradykinin receptor antagonist with long-lasting oral activity in animal models of inflammatory hyperalgesia. Br. J. Pharmacol. 2000, 129, 77–86. [Google Scholar] [CrossRef]
- Dziadulewicz, E.K.; Ritchie, T.J.; Hallett, A.; Snell, C.R.; Davies, J.W.; Wrigglesworth, R.; Dunstan, A.R.; Bloomfield, G.C.; Drake, G.S.; McIntyre, P.; et al. Nonpeptide bradykinin B2 receptor antagonists: Conversion of rodent-selective bradyzide analogues into potent, orally-active human bradykinin B2 receptor antagonists. J. Med. Chem. 2002, 45, 2160–2172. [Google Scholar] [CrossRef]
- Lesage, A.S.; Loenders, B.; Knolle, J. PHA-022121, a first in class oral bradykinin B2 receptor antagonist in clinical development: Proof of concept study in a translational monkey bradykinin challenge model. J. Allergy Clin. Immunol. 2020, 145, AB346, (Abstract). [Google Scholar] [CrossRef]
- Derendorf, H.; Lesage, A.; Crabbé, R.; Lu, P.; Groen, K.; Rodriguez, M.; Leal, N.; Knolle, J. Bradykinin challenge provides surrogate endpoints for hereditary angioedema treatment using bradykinin B2-recepror antagonists. Ann. Allergy Asthma Immunol. 2020, 125, S21, (Abstract). [Google Scholar] [CrossRef]
- Lu, P.; Lesage, A.; Crabbé, R.; Groen, K.; Gibson, C.; Walther, N.; Knolle, J. PHA-022121, A selective bradykinin B2-receptor antagonist, is safe and shows rapid oral bioavailability in humans. Ann. Allergy Asthma Immunol. 2020, 125, S21, (Abstract). [Google Scholar] [CrossRef]
- Jean, M.; Gera, L.; Charest-Morin, X.; Marceau, F.; Bachelard, H. In Vivo Effects of Bradykinin B2 receptor agonists with varying susceptibility to peptidases. Front. Pharmacol. 2016, 6, 306. [Google Scholar] [CrossRef]
- Aramori, I.; Zenkoh, J.; Morikawa, N.; Asano, M.; Hatori, C.; Sawai, H.; Kayakiri, H.; Satoh, S.; Inoue, T.; Abe, Y.; et al. Nonpeptide mimic of bradykinin with long-acting properties at the bradykinin B2 receptor. Mol. Pharmacol. 1997, 52, 16–20. [Google Scholar] [CrossRef]
- Sawada, Y.; Kayakiri, H.; Abe, Y.; Mizutani, T.; Inamura, N.; Asano, M.; Hatori, C.; Aramori, I.; Oku, T.; Tanaka, H. Discovery of the first non-peptide full agonists forthe human bradykinin B2 receptor incorporating 4-(2-picolyloxy)quinoline and 1-(2-picolyl)benzimidazole frameworks. J. Med. Chem. 2004, 47, 2853–2863. [Google Scholar] [CrossRef]
- Bélanger, S.; Bovenzi, V.; Côté, J.; Neugebauer, W.; Amblard, M.; Martinez, J.; Lammek, B.; Savard, M.; Gobeil, F., Jr. Structure-activity relationships of novel peptide agonists of the human bradykinin B2 receptor. Peptides 2009, 30, 777–787. [Google Scholar] [CrossRef]
- Sharif, N.A.; Katoli, P.; Scott, D.; Li, L.; Kelly, C.; Xu, S.; Husain, S.; Toris, C.; Crosson, C. FR-190997, a nonpeptide bradykinin B2-receptor partial agonist, is a potent and efficacious intraocular pressure lowering agent in ocular hypertensive cynomolgus monkeys. Drug Dev. Res. 2014, 75, 211–323. [Google Scholar] [CrossRef]
- Rassias, G.; Leonardi, S.; Rigopoulou, D.; Vachlioti, E.; Afratis, K.; Piperigkou, Z.; Koutsakis, C.; Karamanos, N.K.; Gavras, H.; Papaioannou, D. Potent antiproliferative activity of bradykinin B2 receptor selective agonist FR-190997 and analogue structures thereof: A paradox resolved? Eur. J. Med. Chem. 2021, 210, 112948. [Google Scholar] [CrossRef]
- Gera, L.; Bawolak, M.-T.; Roy, C.; Lodge, R.; Marceau, F. Design of fluorescent bradykinin analogs: Application to imaging of B2 receptor-mediated agonist endocytosis and trafficking and angiotensin-converting enzyme. J. Pharmacol. Exp. Ther. 2011, 337, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Hecquet, C.; Tan, F.; Marcic, B.M.; Erdös, E.G. Human bradykinin B2 receptor is activated by kallikrein and other serine proteases. Mol. Pharmacol. 2000, 58, 828–836. [Google Scholar] [CrossRef]
- Charest-Morin, X.; Raghavan, A.; Charles, M.L.; Kolodka, T.; Bouthillier, J.; Jean, M.; Robbins, M.S.; Marceau, F. Pharmacological effects of recombinant human tissue kallikrein on bradykinin B2 receptors. Pharmacol. Res. Perspect. 2015, 3, e00119. [Google Scholar] [CrossRef]
- Jean, M.; Raghavan, A.; Charles, M.L.; Robbins, M.S.; Wagner, E.; Rivard, G.É.; Charest-Morin, X.; Marceau, F. The isolated human umbilical vein as a bioassay for kinin-generating proteases: An in vitro model for therapeutic angioedema agents. Life Sci. 2016, 155, 180–188. [Google Scholar] [CrossRef]
- Yang, H.Y.; Erdös, E.G.; Levin, Y. A dipeptidyl carboxypeptidase that converts angiotensin I and inactivates bradykinin. Biochim. Biophys. Acta 1970, 214, 374–376. [Google Scholar] [CrossRef]
- Gavras, I. Bradykinin-mediated effects of ACE inhibition. Kidney Int. 1992, 42, 1020–1029. [Google Scholar] [CrossRef]
- Brown, N.J.; Gainer, J.V.; Murphey, L.J.; Vaughan, D.E. Bradykinin stimulates tissue plasminogen activator release from human forearm vasculature through B2 receptor-dependent, NO synthase-independent, and cyclooxygenase-independent path- way. Circulation 2000, 102, 2190–2196. [Google Scholar] [CrossRef]
- Pretorius, M.; Rosenbaum, D.; Vaughan, D.E.; Brown, N.J. Angiotensin converting enzyme inhibition increases human vascular-type plasminogen activator release through endogenous bradykinin. Circulation 2003, 107, 579–585. [Google Scholar] [CrossRef]
- Griol-Charhbili, V.; Messadi-Laribi, E.; Bascands, J.L.; Heudes, D.; Meneton, P.; Giudicelli, J.F.; Alhenc-Gelas, F.; Richer, C. Role of tissue kallikrein in the cardioprotective effects of ischemic and pharmacological preconditioning in myocardial ischemia. FASEB J. 2005, 19, 1172–1174. [Google Scholar] [CrossRef]
- Kakoki, M.; McGarrah, R.W.; Kim, H.S.; Smithies, O. Bradykinin B1 and B2 receptors both have protective roles in renal ischemia/reperfusion injury. Proc. Natl. Acad. Sci. USA 2007, 104, 7576–7581. [Google Scholar] [CrossRef] [PubMed]
- Charest-Morin, X.; Roy, C.; Fortin, E.J.; Bouthillier, J.; Marceau, F. Pharmacological evidence of bradykinin regeneration from extended sequences that behave as peptidase-activated B2 receptor agonists. Front. Pharmacol. 2014, 5, 32. [Google Scholar] [CrossRef]
- Charest-Moxin, X.; Bachelard, H.; Jean, M.; Marceau, F. Species-specific pharmacology of maximakinin, an amphibian homologue of bradykinin: Putative prodrug activity at the human B2 receptor and peptidase resistance in rats. PeerJ 2017, 5, e2911. [Google Scholar] [CrossRef]
- Bachelard, H.; Charest-Morin, X.; Marceau, F. D-Arg0-Bradykinin-Arg-Arg, a latent vasoactive bradykinin B2 receptor agonist metabolically activated by carboxypeptidases. Front. Pharmacol. 2018, 9, 273. [Google Scholar] [CrossRef]
- Kahn, R.; Hellmark, T.; Leeb-Lundberg, L.M.; Akbari, N.; Todiras, M.; Olofsson, T.; Wieslander, J.; Christensson, A.; Westman, K.; Bader, M.; et al. Neutrophil-derived proteinase 3 induces kallikrein-independent release of a novel vasoactive kinin. J. Immunol. 2009, 182, 7906–7915. [Google Scholar] [CrossRef]
- Bawolak, M.-T.; Roy, C.; Gera, L.; Marceau, F. Prolonged signalling and trafficking of the bradykinin B2, receptor stimulated with the amphibian peptide maximakinin: Insight into the endosomal inactivation of kinins. Pharmacol. Res. 2012, 65, 247–253. [Google Scholar] [CrossRef]
- Regoli, D.; Barabé, J.; Park, W.K. Receptors for bradykinin in rabbit aortae. Can. J. Physiol. Pharmacol. 1977, 55, 855–867. [Google Scholar] [CrossRef]
- Regoli, D.; Marceau, F.; Barabé, J. De novo formation of vascular receptors for bradykinin. Can. J. Physiol. Pharmacol. 1978, 56, 674–677. [Google Scholar] [CrossRef]
- Bouthillier, J.; Deblois, D.; Marceau, F. Studies on the induction of pharmacological responses to des-Arg9-bradykinin in vitro and in vivo. Br. J. Pharmacol. 1987, 92, 257–264. [Google Scholar] [CrossRef]
- Marceau, F.; Larrivée, J.-F.; Bouthillier, B.; Bachvarova, M.; Houle, S.; Bachvarov, D.R. Effect of endogenous kininjs, prostanoids, and NO on kinin B1 and B2 receptor expression in the rabbit. Am. J. Physiol. 1999, 46, R1568–R1578. [Google Scholar]
- Larrivée, J.-F.; Bachvarov, D.R.; Houle, F.; Landry, J.; Huot, J.; Marceau, F. Role of the mitogen-activated protein kinases in the expression of the kinin B1 receptors induced by tissue injury. J. Immunol. 1998, 160, 1419–1426. [Google Scholar]
- Sabourin, T.; Morissette, G.; Bouthillier, J.; Levesque, L.; Marceau, F. Expression of kinin B1 receptor in fresh or cultured rabbit aortic smooth muscle: Role of NF-κB. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H227–H237. [Google Scholar] [CrossRef] [PubMed]
- Moreau, M.E.; Bawolak, M.-T.; Morissette, G.; Adam, A.; Marceau, F. Role of nuclear factor-κB and protein kinase C signaling in the expression of the kinin B1 receptor in human vascular smooth muscle cells. Mol. Pharmacol. 2007, 71, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Koumbadinga, G.A.; Désormeaux, A.; Adam, A.; Marceau, F. Effect of interferon-γ on inflammatory cytokine-induced bradykinin B1 receptor expression in human vascular cells. Eur. J. Pharmacol. 2010, 647, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Kilstein, Y.; Nowak, W.; Errasti, A.E.; Feás, A.A.; Armesto, A.R.; Pelorosso, F.G.; Rothlin, R.P. Involvement of extracellular signal-regulated kinase 5 in kinin B1 receptor upregulation in isolated human umbilical veins. J. Pharmacol. Exp. Ther. 2016, 357, 114–124. [Google Scholar] [CrossRef]
- Menke, J.G.; Borkowski, J.A.; Bierilo, K.K.; MacNeil, T.; Derrick, A.W.; Schneck, K.A.; Ransom, R.W.; Strader, C.D.; Linemeyer, D.L.; Hess, J.F. Expression cloning of a human B1 bradykinin receptor. J. Biol. Chem. 1994, 269, 21583–21586. [Google Scholar] [CrossRef]
- Sardi, S.P.; Pérez, H.; Antúnez, P.; Rothlin, R.P. Bradykinin B1 receptors in human umbilical vein. Eur. J. Pharmacol. 1997, 321, 33–38. [Google Scholar] [CrossRef]
- Nowak, W.; Goldschmidt, E.D.; Falcioni, A.G.; Pugliese, M.I.; Errasti, A.E.; Pelorosso, F.G.; Daray, F.M.; Gago, J.E.; Rothlin, R.P. Functional evidence of des-Arg10-kallidin enzymatic inactivating pathway in isolated human umbilical vein. Naunyn Schmiedebergs Arch. Pharmacol. 2007, 375, 221–229. [Google Scholar] [CrossRef]
- Drapeau, G.; deBlois, D.; Marceau, F. Hypotensive effects of Lys-des-Arg9-bradykinin and metabolically protected agonists of B1 receptors for kinins. J. Pharmacol. Exp. Ther. 1991, 259, 997–1003. [Google Scholar]
- Côté, J.; Savard, M.; Bovenzi, V.; Bélanger, S.; Morin, J.; Neugebauer, W.; Larouche, A.; Dubuc, C.; Gobeil, F., Jr. Novel kinin B1 receptor agonists with improved pharmacological profiles. Peptides 2009, 30, 788–795. [Google Scholar] [CrossRef]
- Houle, S.; Landry, M.; Audet, R.; Bouthillier, J.; Bachvarov, D.R.; Marceau, F. Effect of allelic polymorphism of the B1 and B2 receptor genes on the contractile responses of the human umbilical vein to kinins. J. Pharmacol. Exp. Ther. 2000, 294, 45–51. [Google Scholar]
- Sardi, S.P.; Rey-Ares, V.; Pujol-Lereis, V.A.; Serrano, S.A.; Rothlin, R.P. Further pharmacological evidence of nuclear factor-κB pathway involvement in bradykinin B1 receptor-sensitized responses in human umbilical vein. J. Pharmacol. Exp. Ther. 2002, 301, 975–980. [Google Scholar] [CrossRef]
- Gera, L.; Roy, C.; Charest-Morin, X.; Marceau, F. Vasopeptidase-activated latent ligands of the histamine receptor-1. Int. Immunopharmacol. 2013, 17, 677–683. [Google Scholar] [CrossRef][Green Version]





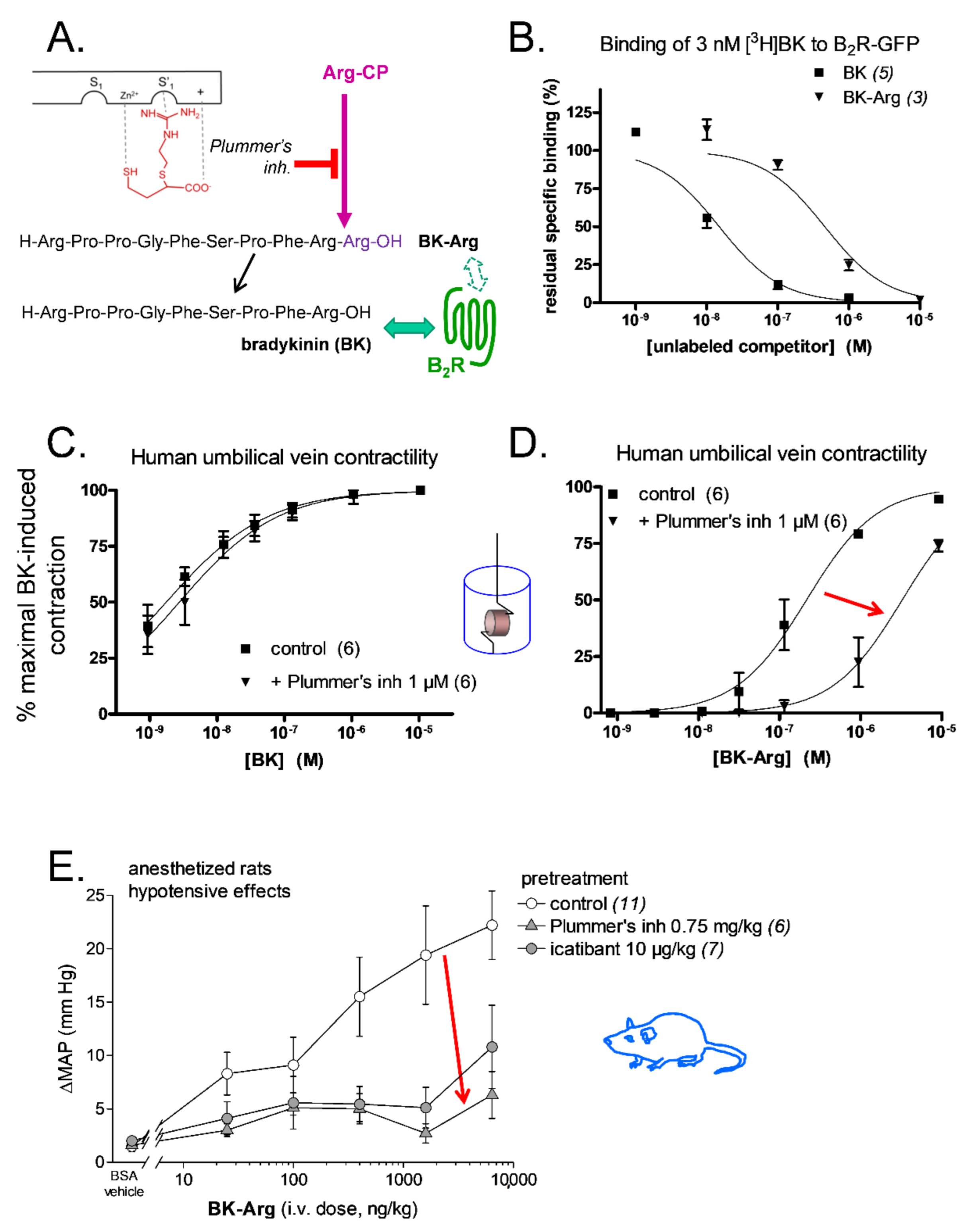
| Peptide | Loss of Affinity vs. BK, Competition of [3H]BK Binding to Recombinant B2R | Effect of Peptidase Inhibitor on Apparent Potency, hUV Contractility Assay | In Vivo Validation: Inhibitor Modulation of Hypotensive Effect in Rats | Ref. | |
|---|---|---|---|---|---|
| Plummer’s Inhibitor | Enalaprilat | ||||
| BK | - | none | none | Plummer’s inh ↔ enalaprilat ↑ | [62,73,89]; Figure 5C |
| Met-Lys-BK-Ser-Ser | >100-fold ↓ | NT | 12-fold ↓ | NT | [62] |
| BK-Arg | 29-fold ↓ | 15-fold ↓ | NT | Plummer’s inh ↓ enalaprilat ↑ | [73,89]; Figure 5D |
| BK-Ser-Tyr | 102-fold ↓ | NT | 3.8-fold ↓ | NT | [89] |
| BK-His-Leu | 363-fold ↓ | NT | 19-fold ↓ | enalaprilat ↔ | [73,89] |
| BK-Ala-Pro | 490-fold ↓ | NT | 11-fold ↓ | enalaprilat ↑ | [73,89] |
| d-Arg-BK-Arg-Arg | 61-fold ↓ | 2-fold ↓ | 3.5-fold ↓ | Plummer’s inh ↓ enalaprilat ↔ | [91] |
| Maximakinin b | human B2R: 1500-fold ↓ rat B2R: 6-fold ↓ | NT c | NT | enalaprilat ↔ | [90] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marceau, F.; Bachelard, H. A Robust Bioassay of the Human Bradykinin B2 Receptor That Extends Molecular and Cellular Studies: The Isolated Umbilical Vein. Pharmaceuticals 2021, 14, 177. https://doi.org/10.3390/ph14030177
Marceau F, Bachelard H. A Robust Bioassay of the Human Bradykinin B2 Receptor That Extends Molecular and Cellular Studies: The Isolated Umbilical Vein. Pharmaceuticals. 2021; 14(3):177. https://doi.org/10.3390/ph14030177
Chicago/Turabian StyleMarceau, François, and Hélène Bachelard. 2021. "A Robust Bioassay of the Human Bradykinin B2 Receptor That Extends Molecular and Cellular Studies: The Isolated Umbilical Vein" Pharmaceuticals 14, no. 3: 177. https://doi.org/10.3390/ph14030177
APA StyleMarceau, F., & Bachelard, H. (2021). A Robust Bioassay of the Human Bradykinin B2 Receptor That Extends Molecular and Cellular Studies: The Isolated Umbilical Vein. Pharmaceuticals, 14(3), 177. https://doi.org/10.3390/ph14030177