
MERTK Inhibition: Potential as a Treatment Strategy in EGFR Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer



Abstract

1. Introduction
2. Acquired EGFR-TKI Resistance in Lung Cancer
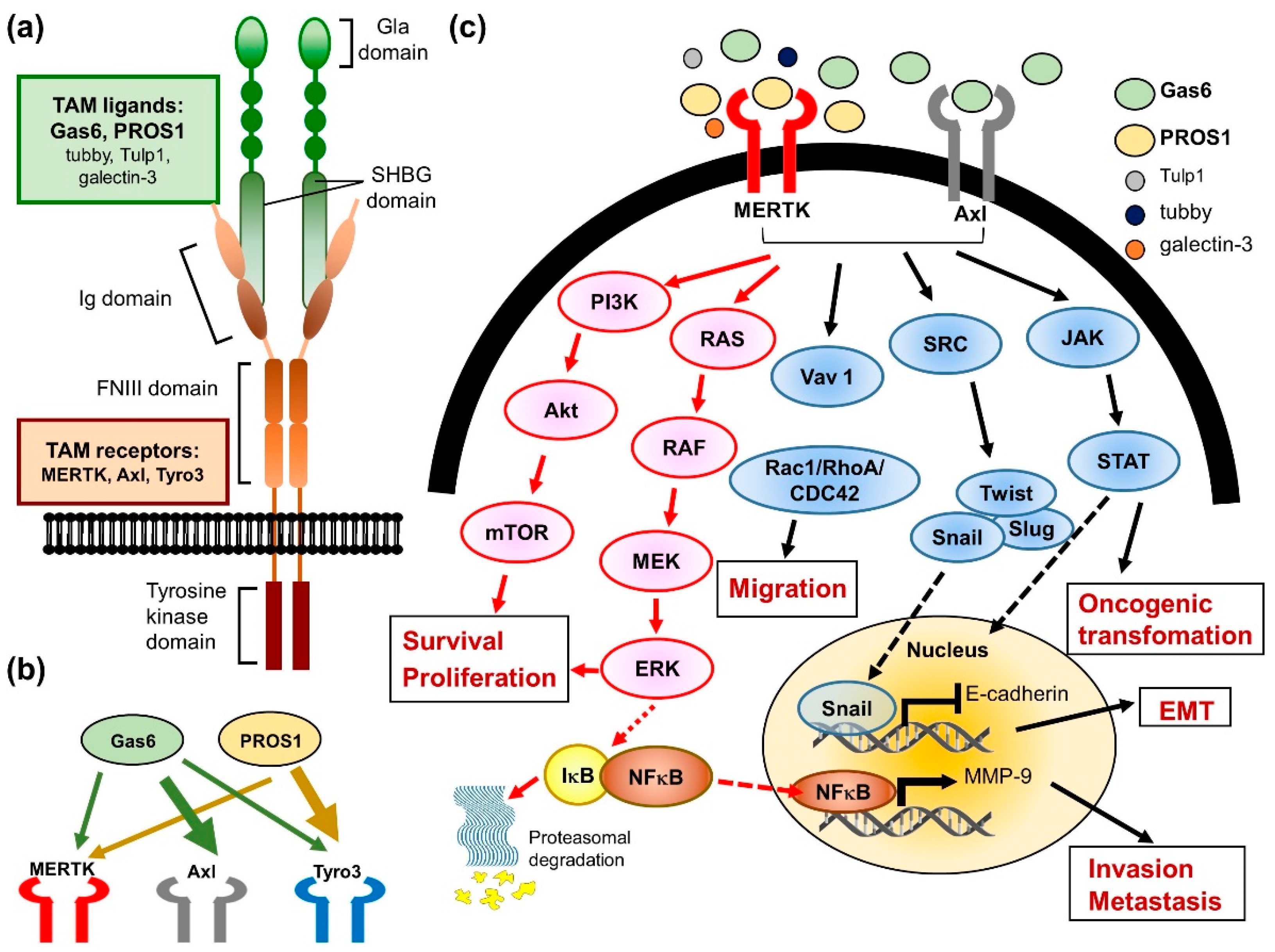
3. Biological Function and Regulation of MERTK
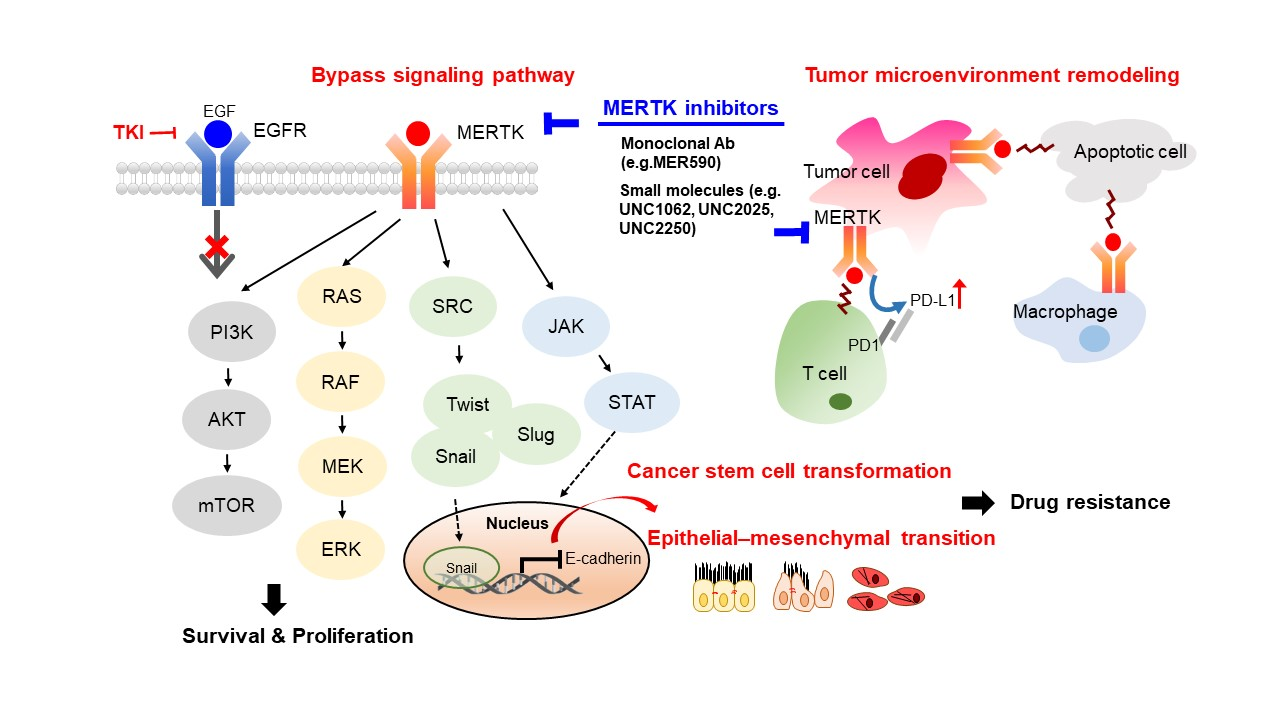
4. The Role of MERTK in EGFR-TKI Resistance
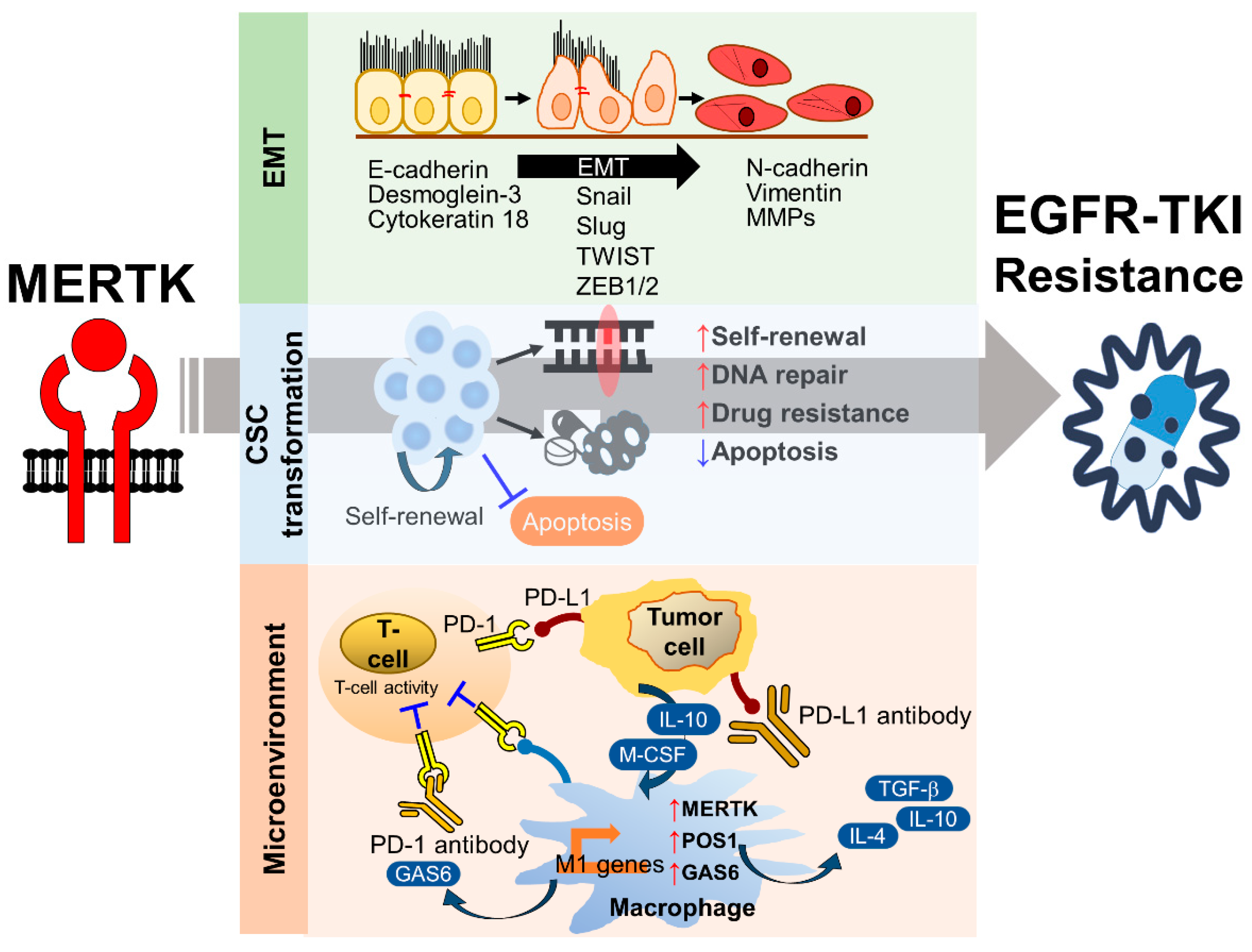
4.1. The Role of MERTK in EMT-Associated EGFR-TKI Resistance
4.1.1. TGF-β-Induced EMT and EGFR-TKI Resistance
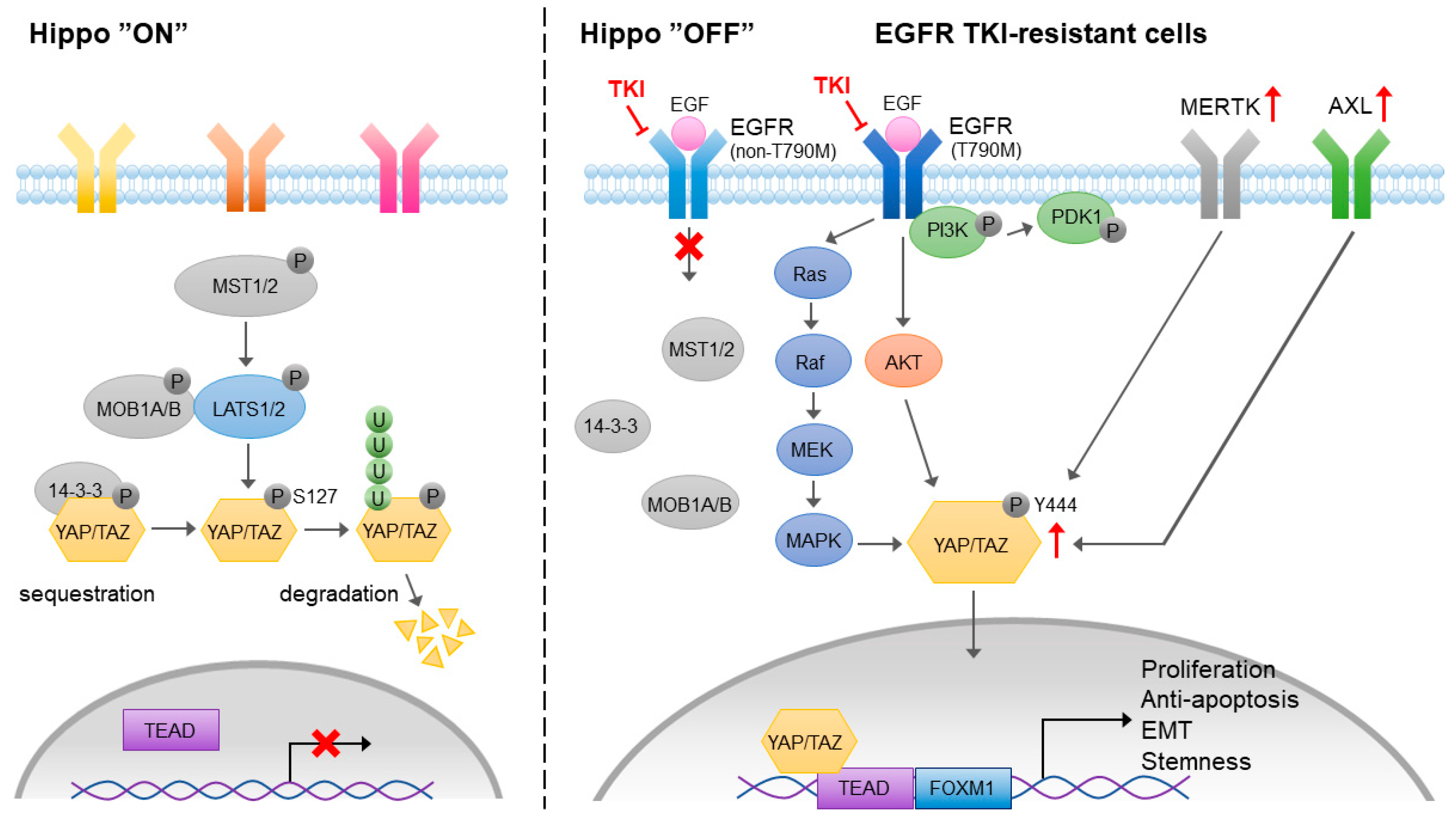
4.1.2. Hippo Pathway-Mediated EMT and EGFR-TKI Resistance
4.2. The Role of MERTK in the Cancer Stem Cell Phenotype
4.3. The Role of MERTK in Microenvironment Remodeling
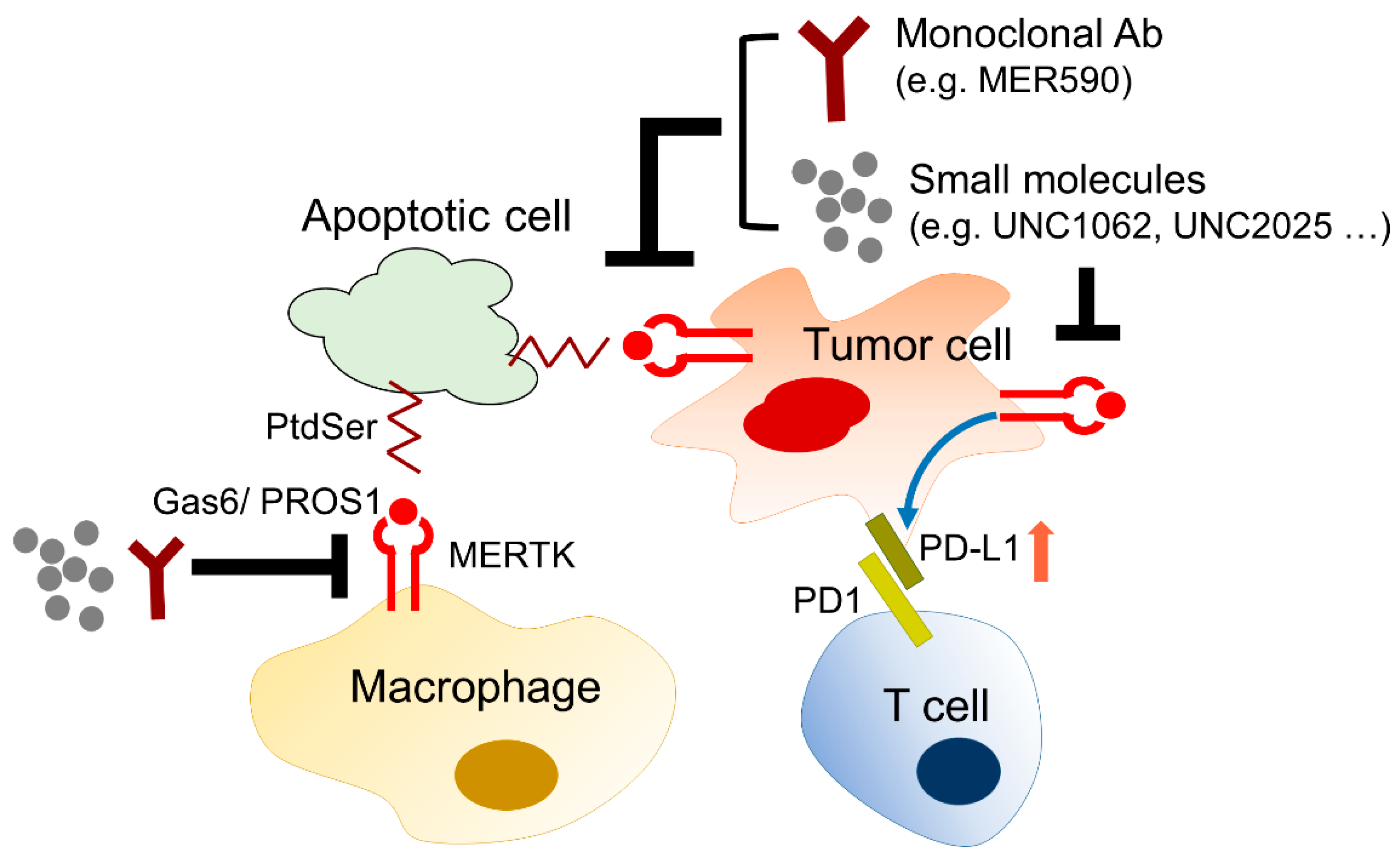
4.3.1. The Role of MERTK in Immune Checkpoint Inhibitor Therapy
The Regulation of PD-L1 Expression by MERTK
The Regulation of MERTK by PD-L1-Mediated EGFR-TKI Resistance
4.3.2. The Role of MERTK in Efferocytosis
5. Development of Biological Agents and Small Molecules Targeting MERTK
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung Cancer Statistics. Adv. Exp. Med. Biol. 2016, 893, 1–19. [Google Scholar]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Garshell, J.; Miller, D.; Altekruse, S.F.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; et al. (Eds.) SEER Cancer Statistics Review, 1975–2012; National Cancer Institute: Bethesda, MD, USA, 2015. Available online: http://seer.cancer.gov/csr/1975_2012/ (accessed on 31 January 2021).
- Ghafoor, Q.; Baijal, S.; Taniere, P.; O’Sullivan, B.; Evans, M.; Middleton, G. Epidermal Growth Factor Receptor (EGFR) Kinase Inhibitors and Non-Small Cell Lung Cancer (NSCLC)—Advances in Molecular Diagnostic Techniques to Facilitate Targeted Therapy. Pathol. Oncol. Res. 2017, 24, 723–731. [Google Scholar] [CrossRef]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef]
- Maione, P.; Sacco, P.C.; Sgambato, A.; Casaluce, F.; Rossi, A.; Gridelli, C. Overcoming resistance to targeted therapies in NSCLC: Current approaches and clinical application. Ther. Adv. Med. Oncol. 2015, 7, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Arcila, M.E.; Chmielecki, J.; Ladanyi, M.; Miller, V.A.; Pao, W. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin. Cancer Res. 2011, 17, 5530–5537. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef] [PubMed]
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef]
- Jotte, R.M.; Spigel, D.R. Advances in molecular-based personalized non-small-cell lung cancer therapy: Targeting epidermal growth factor receptor and mechanisms of resistance. Cancer Med. 2015, 4, 1621–1632. [Google Scholar] [CrossRef]
- Graham, D.K.; Dawson, T.L.; Mullaney, D.L.; Snodgrass, H.R.; Earp, H.S. Cloning and mRNA expression analysis of a novel human protooncogene, c-mer. Cell Growth Differ. 1994, 5, 647–657. [Google Scholar]
- Sather, S.; Kenyon, K.D.; Lefkowitz, J.B.; Liang, X.; Varnum, B.C.; Henson, P.M.; Graham, D.K. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood 2007, 109, 1026–1033. [Google Scholar] [CrossRef]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef]
- Tajbakhsh, A.; Rezaee, M.; Kovanen, P.T.; Sahebkar, A. Efferocytosis in atherosclerotic lesions: Malfunctioning regulatory pathways and control mechanisms. Pharmacol. Ther. 2018, 188, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, S.; Liu, W.; Yang, Y.; Zhang, L.; Zhang, S.; Jiang, Z.; Yang, Z.; Zhu, X. Targeted Next-Generation Sequencing Reveals a Novel Frameshift Mutation in the MERTK Gene in a Chinese Family with Retinitis Pigmentosa. Genet. Test. Mol. Biomark. 2018, 22, 165–169. [Google Scholar] [CrossRef]
- Kim, W.J.; Park, H.J.; Choi, Y.J.; Kwon, E.Y.; Kim, B.M.; Lee, J.H.; Chang, J.H.; Lee Kang, J.; Choi, J.H. Association between Genetic Variations of MERTK and Chronic Obstructive Pulmonary Disease in Koreans. J. Korean Med. Sci. 2018, 33, e56. [Google Scholar] [CrossRef]
- Branchford, B.R.; Stalker, T.J.; Law, L.; Acevedo, G.; Sather, S.; Brzezinski, C.; Wilson, K.M.; Minson, K.; Lee-Sherick, A.B.; Davizon-Castillo, P.; et al. The small-molecule MERTK inhibitor UNC2025 decreases platelet activation and prevents thrombosis. J. Thromb. Haemost. 2018, 16, 352–363. [Google Scholar] [CrossRef]
- Triantafyllou, E.; Pop, O.T.; Possamai, L.A.; Wilhelm, A.; Liaskou, E.; Singanayagam, A.; Bernsmeier, C.; Khamri, W.; Petts, G.; Dargue, R.; et al. MerTK expressing hepatic macrophages promote the resolution of inflammation in acute liver failure. Gut 2018, 67, 333–347. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; De Michieli, F.; Paschetta, E.; Pinach, S.; Saba, F.; Bongiovanni, D.; Framarin, L.; Berrutti, M.; Leone, N.; et al. MERTK rs4374383 variant predicts incident nonalcoholic fatty liver disease and diabetes: Role of mononuclear cell activation and adipokine response to dietary fat. Hum. Mol. Genet. 2017, 26, 1747–1758. [Google Scholar] [CrossRef] [PubMed]
- Cummings, C.T.; Deryckere, D.; Earp, H.S.; Graham, D.K. Molecular pathways: MERTK signaling in cancer. Clin. Cancer Res. 2013, 19, 5275–5280. [Google Scholar] [CrossRef]
- Png, K.J.; Halberg, N.; Yoshida, M.; Tavazoie, S.F. A microRNA regulon that mediates endothelial recruitment and metastasis by cancer cells. Nature 2011, 481, 190–194. [Google Scholar] [CrossRef]
- Faltermeier, C.M.; Drake, J.M.; Clark, P.M.; Smith, B.A.; Zong, Y.; Volpe, C.; Mathis, C.; Morrissey, C.; Castor, B.; Huang, J.; et al. Functional screen identifies kinases driving prostate cancer visceral and bone metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, E172–E181. [Google Scholar] [CrossRef] [PubMed]
- Vouri, M.; Hafizi, S. TAM Receptor Tyrosine Kinases in Cancer Drug Resistance. Cancer Res. 2017, 77, 2775–2778. [Google Scholar] [CrossRef]
- Verma, A.; Warner, S.L.; Vankayalapati, H.; Bearss, D.J.; Sharma, S. Targeting Axl and Mer kinases in cancer. Mol. Cancer Ther. 2011, 10, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Komurov, K.; Padron, D.; Cheng, T.; Roth, M.; Rosenblatt, K.P.; White, M.A. Comprehensive mapping of the human kinome to epidermal growth factor receptor signaling. J. Biol. Chem. 2010, 285, 21134–21142. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Kondo, K.; Yamaguchi, M.; Richmond, G.; Hutchison, M.; Wakeling, A.; Averbuch, S.; Wadsworth, P. Distribution and function of EGFR in human tissue and the effect of EGFR tyrosine kinase inhibition. Anticancer Res. 2003, 23, 3639–3650. [Google Scholar] [PubMed]
- Wang, K.; Yamamoto, H.; Chin, J.R.; Werb, Z.; Vu, T.H. Epidermal growth factor receptor-deficient mice have delayed primary endochondral ossification because of defective osteoclast recruitment. J. Biol. Chem. 2004, 279, 53848–53856. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.C.; Threadgill, D.W. Generation and validation of mice carrying a conditional allele of the epidermal growth factor receptor. Genesis 2009, 47, 85–92. [Google Scholar] [CrossRef]
- Schreier, B.; Rabe, S.; Schneider, B.; Bretschneider, M.; Rupp, S.; Ruhs, S.; Neumann, J.; Rueckschloss, U.; Sibilia, M.; Gotthardt, M.; et al. Loss of epidermal growth factor receptor in vascular smooth muscle cells and cardiomyocytes causes arterial hypotension and cardiac hypertrophy. Hypertension 2013, 61, 333–340. [Google Scholar] [CrossRef]
- Rayego-Mateos, S.; Morgado-Pascual, J.L.; Rodrigues-Diez, R.R.; Rodrigues-Diez, R.; Falke, L.L.; Mezzano, S.; Ortiz, A.; Egido, J.; Goldschmeding, R.; Ruiz-Ortega, M. Connective tissue growth factor induces renal fibrosis via epidermal growth factor receptor activation. J. Pathol. 2018, 244, 227–241. [Google Scholar] [CrossRef]
- Ghatge, M.; Nair, J.; Sharma, A.; Vangala, R.K. Integrative gene ontology and network analysis of coronary artery disease associated genes suggests potential role of ErbB pathway gene EGFR. Mol. Med. Rep. 2018, 17, 4253–4264. [Google Scholar] [CrossRef]
- Spindler, V.; Waschke, J. Pemphigus—A Disease of Desmosome Dysfunction Caused by Multiple Mechanisms. Front. Immunol. 2018, 9, 136. [Google Scholar] [CrossRef]
- Wang, H.; Yang, T.; Wang, T.; Hao, N.; Shen, Y.; Wu, Y.; Yuan, Z.; Chen, L.; Wen, F. Phloretin attenuates mucus hypersecretion and airway inflammation induced by cigarette smoke. Int. Immunopharmacol. 2018, 55, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Yewale, C.; Baradia, D.; Vhora, I.; Patil, S.; Misra, A. Epidermal growth factor receptor targeting in cancer: A review of trends and strategies. Biomaterials 2013, 34, 8690–8707. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Hiroki, K.; Yamashita, Y. The role of epidermal growth factor receptor in cancer metastasis and microenvironment. Biomed. Res. Int. 2013, 2013, 546318. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Yatabe, Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J. 2010, 277, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Rajaram, P.; Chandra, P.; Ticku, S.; Pallavi, B.K.; Rudresh, K.B.; Mansabdar, P. Epidermal growth factor receptor: Role in human cancer. Indian J. Dent. Res. 2017, 28, 687–694. [Google Scholar]
- Harris, R.C.; Chung, E.; Coffey, R.J. EGF receptor ligands. EGF Recept. Fam. 2003, 284, 2–13. [Google Scholar]
- Begley, M.J.; Yun, C.H.; Gewinner, C.A.; Asara, J.M.; Johnson, J.L.; Coyle, A.J.; Eck, M.J.; Apostolou, I.; Cantley, L.C. EGF-receptor specificity for phosphotyrosine-primed substrates provides signal integration with Src. Nat. Struct. Mol. Biol. 2015, 22, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Rappoport, J.Z. Interdependent epidermal growth factor receptor signalling and trafficking. Int. J. Biochem. Cell Biol. 2014, 51, 23–28. [Google Scholar] [CrossRef]
- Zandi, R.; Larsen, A.B.; Andersen, P.; Stockhausen, M.T.; Poulsen, H.S. Mechanisms for oncogenic activation of the epidermal growth factor receptor. Cell. Signal. 2007, 19, 2013–2023. [Google Scholar] [CrossRef]
- Komotar, R.J.; Starke, R.M.; Connolly, E.S.; Sisti, M.B. Alteration in NFKBIA and EGFR in glioblastoma multiforme. Neurosurgery 2011, 68, N14–N15. [Google Scholar] [CrossRef]
- Tong, L.; Yang, X.X.; Liu, M.F.; Yao, G.Y.; Dong, J.Y.; Ye, C.S.; Li, M. Mutational analysis of key EGFR pathway genes in Chinese breast cancer patients. Asian Pac. J. Cancer Prev. 2012, 13, 5599–5603. [Google Scholar] [CrossRef]
- Lv, N.; Xie, X.; Ge, Q.; Lin, S.; Wang, X.; Kong, Y.; Shi, H.; Xie, X.; Wei, W. Epidermal growth factor receptor in breast carcinoma: Association between gene copy number and mutations. Diagn. Pathol. 2011, 6, 118. [Google Scholar] [CrossRef]
- Metro, G.; Crino, L. Advances on EGFR mutation for lung cancer. Transl. Lung Cancer Res. 2012, 1, 5–13. [Google Scholar]
- Purba, E.R.; Saita, E.I.; Maruyama, I.N. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”. Cells 2017, 6, 13. [Google Scholar] [CrossRef]
- Tosoian, J.J.; Mamawala, M.; Epstein, J.I.; Landis, P.; Wolf, S.; Trock, B.J.; Carter, H.B. Intermediate and longer-term outcomes from a prospective active-surveillance program for favorable-risk prostate cancer. J. Clin. Oncol. 2015, 33, 3379–3385. [Google Scholar] [CrossRef]
- Yang, Z.; Hackshaw, A.; Feng, Q.; Fu, X.; Zhang, Y.; Mao, C.; Tang, J. Comparison of gefitinib, erlotinib and afatinib in non-small cell lung cancer: A meta-analysis. Int. J. Cancer 2017, 140, 2805–2819. [Google Scholar] [CrossRef]
- Ercan, D.; Choi, H.G.; Yun, C.H.; Capelletti, M.; Xie, T.; Eck, M.J.; Gray, N.S.; Janne, P.A. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin. Cancer Res. 2015, 21, 3913–3923. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, C.; Gasper, H.; Sahin, K.B.; Tang, M.; Kulasinghe, A.; Adams, M.N.; Richard, D.J.; O’Byrne, K.J. Epidermal Growth Factor Receptor (EGFR)-Mutated Non-Small-Cell Lung Cancer (NSCLC). Pharmaceuticals 2020, 13, 273. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Franchina, T.; Ricciardi, G.R.R.; Smiroldo, V.; Picciotto, M.; Zanghi, M.; Rolfo, C.; Adamo, V. Third generation EGFR TKIs in EGFR-mutated NSCLC: Where are we now and where are we going. Crit. Rev. Oncol. Hematol. 2017, 117, 38–47. [Google Scholar] [CrossRef]
- Planchard, D. Adjuvant Osimertinib in EGFR-Mutated Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 1780–1782. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.A.; Mok, T.S.; Han, J.Y.; Ahn, M.J.; Delmonte, A.; Ramalingam, S.S.; Kim, S.W.; Shepherd, F.A.; Laskin, J.; He, Y.; et al. Osimertinib versus platinum-pemetrexed for patients with EGFR T790M advanced NSCLC and progression on a prior EGFR-tyrosine kinase inhibitor: AURA3 overall survival analysis. Ann. Oncol. 2020, 31, 1536–1544. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Tumbrink, H.L.; Heimsoeth, A.; Sos, M.L. The next tier of EGFR resistance mutations in lung cancer. Oncogene 2020, 40, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kwok, H.F. Current Strategies for Treating NSCLC: From Biological Mechanisms to Clinical Treatment. Cancers 2020, 12, 1587. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Chen, X.; Zhou, F. EGFR-mutant NSCLC: Emerging novel drugs. Curr. Opin. Oncol. 2021, 33, 87–94. [Google Scholar] [CrossRef]
- Chen, J.A.; Riess, J.W. Advances in targeting acquired resistance mechanisms to epidermal growth factor receptor tyrosine kinase inhibitors. J. Thorac. Dis. 2020, 12, 2859–2876. [Google Scholar] [CrossRef]
- Shah, R.; Lester, J.F. Tyrosine Kinase Inhibitors for the Treatment of EGFR Mutation-Positive Non-Small-Cell Lung Cancer: A Clash of the Generations. Clin. Lung Cancer 2020, 21, e216–e228. [Google Scholar] [CrossRef] [PubMed]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar]
- Lai, C.; Lemke, G. An extended family of protein-tyrosine kinase genes differentially expressed in the vertebrate nervous system. Neuron 1991, 6, 691–704. [Google Scholar] [CrossRef]
- Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076. [Google Scholar] [CrossRef]
- Nguyen, K.Q.; Tsou, W.I.; Kotenko, S.; Birge, R.B. TAM receptors in apoptotic cell clearance, autoimmunity, and cancer. Autoimmunity 2013, 46, 294–297. [Google Scholar] [CrossRef]
- Van der Meer, J.H.; van der Poll, T.; van’t Veer, C. TAM receptors, Gas6, and protein S: Roles in inflammation and hemostasis. Blood 2014, 123, 2460–2469. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Leighton, J.A.; Ghosh, S. Tyro3, Axl, and Mertk receptor signaling in inflammatory bowel disease and colitis-associated cancer. Inflamm. Bowel Dis. 2014, 20, 1472–1480. [Google Scholar] [CrossRef]
- Nguyen, K.Q.; Tsou, W.I.; Kotenko, S.; Birge, R.B. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar]
- Ji, R.; Meng, L.; Li, Q.; Lu, Q. TAM receptor deficiency affects adult hippocampal neurogenesis. Metab. Brain Dis. 2015, 30, 633–644. [Google Scholar] [CrossRef]
- Pierce, A.M.; Keating, A.K. TAM receptor tyrosine kinases: Expression, disease and oncogenesis in the central nervous system. Brain Res. 2014, 1542, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.L.; Caricchio, R.; Abraham, V.; Camenisch, T.D.; Jennette, J.C.; Roubey, R.A.; Earp, H.S.; Matsushima, G.; Reap, E.A. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J. Exp. Med. 2002, 196, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.S.; McMahon, E.J.; Pop, S.M.; Reap, E.A.; Caricchio, R.; Cohen, P.L.; Earp, H.S.; Matsushima, G.K. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 2001, 411, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Hanayama, R.; Kawane, K. Autoimmunity and the clearance of dead cells. Cell 2010, 140, 619–630. [Google Scholar] [CrossRef]
- Caberoy, N.B.; Alvarado, G.; Bigcas, J.L.; Li, W. Galectin-3 is a new MerTK-specific eat-me signal. J. Cell. Physiol. 2012, 227, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Caberoy, N.B.; Zhou, Y.; Li, W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 2010, 29, 3898–3910. [Google Scholar] [CrossRef] [PubMed]
- Keating, A.K.; Salzberg, D.B.; Sather, S.; Liang, X.; Nickoloff, S.; Anwar, A.; Deryckere, D.; Hill, K.; Joung, D.; Sawczyn, K.K.; et al. Lymphoblastic leukemia/lymphoma in mice overexpressing the Mer (MerTK) receptor tyrosine kinase. Oncogene 2006, 25, 6092–6100. [Google Scholar] [CrossRef]
- Brandao, L.N.; Winges, A.; Christoph, S.; Sather, S.; Migdall-Wilson, J.; Schlegel, J.; McGranahan, A.; Gao, D.; Liang, X.; Deryckere, D.; et al. Inhibition of MerTK increases chemosensitivity and decreases oncogenic potential in T-cell acute lymphoblastic leukemia. Blood Cancer J. 2013, 3, e101. [Google Scholar] [CrossRef]
- Schlegel, J.; Sambade, M.J.; Sather, S.; Moschos, S.J.; Tan, A.C.; Winges, A.; DeRyckere, D.; Carson, C.C.; Trembath, D.G.; Tentler, J.J.; et al. MERTK receptor tyrosine kinase is a therapeutic target in melanoma. J. Clin. Invest. 2013, 123, 2257–2267. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Carrera-Silva, E.A.; Bosurgi, L.; Ghosh, S. TAM receptor signaling in immune homeostasis. Annu. Rev. Immunol. 2015, 33, 355–391. [Google Scholar] [CrossRef] [PubMed]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef]
- Cook, R.S.; Jacobsen, K.M.; Wofford, A.M.; DeRyckere, D.; Stanford, J.; Prieto, A.L.; Redente, E.; Sandahl, M.; Hunter, D.M.; Strunk, K.E.; et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J. Clin. Investig. 2013, 123, 3231–3242. [Google Scholar] [CrossRef]
- Coussens, L.M.; Zitvogel, L.; Palucka, A.K. Neutralizing tumor-promoting chronic inflammation: A magic bullet? Science 2013, 339, 286–291. [Google Scholar] [CrossRef]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Karnevi, E.; Andersson, R.; Rosendahl, A.H. Tumour-educated macrophages display a mixed polarisation and enhance pancreatic cancer cell invasion. Immunol. Cell Biol. 2014, 92, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Schoumacher, M.; Burbridge, M. Key Roles of Axl and MER Receptor Tyrosine Kinases in Resistance to Multiple Anticancer Therapies. Curr. Oncol. Rep. 2017, 19, 19. [Google Scholar] [CrossRef]
- Frejno, M.; Zenezini Chiozzi, R.; Wilhelm, M.; Koch, H.; Zheng, R.; Klaeger, S.; Ruprecht, B.; Meng, C.; Kramer, K.; Jarzab, A.; et al. Pharmacoproteomic characterisation of human colon and rectal cancer. Mol. Syst. Biol. 2017, 13, 951. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Kohler, R.; Tang, F.; Hynx, D.; Wang, Y.; Orso, F.; Pretre, V.; Ritschard, R.; Hirschmann, P.; Cron, P.; et al. mTORC1/autophagy-regulated MerTK in mutant BRAFV600 melanoma with acquired resistance to BRAF inhibition. Oncotarget 2017, 8, 69204–69218. [Google Scholar] [CrossRef][Green Version]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the Axl kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Strategies to Overcome Bypass Mechanisms Mediating Clinical Resistance to EGFR Tyrosine Kinase Inhibition in Lung Cancer. Mol. Cancer Ther. 2017, 16, 265–272. [Google Scholar]
- Xie, S.; Li, Y.; Li, X.; Wang, L.; Yang, N.; Wang, Y.; Wei, H. Mer receptor tyrosine kinase is frequently overexpressed in human non-small cell lung cancer, confirming resistance to erlotinib. Oncotarget 2015, 6, 9206–9219. [Google Scholar] [CrossRef]
- Cummings, C.T.; Zhang, W.; Davies, K.D.; Kirkpatrick, G.D.; Zhang, D.; DeRyckere, D.; Wang, X.; Frye, S.V.; Earp, H.S.; Graham, D.K. Small Molecule Inhibition of MERTK Is Efficacious in Non-Small Cell Lung Cancer Models Independent of Driver Oncogene Status. Mol. Cancer Ther. 2015, 14, 2014–2022. [Google Scholar] [CrossRef] [PubMed]
- Linger, R.M.; Cohen, R.A.; Cummings, C.T.; Sather, S.; Migdall-Wilson, J.; Middleton, D.H.; Lu, X.; Baron, A.E.; Franklin, W.A.; Merrick, D.T.; et al. Mer or Axl receptor tyrosine kinase inhibition promotes apoptosis, blocks growth and enhances chemosensitivity of human non-small cell lung cancer. Oncogene 2013, 32, 3420–3431. [Google Scholar] [CrossRef] [PubMed]
- Cummings, C.T.; Linger, R.M.; Cohen, R.A.; Sather, S.; Kirkpatrick, G.D.; Davies, K.D.; DeRyckere, D.; Earp, H.S.; Graham, D.K. Mer590, a novel monoclonal antibody targeting MER receptor tyrosine kinase, decreases colony formation and increases chemosensitivity in non-small cell lung cancer. Oncotarget 2014, 5, 10434–10445. [Google Scholar] [CrossRef]
- Tsou, W.I.; Nguyen, K.Q.; Calarese, D.A.; Garforth, S.J.; Antes, A.L.; Smirnov, S.V.; Almo, S.C.; Birge, R.B.; Kotenko, S.V. Receptor tyrosine kinases, Tyro3, Axl, and MER, demonstrate distinct patterns and complex regulation of ligand-induced activation. J. Biol. Chem. 2014, 289, 25750–25763. [Google Scholar] [CrossRef]
- Lu, Y.; Wan, J.; Yang, Z.; Lei, X.; Niu, Q.; Jiang, L.; Passtoors, W.M.; Zang, A.; Fraering, P.C.; Wu, F. Regulated intramembrane proteolysis of the Axl receptor kinase generates an intracellular domain that localizes in the nucleus of cancer cells. FASEB J. 2017, 31, 1382–1397. [Google Scholar] [CrossRef]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. Taking aim at Mer and Axl receptor tyrosine kinases as novel therapeutic targets in solid tumors. Expert Opin. Ther. Targets 2010, 14, 1073–1090. [Google Scholar] [CrossRef]
- Fraunhoffer, N.A.; Abuelafia, A.M.; Bigonnet, M.; Gayet, O.; Roques, J.; Telle, E.; Santofimia-Castano, P.; Borrello, M.T.; Chuluyan, E.; Dusetti, N.; et al. Evidencing a Pancreatic Ductal Adenocarcinoma Subpopulation Sensitive to the Proteasome Inhibitor Carfilzomib. Clin. Cancer Res. 2020, 26, 5506–5519. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, X.; Ren, S.; Su, C.; Li, C.; Li, W.; Yu, J.; Cheng, N.; Zhou, C. HOTAIR induces EGFR-TKIs resistance in non-small cell lung cancer through epithelial-mesenchymal transition. Lung Cancer 2020, 147, 99–105. [Google Scholar] [CrossRef]
- Tian, H.; Lian, R.; Li, Y.; Liu, C.; Liang, S.; Li, W.; Tao, T.; Wu, X.; Ye, Y.; Yang, X.; et al. Akt-induced lncRNA VAL promotes EMT-independent metastasis through diminishing Trim16-dependent Vimentin degradation. Nat. Commun. 2020, 11, 5127. [Google Scholar] [CrossRef]
- Lee, A.F.; Chen, M.C.; Chen, C.J.; Yang, C.J.; Huang, M.S.; Liu, Y.P. Reverse epithelial-mesenchymal transition contributes to the regain of drug sensitivity in tyrosine kinase inhibitor-resistant non-small cell lung cancer cells. PLoS ONE 2017, 12, e0180383. [Google Scholar] [CrossRef]
- Zhou, J.; Hu, Q.; Zhang, X.; Zheng, J.; Xie, B.; Xu, Z.; Zhang, W. Sensitivity to chemotherapeutics of NSCLC cells with acquired resistance to EGFR-TKIs is mediated by T790M mutation or epithelial-mesenchymal transition. Oncol. Rep. 2018, 39, 1783–1792. [Google Scholar] [CrossRef]
- Du, W.; Sun, L.; Liu, T.; Zhu, J.; Zeng, Y.; Zhang, Y.; Wang, X.; Liu, Z.; Huang, J.A. The miR6253p/Axl axis induces nonT790M acquired resistance to EGFRTKI via activation of the TGF-β/Smad pathway and EMT in EGFRmutant nonsmall cell lung cancer. Oncol. Rep. 2020, 44, 185–195. [Google Scholar]
- Jung, J.; Lee, Y.J.; Choi, Y.H.; Park, E.M.; Kim, H.S.; Kang, J.L. Gas6 Prevents Epithelial-Mesenchymal Transition in Alveolar Epithelial Cells via Production of PGE2, PGD2 and Their Receptors. Cells 2019, 8, 643. [Google Scholar] [CrossRef]
- Feldker, N.; Ferrazzi, F.; Schuhwerk, H.; Widholz, S.A.; Guenther, K.; Frisch, I.; Jakob, K.; Kleemann, J.; Riegel, D.; Bonisch, U.; et al. Genome-wide cooperation of EMT transcription factor ZEB1 with YAP and AP-1 in breast cancer. EMBO J. 2020, 39, e103209. [Google Scholar] [CrossRef]
- Keller, M.; Dubois, F.; Teulier, S.; Martin, A.P.J.; Levallet, J.; Maille, E.; Brosseau, S.; Elie, N.; Hergovich, A.; Bergot, E.; et al. NDR2 kinase contributes to cell invasion and cytokinesis defects induced by the inactivation of RASSF1A tumor-suppressor gene in lung cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 158. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.B.; Sun, H.; Robichaux, J.; Pfeifer, M.; McDermott, U.; Travers, J.; Diao, L.; Xi, Y.; Tong, P.; Shen, L.; et al. A YAP/FOXM1 axis mediates EMT-associated EGFR inhibitor resistance and increased expression of spindle assembly checkpoint components. Sci. Transl. Med. 2020, 12, eaaz4589. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.F.; Tseng, Y.C.; Nguyen, P.A.; Li, Y.C.; Ho, C.C.; Wu, C.W. Enhanced YAP expression leads to EGFR TKI resistance in lung adenocarcinomas. Sci. Rep. 2018, 8, 271. [Google Scholar] [CrossRef]
- Xia, H.; Dai, X.; Yu, H.; Zhou, S.; Fan, Z.; Wei, G.; Tang, Q.; Gong, Q.; Bi, F. EGFR- PI3K -PDK1 pathway regulates YAP signaling in hepatocellular carcinoma: The mechanism and its implications in targeted therapy. Cell Death Dis. 2018, 9, 269. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhu, G.Y.; Cao, D.; Pan, H.; Li, Y.W. Gossypol overcomes EGFR-TKIs resistance in non-small cell lung cancer cells by targeting YAP/TAZ and EGFR(L858R/T790M). Biomed. Pharmacother. 2019, 115, 108860. [Google Scholar] [CrossRef]
- Kaowinn, S.; Yawut, N.; Koh, S.S.; Chung, Y.H. Cancer upregulated gene (CUG)2 elevates YAP1 expression, leading to enhancement of epithelial-mesenchymal transition in human lung cancer cells. Biochem. Biophys. Res. Commun. 2019, 511, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Azad, T.; Nouri, K.; Janse van Rensburg, H.J.; Maritan, S.M.; Wu, L.; Hao, Y.; Montminy, T.; Yu, J.; Khanal, P.; Mulligan, L.M.; et al. A gain-of-functional screen identifies the Hippo pathway as a central mediator of receptor tyrosine kinases during tumorigenesis. Oncogene 2020, 39, 334–355. [Google Scholar] [CrossRef] [PubMed]
- Saab, S.; Chang, O.S.; Nagaoka, K.; Hung, M.C.; Yamaguchi, H. The potential role of YAP in Axl-mediated resistance to EGFR tyrosine kinase inhibitors. Am. J. Cancer Res. 2019, 9, 2719–2729. [Google Scholar] [PubMed]
- Ghiso, E.; Migliore, C.; Ciciriello, V.; Morando, E.; Petrelli, A.; Corso, S.; De Luca, E.; Gatti, G.; Volante, M.; Giordano, S. YAP-Dependent Axl Overexpression Mediates Resistance to EGFR Inhibitors in NSCLC. Neoplasia 2017, 19, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Lv, H.; Zhong, D.S.; Zhou, Q.H. EGFR-TKI resistance and MAP17 are associated with cancer stem cell like properties. Oncol. Lett. 2018, 15, 6655–6665. [Google Scholar] [CrossRef]
- Hu, F.; Li, C.; Zheng, X.; Zhang, H.; Shen, Y.; Zhou, L.; Yang, X.; Han, B.; Zhang, X. Lung adenocarcinoma resistance to therapy with EGFRtyrosine kinase inhibitors is related to increased expression of cancer stem cell markers SOX2, OCT4 and NANOG. Oncol. Rep. 2020, 43, 727–735. [Google Scholar]
- Kunimasa, K.; Nagano, T.; Shimono, Y.; Dokuni, R.; Kiriu, T.; Tokunaga, S.; Tamura, D.; Yamamoto, M.; Tachihara, M.; Kobayashi, K.; et al. Glucose metabolism-targeted therapy and withaferin A are effective for epidermal growth factor receptor tyrosine kinase inhibitor-induced drug-tolerant persisters. Cancer Sci. 2017, 108, 1368–1377. [Google Scholar] [CrossRef]
- Kurppa, K.J.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.H.; Xie, Y.; Lim, K.; Cejas, P.; et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 2020, 37, 104–122.e12. [Google Scholar] [CrossRef]
- Togashi, Y.; Hayashi, H.; Terashima, M.; de Velasco, M.A.; Sakai, K.; Fujita, Y.; Tomida, S.; Nakagawa, K.; Nishio, K. Inhibition of beta-Catenin enhances the anticancer effect of irreversible EGFR-TKI in EGFR-mutated non-small-cell lung cancer with a T790M mutation. J. Thorac. Oncol. 2015, 10, 93–101. [Google Scholar] [CrossRef]
- Sun, J.; Li, G.; Liu, Y.; Ma, M.; Song, K.; Li, H.; Zhu, D.; Tang, X.; Kong, J.; Yuan, X. Targeting histone deacetylase SIRT1 selectively eradicates EGFR TKI-resistant cancer stem cells via regulation of mitochondrial oxidative phosphorylation in lung adenocarcinoma. Neoplasia 2020, 22, 33–46. [Google Scholar] [CrossRef]
- Jia, Z.; Zhang, Y.; Yan, A.; Wang, M.; Han, Q.; Wang, K.; Wang, J.; Qiao, C.; Pan, Z.; Chen, C.; et al. 1,25-dihydroxyvitamin D3 signaling-induced decreases in IRX4 inhibits NANOG-mediated cancer stem-like properties and gefitinib resistance in NSCLC cells. Cell Death Dis. 2020, 11, 670. [Google Scholar] [CrossRef]
- Weng, C.H.; Chen, L.Y.; Lin, Y.C.; Shih, J.Y.; Lin, Y.C.; Tseng, R.Y.; Chiu, A.C.; Yeh, Y.H.; Liu, C.; Lin, Y.T.; et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 2019, 38, 455–468. [Google Scholar] [CrossRef]
- Chang, Y.F.; Lim, K.H.; Chiang, Y.W.; Sie, Z.L.; Chang, J.; Ho, A.S.; Cheng, C.C. STAT3 induces G9a to exacerbate HER3 expression for the survival of epidermal growth factor receptor-tyrosine kinase inhibitors in lung cancers. BMC Cancer 2019, 19, 959. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Ma, Y.; Bi, J.W.; Xiong, Y.; Lv, C.; Li, S.; Wu, N.; Yang, Y. Shisa3 brakes resistance to EGFR-TKIs in lung adenocarcinoma by suppressing cancer stem cell properties. J. Exp. Clin. Cancer Res. 2019, 38, 481. [Google Scholar] [CrossRef]
- Wang, Y.; Moncayo, G.; Morin, P., Jr.; Xue, G.; Grzmil, M.; Lino, M.M.; Clement-Schatlo, V.; Frank, S.; Merlo, A.; Hemmings, B.A. Mer receptor tyrosine kinase promotes invasion and survival in glioblastoma multiforme. Oncogene 2013, 32, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Eom, H.; Eom, H.; Kaushik, N.; Yoo, K.C.; Shim, J.K.; Kwon, M.; Choi, M.Y.; Yoon, T.; Kang, S.G.; Lee, S.J. MerTK mediates STAT3-kRas/Src-signaling axis for glioma stem cell maintenance. Artif. Cells Nanomed. Biotechnol. 2018, 46 (Suppl. 2), 87–95. [Google Scholar] [CrossRef]
- Jung, Y.; Decker, A.M.; Wang, J.; Lee, E.; Kana, L.A.; Yumoto, K.; Cackowski, F.C.; Rhee, J.; Carmeliet, P.; Buttitta, L.; et al. Endogenous Gas6 and Mer receptor signaling regulate prostate cancer stem cells in bone marrow. Oncotarget 2016, 7, 25698–25711. [Google Scholar] [CrossRef]
- Cackowski, F.C.; Eber, M.R.; Rhee, J.; Decker, A.M.; Yumoto, K.; Berry, J.E.; Lee, E.; Shiozawa, Y.; Jung, Y.; Aguirre-Ghiso, J.A.; et al. Mer Tyrosine Kinase Regulates Disseminated Prostate Cancer Cellular Dormancy. J. Cell. Biochem. 2017, 118, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. N. Engl. J. Med. 2018, 379, 2342–2350. [Google Scholar] [CrossRef] [PubMed]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Lastwika, K.J.; Wilson, W., 3rd; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the Akt-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef]
- Minchom, A.; Thavasu, P.; Ahmad, Z.; Stewart, A.; Georgiou, A.; O’Brien, M.E.R.; Popat, S.; Bhosle, J.; Yap, T.A.; de Bono, J.; et al. A study of PD-L1 expression in kRas mutant non-small cell lung cancer cell lines exposed to relevant targeted treatments. PLoS ONE 2017, 12, e0186106. [Google Scholar] [CrossRef]
- Han, J.J.; Kim, D.W.; Koh, J.; Keam, B.; Kim, T.M.; Jeon, Y.K.; Lee, S.H.; Chung, D.H.; Heo, D.S. Change in PD-L1 Expression After Acquiring Resistance to Gefitinib in EGFR-Mutant Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2016, 17, 263–270.e2. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Wang, R.; Zhang, X.; Ma, Y.; Zhong, L.; Li, K.; Nishiyama, A.; Arai, S.; Yano, S.; Wang, W. EGFR-TKI resistance promotes immune escape in lung cancer via increased PD-L1 expression. Mol. Cancer 2019, 18, 165. [Google Scholar] [CrossRef] [PubMed]
- Kasikara, C.; Kumar, S.; Kimani, S.; Tsou, W.I.; Geng, K.; Davra, V.; Sriram, G.; Devoe, C.; Nguyen, K.N.; Antes, A.; et al. Phosphatidylserine Sensing by TAM Receptors Regulates Akt-Dependent Chemoresistance and PD-L1 Expression. Mol. Cancer Res. 2017, 15, 753–764. [Google Scholar] [CrossRef]
- Lee-Sherick, A.B.; Jacobsen, K.M.; Henry, C.J.; Huey, M.G.; Parker, R.E.; Page, L.S.; Hill, A.A.; Wang, X.; Frye, S.V.; Earp, H.S.; et al. MERTK inhibition alters the PD-1 axis and promotes anti-leukemia immunity. JCI Insight 2018, 3, e97941. [Google Scholar] [CrossRef] [PubMed]
- Kasikara, C.; Davra, V.; Calianese, D.; Geng, K.; Spires, T.E.; Quigley, M.; Wichroski, M.; Sriram, G.; Suarez-Lopez, L.; Yaffe, M.B.; et al. Pan-TAM Tyrosine Kinase Inhibitor BMS-777607 Enhances Anti-PD-1 mAb Efficacy in a Murine Model of Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 2669–2683. [Google Scholar] [CrossRef]
- Yang, C.Y.; Liao, W.Y.; Ho, C.C.; Chen, K.Y.; Tsai, T.H.; Hsu, C.L.; Su, K.Y.; Chang, Y.L.; Wu, C.T.; Hsu, C.C.; et al. Association between programmed death-ligand 1 expression, immune microenvironments, and clinical outcomes in epidermal growth factor receptor mutant lung adenocarcinoma patients treated with tyrosine kinase inhibitors. Eur. J. Cancer 2020, 124, 110–122. [Google Scholar] [CrossRef]
- Escors, D.; Gato-Canas, M.; Zuazo, M.; Arasanz, H.; Garcia-Granda, M.J.; Vera, R.; Kochan, G. The intracellular signalosome of PD-L1 in cancer cells. Signal. Transduct. Target. Ther. 2018, 3, 26. [Google Scholar] [CrossRef]
- Tung, J.N.; Lin, P.L.; Wang, Y.C.; Wu, D.W.; Chen, C.Y.; Lee, H. PD-L1 confers resistance to EGFR mutation-independent tyrosine kinase inhibitors in non-small cell lung cancer via upregulation of YAP1 expression. Oncotarget 2018, 9, 4637–4646. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zeng, Y.; Liu, T.; Du, W.; Zhu, J.; Liu, Z.; Huang, J.A. The canonical TGF-β/Smad signalling pathway is involved in PD-L1-induced primary resistance to EGFR-TKIs in EGFR-mutant non-small-cell lung cancer. Respir. Res. 2019, 20, 164. [Google Scholar] [CrossRef]
- Du, W.; Zhu, J.; Zeng, Y.; Liu, T.; Zhang, Y.; Cai, T.; Fu, Y.; Zhang, W.; Zhang, R.; Liu, Z.; et al. KPNB1-mediated nuclear translocation of PD-L1 promotes non-small cell lung cancer cell proliferation via the Gas6/MerTK signaling pathway. Cell Death Differ. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Xiao, F.; Liu, N.; Ma, X.; Qin, J.; Liu, Y.; Wang, X. M2 macrophages reduce the effect of gefitinib by activating Akt/mTOR in gefitinib-resistant cell lines HCC827/GR. Thorac. Cancer 2020, 11, 3289–3298. [Google Scholar] [CrossRef]
- Hwang, I.; Kim, J.W.; Ylaya, K.; Chung, E.J.; Kitano, H.; Perry, C.; Hanaoka, J.; Fukuoka, J.; Chung, J.Y.; Hewitt, S.M. Tumor-associated macrophage, angiogenesis and lymphangiogenesis markers predict prognosis of non-small cell lung cancer patients. J. Transl. Med. 2020, 18, 443. [Google Scholar] [CrossRef]
- Trus, E.; Basta, S.; Gee, K. Who’s in charge here? Macrophage colony stimulating factor and granulocyte macrophage colony stimulating factor: Competing factors in macrophage polarization. Cytokine 2020, 127, 154939. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Reinoso, V.; Baek, D.Y.; Kurutz, A.; Rubin, J.R.; Koh, A.J.; McCauley, L.K.; Roca, H. Unique Pro-Inflammatory Response of Macrophages during Apoptotic Cancer Cell Clearance. Cells 2020, 9, 429. [Google Scholar] [CrossRef] [PubMed]
- Giroud, P.; Renaudineau, S.; Gudefin, L.; Calcei, A.; Menguy, T.; Rozan, C.; Mizrahi, J.; Caux, C.; Duong, V.; Valladeau-Guilemond, J. Expression of TAM-R in Human Immune Cells and Unique Regulatory Function of MerTK in IL-10 Production by Tolerogenic DC. Front. Immunol. 2020, 11, 564133. [Google Scholar] [CrossRef]
- Lai, Y.S.; Putra, R.; Aui, S.P.; Chang, K.T. M2C Polarization by Baicalin Enhances Efferocytosis via Upregulation of MERTK Receptor. Am. J. Chin. Med. 2018, 46, 1899–1914. [Google Scholar] [CrossRef]
- Yang, F.; Wei, Y.; Han, D.; Li, Y.; Shi, S.; Jiao, D.; Wu, J.; Zhang, Q.; Shi, C.; Yang, L.; et al. Interaction with CD68 and Regulation of Gas6 Expression by Endosialin in Fibroblasts Drives Recruitment and Polarization of Macrophages in Hepatocellular Carcinoma. Cancer Res. 2020, 80, 3892–3905. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wu, S.; Liu, Q.; Xie, J.; Zhang, J.; Han, D.; Lu, Q.; Lu, Q. Mertk deficiency affects macrophage directional migration via disruption of cytoskeletal organization. PLoS ONE 2015, 10, e0117787. [Google Scholar] [CrossRef]
- Zheng, G.; Li, L.F.; Zhang, Y.; Qu, L.; Wang, W.; Li, M.; Yu, S.; Zhou, M.; Luo, Y.; Sun, Y.; et al. MERTK is a host factor that promotes classical swine fever virus entry and antagonizes innate immune response in PK-15 cells. Emerg. Microbes Infect. 2020, 9, 571–581. [Google Scholar] [CrossRef]
- Caetano, M.S.; Younes, A.I.; Barsoumian, H.B.; Quigley, M.; Menon, H.; Gao, C.; Spires, T.; Reilly, T.P.; Cadena, A.P.; Cushman, T.R.; et al. Triple Therapy with MerTK and PD1 Inhibition Plus Radiotherapy Promotes Abscopal Antitumor Immune Responses. Clin. Cancer Res. 2019, 25, 7576–7584. [Google Scholar] [CrossRef]
- Rogers, A.E.; Le, J.P.; Sather, S.; Pernu, B.M.; Graham, D.K.; Pierce, A.M.; Keating, A.K. Mer receptor tyrosine kinase inhibition impedes glioblastoma multiforme migration and alters cellular morphology. Oncogene 2012, 31, 4171–4181. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Wallet, M.A.; Yi, Z.; Huang, Y.; Henderson, M.; Mathews, C.E.; Earp, H.S.; Matsushima, G.; Baldwin, A.S., Jr.; Tisch, R.M. Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-kappaB activation in dendritic cells. Blood 2007, 109, 653–660. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, H.; Fang, L.; Ding, P.; Xu, K.; Yang, Q.; Liu, R. MerTK Does Not Mediate Phagocytosis of Staphylococcus aureus but Attenuates Inflammation Induced by Staphylococcal Lipoteichoic Acid Through Blocking NF-kappaB Activation. Inflammation 2017, 40, 1543–1552. [Google Scholar] [CrossRef]
- Zagórska, A.; Través, P.G.; Lew, E.D.; Dransfield, I.; Lemke, G. Diversification of TAM receptor tyrosine kinase function. Nat. Immunol. 2014, 15, 920–928. [Google Scholar] [CrossRef] [PubMed]
- DuBois, J.C.; Ray, A.K.; Davies, P.; Shafit-Zagardo, B. Anti-Axl antibody treatment reduces the severity of experimental autoimmune encephalomyelitis. J. Neuroinflamm. 2020, 17, 324. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, A.; Wang, Y.; Chen, F.; Zhao, Z.; Davra, V.; Suzuki-Inoue, K.; Ozaki, Y.; Birge, R.B.; Lu, Q.; et al. Tyro3, Axl, and Mertk receptors differentially participate in platelet activation and thrombus formation. Cell Commun. Signal. 2018, 16, 98. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, W.; Stashko, M.A.; Deryckere, D.; Cummings, C.T.; Hunter, D.; Yang, C.; Jayakody, C.N.; Cheng, N.; Simpson, C.; et al. UNC1062, a new and potent Mer inhibitor. Eur. J. Med. Chem. 2013, 65, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, D.; Stashko, M.A.; DeRyckere, D.; Hunter, D.; Kireev, D.; Miley, M.J.; Cummings, C.; Lee, M.; Norris-Drouin, J.; et al. Pseudo-cyclization through intramolecular hydrogen bond enables discovery of pyridine substituted pyrimidines as new Mer kinase inhibitors. J. Med. Chem. 2013, 56, 9683–9692. [Google Scholar] [CrossRef]
- Roohullah, A.; Cooper, A.; Lomax, A.J.; Aung, J.; Barge, A.; Chow, L.; McHale, M.; Desai, J.; Whittle, J.R.; Tran, B.; et al. A phase I trial to determine safety and pharmacokinetics of ASLAN002, an oral MET superfamily kinase inhibitor, in patients with advanced or metastatic solid cancers. Invest. New Drugs 2018, 36, 886–894. [Google Scholar] [CrossRef]
- Arkenau, H.T.; Plummer, R.; Molife, L.R.; Olmos, D.; Yap, T.A.; Squires, M.; Lewis, S.; Lock, V.; Yule, M.; Lyons, J.; et al. A phase I dose escalation study of AT9283, a small molecule inhibitor of aurora kinases, in patients with advanced solid malignancies. Ann. Oncol. 2012, 23, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.I.; Krishnamurthi, S.S.; Saleh, M.N.; Gitlitz, B.J.; Borad, M.J.; Gold, P.J.; Chiorean, E.G.; Springett, G.M.; Abbas, R.; Agarwal, S.; et al. Phase I study of bosutinib, a Src/Abl tyrosine kinase inhibitor, administered to patients with advanced solid tumors. Clin. Cancer Res. 2012, 18, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Taverna, J.A.; Hung, C.N.; DeArmond, D.T.; Chen, M.; Lin, C.L.; Osmulski, P.A.; Gaczynska, M.E.; Wang, C.M.; Lucio, N.D.; Chou, C.W.; et al. Single-Cell Proteomic Profiling Identifies Combined Axl and JAK1 Inhibition as a Novel Therapeutic Strategy for Lung Cancer. Cancer Res. 2020, 80, 1551–1563. [Google Scholar] [CrossRef]
- Leighl, N.B.; Tsao, M.S.; Liu, G.; Tu, D.; Ho, C.; Shepherd, F.A.; Murray, N.; Goffin, J.R.; Nicholas, G.; Sakashita, S.; et al. A phase I study of foretinib plus erlotinib in patients with previously treated advanced non-small cell lung cancer: Canadian cancer trials group IND.196. Oncotarget 2017, 8, 69651–69662. [Google Scholar] [CrossRef]
- He, A.R.; Cohen, R.B.; Denlinger, C.S.; Sama, A.; Birnbaum, A.; Hwang, J.; Sato, T.; Lewis, N.; Mynderse, M.; Niland, M.; et al. First-in-Human Phase I Study of Merestinib, an Oral Multikinase Inhibitor, in Patients with Advanced Cancer. Oncologist 2019, 24, e930–e942. [Google Scholar] [CrossRef]
- Wang, J.; Mirzapoiazova, T.; Carol Tan, Y.H.; Pang, K.M.; Pozhitkov, A.; Wang, Y.; Wang, Y.; Mambetsariev, B.; Wang, E.; Nasser, M.W.; et al. Inhibiting crosstalk between MET signaling and mitochondrial dynamics and morphology: A novel therapeutic approach for lung cancer and mesothelioma. Cancer Biol. Ther. 2018, 19, 1023–1032. [Google Scholar] [CrossRef]
- Padda, S.; Neal, J.W.; Wakelee, H.A. MET inhibitors in combination with other therapies in non-small cell lung cancer. Transl. Lung Cancer Res. 2012, 1, 238–253. [Google Scholar] [PubMed]
- Yan, D.; Parker, R.E.; Wang, X.; Frye, S.V.; Earp, H.S., 3rd; DeRyckere, D.; Graham, D.K. MERTK Promotes Resistance to Irreversible EGFR Tyrosine Kinase Inhibitors in Non-small Cell Lung Cancers Expressing Wild-type EGFR Family Members. Clin. Cancer Res. 2018, 24, 6523–6535. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, L.; Bahleda, R.; Tolaney, S.M.; Kwak, E.L.; Cleary, J.M.; Pandya, S.S.; Hollebecque, A.; Abbas, R.; Ananthakrishnan, R.; Berkenblit, A.; et al. Phase I study of neratinib in combination with temsirolimus in patients with human epidermal growth factor receptor 2-dependent and other solid tumors. J. Clin. Oncol. 2014, 32, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Okura, N.; Nishioka, N.; Yamada, T.; Taniguchi, H.; Tanimura, K.; Katayama, Y.; Yoshimura, A.; Watanabe, S.; Kikuchi, T.; Shiotsu, S.; et al. ONO-7475, a Novel Axl Inhibitor, Suppresses the Adaptive Resistance to Initial EGFR-TKI Treatment in EGFR-Mutated Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 2244–2256. [Google Scholar] [CrossRef]
- Clémenson, C.; Chargari, C.; Liu, W.; Mondini, M.; Ferté, C.; Burbridge, M.F.; Cattan, V.; Jacquet-Bescond, A.; Deutsch, E. The MET/Axl/FGFR Inhibitor S49076 Impairs Aurora B Activity and Improves the Antitumor Efficacy of Radiotherapy. Mol. Cancer Ther. 2017, 16, 2107–2119. [Google Scholar] [CrossRef]
- Kerkemeyer, K.L.S.; Lai, F.Y.X.; Mar, A. Lichen planus pemphigoides during therapy with tislelizumab and sitravatinib in a patient with metastatic lung cancer. Australas. J. Dermatol. 2020, 61, 180–182. [Google Scholar] [CrossRef]
- Fiedler, W.; Giaccone, G.; Lasch, P.; van der Horst, I.; Brega, N.; Courtney, R.; Abbattista, A.; Shalinsky, D.R.; Bokemeyer, C.; Boven, E. Phase I trial of SU14813 in patients with advanced solid malignancies. Ann. Oncol. 2011, 22, 195–201. [Google Scholar] [CrossRef]
- Yoh, K.; Seto, T.; Satouchi, M.; Nishio, M.; Yamamoto, N.; Murakami, H.; Nogami, N.; Matsumoto, S.; Kohno, T.; Tsuta, K.; et al. Vandetanib in patients with previously treated RET-rearranged advanced non-small-cell lung cancer (LURET): An open-label, multicentre phase 2 trial. Lancet Respir. Med. 2017, 5, 42–50. [Google Scholar] [CrossRef]
- Crittenden, M.R.; Baird, J.; Friedman, D.; Savage, T.; Uhde, L.; Alice, A.; Cottam, B.; Young, K.; Newell, P.; Nguyen, C.; et al. Mertk on tumor macrophages is a therapeutic target to prevent tumor recurrence following radiation therapy. Oncotarget 2016, 7, 78653–78666. [Google Scholar] [CrossRef]
- Minson, K.A.; Smith, C.C.; DeRyckere, D.; Libbrecht, C.; Lee-Sherick, A.B.; Huey, M.G.; Lasater, E.A.; Kirkpatrick, G.D.; Stashko, M.A.; Zhang, W.; et al. The MERTK/FLT3 inhibitor MRX-2843 overcomes resistance-conferring FLT3 mutations in acute myeloid leukemia. JCI Insight 2016, 1, e85630. [Google Scholar] [CrossRef]
- Illig, C.R.; Manthey, C.L.; Wall, M.J.; Meegalla, S.K.; Chen, J.; Wilson, K.J.; Ballentine, S.K.; Desjarlais, R.L.; Schubert, C.; Crysler, C.S.; et al. Optimization of a potent class of arylamide colony-stimulating factor-1 receptor inhibitors leading to anti-inflammatory clinical candidate 4-cyano-N-[2-(1-cyclohexen-1-yl)-4-[1-[(dimethylamino)acetyl]-4-piperidinyl]pheny l]-1H-imidazole-2-carboxamide (JNJ-28312141). J. Med. Chem. 2011, 54, 7860–7883. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Target(s) | Experimental Models | Outcomes |
|---|---|---|---|
| Mer590 1 | MERTK/Tyro3 | Cell culture | Reduction of MERTK protein levels, decrease in colony formation and chemosensitivity [95] |
| UNC1062 | MERTK/Axl/Tyro3 | Cell culture | Inhibition of A549 colony formation [163] |
| UNC2025 | MERTK/Axl/Tyro3 | Cell culture/ subcutaneous xenograft in mice | Inhibition of colony formation and tumor growth using multiple NSCLC cell lines [93] |
| UNC2250 | MERTK/Axl/Tyro3 | Cell culture | Inhibition of Colo699 colony formation [164] |
| Name | Primary Target(s) 1 | Other Target(s) 2 | Development Phase (Trials Registry Number 3) | Tumor Type 1 |
|---|---|---|---|---|
| ASLAN002/BMS-777607 | MET and RON | Axl, Flt3, MERTK and Tyro3 | Phase I (NCT01721148) | Advanced or Metastatic Solid Tumours [165] |
| AT9283 | Aurora kinases | GSK3β, FGFR2, JAK2/3, MERTK, RET, RSK2/3, VEGFR3 (Flt4), Tyk2 and Yes | Phase I (NCT00443976) Phase I (NCT00985868) | Advanced or metastatic solid tumors [166] Relapsed and refractory solid tumors |
| Bemcentinib/BGB324/R428 | Axl | MERTK and Tyro3 | Phase II (NCT03184571) | Advanced NSCLC |
| Bosutinib/SKI-606/PF-5208763 | Src and Abl | MERTK, Axl, Tyro3, EphA2, Kit, Ste20, Tec and Trk | Phase I (NCT03023319) Phase I (NCT01001936) Phase II (NCT03297606) | NSCLC [167] Advanced malignant solid tumors Advanced solid tumors |
| Crizotinib/PF-2341066 | ALK, MET and ROS1 | MERTK, Axl, Tyro3, and RON | Multiple trials | MET+, ROS1+ or ALK+ advanced NSCLC |
| Dubermatinib/TP-0903 | Axl | MERTK, Tyro3, AURKA, Flt3, AURKB, JAK2 and Abl | Phase I (NCT02729298) | EGFR+ NSCLC [168] |
| Foretinib/GSK1363089/XL880/EXEL-2880 | MET and VEGFR2 | MERTK, Axl, Tyro3, RON, PDGFR, KDR, Kit, Flt3 and TIE2 | Phase II (NCT02034097) Phase I (NCT00742261) Phase I (NCT01068587) Phase II (IND.196) | NSCLC Solid Tumors Advanced or Metastatic NSCLC Advanced or Metastatic NSCLC [169] |
| INCB081776 | Axl and MERTK | Phase I (NCT03522142) | Advanced solid tumors | |
| Merestinib/LY2801653 | MET | MERTK, Axl, Tyro3, MET, Flt3, RON, ROS1, Tek, DDR1/2, MKNK1/2 | Phase I (NCT03027284) Phase II (NCT02920996) Phase I (NCT02745769) | Advanced or metastatic cancers [170] NSCLC Advanced cancers |
| Glesatinib/MGCD265 | MET and VEGFR2 | MERTK, Axl, Tyro3, Tek and RON | Phase II (NCT02544633) Phase I (NCT00975767) | NSCLC [171] Advanced NSCLC |
| MK2461 | MET | MERTK, RON, Flt1, Flt3 and PDGFR | Phase I/II (NCT00496353) Phase I (NCT00518739) | Advanced solid tumors [172] Advanced cancers |
| MRX-2843 | MERTK and Flt3 | Axl and Tyro3 | Phase I (NCT03510104) | Advanced and metastatic solid tumors [173] |
| Neratinib/HKI-272 | HER2 and EGFR | MERTK, Axl and Tyro3 | Phase II (NCT01827267) Phase II (NCT00266877) | HER2-activating mutations in NSCLC [174] Advanced NSCLC |
| ONO-7475 | Axl and MERTK | Phase I (NCT03730337) | Advanced or metastatic solid tumors [175] | |
| S49076 | MET | Axl, MERTK and FGFR | Phase I (ISRCTN00759419) | NSCLC [176] |
| Sitravatinib/MGCD516 | spectrum-selective RTKs | MERTK, Axl, Tyro3, VEGFR2, PDGFR, Kit and MET | Phase I (NCT02219711) Phase III (NCT03906071) Phase II (NCT02954991) Phase I (NCT03666143) Phase II (NCT02664935) | Advanced cancers [177] Metastatic non-squamous NSCLC NSCLC Advanced solid tumors. NSCLC |
| SU-14813 | multi-targeted RTKs | MERTK, Axl, Tyro3, FLT3, VEGFR, PDGFR, Kit | Phase I (NCT00982267) | Advanced solid tumors [178] |
| Vandetanib/ZD6474 | multi-targeted RTKs | MERTK, Axl, Tyro3, VEGFR2, VEGFR3, EGFR and RET | Multiple trials | NSCLC [179] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-J.; Liu, Y.-P. MERTK Inhibition: Potential as a Treatment Strategy in EGFR Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer. Pharmaceuticals 2021, 14, 130. https://doi.org/10.3390/ph14020130
Chen C-J, Liu Y-P. MERTK Inhibition: Potential as a Treatment Strategy in EGFR Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer. Pharmaceuticals. 2021; 14(2):130. https://doi.org/10.3390/ph14020130
Chicago/Turabian StyleChen, Chao-Ju, and Yu-Peng Liu. 2021. "MERTK Inhibition: Potential as a Treatment Strategy in EGFR Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer" Pharmaceuticals 14, no. 2: 130. https://doi.org/10.3390/ph14020130
APA StyleChen, C.-J., & Liu, Y.-P. (2021). MERTK Inhibition: Potential as a Treatment Strategy in EGFR Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer. Pharmaceuticals, 14(2), 130. https://doi.org/10.3390/ph14020130