1. Introduction
Zeta-chain-associated protein kinase 70 kDa (ZAP-70) is a non-receptor tyrosine kinase belonging to the Syk family mainly expressed in T lymphocytes and NK cells. After the activation of the T cell receptor (TCR) by an antigen, ZAP-70 is recruited by the ITAM motifs of CD3Z chains of the TCR complex and activated. Upon phosphorylation, ZAP-70 phosphorylates LAT and SLP-76, which are responsible for the proliferation and survival of T lymphocytes [
1,
2]. Given this, ZAP-70 plays a pivotal role in the regulation and adaptive immune signaling of T cells, thus it has been identified as a promising therapeutic target for some diseases like peripheral T cell lymphoma (PTCL). Despite its significance in T cell activation, few compounds have been reported as ZAP-70 inhibitors and they have low affinity [
3] or are unselective [
2]. Irreversible inhibitor RDN009 has been recently described [
4] as a good strategy to improve selectivity, albeit their pharmacokinetic properties need to be improved.
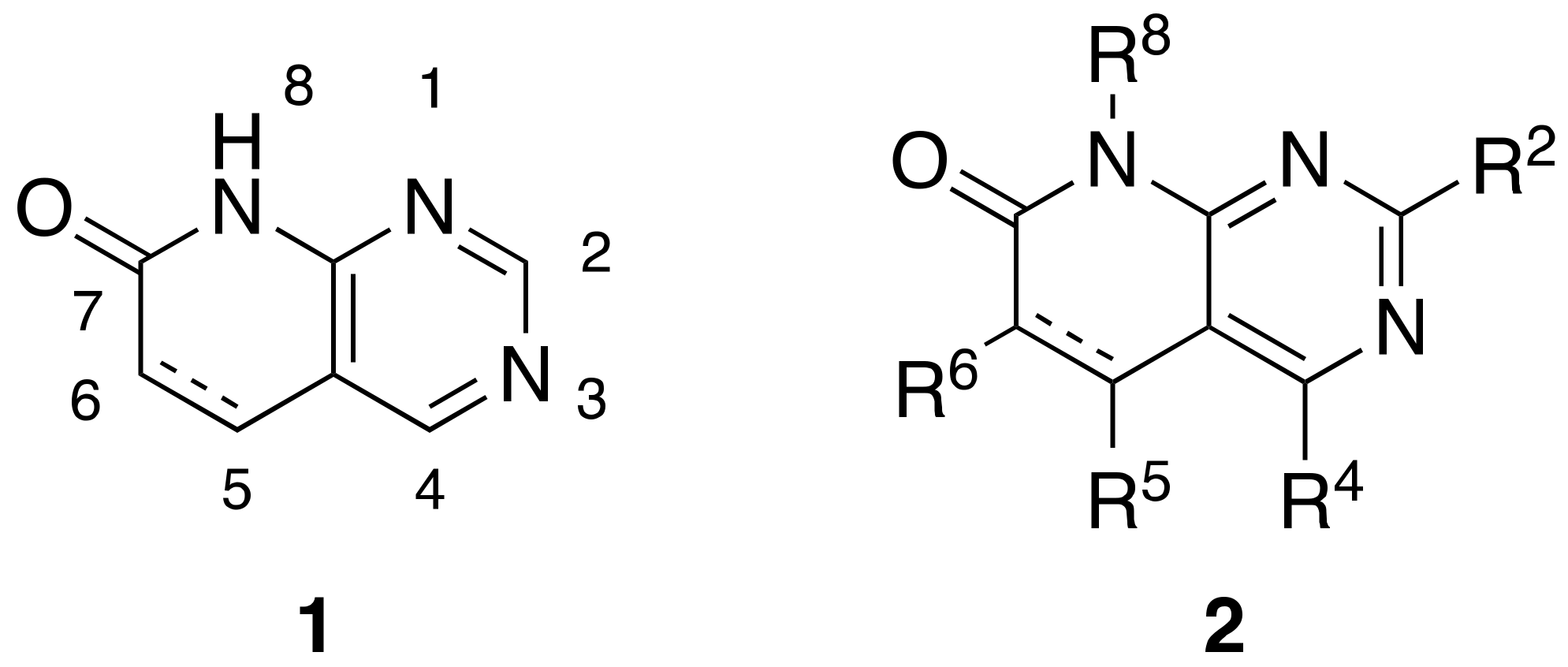
Pyrido[2,3-
d]pyrimidin-7(8
H)-ones
1 are
ortho-fused bicyclic heterocycles consisting of pyridone and pyrimidine rings. This kind of structure presents up to five diversity centers (R
2, R
4, R
5, R
6, and R
8) and two possible degrees of unsaturation between C5 and C6 (
Figure 1). Functionalized pyrido[2,3-
d]pyrimidines
2 are considered
privileged heterocyclic scaffolds for drug discovery due to their well-known activity as tyrosine kinase inhibitors (TKI) [
5,
6]. In this context, our group has previously synthesized multiple compounds with activities in the nanomolar range as BCR inhibitors for B lymphoid malignancies or DDR2 inhibitors for the treatment of lung cancer, among others [
7].
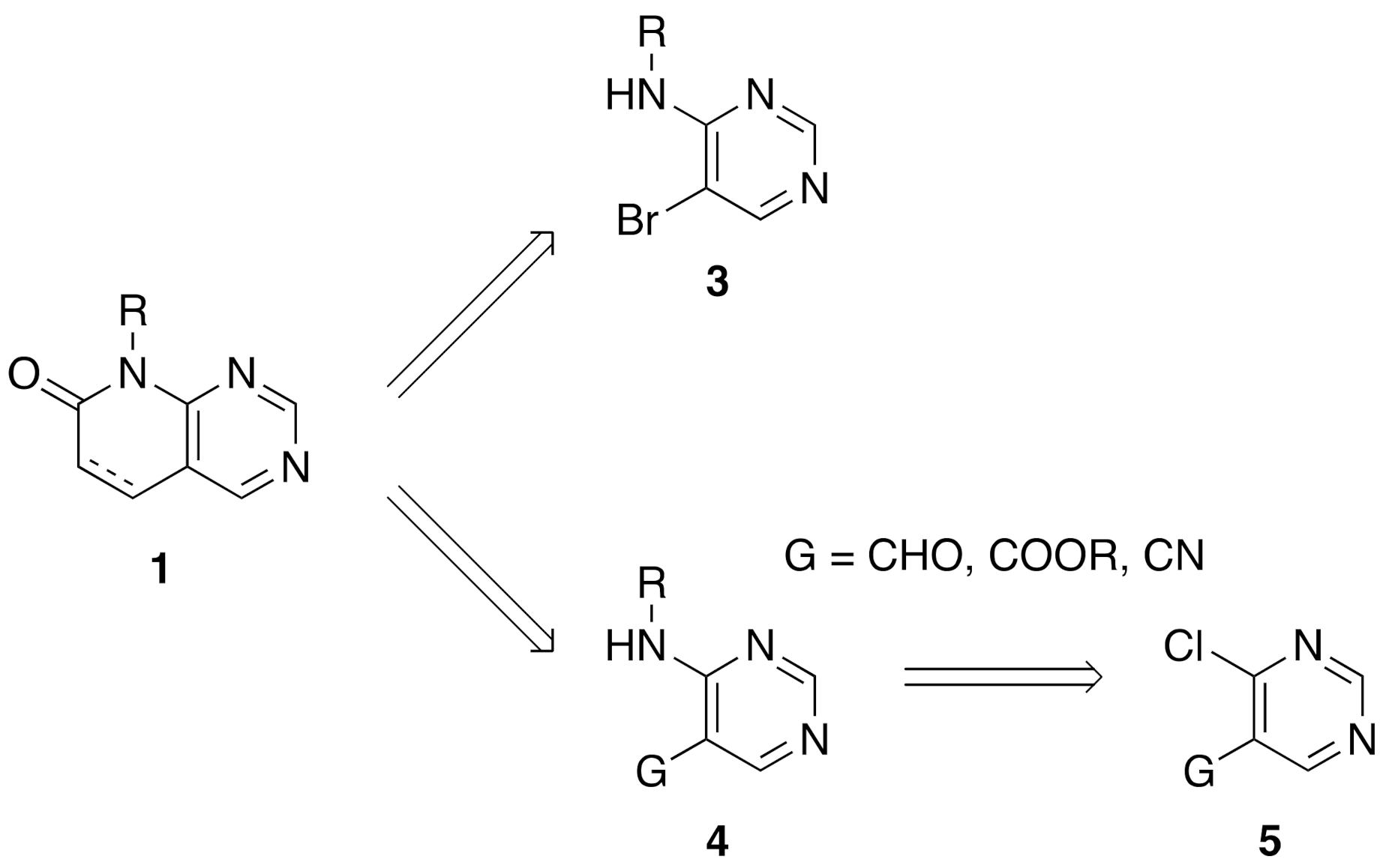
From a synthetic point of view, most of the research groups currently working with such a scaffold construct the molecule either from a preformed pyrimidine or from a preformed pyridone ring. In the first strategy, two approaches have been described to synthesize a pyrido[2,3-
d]pyrimidin-7(8
H)-one
1: starting from an adequately substituted 4-amino-5-bromopyrimidine
3 or starting from an
N-substituted pyrimidine-4-amine
4, which contains a carbon functional group G (CHO, COOR, or CN) at position C5 of the pyrimidine ring. This latter compound is usually obtained from the corresponding 4-chloro-substituted pyrimidine
5 (
Figure 2) [
8].
One of the major drawbacks of these two synthetic approaches is the difficulty of introducing substituents at the C4 position of the final pyrido[2,3-
d]pyrimidin-7(8
H)-one
1 either because they could be involved in a possible secondary reaction during the formation of the pyridone ring or because they can even preclude the construction of such a ring due to the steric hindrance that such a substituent could produce. These difficulties have reduced the chemical diversity of substituents present at C4 of the resulting pyridopyrimidine to hydrogen, amino, or short alkyl groups. Only the introduction of a chlorine atom at the starting pyrimidine
4 allows the construction of the corresponding 4-chloro-substituted pyrido[2,3-
d]pyrimidin-7(8
H)-one
1 that could allow the further introduction of diversity at such a position [
9].
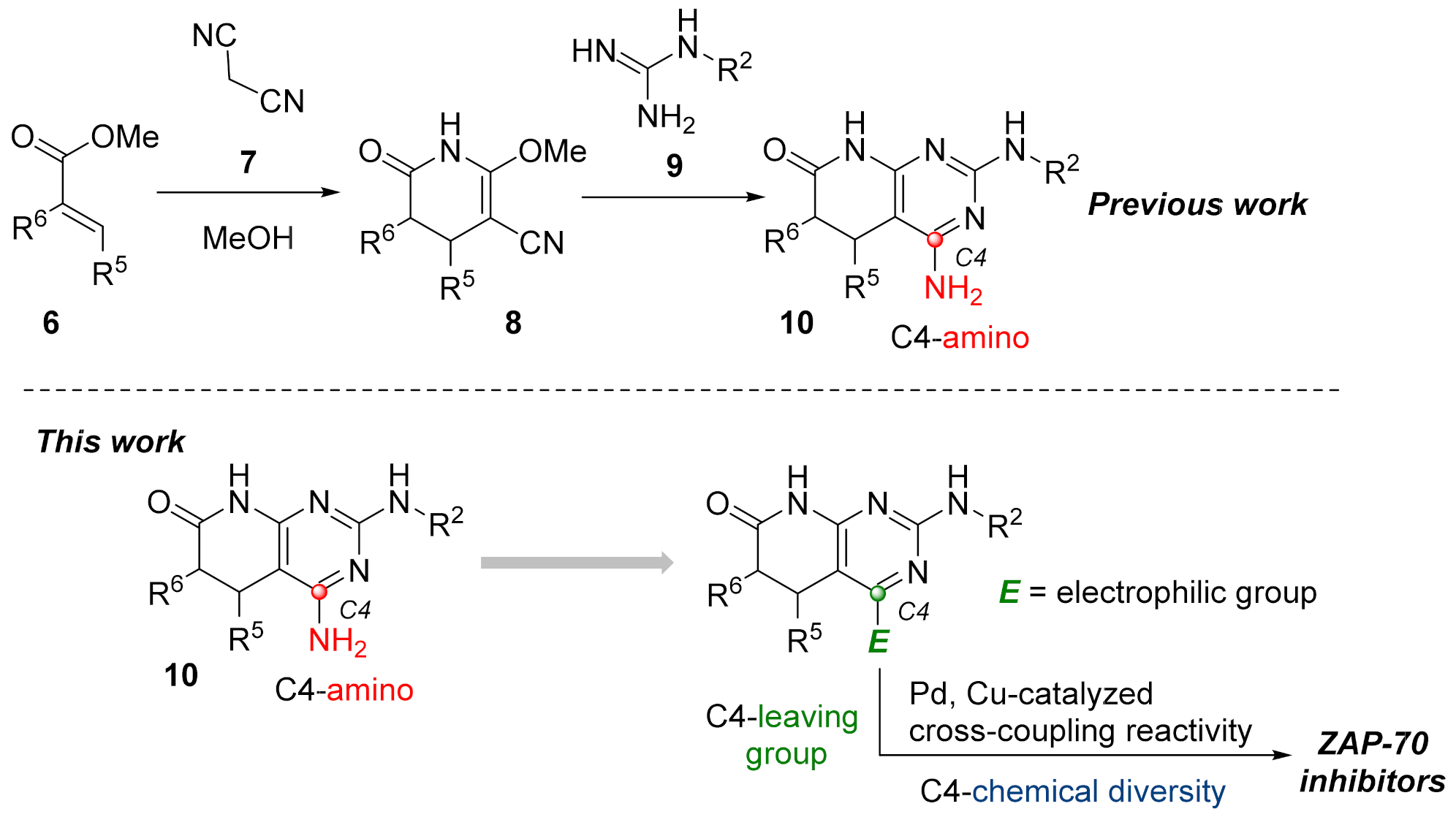
As for the second strategy, the construction of structures
2 from a pyridone ring, there are clearly fewer protocols and references, but our group has large experience in the synthesis of 4-amino-5,6-dihydropyrido[2,3-
d]pyrimidin-7-(8
H)-ones (
10) by cyclization of guanidine systems like
9 with 2-methoxy-6-oxo-1,4,5,6-tetrahydropyridine-3-carbonitriles (
8), which in turn are obtained by means of a Michael-type reaction between malononitrile (
7) and an α,β-unsaturated ester of general structure
6 in NaOMe/MeOH media (
Figure 3 Left). We also described a protocol to obtain 4-oxo-5,6-dihydropyrido[2,3-
d]pyrimidin-7-(8
H)-ones via the corresponding Michael adduct between an α,β-unsaturated ester
6 and methyl cyanoacetate [
10].
The accessible chemical diversity at the C4 position of pyrido[2,3-
d]pyrimidin-7-(8
H)-ones using the preceding synthetic approaches is certainly limited as we showed in a recent review [
11]. Depending on the degree of unsaturation between C5 and C6, the most described C4 substituent differs, being hydrogen (79% of reported structures) and carbon substituents (18%) when having a double bond, and oxygen (mainly as a carbonyl group, 63%) or alkyl groups (26%, mainly methyl) when having a C5-C6 single bond. Most of the molecules containing an amino group in the C4 position have been described by our research group using the strategy included in the left of
Figure 3 or similar.
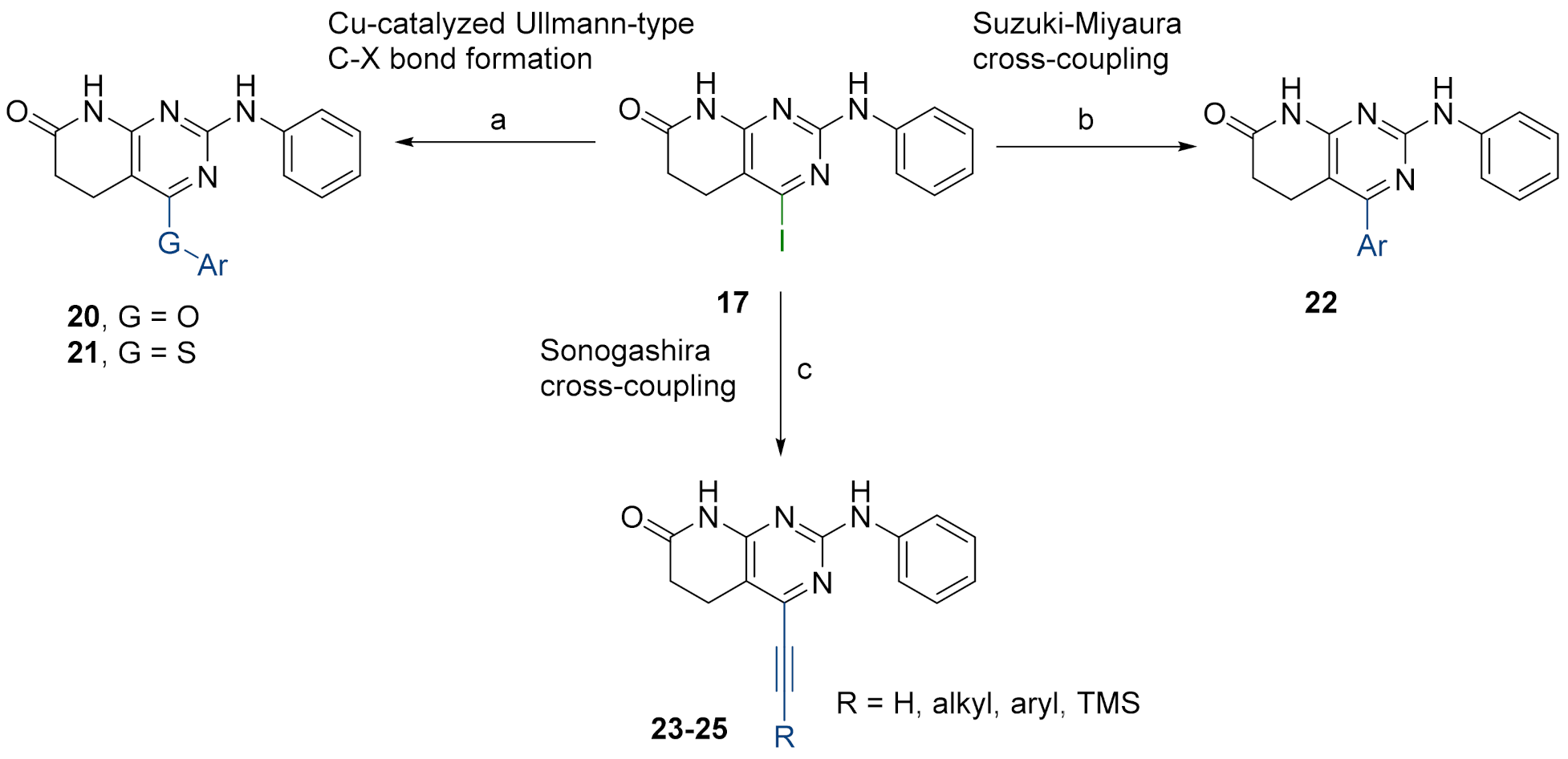
Such limited chemical diversity and the need to explore new tyrosine kinase targets taking advantage of our methodology impelled us to study the introduction of new substituents at C4 to evaluate the potential biological activity of the resulting derivatives on ZAP-70. Hence, and given the great impact that transition metal-catalyzed coupling reactivity have demonstrated in the synthesis of pharmaceuticals [
12], we sought the introduction of an electrophilic manifold at the C4 position capable of engaging in either Pd and/or Cu cross-coupling reactions as a key point to rapidly enlarge the number of possible substitutions at such a position (
Figure 3 right).
3. Materials and Methods
3.1. General Considerations
All solvents and chemicals were reagent grade. Unless otherwise mentioned, all solvents and chemicals were purchased from commercial suppliers (Sigma Aldrich, Fluorochem, Apollo scientific, Activate scientific, Alfa Aesar, and Enamine) and used without further purification.
1H,
13C, and
19F NMR spectra were recorded on a Varian 400-MR spectrometer (
1H NMR at 400 MHz,
13C NMR at 100.5 MHz, and
19F NMR at 376 MHz). Chemical shifts were reported in parts per million (δ) and are referenced to the residual signal of the solvent DMSO-
d6 2.50 ppm or tetramethylsilane (TMS) 0 ppm in
1H NMR spectra and to the residual signal of the solvent DMSO-
d6 39.5 ppm in
13C NMR. Coupling constants are reported in Hertz (Hz). Standard and peak multiplicities are designed as follows: s, singlet; d, doublet; dd, doublet of doublets; t, triplet; q, quartet; p, quintet; br, broad signal. “*” means interchangeable assignment. IR spectra were recorded in a Thermo Scientific Nicolet iS10 FTIR spectrophotometer with Smart iTr. Wavenumbers (ν) are reported in cm
−1. MS data (m/z (%), EI, 70 eV) were obtained by using an Agilent Technologies 5975. HRMS data were obtained by using a X500B (SCIEX) QTOF high-resolution mass spectrometer (ESI mode). Elemental microanalyses were obtained on a EuroVector Instruments Euro EA 3000 elemental analyzer. All microwave irradiation experiments were carried out in a dedicated Biotage-Initiator microwave apparatus, operating at a frequency of 2.45 GHz with continuous irradiation power from 0 to 400 W with the utilization of the standard absorbance level of 400 W maximum power. Reactions were carried out in glass tubes, sealed with aluminum/Teflon crimp tops, which can be exposed up to 250 °C and 20 bar internal pressure. Temperature was measured with an IR sensor on the outer surface of the process vial. After the irradiation period, the reaction vessel was cooled rapidly (60–120 s) to ambient temperature by air-jet cooling. Synthesis and spectroscopic data for all compounds are described in the
Supplementary Materials.
3.2. General Procedure for the Synthesis of Structures 18
The intermediate 4-((1H-benzo[d][1-3]triazol-1-yl)oxy)-2-(phenylamino)-4,5,6,8-tetrahydropyrido[2,3-d]pyrimidin-7(8H)-one (15) (100 mg, 0.267 mmol) was suspended in ACN (20 mL), 3 equivalents of the corresponding amine (0.803 mmol) were added to the suspension, and the mixture was heated at 140 °C under microwave irradiation for 6 h. Then, water was added to the residue and the solid was collected by filtration and washed with water, ethanol, and diethyl ether in order to afford the corresponding spectroscopically pure product.
4-((3-Morpholinopropyl)amino)-2-(phenylamino)-5,6-dihydropyrido[2,3-d]pyrimidin-7(8H)-one (18a)
As above but carried out by using 4-((1H-benzo[d][1-3]triazol-1-yl)oxy)-2-(phenylamino)-4,5,6,8-tetrahydropyrido[2,3-d]pyrimidin-7(8H)-one (15) (100 mg, 0.267 mmol) and N-(3-aminopropyl)morpholine (117 μL, 0.803 mmol). The mixture was heated at 140 °C for 6h under microwave irradiation. In total, 54.1 mg (0.141 mmol, 54%) of spectroscopically pure 4-((3-morpholinopropyl)amino)-2-(phenylamino)-5,6-dihydropyrido[2,3-d]pyrimidin-7(8H)-one (18a) were obtained as an orangish solid. mp: >250 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 9.96 (s, 1H, N8-H), 8.73 (s, 1H, N9-H), 7.85–7.79 (m, 2H, C11-H), 7.22–7.16 (m, 2H, C12-H), 6.88–6.81 (m, 1H, C13-H), 6.67 (t, J = 5.5 Hz, 1H, N14-H), 3.56 (t, J = 4.6 Hz, 4H, C20-H), 3.41 (q, J = 6.7 Hz, 2H, C15-H), 2.60–2.51 (m, 4H, C5-H, C6-H), 2.40–2.29 (m, 6H, C19-H, C17-H), 1.74 (p, J = 7.1 Hz, 2H, C16-H). 13C-NMR (100.5 MHz, DMSO-d6) δ (ppm): 171.1 (C7), 160.1 (C4), 157.9 (C2), 155.5 (C8a), 141.5 (C10), 128.2 (C12), 120.1 (C13), 118.2 (C11), 85.9 (C4a), 66.2 (C20), 56.4 (C17), 53.4 (C19), 39.1 (C15), 30.3 (C6), 26.0 (C16), 17.1 (C5). IR (KBr) ν (cm−1): 3429, 3287, 3204, 2917, 2861, 1635, 1601, 1579, 1548, 1444, 1375, 1245, 1119, 752. OEA calculated for C20H26N6O2: C: 62.81%, H: 6.85%, N: 21.97%; found: C: 62.46%, H: 6.67%, N: 21.57%. HRMS (APCI-FIA-TOF) (m/z) calculated for C20H26N6O2: 382.2117, [M]+, found: 383.2187, [M+H]+.
3.3. General Procedure for the Synthesis of Structures 19
Intermediate 7-oxo-2-(phenylamino)-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidin-4-yl trifluoromethanesulfonate (16) (1 eq), cesium carbonate (1.2 eq), palladium(II)acetate (0.1 eq), XPhos (0.15 eq), and the corresponding aniline (1.1 eq) were introduced under argon atmosphere into a Schlenk tube. After that, anhydrous toluene (1 mL) was added and the mixture was heated overnight at 100 °C. Then, water (30 mL) was added to the residue and the solid was collected by filtration and washed with water, ethanol, and cyclohexane in order to afford the spectroscopically pure product.
2-(Phenylamino)-4-((3,4,5-trimethoxyphenyl)amino)-5,6-dihydropyrido[2,3-d]pyrimidin-7(8H)-one (19a)
As above but carried out by using 7-oxo-2-(phenylamino)-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidin-4-yl trifluoromethanesulfonate (16) (77.66 mg, 0.20 mmol) and 3,4,5-trimethoxyaniline (40.30 mg, 0.22 mmol). In total, 79.2 mg (0.187 mmol, 94%) of spectroscopically pure 2-(phenylamino)-4-((3,4,5-trimethoxyphenyl)amino)-5,6-dihydropyrido[2,3-d]pyrimidin-7(8H)-one (19a) were obtained as a pale brown solid. mp: >250 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.17 (s, 1H, N8-H), 8.87 (s, 1H, N9-H), 8.29 (s, 1H, N14-H), 7.75–7.72 (m, 2H, C11-H), 7.13–7.09 (m, 2H, C12-H), 6.90 (s, 2H, C16-H), 6.86–6.82 (m, 1H, C13-H), 3.69 (s, 6H, C19-H), 3.65 (s, 3H, C20-H), 2.77 (dd, J = 8.3, 7.0 Hz, 2H, C5-H), 2.56 (dd, J = 8.2, 7.0 Hz, 2H, C6-H). 13C-NMR (100.5 MHz, DMSO-d6) δ (ppm): 171.2 (C7), 158.1 (C4), 157.5 (C2), 157.0 (C8a), 152.4 (C17), 141.0 (C10), 135.9 (C15), 133.1 (C18), 128.1 (C12), 120.5 (C13), 118.6 (C11), 100.2 (C16), 87.9 (C4a), 60.1 (C20), 55.6 (C19), 30.3 (C6), 17.3 (C5). IR (KBr) ν (cm−1): 3350, 3288, 3206, 3138, 2959, 2927, 2851, 1677, 1601, 1580, 1503, 1443, 1239, 1221, 1129, 996, 745. HRMS (APCI-FIA-TOF) (m/z) calculated for C22H23N5O4: 421.1750, [M]+, found: 422.1820, [M+H]+.
3.4. General Procedure for the Synthesis of Structures 20 and 21
Intermediate 4-iodo-2-(phenylamino)-5,6-dihydropyrido[2,3-d]pyrimidin-7(8H)-one (17) (1 eq), tripotassium phosphate (2 eq), copper(I) iodide (0.05 eq), 2-picolinic acid (0.1 eq), and the corresponding phenol or thiophenol (1.2 eq) were introduced under argon atmosphere into a Schlenk tube. After that, anhydrous DMSO (1.4 mL) was added and the mixture was heated overnight at 80 °C. Then, water (30 mL) was added to the residue and the solid was collected by filtration and washed with more water, ethanol, and diethyl ether to afford the spectroscopically pure product.
4-Phenoxy-2-(phenylamino)-5,6-dihydropyrido[2,3-d]pyrimidin-7(8H)-one (20a)
As above but carried out by using 4-iodo-2-(phenylamino)-5,6-dihydropyrido[2,3-d]pyrimidin-7(8H)-one (17) (73.23 mg, 0.20 mmol) and phenol (22.59 mg, 0,24 mmol). The mixture reaction was heated at 80 °C for 48h. In total, 41.7 mg (0.125 mmol, 64%) of spectroscopically pure 4-phenoxy-2-(phenylamino)-5,6-dihydropyrido[2,3-d]pyrimidin-7(8H)-one (20a) were obtained as a greyish solid. mp: >250 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.60 (s, 1H, N8-H), 9.20 (s, 1H, N9-H), 7.53–7.41 (m, 4H, C11-H, C17-H), 7.33–7.24 (m, 1H, C18-H), 7.26–7.17 (m, 2H, C16-H), 7.06–6.98 (m, 2H, C12-H), 6.85–6.76 (m, 1H, C13-H), 2.85 (dd, J = 8.2, 7.1 Hz, 2H, C5-H), 2.61 (dd, J = 8.2, 7.1 Hz, 2H, C6-H). 13C-NMR (100.5 MHz, DMSO-d6) δ (ppm): 171.5 (C7), 165.9 (C4*), 159.6 (C8a*), 157.4 (C2), 153.0 (C15), 140.4 (C10), 129.5 (C17), 128.0 (C12), 125.0 (C18), 121.9 (C16), 120.8 (C13), 118.2 (C11), 89.9 (C4a), 30.2 (C6), 16.4 (C5). IR (ATR) ν (cm−1): 3284, 3203, 3138, 2968, 1683, 1618, 1577, 1549, 1441, 1401, 1349, 1236, 1199, 750, 688. HRMS (APCI-FIA-TOF) (m/z) calculated for C19H16N4O2: 332.1273, [M]+, found: 333.1345, [M+H]+.
3.5. General Procedure for the Synthesis of Structures 22
Intermediate 4-iodo-2-(phenylamino)-5,6-dihydropyrido[2,3-d]pyrimidin-7(8H)-one (17) (1 eq), cesium carbonate (2.5 eq), tetrakis(triphenylphosphine)palladium(0) (2% molar), and the corresponding boronic acid (1.4 eq) were introduced under argon atmosphere into a Schlenk tube. After that, a deoxygenated mixture of 1,4-dioxane/water (10:1) (1.5 mL) was added and the resultant reaction mixture was heated overnight at 90 °C. Then, water was added (20 mL) to the residue and the solid appeared was collected by filtration and washed with more water, ethanol, and diethyl ether to afford the spectroscopically pure product.
4-Phenyl-2-(phenylamino)-5,6-dihydropyrido[2,3-d]pyrimidin-7(8H)-one (22a)
As above but carried out by using 4-iodo-2-(phenylamino)-5,6-dihydropyrido[2,3-d]pyrimidin-7(8H)-one (17) (73.23 mg, 0.20 mmol) and phenylboronic acid (34.14 mg, 0.28 mmol). 37.4 mg (60%, 0.118 mmol) of spectroscopically pure 4-phenyl-2-(phenylamino)-5,6-dihydropyrido[2,3-d]pyrimidin-7(8H)-one (22a) were obtained as a dark grey solid. mp: >250 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.72 (s, 1H, N8-H), 9.41 (s, 1H, N9-H), 7.90–7.81 (m, 2H, C11-H), 7.65–7.59 (m, 2H, C15-H), 7.54–7.46 (m, 3H, C16-H, C17-H), 7.26–7.20 (m, 2H, C12-H), 6.93–6.87 (m, 1H, C13-H), 2.85 (dd, J = 8.4, 6.6 Hz, 2H, C5-H), 2.53–2.50 (m, 2H, C6-H). 13C-NMR (100.5 MHz, DMSO-d6) δ (ppm): 171.6 (C7), 163.2 (C4), 158.9 (C8a), 158.1 (C2), 140.9 (C10), 137.7 (C14), 129.0 (C17), 128.6 (C15), 128.3 (C12), 128.1 (C16), 120.8 (C13), 118.5 (C11), 103.4 (C4a), 30.7 (C6), 20.5 (C5). IR (ATR) ν (cm−1): 3281, 3198, 3141, 3047, 2904, 1686, 1592, 1544, 1435, 1343, 1303, 1217, 815, 749, 689. HRMS (APCI-FIA-TOF) (m/z) calculated for C19H16N4O: 316.1324, [M]+, found: 317.1392, [M+H]+.
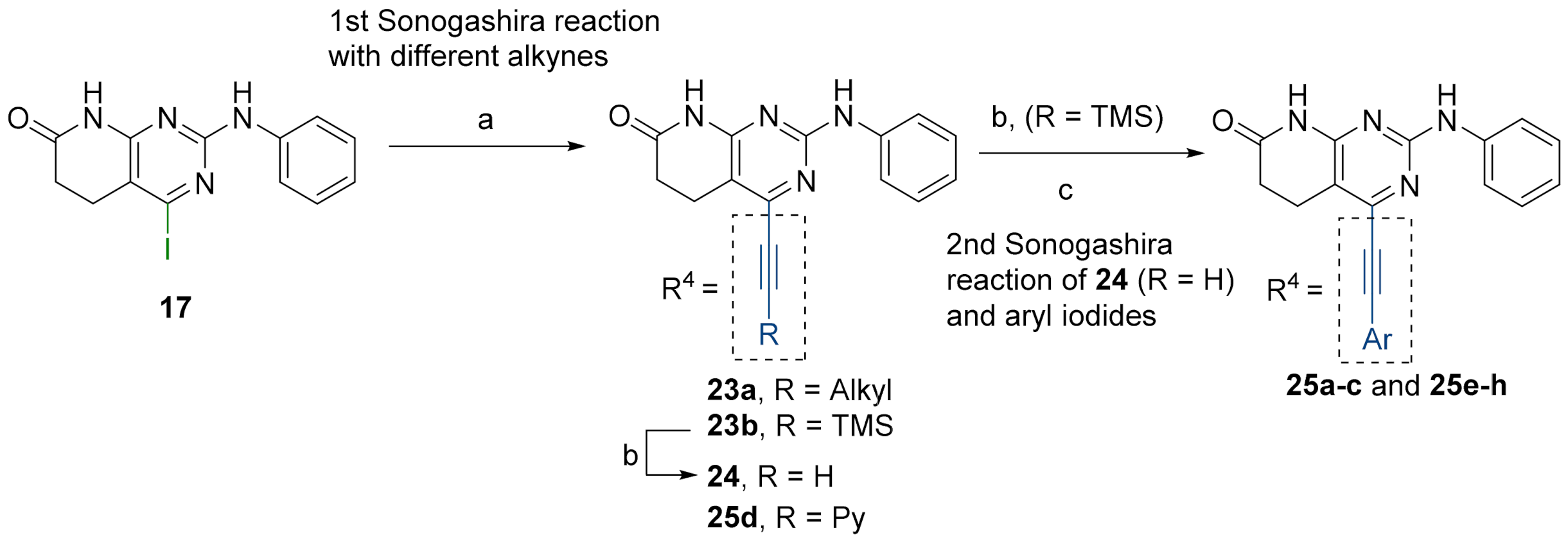
3.6. General Procedure for the Synthesis of Structures 23
Intermediate 4-iodo-2-(phenylamino)-5,6-dihydropyrido[2,3-d]pyrimidin-7(8H)-one (17) (1 eq), bis(triphenylphosphine)palladium dichloride (1.5% molar), copper(I) iodide (3% molar), and the corresponding alkyne (1.5 eq) were introduced under argon atmosphere into a Schlenk tube. Then, triethylamine (1.5 mL) was added and the Schlenk tube was sealed. The resulting reaction mixture was heated at 65 °C overnight under vigorous stirring. The solvent was evaporated in vacuo and the resulting mixture was suspended in water (50 mL) and the spectroscopically pure product was collected by filtration after being washed with more water, ethanol, and diethyl ether.
2-(Phenylamino)-4-((trimethylsilyl)ethynyl)-5,6-dihydropyrido[2,3-d]pyrimidin-7(8H)-one (23b)
As above but carried out by using 4-iodo-2-(phenylamino)-5,6-dihydropyrido[2,3-d]pyrimidin-7(8H)-one (17) (300.0 mg, 0.82 mmol) and ethynyltrimethylsilane (170 μL, 1.23 mmol). In total, 218.3 mg (0.649 mmol, 79%) of spectroscopically pure 2-(phenylamino)-4-((trimethylsilyl)ethynyl)-5,6-dihydropyrido[2,3-d]pyrimidin-7(8H)-one (23b) were obtained as a pale green solid. mp: >250 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.80 (s, 1H, N8-H), 9.51 (s, 1H, N9-H), 7.83–7.71 (m, 2H, C11-H), 7.34–7.16 (m, 2H, C12-H), 7.01–6.87 (m, 1H, C13-H), 2.86 (dd, J = 8.3, 6.9 Hz, 2H, C5-H), 2.59 (dd, J = 8.3, 6.9 Hz, 2H, C6-H), 0.26 (s, 9H, C17-H). 13C-NMR (100.5 MHz, DMSO-d6) δ (ppm): 171.4 (C7), 158.5 (C8a), 158.2 (C2), 146.3 (C4), 140.5 (C10), 128.4 (C12), 121.1 (C13), 118.5 (C11), 107.9 (C4a), 100.7 (C15), 100.5 (C14), 30.1 (C6), 19.9 (C5), -0.5 (C17). IR (ATR) ν (cm−1): 3283, 3144, 2957, 1685, 1592, 1567, 1547, 1500, 1437, 1335, 1210, 1063, 1020, 839, 744. HRMS (APCI-FIA-TOF) (m/z) calculated for C18H20N4OSi: 336.1406, [M]+, found: 337.1475, [M+H]+.
Kinase inhibition profile. The kinase inhibition profile of compounds was evaluated at Reaction Biology (
https://www.reactionbiology.com/) (accessed on 10 November 2021) by measuring residual activity values at a concentration of 10 µM of the test compound in singlicate in front of the several kinases including Btk, Lyn, Syk aa1-635, and Zap70 (see the
Supplementary Materials for the complete list of targets) using the following protocol: The compounds were dissolved to 1 × 10
−3 M stock solutions in 100% DMSO. Subsequently, 100 μL of each stock solution were transferred into wells A3-F12 of a microtiter plate (“master plate”). Wells A1-F2 were filled with 100 μL of 100% DMSO as controls. In total, 5 × 10 μL of the master plate were aliquoted into 5 copy plates, which were stored at −20 °C until use. For the testing of each group of up to 8 kinases, one copy plate was used. In the process, 90 μL of H
2O were added to each well of a copy plate. To minimize precipitation, the H
2O was added to each well only a few minutes before the transfer of the compound solutions into the assay plates. The plate was shaken thoroughly, resulting in a “compound dilution plate” with a compound concentration of 1 × 10
−4 M/10 % DMSO. This plate was used for the transfer of 5 μL of compound solution into the assay plates. The final volume of the assay was 50 μL. All compounds were tested at 1 × 10
−5 M in singlicate. The final DMSO concentration in the reaction cocktails was 1% in all cases. The compound dilution plates were disposed of at the end of each working day.
A radiometric protein kinase assay (33PanQinase® Activity Assay) was used for measuring the kinase activity of the corresponding protein kinases. All kinase assays were performed in 96-well FlashPlatesTM from Perkin Elmer (Boston, MA, USA) in a 50 μL reaction volume. The reaction cocktail was pipetted in 4 steps in the following order: 10 μL of non-radioactive ATP solution (in H2O); 25 μL of assay buffer/ [γ-33P]-ATP mixture; 5 μL of test sample in 10% DMSO; and 10 μL of enzyme/substrate mixture. The assay for all protein kinases contained 70 mM HEPES-NaOH pH 7.5, 3 mM MgCl2, 3 mM MnCl2, 3 μM Na-orthovanadate, 1.2 mM DTT, ATP (variable amounts, corresponding to the apparent ATP-Km of the respective kinase), [γ-33P]-ATP (approximately 8 × 105 cpm per well), protein kinase (variable amounts), and substrate (variable amounts). The protein kinase reaction cocktails were incubated at 30 °C for 60 min. The reaction was stopped with 50 μL of 2 % (v/v) H3PO4, plates were aspirated, and washed two times with 200 μL of 0.9 % (w/v) NaCl. All assays were performed with a BeckmanCoulter Biomek 2000/SL robotic system. Incorporation of 33Pi (counting of “cpm”) was determined with a microplate scintillation counter (Microbeta, Wallac). All protein kinase assays were performed with a BeckmanCoulter Core robotic system.
For each kinase, the median value of the cpm of six wells of column 1 of each assay plate was defined as the “low control” (
n = 6). This value reflects unspecific binding of radioactivity to the plate in the absence of a protein kinase but in the presence of the substrate. Additionally, for each kinase, the median value of the cpm of six wells of column 2 of each assay plate was taken as the “high control”, i.e., full activity in the absence of any inhibitor (
n = 6). The difference between the high and low control of each enzyme was taken as 100% activity. As part of the data evaluation the low control of each kinase was subtracted from the high control value as well as from their corresponding “compound values”. The residual activity (in %) for each compound well was calculated by using the following formula:
As a parameter for assay quality, the Z´-factor30 for the low and high controls of each assay plate (n = 8) was used. Reaction Biology´s criterion for repetition of an assay plate is a Z´-factor below 0.4.31 Z´-factors did not drop below 0.51, indicating an excellent assay quality.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



















































