
An Angiopep2-PAPTP Construct Overcomes the Blood-Brain Barrier. New Perspectives against Brain Tumors

 ,
,  , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. Results
2.1. Preliminary Studies
2.2. Synthesis of PAPTPL-I, TAT-PAPTP, and An2-PAPTP
2.2.1. Synthesis of PAPTPL-I
2.2.2. Synthesis of TAT-PAPTPL
2.2.3. Synthesis of An2-PAPTPL
2.3. Cytotoxicity of PAPTP-Linker (PAPTPL)
2.4. Aqueous Solubility
2.5. Stability in Blood
2.6. Brain Delivery of TAT-PAPTP
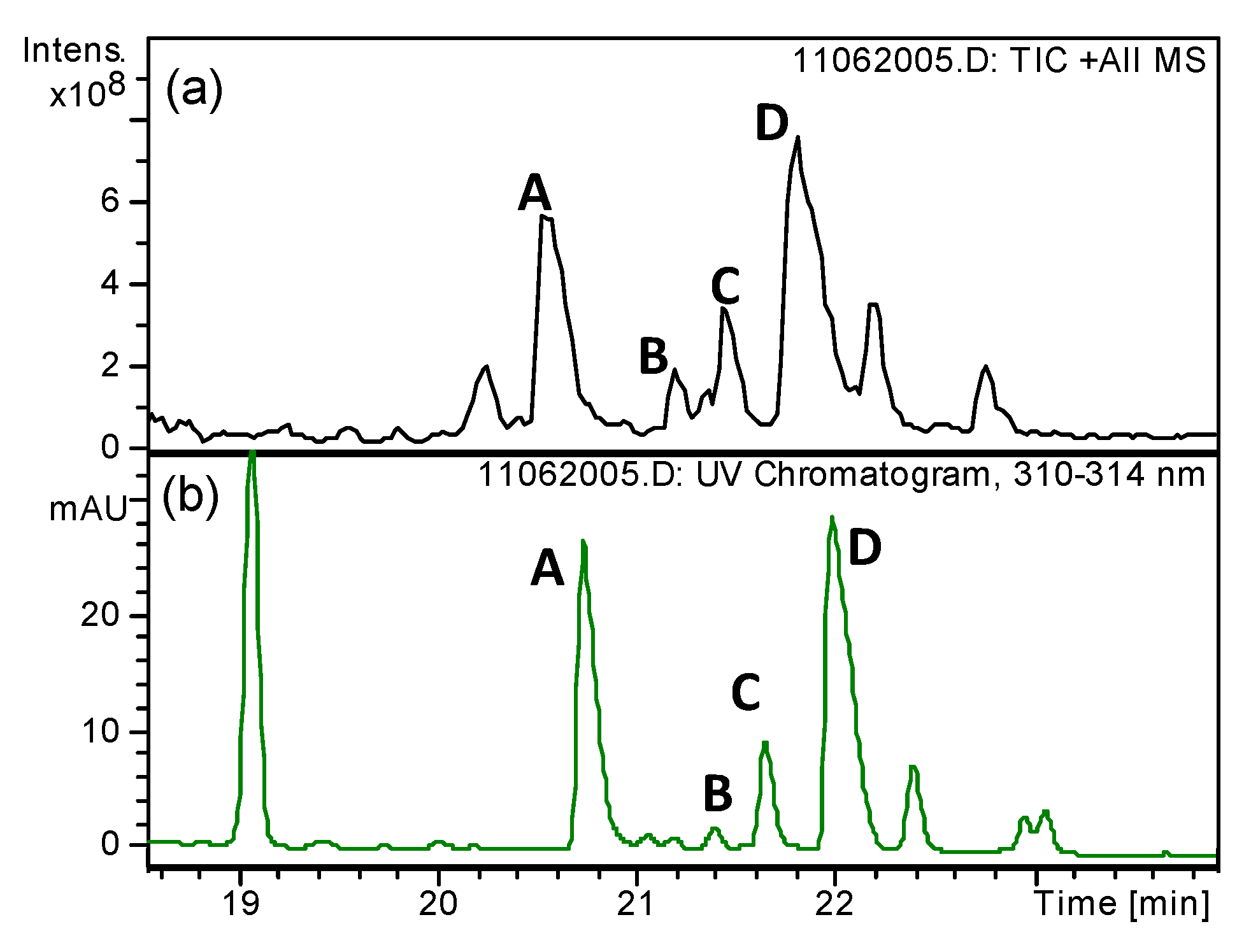
2.7. Tissue Distribution of An2-PAPTP
2.7.1. Brain Delivery of An2-PAPTP
2.7.2. Liver Metabolism of An2-PAPTP
2.7.3. Levels of An2-PAPTP and Metabolites in Other Organs
2.8. Angiopep2-PAPTP Does Not Disrupt Membranes
3. Discussion
4. Materials and Methods
4.1. Chemicals and Instrumentation
4.1.1. Nuclear Magnetic Resonance (NMR) Analysis
4.1.2. Preparative High-Performance Liquid Chromatography (HPLC)
4.1.3. HPLC-UV (Ultraviolet) Analysis
4.1.4. HPLC-UV/Electrospray Ionization–Mass Spectrometry (ESI-MS) Analysis
4.1.5. High-Resolution Mass Spectrometry (HRMS) Analysis
4.2. Synthesis of TAT-PAPTP and An2-PAPTP
4.2.1. Synthesis of Intermediate 4
Synthesis of 4-(3-chloropropyl)phenol (1)
Synthesis of tert-butyl 3-methyl-2-(((4-nitrophenoxy)carbonyl)amino)pentanoate (2)
Synthesis of tert-butyl 2-(((4-(3-chloropropyl)phenoxy)carbonyl)amino)-3-methylpentanoate (3)
Synthesis of tert-butyl 2-(((4-(3-iodopropyl)phenoxy)carbonyl)amino)-3-methylpentanoate (4)
4.2.2. Synthesis of Intermediate PAPTPL-I
Synthesis of 4-hydroxy-7H-furo[3,2-g]chromen-7-one (5)
Synthesis of 4-(4-chlorobutoxy)-7H-furo[3,2-g]chromen-7-one (6)
Synthesis of 4-(4-iodobutoxy)-7H-furo[3,2-g]chromen-7-one (7)
Synthesis of 4-(4-(4-(3-hydroxypropyl)phenoxy)butoxy)-7H-furo[3,2-g]chromen-7-one (8)
Synthesis of 4-(4-(4-(3-chloropropyl)phenoxy)butoxy)-7H-furo[3,2-g]chromen-7-one (9)
Synthesis of 4-(4-(4-(3-iodopropyl)phenoxy)butoxy)-7H-furo[3,2-g]chromen-7-one (10)
Synthesis of (4-hydroxyphenyl)(3-(4-(4-((7-oxo-7H-furo[3,2-g]chromen-4-yl)oxy)butoxy) phenyl)propyl) diphenylphosphonium iodide (11)
Synthesis of (4-(3-(4-(((1-(tert-butoxy)-3-methyl-1-oxopentan-2-yl)carbamoyl)oxy)phenyl) propoxy)phenyl)(3-(4-(4-((7-oxo-7H-furo[3,2-g]chromen-4-yl)oxy)butoxy)phenyl)propyl)diphenylphosphonium 2,2,2-trifluoroacetate (12)
Synthesis of (4-(3-(4-(((1-carboxy-2-methylbutyl)carbamoyl)oxy)phenyl)propoxy)phenyl) (3-(4-(4-((7-oxo-7H-furo[3,2-g]chromen-4-yl)oxy)butoxy)phenyl)propyl)diphenylphosphonium 2,2,2-trifluoroacetate (PAPTPL-I)
4.2.3. Peptide Synthesis
4.2.4. Synthesis of TAT-PAPTP
4.2.5. Synthesis of An2-PAPTP
4.3. Synthesis of PAPTPL
4.4. Solubility Determinations
4.5. Cell Cultures
4.6. MTS Assay
4.7. Quantification of PAPTPL and Peptide-PAPTP Conjugates in Tissues
4.7.1. Recovery of PAPTPL
4.7.2. Recovery of TAT-PAPTP from Brain
4.7.3. Recovery of An2-PAPTP
4.8. Stability in Blood
4.9. In Vivo Experiments
4.10. Planar Bilayer Experiments with An2-PAPTP and TAT-PAPTP
4.11. Haemolysis Assays
4.12. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| An2 | Angiopep2 |
| ACN | acetonitrile |
| AUC | area under the curve |
| BBB | blood-brain barrier |
| bw | body weight |
| CD | circular dichroism |
| CLL | chronic lymphocytic leukemia |
| CNS | central nervous system |
| CPP | cell-penetrating peptide |
| DCM | dichloromethane |
| DIPEA | N,N-diisopropylethylamine |
| DMAP | 4-dimethylaminopyridine |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMF | N,N-dimethylformamide |
| DMSO | dimethylsulfoxide |
| DOX | doxorubicin |
| DTX | docetaxel |
| ESI | electrospray ionization |
| EtOAc | ethyl acetate |
| Et2O | diethyl ether |
| Fmoc | fluoren-9-ylmethoxycarbonyl |
| HATU | 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate |
| HBTU | 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate |
| HOBt | 1-hydroxybenzotriazole |
| HPLC | high-performance liquid chromatography |
| HRMS | high-resolution mass spectrometry |
| IMM | inner mitochondrial membrane |
| i.p. | intraperitoneal |
| i.v. | intravenous |
| Kv1.3 | voltage-dependent potassium channel 1.3 |
| LRP-1 | low-density lipoprotein receptor related Protein-1 |
| MDR | multiple drug resistance |
| MS | mass spectrometry |
| MTS | 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium |
| PBS | phosphate-buffered saline |
| PDAC | pancreatic ductal adenocarcinoma |
| PTX | paclitaxel |
| r.t. | room temperature |
| SEM | standard error of the mean |
| TEA | triethylamine |
| TFA | trifluoroacetic acid |
| TIPS | triisopropyl silane |
| TPP | triphenylphosphonium |
| WT | wild type |
References
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Lopez, M.; Joseph, J.; Zielonka, J.; Dwinell, M.B. A review of the basics of mitochondrial bioenergetics, metabolism, and related signaling pathways in cancer cells: Therapeutic targeting of tumor mitochondria with lipophilic cationic compounds. Redox Biol. 2018, 14, 316–327. [Google Scholar] [CrossRef]
- Ralph, S.J.; Rodríguez-Enríquez, S.; Neuzil, J.; Saavedra, E.; Moreno-Sánchez, R. The causes of cancer revisited: “Mitochondrial malignancy” and ROS-induced oncogenic transformation—Why mitochondria are targets for cancer therapy. Mol. Asp. Med. 2010, 31, 145–170. [Google Scholar] [CrossRef]
- Bing, Y.; Dong, L.; Neuzil, J. Mitochondria: An intriguing target for killing tumour-initiating cells. Mitochondrion 2016, 26, 86–93. [Google Scholar] [CrossRef]
- Denisenko, T.V.; Gorbunova, A.S.; Zhivotovsky, B. Mitochondrial involvement in migration, invasion and metastasis. Front. Cell Dev. Biol. 2019, 7, 355. [Google Scholar] [CrossRef] [PubMed]
- Jeena, M.; Kim, S.; Jin, S.; Ryu, J.-H. Recent progress in mitochondria-targeted drug and drug-free agents for cancer therapy. Cancers 2019, 12, 4. [Google Scholar] [CrossRef]
- Leanza, L.; Romio, M.; Becker, K.A.; Azzolini, M.; Trentin, L.; Manago, A.; Venturini, E.; Zaccagnino, A.; Mattarei, A.; Carraretto, L.; et al. Direct pharmacological targeting of a mitochondrial ion channel selectively kills tumor cells in vivo. Cancer Cell 2017, 31, 516–531. [Google Scholar] [CrossRef] [PubMed]
- Hao, B.; Chen, Z.-W.; Zhou, X.-J.; Zimin, P.I.; Miljanich, G.P.; Wulff, H.; Wang, Y.-X. Identification of phase-I metabolites and chronic toxicity study of the Kv1.3 blocker PAP-1 (5-(4-phenoxybutoxy)psoralen) in the rat. Xenobiotica Fate Foreign Compd. Biol. Syst. 2010, 41, 198–211. [Google Scholar] [CrossRef]
- Schmitz, A.; Sankaranarayanan, A.; Azam, P.; Schmidt-Lassen, K.; Homerick, D.; Hänsel, W.; Wulff, H. Design of PAP-1, a selective small molecule Kv1.3 blocker, for the suppression of effector memory T cells in autoimmune diseases. Mol. Pharmacol. 2005, 68, 1254–1270. [Google Scholar] [CrossRef] [PubMed]
- Szabò, I.; Bock, J.; Jekle, A.; Soddemann, M.; Adams, C.; Lang, F.; Zoratti, M.; Gulbins, E. A novel potassium channel in lymphocyte mitochondria. J. Biol. Chem. 2005, 280, 12790–12798. [Google Scholar] [CrossRef] [PubMed]
- Peruzzo, R.; Mattarei, A.; Azzolini, M.; Becker-Flegler, K.A.; Romio, M.; Rigoni, G.; Carrer, A.; Biasutto, L.; Parrasia, S.; Kadow, S.; et al. Insight into the mechanism of cytotoxicity of membrane-permeant psoralenic Kv1.3 channel inhibitors by chemical dissection of a novel member of the family. Redox Biol. 2020, 37, 101705. [Google Scholar] [CrossRef]
- Smith, R.A.J.; Hartley, R.C.; Murphy, M.P. Mitochondria-targeted small molecule therapeutics and probes. Antioxid. Redox Signal. 2011, 15, 3021–3038. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Li, J.; Xiao, Y.; Fu, B.; Qin, Z. TPP-based mitocans: A potent strategy for anticancer drug design. RSC Med. Chem. 2020, 11, 858–875. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Novillo, C.; Capera, J.; Colomer-Molera, M.; Condom, E.; Ferreres, J.C.; Felipe, A. Implication of voltage-gated potassium channels in neoplastic cell proliferation. Cancers 2019, 11, 287. [Google Scholar] [CrossRef] [PubMed]
- Beeton, C.; Wulff, H.; Standifer, N.E.; Azam, P.; Mullen, K.M.; Pennington, M.W.; Kolski-Andreaco, A.; Wei, E.; Grino, A.; Counts, D.R.; et al. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc. Natl. Acad. Sci. USA 2006, 103, 17414–17419. [Google Scholar] [CrossRef]
- Teisseyre, A.; Palko-Łabuz, A.; Sroda-Pomianek, K.; Michalak, K. Voltage-gated potassium channel Kv1.3 as a target in therapy of cancer. Front. Oncol. 2019, 9, 933. [Google Scholar] [CrossRef]
- Venturini, E.; Leanza, L.; Azzolini, M.; Kadow, S.; Mattarei, A.; Weller, M.; Tabatabai, G.; Edwards, M.J.; Zoratti, M.; Paradisi, C.; et al. Targeting the potassium channel Kv1.3 kills glioblastoma cells. Neurosignals 2017, 25, 26–38. [Google Scholar] [CrossRef]
- Chen, Y.; Nguyen, H.M.; Maezawa, I.; Jin, L.; Wulff, H. Inhibition of the potassium channel Kv1.3 reduces infarction and inflammation in ischemic stroke. Ann. Clin. Transl. Neurol. 2017, 5, 147–161. [Google Scholar] [CrossRef]
- Di Lucente, J.; Nguyen, H.M.; Wulff, H.; Jin, L.-W.; Maezawa, I. The voltage-gated potassium channel Kv1.3 is required for microglial pro-inflammatory activation in vivo. Glia 2018, 66, 1881–1895. [Google Scholar] [CrossRef]
- Grimaldi, A.; D’Alessandro, G.; Di Castro, M.A.; Lauro, C.; Singh, V.; Pagani, F.; Sforna, L.; Grassi, F.; Di Angelantonio, S.; Catacuzzeno, L.; et al. Kv1.3 activity perturbs the homeostatic properties of astrocytes in glioma. Sci. Rep. 2018, 8, 7654. [Google Scholar] [CrossRef]
- Wulff, H.; Nguyen, H.M.; Di Lucente, J.; Jenkins, D.P.; Singh, V.; Hilt, S.; Kim, K.; Rangaraju, S.; Levey, A.I.; Wulff, H.; et al. Kv1.3 inhibition as a potential microglia-targeted therapy for Alzheimer’s disease: Preclinical proof of concept. Brain J. Neurol. 2017, 141, 596–612. [Google Scholar] [CrossRef]
- DiChiara, M.; Amata, B.; Turnaturi, R.; Marrazzo, A.; Amata, E. Tuning properties for blood–brain barrier permeation: A statistics-based analysis. ACS Chem. Neurosci. 2019, 11, 34–44. [Google Scholar] [CrossRef]
- Gupta, M.; Lee, H.J.; Barden, C.J.; Weaver, D.F. The Blood-Brain Barrier (BBB) score. J. Med. Chem. 2019, 62, 9824–9836. [Google Scholar] [CrossRef]
- Roy, D.; Hinge, V.K.; Kovalenko, A. To pass or not to pass: Predicting the blood–brain barrier permeability with the 3D-RISM-KH molecular solvation theory. ACS Omega 2019, 4, 16774–16780. [Google Scholar] [CrossRef] [PubMed]
- Shityakov, S.; Neuhaus, W.; Dandekar, T.; Förster, C. Analysing molecular polar surface descriptors to predict blood-brain barrier permeation. Int. J. Comput. Biol. Drug Des. 2013, 6, 146–156. [Google Scholar] [CrossRef]
- Ghosh, D.; Peng, X.; Leal, J.; Mohanty, R.P. Peptides as drug delivery vehicles across biological barriers. J. Pharm. Investig. 2017, 48, 89–111. [Google Scholar] [CrossRef]
- Jafari, B.; Pourseif, M.M.; Barar, J.; Rafi, M.A.; Omidi, Y. Peptide-mediated drug delivery across the blood-brain barrier for targeting brain tumors. Expert Opin. Drug Deliv. 2019, 16, 583–605. [Google Scholar] [CrossRef]
- Raucher, D. Tumor targeting peptides: Novel therapeutic strategies in glioblastoma. Curr. Opin. Pharmacol. 2019, 47, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Navarro, M.; Teixidó, M.; Giralt, E. Jumping hurdles: Peptides able to overcome biological barriers. Acc. Chem. Res. 2017, 50, 1847–1854. [Google Scholar] [CrossRef]
- Varnamkhasti, B.S.; Jafari, S.; Taghavi, F.; Alaei, L.; Izadi, Z.; Lotfabadi, A.; Dehghanian, M.; Jaymand, M.; Derakhshankhah, H.; Saboury, A.A. Cell-penetrating peptides: As a promising theranostics strategy to circumvent the blood-brain barrier for CNS diseases. Curr. Drug Deliv. 2020, 17, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Vale, N.; Duarte, D.; Silva, S.; Correia, A.S.; Costa, B.; Gouveia, M.J.; Ferreira, A. Cell-penetrating peptides in oncologic pharmacotherapy: A review. Pharmacol. Res. 2020, 162, 105231. [Google Scholar] [CrossRef]
- Qin, Y.; Zhang, Q.; Chen, H.; Yuan, W.; Kuai, R.; Xie, F.; Zhang, L.; Wang, X.; Zhang, Z.; Liu, J.; et al. Comparison of four different peptides to enhance accumulation of liposomes into the brain. J. Drug Target. 2011, 20, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Stalmans, S.; Bracke, N.; Wynendaele, E.; Gevaert, B.; Peremans, K.; Burvenich, C.; Polis, I.; De Spiegeleer, B. Cell-penetrating peptides selectively cross the blood-brain barrier in vivo. PLoS ONE 2015, 10, e0139652. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, Y.; Currie, J.-C.; Demeule, M.; Régina, A.; Che, C.; Abulrob, A.; Fatehi, R.; Sartelet, H.; Gabathuler, R.; Castaigne, J.-P.; et al. Transport characteristics of a novel peptide platform for CNS therapeutics. J. Cell. Mol. Med. 2010, 14, 2827–2839. [Google Scholar] [CrossRef] [PubMed]
- Demeule, M.; Régina, A.; Ché, C.; Poirier, J.; Nguyen, T.; Gabathuler, R.; Castaigne, J.-P.; Béliveau, R. Identification and design of peptides as a new drug delivery system for the brain. J. Pharmacol. Exp. Ther. 2007, 324, 1064–1072. [Google Scholar] [CrossRef]
- Eiselt, E.; Otis, V.; Belleville, K.; Yang, G.; Larocque, A.; Regina, A.; Demeule, M.; Sarret, P.; Gendron, L. Use of a noninvasive brain-penetrating peptide-drug conjugate strategy to improve the delivery of opioid pain relief medications to the brain. J. Pharmacol. Exp. Ther. 2020, 374, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Kurzrock, R.; Gabrail, N.; Chandhasin, C.; Moulder, S.; Smith, C.; Brenner, A.; Sankhala, K.; Mita, A.; Elian, K.; Bouchard, D.; et al. Safety, pharmacokinetics, and activity of GRN1005, a novel conjugate of angiopep-2, a peptide facilitating brain penetration, and paclitaxel, in patients with advanced solid tumors. Mol. Cancer Ther. 2012, 11, 308–316. [Google Scholar] [CrossRef]
- Demeule, M.; Currie, J.-C.; Bertrand, Y.; Ché, C.; Nguyen, T.; Régina, A.; Gabathuler, R.; Castaigne, J.-P.; Béliveau, R. Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector angiopep-2. J. Neurochem. 2008, 106, 1534–1544. [Google Scholar] [CrossRef]
- O’Sullivan, C.C.; Lindenberg, M.; Bryla, C.; Patronas, N.J.; Peer, C.J.; Amiri-Kordestani, L.; Davarpanah, N.; Gonzalez, E.; Burotto, M.; Choyke, P.L.; et al. ANG1005 for breast cancer brain metastases: Correlation between 18F-FLT-PET after first cycle and MRI in response assessment. Breast Cancer Res. Treat. 2016, 160, 51–59. [Google Scholar] [CrossRef]
- Kumthekar, P.; Tang, S.-C.; Brenner, A.J.; Kesari, S.; Piccioni, D.E.; Anders, C.K.; Carrillo, J.A.; Chalasani, P.; Kabos, P.; Puhalla, S.L.; et al. ANG1005, a brain-penetrating peptide–drug conjugate, shows activity in patients with breast cancer with leptomeningeal carcinomatosis and recurrent brain metastases. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 2789–2799. [Google Scholar] [CrossRef]
- Vacondio, F.; Silva, C.; Mor, M.; Testa, B. Qualitative structure-metabolism relationships in the hydrolysis of carbamates. Drug Metab. Rev. 2010, 42, 551–589. [Google Scholar] [CrossRef]
- Shytikov, D.; Balva, O.; Debonneuil, E.; Glukhovskiy, P.; Pishel, I.M. Aged mice repeatedly injected with plasma from young mice: A survival study. BioRes. Open Access 2014, 3, 226–232. [Google Scholar] [CrossRef]
- Villeda, S.A.; Plambeck, K.E.; Middeldorp, J.; Castellano, J.M.; Mosher, K.I.; Luo, J.; Smith, L.K.; Bieri, G.; Lin, K.; Berdnik, D.; et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat. Med. 2014, 20, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Kourie, J.I.; Shorthouse, A.A. Properties of cytotoxic peptide-formed ion channels. Am. J. Physiol. Cell Physiol. 2000, 278, 1063–1087. [Google Scholar] [CrossRef]
- Hanke, W.; Methfessel, C.; Wilmsen, H.U.; Katz, E.; Jung, G.; Boheim, G. Melittin and a chemically modified trichotoxin form alamethicin-type multi-state pores. Biochim. Biophys. Acta 1983, 727, 108–114. [Google Scholar] [CrossRef]
- Kelso, G.F.; Porteous, C.M.; Hughes, G.; Ledgerwood, E.C.; Gane, A.M.; Smith, R.A.J.; Murphy, M.P. Prevention of mitochondrial oxidative damage using targeted antioxidants. Ann. N. Y. Acad. Sci. 2002, 959, 263–274. [Google Scholar] [CrossRef]
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef] [PubMed]
- Oliver, D.M.A.; Reddy, P.H. Small molecules as therapeutic drugs for Alzheimer’s disease. Mol. Cell. Neurosci. 2019, 96, 47–62. [Google Scholar] [CrossRef]
- Smith, R.A.J.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Zhu, Z.; Leung, C.W.T.; Xi, W.; Su, L.; Chen, G.; Qin, A.; Tang, B.Z.; Zhenfeng, Z. Long-term two-photon neuroimaging with a photostable AIE luminogen. Biomed. Opt. Express 2015, 6, 1477–1486. [Google Scholar] [CrossRef]
- McManus, M.J.; Murphy, M.P.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef]
- Young, M.L.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ inhibits memory loss, neuropathology, and extends lifespan in aged 3xTg-AD mice. Mol. Cell. Neurosci. 2019, 101, 103409. [Google Scholar] [CrossRef]
- El-Andaloussi, S.; Järver, P.; Johansson, H.J.; Langel, U. Cargo-dependent cytotoxicity and delivery efficacy of cell-penetrating peptides: A comparative study. Biochem. J. 2007, 407, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Genrikhs, E.E.; Stelmashook, E.V.; Alexandrova, O.P.; Novikova, S.V.; Voronkov, D.N.; Glibka, Y.A.; Skulachev, V.P.; Isaev, N.K. The single intravenous administration of mitochondria-targeted antioxidant SkQR1 after traumatic brain injury attenuates neurological deficit in rats. Brain Res. Bull. 2019, 148, 100–108. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, H.; Shen, R.; Fang, J.; Yang, Y.; Dai, W.; Zhu, Y.; Zhou, M. Mitochondrial-targeted antioxidant MitoQ provides neuroprotection and reduces neuronal apoptosis in experimental traumatic brain injury possibly via the Nrf2-ARE pathway. Am. J. Transl. Res. 2018, 10, 1887–1899. [Google Scholar]
- Isaev, N.K.; Stelmashook, E.V.; Genrikhs, E.E.; Korshunova, G.A.; Sumbatyan, N.V.; Kapkaeva, M.R.; Skulachev, V.P. Neuroprotective properties of mitochondria-targeted antioxidants of the SkQ-type. Rev. Neurosci. 2016, 27, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Silachev, D.N.; Plotnikov, E.Y.; Zorova, L.D.; Pevzner, I.B.; Sumbatyan, N.V.; Korshunova, G.A.; Vladimirivich, G.M.; Pirogov, Y.A.; Skulachev, V.P.; Zorov, D.B. Neuroprotective effects of mitochondria-targeted plastoquinone and thymoquinone in a rat model of brain ischemia/reperfusion injury. Molecules 2015, 20, 14487–14503. [Google Scholar] [CrossRef]
- Porteous, C.M.; Menon, D.K.; Aigbirhio, F.I.; Smith, R.A.; Murphy, M.P. P-glycoprotein (Mdr1a/1b) and breast cancer resistance protein (Bcrp) decrease the uptake of hydrophobic alkyl triphenylphosphonium cations by the brain. Biochim. Biophys. Acta 2013, 1830, 3458–3465. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pinho, B.R.; Duarte, A.I.; Canas, P.M.; Moreira, P.I.; Murphy, M.P.; Oliveira, J.M.A. The interplay between redox signalling and proteostasis in neurodegeneration: In vivo effects of a mitochondria-targeted antioxidant in Huntington’s disease mice. Free Radic. Biol. Med. 2020, 146, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Thomas, F.C.; Taskar, K.; Rudraraju, V.; Goda, S.; Thorsheim, H.R.; Gaasch, J.A.; Mittapalli, R.K.; Palmieri, D.; Steeg, P.S.; Lockman, P.R.; et al. Uptake of ANG1005, a novel paclitaxel derivative, through the blood-brain barrier into brain and experimental brain metastases of breast cancer. Pharm. Res. 2009, 26, 2486–2494. [Google Scholar] [CrossRef]
- Ché, C.; Yang, G.; Thiot, C.; Lacoste, M.-C.; Currie, J.-C.; Demeule, M.; Régina, A.; Béliveau, R.; Castaigne, J.-P. New angiopep-modified doxorubicin (ANG1007) and etoposide (ANG1009) chemotherapeutics with increased brain penetration. J. Med. Chem. 2010, 53, 2814–2824. [Google Scholar] [CrossRef] [PubMed]
- Kadari, A.; Pooja, D.; Gora, R.H.; Gudem, S.; Kolapalli, V.R.M.; Kulhari, H.; Sistla, R. Design of multifunctional peptide collaborated and docetaxel loaded lipid nanoparticles for antiglioma therapy. Eur. J. Pharm. Biopharm. 2018, 132, 168–179. [Google Scholar] [CrossRef]
- Qin, Y.; Chen, H.; Zhang, Q.; Wang, X.; Yuan, W.; Kuai, R.; Tang, J.; Zhang, L.; Zhang, Z.; Zhang, Q.; et al. Liposome formulated with TAT-modified cholesterol for improving brain delivery and therapeutic efficacy on brain glioma in animals. Int. J. Pharm. 2011, 420, 304–312. [Google Scholar] [CrossRef]
- Aguiar, L.; Biosca, A.; Lantero, E.; Gut, J.; Vale, N.; Rosenthal, P.J.; Nogueira, F.; Andreu, D.; Fernàndez-Busquets, X.; Gomes, P. Coupling the antimalarial cell penetrating peptide TP10 to classical antimalarial drugs primaquine and chloroquine produces strongly hemolytic conjugates. Molecules 2019, 24, 4559. [Google Scholar] [CrossRef]
- Li, Q.; Xu, M.; Cui, Y.; Huang, C.; Sun, M.-J. Arginine-rich membrane-permeable peptides are seriously toxic. Pharmacol. Res. Perspect. 2017, 5, e00334. [Google Scholar] [CrossRef]
- Rahn, J.J.; Lun, X.; Jorch, S.K.; Hao, X.; Venugopal, C.; Vora, P.; Ahn, B.Y.; Babes, L.; AlShehri, M.M.; Cairncross, J.G.; et al. Development of a peptide-based delivery platform for targeting malignant brain tumors. Biomaterials 2020, 252, 120105. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Shinohara, T.; Adachi, Y.; Asami, T.; Ohtaki, T. A novel LRP1-binding peptide L57 that crosses the blood brain barrier. Biochem. Biophys. Rep. 2017, 12, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Jiang, Y.; Meng, F.; Deng, C.; Cheng, R.; Zhang, J.; Feijen, J.; Zhong, Z. Highly efficacious and specific anti-glioma chemotherapy by tandem nanomicelles co-functionalized with brain tumor-targeting and cell-penetrating peptides. J. Control. Release 2018, 278, 1–8. [Google Scholar] [CrossRef]
- Behrendt, R.; White, P.; Offer, J. Advances in fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016, 22, 4–27. [Google Scholar] [CrossRef]
- Wade, J.D.; Mathieu, M.N.; Macris, M.; Tregear, G.W. Base-induced side reactions in fmoc-solid phase peptide synthesis: Minimization by use of piperazine as Nα-deprotectionreagent. Int. J. Pept. Res. Ther. 2000, 7, 107–112. [Google Scholar] [CrossRef]
- Lakkadwala, S.; Singh, J. Co-delivery of doxorubicin and erlotinib through liposomal nanoparticles for glioblastoma tumor regression using an in vitro brain tumor model. Colloids Surf. B Biointerfaces 2019, 173, 27–35. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time of Treatment (min) | Psoralenic Species (nmol/g Tissue) | Psoralenic Species (% of Injected Dose) |
|---|---|---|
| 15 | 0.3 ± 0.1 | 0.10 ± 0.04 |
| 30 | 0.4 ± 0.6 | 0.2 ± 0.2 |
| Time of Treatment (min) | PAPTPL | PAPTPL-I | PAPTPL-IT | PAPTPL-ITF |
|---|---|---|---|---|
| nmol/g fresh tissue | ||||
| 15 | 1.5 ± 0.6 | 0.5 ± 0.3 | 3.6 ± 1.3 | 2.2 ± 0.6 |
| 30 | 1.1 ± 0.3 | 0.2 in one animal out of four | 3.6 ± 2.6 | 1.7 ± 0.6 |
| 60 | 0.4 ± 0.2 | 5.4 ± 1.6 | 7.5 ± 2.1 | 4.5 ± 1.0 |
| % administered dose | ||||
| 15 | 1.2 ± 0.5 | 0.4 ± 0.2 | 2.8 ± 1.0 | 1.7 ± 1.0 |
| 30 | 0.9 ± 0.2 | 0.03 in one animal out of four | 2.7 ± 1.8 | 1.2 ± 0.5 |
| 60 | 0.4 ± 0.1 | 5.0 ± 1.8 | 6.2 ± 1.6 | 3.9 ± 1.0 |
| Time of Treatment (min) | Psoralenic Species (nmol/g Tissue) | Psoralenic Species (% of Injected Dose) |
|---|---|---|
| 15 | 23 ± 3 | 20 ± 3 |
| 30 | 17 ± 2 | 16 ± 1 |
| 60 | 11 ± 3 | 12 ± 1 |
| Time of Treatment (min) | Psoralenic Species (nmol/g Tissue) | Psoralenic Species (% of Administered Dose) |
|---|---|---|
| SPLEEN | ||
| 15 | 28 ± 9 | 1.2 ± 0.3 |
| 30 | 4.3 ± 1.4 | 0.27 ± 0.09 |
| 60 | 11 ± 3 | 0.8 ± 0.2 |
| KIDNEY | ||
| 15 | 5 ± 4 | 1.2 ± 0.8 |
| 30 | 1.3 ± 0.2 | 0.32 ± 0.07 |
| 60 | 1.2 ± 0.1 | 0.31 ± 0.03 |
| HEART | ||
| 15 | 0.2 ± 0.1 | 0.07 ± 0.01 |
| 30 | 0.36 in one animal out of four | 0.07 in one animal out of four |
| 60 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parrasia, S.; Rossa, A.; Varanita, T.; Checchetto, V.; De Lorenzi, R.; Zoratti, M.; Paradisi, C.; Ruzza, P.; Mattarei, A.; Szabò, I.; et al. An Angiopep2-PAPTP Construct Overcomes the Blood-Brain Barrier. New Perspectives against Brain Tumors. Pharmaceuticals 2021, 14, 129. https://doi.org/10.3390/ph14020129
Parrasia S, Rossa A, Varanita T, Checchetto V, De Lorenzi R, Zoratti M, Paradisi C, Ruzza P, Mattarei A, Szabò I, et al. An Angiopep2-PAPTP Construct Overcomes the Blood-Brain Barrier. New Perspectives against Brain Tumors. Pharmaceuticals. 2021; 14(2):129. https://doi.org/10.3390/ph14020129
Chicago/Turabian StyleParrasia, Sofia, Andrea Rossa, Tatiana Varanita, Vanessa Checchetto, Riccardo De Lorenzi, Mario Zoratti, Cristina Paradisi, Paolo Ruzza, Andrea Mattarei, Ildikò Szabò, and et al. 2021. "An Angiopep2-PAPTP Construct Overcomes the Blood-Brain Barrier. New Perspectives against Brain Tumors" Pharmaceuticals 14, no. 2: 129. https://doi.org/10.3390/ph14020129
APA StyleParrasia, S., Rossa, A., Varanita, T., Checchetto, V., De Lorenzi, R., Zoratti, M., Paradisi, C., Ruzza, P., Mattarei, A., Szabò, I., & Biasutto, L. (2021). An Angiopep2-PAPTP Construct Overcomes the Blood-Brain Barrier. New Perspectives against Brain Tumors. Pharmaceuticals, 14(2), 129. https://doi.org/10.3390/ph14020129