
Fragment-Based Ligand Discovery Applied to the Mycolic Acid Methyltransferase Hma (MmaA4) from Mycobacterium tuberculosis: A Crystallographic and Molecular Modelling Study

 , ,
, , 
Abstract
1. Introduction
2. Results
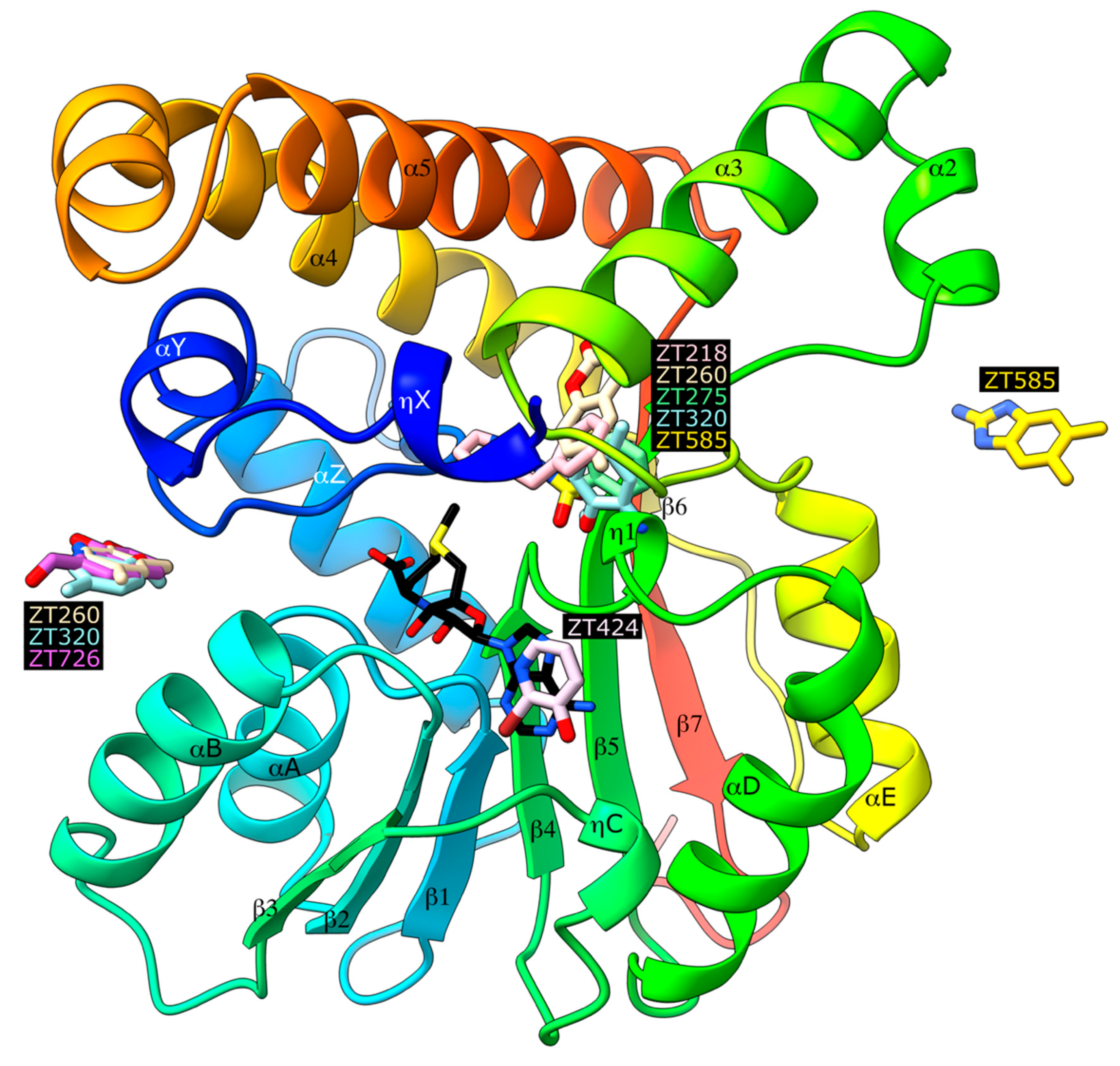
2.1. Crystallographic Structures of Fragment-Bound Hma
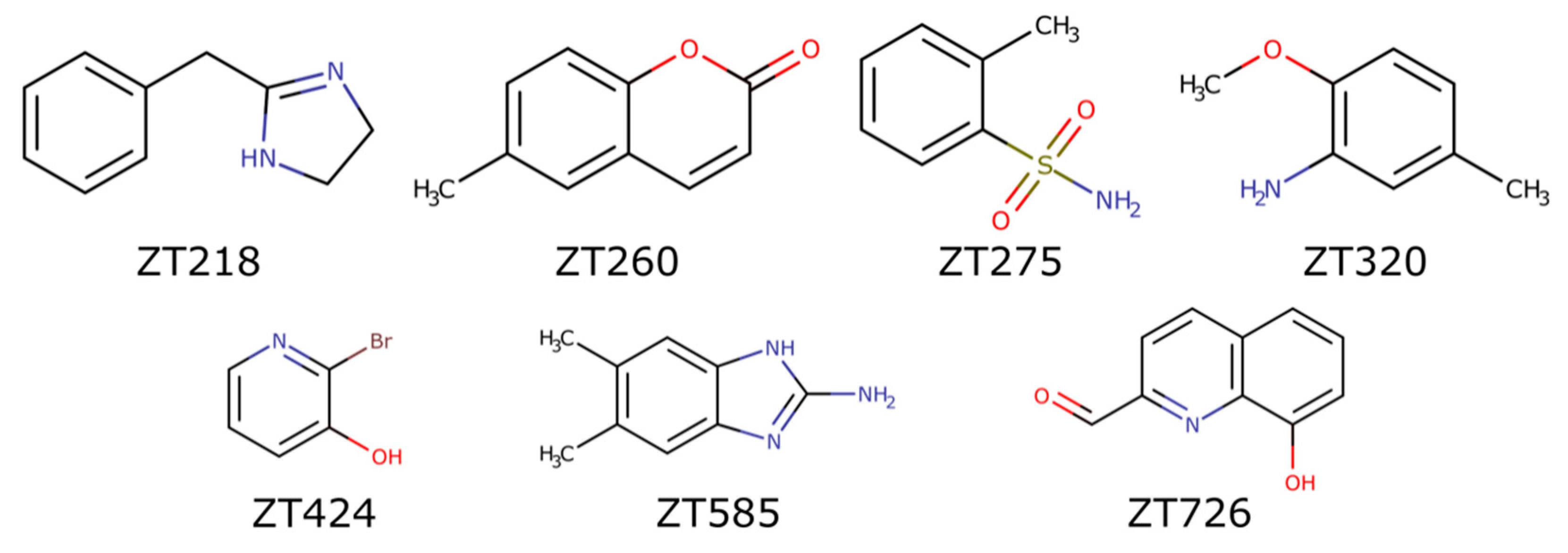
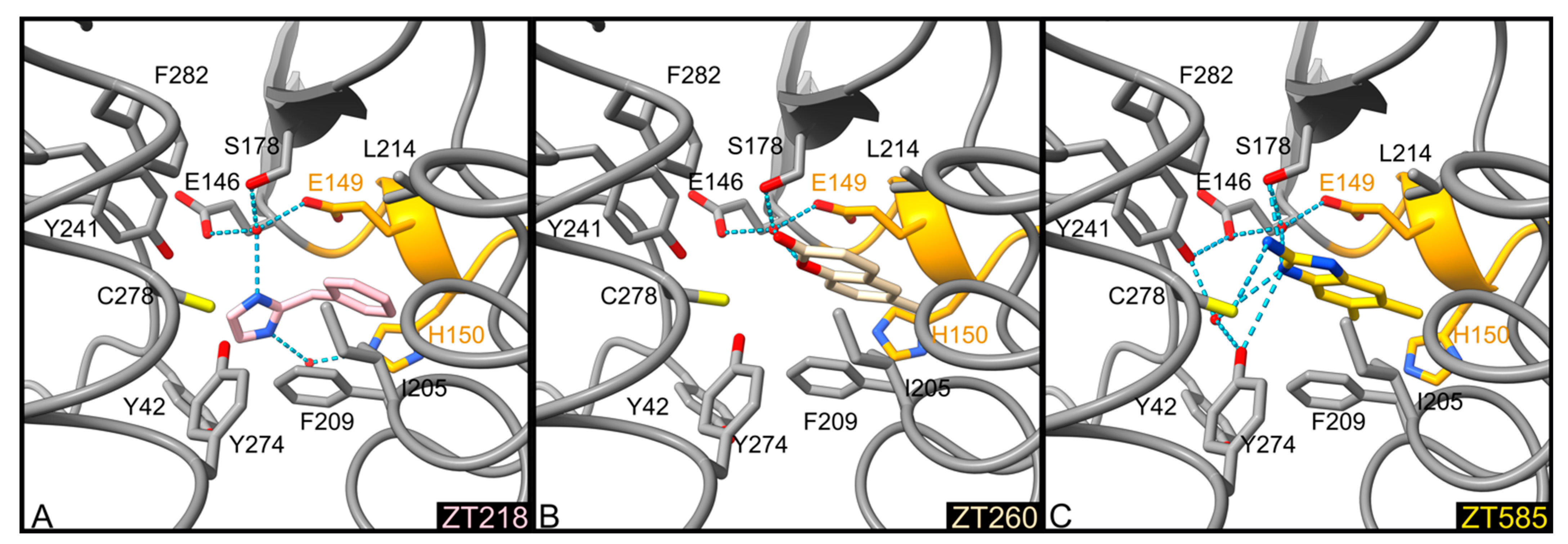
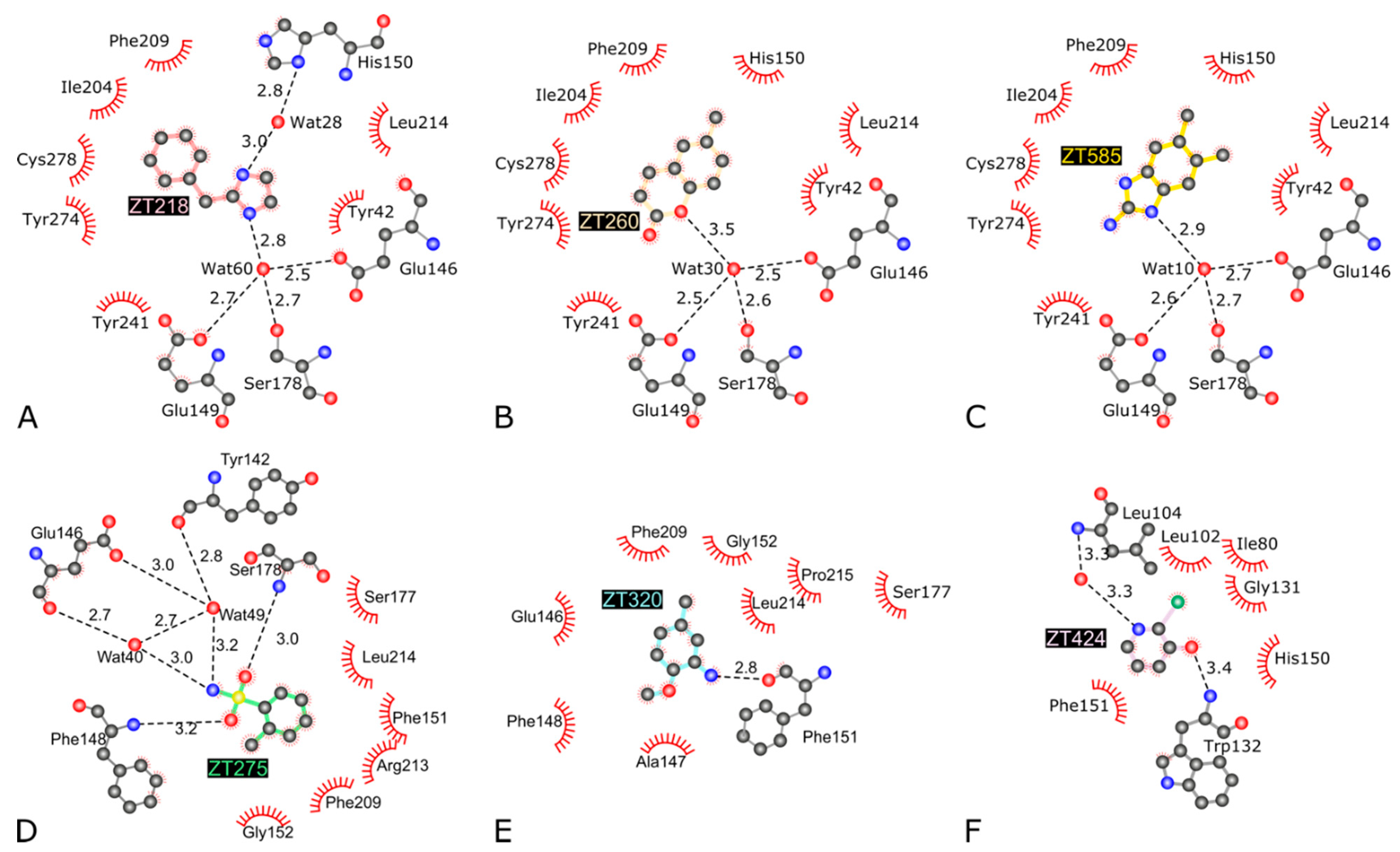
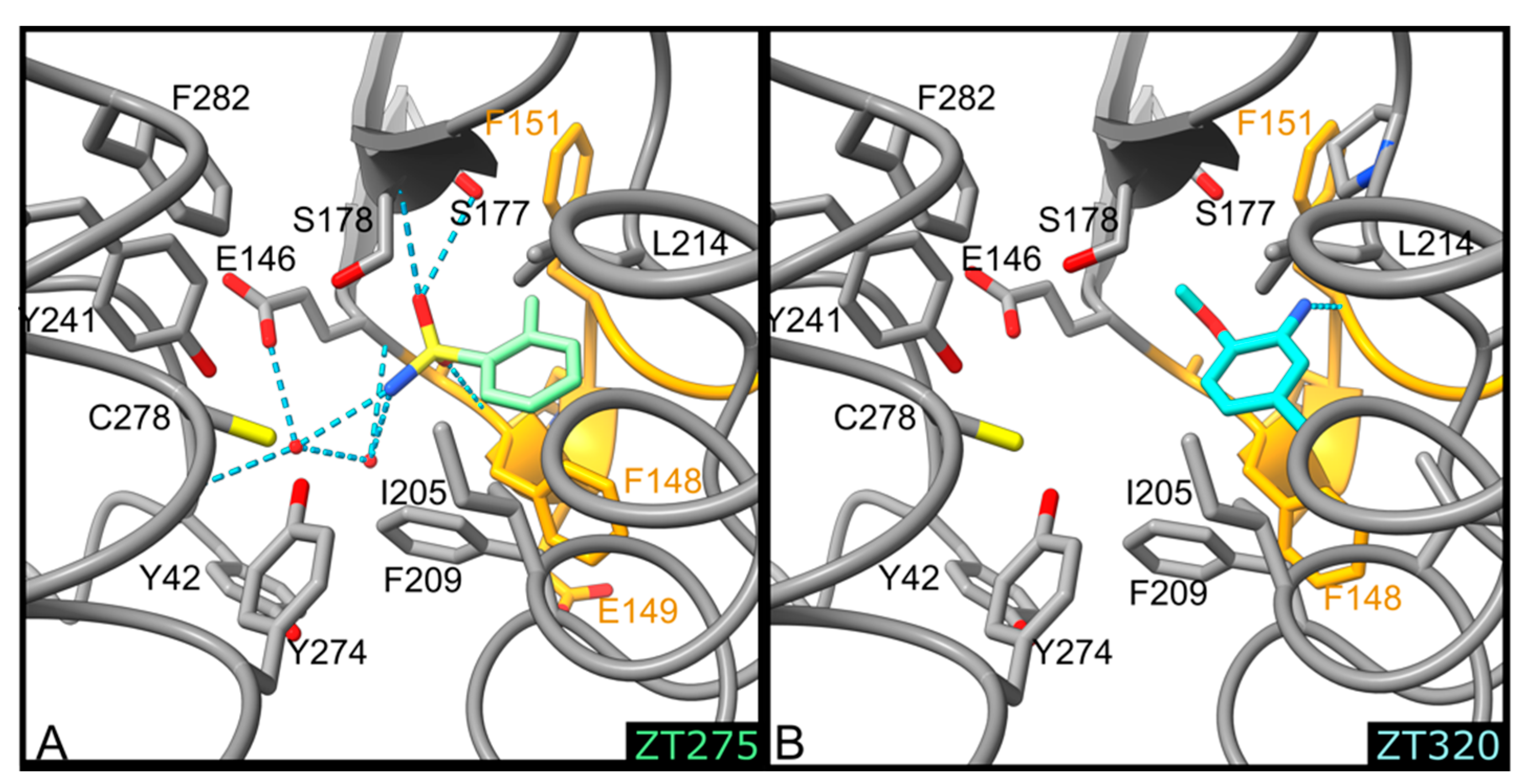
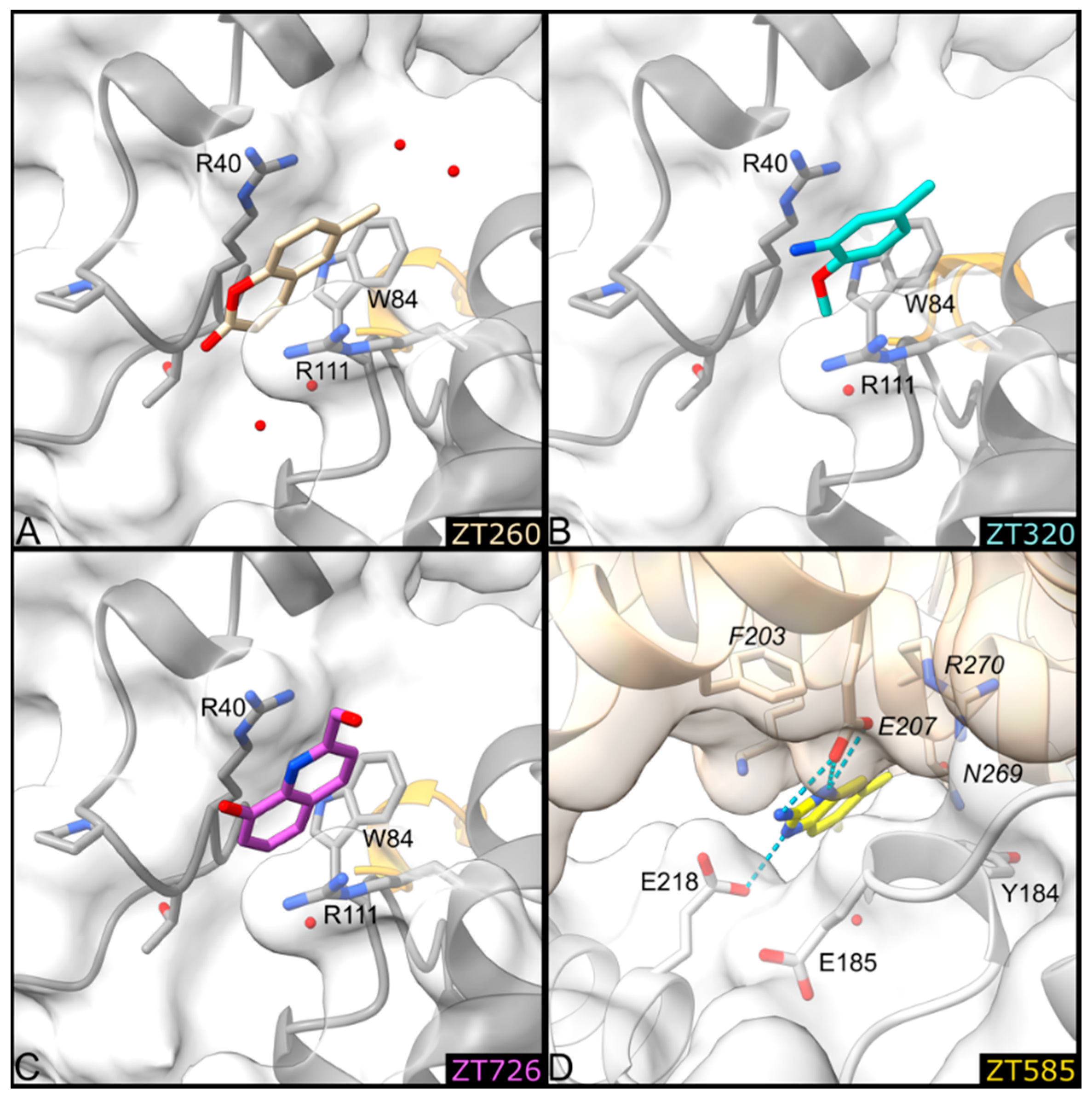
2.1.1. Fragments ZT218, ZT260, ZT275, ZT320, and ZT585 Bind at the Substrate Binding Site
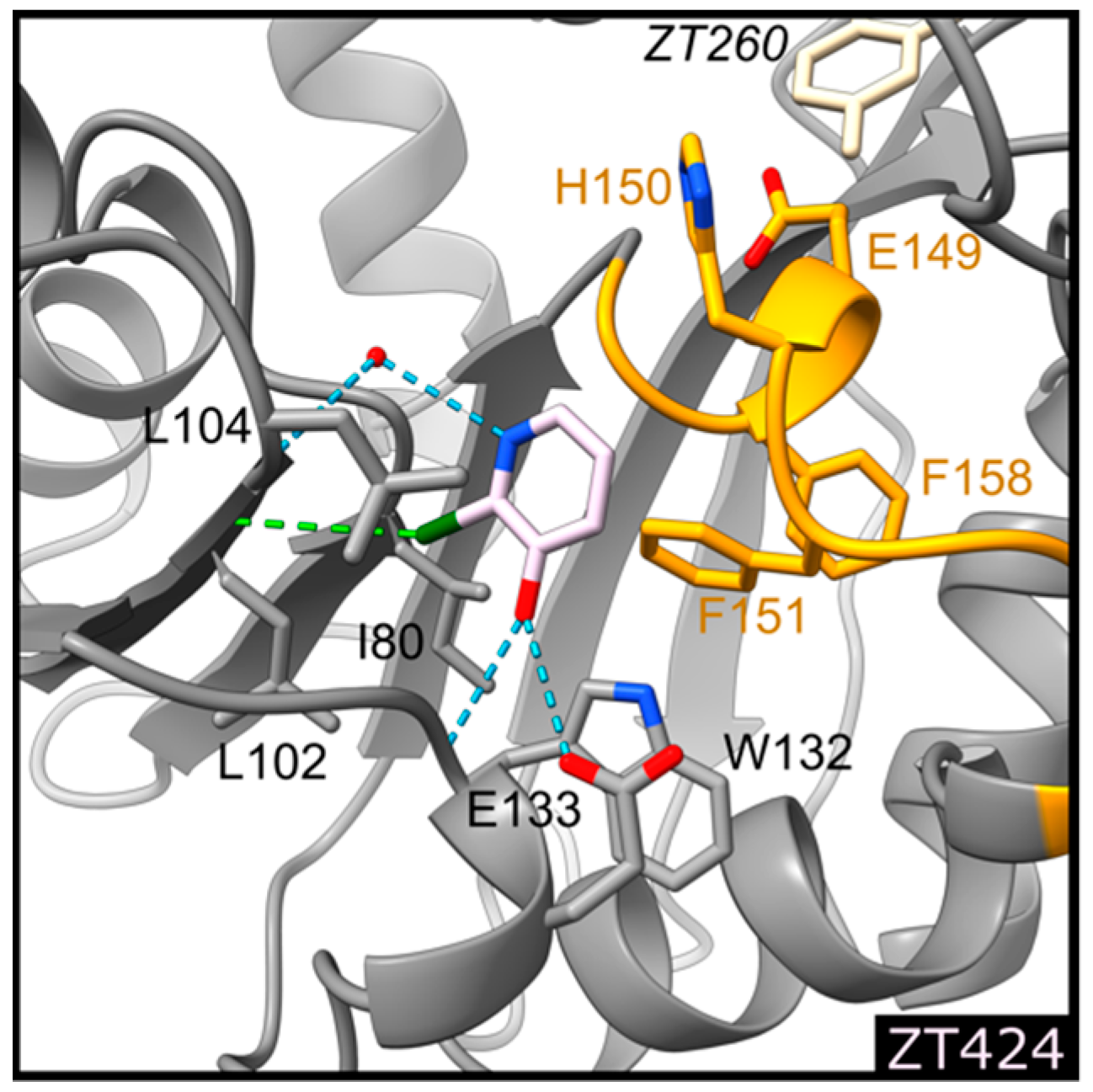
2.1.2. Fragment ZT424 Binds at the Cofactor Adenine Site
2.1.3. Fragments ZT260, ZT320, ZT585, and ZT726 Bind at the Protein Surface
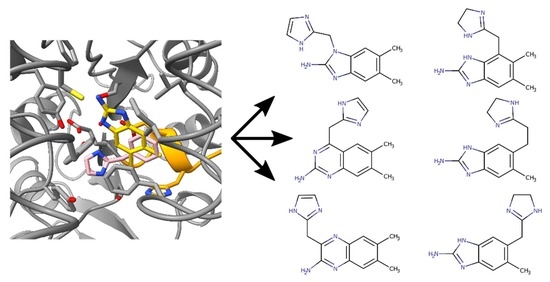
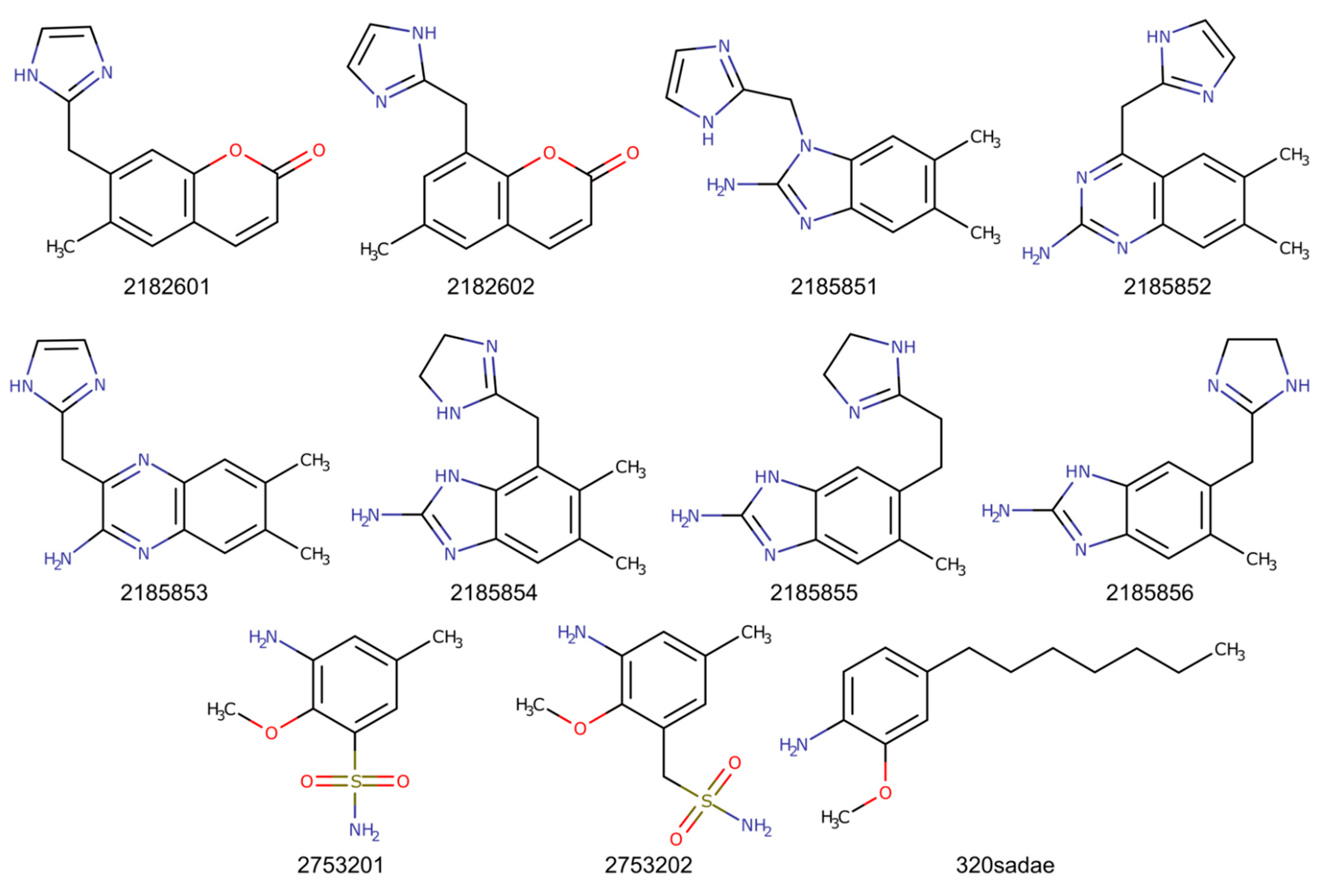
2.2. Chimeric Compounds
2.3. Molecular Dynamics Simulations
2.3.1. Apo-Hma
2.3.2. Hma in the Presence of Fragments or of Chimeric Compounds
2.4. Estimation of Binding Energies for Fragments and Chimeric Compounds
3. Discussion
3.1. Crystallographic Screening
3.2. Molecular Plasticity of Hma
3.3. Computed Binding Energies of Fragments and Chimeric Compounds
4. Materials and Methods
4.1. Expression, Purification, and Crystallisation of Hma
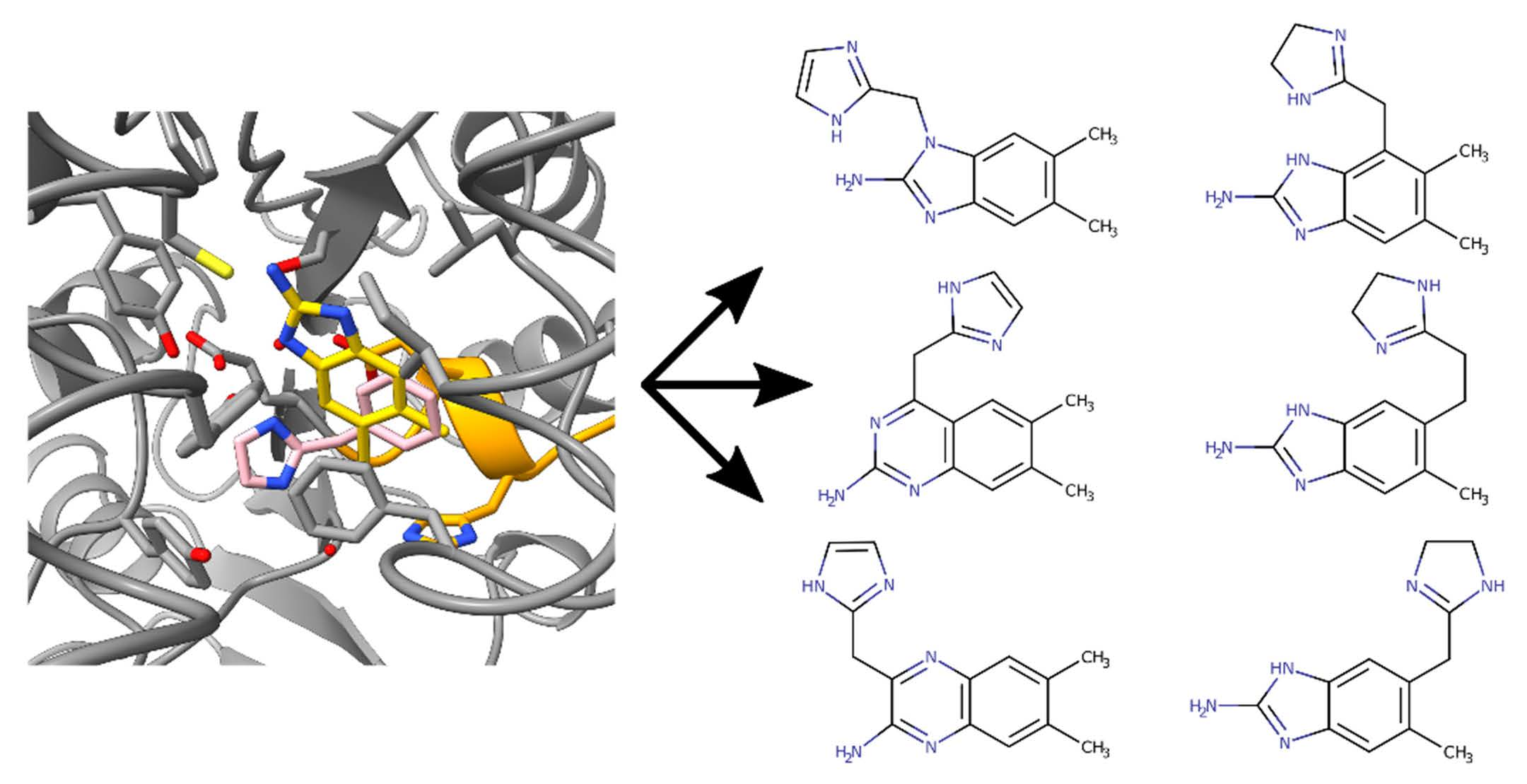
4.2. Fragments
4.3. Crystallographic Screening
4.4. Molecular Dynamics Simulation
4.5. Relative Binding Affinity Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2021; World Health Organization: Geneva, Switzerland, 2021; ISBN 978-92-4-003702-1. [Google Scholar]
- Daffé, M.; Marrakchi, H. Unraveling the Structure of the Mycobacterial Envelope. Microbiol. Spectr. 2019, 7, GPP3-0027-2018. [Google Scholar] [CrossRef] [PubMed]
- Jarlier, V.; Nikaido, H. Mycobacterial Cell Wall: Structure and Role in Natural Resistance to Antibiotics. FEMS Microbiol. Lett. 1994, 123, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Cambier, C.J.; Takaki, K.K.; Larson, R.P.; Hernandez, R.E.; Tobin, D.M.; Urdahl, K.B.; Cosma, C.L.; Ramakrishnan, L. Mycobacteria Manipulate Macrophage Recruitment through Coordinated Use of Membrane Lipids. Nature 2014, 505, 218–222. [Google Scholar] [CrossRef]
- Batt, S.M.; Minnikin, D.E.; Besra, G.S. The Thick Waxy Coat of Mycobacteria, a Protective Layer against Antibiotics and the Host’s Immune System. Biochem. J. 2020, 477, 1983–2006. [Google Scholar] [CrossRef]
- Marrakchi, H.; Lanéelle, M.-A.; Daffé, M. Mycolic Acids: Structures, Biosynthesis, and Beyond. Chem. Biol. 2014, 21, 67–85. [Google Scholar] [CrossRef]
- Jackson, M. The Mycobacterial Cell Envelope—Lipids. Cold Spring Harb. Perspect. Med. 2014, 4, a021105. [Google Scholar] [CrossRef]
- McNeil, M.; Daffé, M.; Brennan, P.J. Location of the Mycolyl Ester Substituents in the Cell Walls of Mycobacteria. J. Biol. Chem. 1991, 266, 13217–13223. [Google Scholar] [CrossRef]
- Abrahams, K.A.; Besra, G.S. Mycobacterial Cell Wall Biosynthesis: A Multifaceted Antibiotic Target. Parasitology 2018, 145, 116–133. [Google Scholar] [CrossRef] [PubMed]
- North, E.J.; Jackson, M.; Lee, R.E. New Approaches to Target the Mycolic Acid Biosynthesis Pathway for the Development of Tuberculosis Therapeutics. Curr. Pharm. Des. 2014, 20, 4357–4378. [Google Scholar] [CrossRef]
- Banerjee, A.; Dubnau, E.; Quémard, A.; Balasubramanian, V.; Um, K.S.; Wilson, T.; Collins, D.; de Lisle, G.; Jacobs, W.R. InhA, a Gene Encoding a Target for Isoniazid and Ethionamide in Mycobacterium tuberculosis. Science 1994, 263, 227–230. [Google Scholar] [CrossRef]
- Quémard, A.; Lanéelle, G.; Lacave, C. Mycolic Acid Synthesis: A Target for Ethionamide in Mycobacteria? Antimicrob. Agents Chemother. 1992, 36, 1316–1321. [Google Scholar] [CrossRef][Green Version]
- Takayama, K.; Wang, L.; David, H.L. Effect of Isoniazid on the In Vivo Mycolic Acid Synthesis, Cell Growth, and Viability of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1972, 2, 29–35. [Google Scholar] [CrossRef]
- Portevin, D.; de Sousa-D’Auria, C.; Houssin, C.; Grimaldi, C.; Chami, M.; Daffé, M.; Guilhot, C. A Polyketide Synthase Catalyzes the Last Condensation Step of Mycolic Acid Biosynthesis in Mycobacteria and Related Organisms. Proc. Natl. Acad. Sci. USA 2004, 101, 314–319. [Google Scholar] [CrossRef]
- Varela, C.; Rittmann, D.; Singh, A.; Krumbach, K.; Bhatt, K.; Eggeling, L.; Besra, G.S.; Bhatt, A. MmpL Genes Are Associated with Mycolic Acid Metabolism in Mycobacteria and Corynebacteria. Chem. Biol. 2012, 19, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.S. The MmaA2 Gene of Mycobacterium tuberculosis Encodes the Distal Cyclopropane Synthase of the α-Mycolic Acid. J. Biol. Chem. 2003, 278, 7844–7849. [Google Scholar] [CrossRef]
- Glickman, M.S.; Cox, J.S.; Jacobs, W.R. A Novel Mycolic Acid Cyclopropane Synthetase Is Required for Cording, Persistence, and Virulence of Mycobacterium tuberculosis. Mol. Cell 2000, 5, 717–727. [Google Scholar] [CrossRef]
- Laval, F.; Haites, R.; Movahedzadeh, F.; Lemassu, A.; Wong, C.Y.; Stoker, N.; Billman-Jacobe, H.; Daffé, M. Investigating the Function of the Putative Mycolic Acid Methyltransferase UmaA: Divergence between the Mycobacterium smegmatis and Mycobacterium tuberculosis Proteins. J. Biol. Chem. 2008, 283, 1419–1427. [Google Scholar] [CrossRef]
- Dubnau, E.; Chan, J.; Raynaud, C.; Mohan, V.P.; Lanéelle, M.-A.; Yu, K.; Quémard, A.; Smith, I.; Daffé, M. Oxygenated Mycolic Acids Are Necessary for Virulence of Mycobacterium tuberculosis in Mice. Mol. Microbiol. 2000, 36, 630–637. [Google Scholar] [CrossRef]
- Dinadayala, P.; Laval, F.; Raynaud, C.; Lemassu, A.; Lanéelle, M.-A.; Lanéelle, G.; Daffé, M. Tracking the Putative Biosynthetic Precursors of Oxygenated Mycolates of Mycobacterium tuberculosis. Structural Analysis of Fatty Acids of a Mutant Strain Devoid of Methoxy- and Ketomycolates. J. Biol. Chem. 2003, 278, 7310–7319. [Google Scholar] [CrossRef] [PubMed]
- Dubnau, E.; Marrakchi, H.; Smith, I.; Daffé, M.; Quémard, A. Mutations in the CmaB Gene are Responsible for the Absence of Methoxymycolic Acid in Mycobacterium bovis BCG Pasteur. Mol. Microbiol. 1998, 29, 1526–1528. [Google Scholar] [PubMed]
- Yuan, Y.; Crane, D.C.; Musser, J.M.; Sreevatsan, S.; Barry, C.E. MMAS-1, the Branch Point Between Cis- and Trans-Cyclopropane-Containing Oxygenated Mycolates in Mycobacterium tuberculosis. J. Biol. Chem. 1997, 272, 10041–10049. [Google Scholar] [CrossRef]
- Felipe, L.A.; Osman, F.; Marti, M.A.; Turjanski, A.G. Structural and mechanistic comparison of the Cylopropane Mycolic Acid Synthases (CMAS) protein of Mycobacterium tuberculosis. Biochem. Biophys. Res. Commun. 2018, 598, 288–295. [Google Scholar] [CrossRef]
- Barkan, D.; Liu, Z.; Sacchettini, J.C.; Glickman, M.S. Mycolic Acid Cyclopropanation Is Essential for Viability, Drug Resistance, and Cell Wall Integrity of Mycobacterium tuberculosis. Chem. Biol. 2009, 16, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Barkan, D.; Hedhli, D.; Yan, H.-G.; Huygen, K.; Glickman, M.S. Mycobacterium tuberculosis Lacking all Mycolic Acid Cyclopropanation is Viable but Highly Attenuated and Hyperinflammatory in Mice. Infect. Immun. 2012, 80, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Vaubourgeix, J.; Bardou, F.; Boissier, F.; Julien, S.; Constant, P.; Ploux, O.; Daffé, M.; Quémard, A.; Mourey, L. S-Adenosyl-N-Decyl-Aminoethyl, a Potent Bisubstrate Inhibitor of Mycobacterium tuberculosis Mycolic Acid Methyltransferases. J. Biol. Chem. 2009, 284, 19321–19330. [Google Scholar] [CrossRef] [PubMed]
- Laval, F.; Lanéelle, M.A.; Déon, C.; Monsarrat, B.; Daffé, M. Accurate Molecular Mass Determination of Mycolic Acids by MALDI-TOF Mass Spectrometry. Anal. Chem. 2001, 73, 4537–4544. [Google Scholar] [CrossRef]
- Dao, D.N.; Sweeney, K.; Hsu, T.; Gurcha, S.S.; Nascimento, I.P.; Roshevsky, D.; Besra, G.S.; Chan, J.; Porcelli, S.A.; Jacobs, W.R., Jr. Mycolic Acid Modification by the MmaA4 Gene of M. tuberculosis Modulates IL-12 Production. PLoS Pathog. 2008, 4, e1000081. [Google Scholar] [CrossRef]
- Peyron, P.; Vaubourgeix, J.; Poquet, Y.; Levillain, F.; Botanch, C.; Bardou, F.; Daffé, M.; Emile, J.; Marchou, B.; Cardona, P.; et al. Foamy Macrophages from Tuberculous Patients’ Granulomas Constitute a Nutrient-Rich Reservoir for M. tuberculosis Persistence. PLoS Pathog. 2008, 4, e1000204. [Google Scholar] [CrossRef]
- Boissier, F.; Bardou, F.; Guillet, V.; Uttenweiler-Joseph, S.; Daffé, M.; Quémard, A.; Mourey, L. Further Insight into S-Adenosylmethionine-Dependent Methyltransferases: Structural Characterization of Hma, an Enzyme Essential for the Biosynthesis of Oxygenated Mycolic Acids in Mycobacterium tuberculosis. J. Biol. Chem. 2006, 281, 4434–4445. [Google Scholar] [CrossRef]
- Pearce, N.M.; Krojer, T.; Bradley, A.R.; Collins, P.; Nowak, R.P.; Talon, R.; Marsden, B.D.; Kelm, S.; Shi, J.; Deane, C.M.; et al. A Multi-Crystal Method for Extracting Obscured Crystallographic States from Conventionally Uninterpretable Electron Density. Nat. Commun. 2017, 8, 15123. [Google Scholar] [CrossRef]
- Pearce, N.M.; Krojer, T.; von Delft, F. Proper Modelling of Ligand Binding Requires an Ensemble of Bound and Unbound States. Acta Crystallogr. Sect. D Biol. Crystallogr. 2017, 73, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Smith, C.V.; Glickman, M.S.; Jacobs, W.R.; Sacchettini, J.C. Crystal Structures of Mycolic Acid Cyclopropane Synthases from Mycobacterium tuberculosis. J. Biol. Chem. 2002, 277, 11559–11569. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Sirimulla, S.; Bailey, J.B.; Vegesna, R.; Narayan, M. Halogen Interactions in Protein–Ligand Complexes: Implications of Halogen Bonding for Rational Drug Design. J. Chem. Inf. Model. 2013, 53, 2781–2791. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Maveyraud, L.; Mourey, L. Protein X-ray Crystallography and Drug Discovery. Molecules 2020, 25, 1030. [Google Scholar] [CrossRef] [PubMed]
- Price, A.J.; Howard, S.; Cons, B.D. Fragment-Based Drug Discovery and its Application to Challenging Drug Targets. Essays Biochem. 2017, 61, 475–484. [Google Scholar] [CrossRef]
- Caliandro, R.; Belviso, D.B.; Aresta, B.M.; de Candia, M.; Altomare, C.D. Protein Crystallography and Fragment-Based Drug Design. Future Med. Chem. 2013, 5, 1121–1140. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Greer, J. A Decade of Fragment-Based Drug Design: Strategic Advances and Lessons Learned. Nat. Rev. Drug Discov. 2007, 6, 211–219. [Google Scholar] [CrossRef]
- Hartshorn, M.J.; Murray, C.W.; Cleasby, A.; Frederickson, M.; Tickle, I.J.; Jhoti, H. Fragment-Based Lead Discovery Using X-ray Crystallography. J. Med. Chem. 2005, 48, 403–413. [Google Scholar] [CrossRef]
- Pearce, N.M.; Bradley, A.R.; Krojer, T.; Marsden, B.D.; Deane, C.M.; von Delft, F. Partial-Occupancy Binders Identified by the Pan-Dataset Density Analysis Method Offer New Chemical Opportunities and Reveal Cryptic Binding Sites. Struct. Dyn. 2017, 4, 032104. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, P.J.; Collins, P.M.; Vrzal, L.; Birchall, K.; Arnold, L.H.; Mpamhanga, C.; Coombs, P.J.; Burgess, S.G.; Richards, M.W.; Winter, A.; et al. Characterization of Three Druggable Hot-Spots in the Aurora-A/TPX2 Interaction Using Biochemical, Biophysical, and Fragment-Based Approaches. ACS Chem. Biol. 2017, 12, 2906–2914. [Google Scholar] [CrossRef] [PubMed]
- Huschmann, F.U.; Linnik, J.; Sparta, K.; Ühlein, M.; Wang, X.; Metz, A.; Schiebel, J.; Heine, A.; Klebe, G.; Weiss, M.S.; et al. Structures of Endothiapepsin–Fragment Complexes from Crystallographic Fragment Screening Using a Novel, Diverse and Affordable 96-Compound Fragment Library. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2016, 72, 346–355. [Google Scholar] [CrossRef]
- Xue, Y.; Guo, H.; Hillertz, P. Fragment Screening of RORγt Using Cocktail Crystallography: Identification of Simultaneous Binding of Multiple Fragments. ChemMedChem 2016, 11, 1881–1885. [Google Scholar] [CrossRef]
- Annaraj, P.D.; Kadirvel, P.; Subramanian, A.; Anishetty, S. Free Enzyme Dynamics of CmaA3 and CmaA2 Cyclopropane Mycolic Acid Synthases from Mycobacterium tuberculosis: Insights into Residues with Potential Significance in Cyclopropanation. J. Mol. Graph. Model. 2019, 91, 61–71. [Google Scholar] [CrossRef]
- Guianvarc’h, D.; Guangqi, E.; Drujon, T.; Rey, C.; Wang, Q.; Ploux, O. Identification of Inhibitors of the E. coli Cyclopropane Fatty Acid Synthase from the Screening of a Chemical Library: In Vitro and in Vivo Studies. Biochim. Biophys. Acta Proteins Proteom. 2008, 1784, 1652–1658. [Google Scholar] [CrossRef] [PubMed]
- Goulding, C.W.; Apostol, M.; Anderson, D.H.; Gill, H.S.; Smith, C.V.; Kuo, M.R.; Yang, J.K.; Waldo, G.S.; Suh, S.W.; Chauhan, R.; et al. The TB Structural Genomics Consortium: Providing a Structural Foundation for Drug Discovery. Curr. Drug Targets Infect. Disord. 2002, 2, 121–141. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Vonrhein, C.; Flensburg, C.; Keller, P.; Sharff, A.; Smart, O.; Paciorek, W.; Womack, T.; Bricogne, G. Data Processing and Analysis with the AutoPROC Toolbox. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 293–302. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; Airlie, M.; et al. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef]
- Bricogne, G.; Blanc, E.; Brandl, M.M.; Flensburg, C.C.; Keller, P.; Paciorek, W.; Roversi, P.; Sharff, A.; Smart, O.S.; Vonrhein, C.; et al. BUSTER Version 2.10.3; Global Phasing: Cambridge, UK, 2017. [Google Scholar]
- Emsley, P.; Cowtan, K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- MarvinSketch Version 20.8.0, ChemAxon, 2020. Available online: https://www.chemaxon.com (accessed on 18 August 2021).
- Smart, O.S.; Womack, T.O.; Sharff, A.; Flensburg, C.; Keller, P.; Paciorek, W.; Vonrhein, C.; Bricogne, G. Grade, Version 1.2; Global Phasing Ltd.: Cambridge, UK, 2020; Available online: https://www.globalphasing.com (accessed on 18 August 2021).
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Menchon, G.; Maveyraud, L.; Czaplicki, G. Molecular Dynamics as a Tool for Virtual Ligand Screening. In Computational Drug Discovery and Design; Methods in Molecular Biology; Gore, M., Jagtap, U.B., Eds.; Springer: New York, NY, USA, 2018; pp. 145–178. ISBN 978-1-4939-7756-7. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ZT218 | ZT260 | ZT275 | ZT320 | ZT424 | ZT585 | ZT726 | |
|---|---|---|---|---|---|---|---|
| PDB code | 7Q2B | 7Q2C | 7Q2H | 7Q2D | 7Q2E | 7Q2F | 7Q2G |
| Data Collection | |||||||
| Beamline | ESRF, ID14-4 | ESRF, ID14-4 | ESRF, ID23-1 | ESRF, ID29 | ESRF, ID29 | SOLEIL, PX1 | ESRF, ID14-4 |
| Spacegroup | P3121 | P3121 | P3121 | P3121 | P3121 | P3121 | P3121 |
| Unit cell a, c (Å) | 57.29, 206.00 | 57.11, 205.90 | 56.62, 207.67 | 57.02, 207.35 | 55.77, 207.02 | 57.11, 204.46 | 57.06, 205.93 |
| Resolution range (Å) 1 | 40.21–1.85 (1.96–1.85) | 35.66–1.85 (1.96–1.85) | 49.03–1.75 (1.86–1.75) | 49.38–1.90 (2.02–1.90) | 49.17–2.00 (2.12–2.00) | 49.46–1.85 (1.88–1.85) | 40.10–2.00 (2.12–2.00) |
| No. unique reflections | 34,449 (5438) | 33,875 (5254) | 40,075 (6368) | 31,853 (5032) | 27,163 (4292) | 33,526 (1649) | 27,352 (4359) |
| Completeness (%) | 99.8 (99.7) | 98.4 (96.7) | 99.5 (99.7) | 100.0 (100.0) | 99.6 (99.7) | 98.3 (99.9) | 99.9 (99.9) |
| Redundancy | 6.0 (6.0) | 5.1 (3.7) | 8.9 (8.8) | 11.4 (11.8) | 6.7 (6.5) | 5.9 (5.2) | 7.5 (7.6) |
| <I/σ(I)> | 11.5 (1.8) | 15.1 (1.0) | 13.7 (2.6) | 16.2 (2.6) | 17.3 (2.4) | 7.2 (1.4) | 11.4 (1.3) |
| Rmerge (%) | 8.2 (95.5) | 5.1 (109.3) | 8.9 (75.8) | 8.1 (90.9) | 5.2 (70.8) | 15.8 (116.7) | 9.1 (133.9) |
| CC(1/2) | 99.6 (75.2) | 99.9 (59.1) | 99.6 (89.0) | 99.8 (89.2) | 99.9 (84.2) | 98.3 (53.1) | 99.6 (81.4) |
| Refinement | |||||||
| Resolution range (Å) | 40.25–1.85 | 35.66–1.85 | 49.03–1.75 | 49.38–1.90 | 49.17–2.00 | 49.46–1.85 | 40.10–2.00 |
| No. reflections (work/test) | 29,899/1703 | 28,298/1619 | 35,907/2034 | 28,849/1645 | 26,984/1545 | 30,090/1718 | 20,616/1191 |
| Rwork/Rfree | 0.1668/0.2059 | 0.1792/0.2256 | 0.1782/0.2105 | 0.1856/0.2281 | 0.1847/0.2375 | 0.1894/0.2293 | 0.1926/0.2538 |
| No. of non-hydrogen atoms | 2484 | 2472 | 2525 | 2440 | 2385 | 2480 | 2339 |
| Protein | 2298 | 2310 | 2305 | 2305 | 2281 | 2291 | 2264 |
| Fragment | 12 | 24 | 11 | 20 | 8 | 24 | 13 |
| Solvent | 174 | 138 | 209 | 115 | 96 | 165 | 62 |
| Rms deviations | |||||||
| Bond length (Å) | 0.007 | 0.004 | 0.003 | 0.005 | 0.005 | 0.004 | 0.008 |
| Bond angles (°) | 1.338 | 1.233 | 1.164 | 1.225 | 1.296 | 1.199 | 1.226 |
| Ramachandran plot | |||||||
| Most favoured (%) | 98 | 98 | 98 | 96 | 98 | 98 | 97 |
| Allowed/Outliers (%) | 2/0 | 2/0 | 2/0 | 4/0 | 2/0 | 2/0 | 3/0 |
| Ligand | GBSA | PBSA |
|---|---|---|
| ZT218 | −22.8 ± 2.2 | −11.9 ± 2.6 |
| ZT218 * | −22.2 ± 1.8 | −10.2 ± 2.2 |
| ZT260 | −18.0 ± 1.8 | −12.0 ± 2.1 |
| ZT260 * | −22.2 ± 1.8 | −10.2 ± 2.2 |
| ZT275 | −17.9 ± 2.1 | −7.0 ± 2.1 |
| ZT320 | −23.0 ± 2.1 | −12.8 ± 2.2 |
| ZT424 | −18.9 ± 1.7 | −9.7 ± 1.9 |
| ZT585 | −26.8 ± 1.7 | −14.8 ± 2.0 |
| ZT585 * | −26.2 ± 1.7 | −14.0 ± 2.0 |
| ZT726 | −19.2 ± 1.8 | −11.4 ± 2.1 |
| 2182601 | −29.9 ± 2.5 | −10.7 ± 3.0 |
| 2182601 * | −33.9 ± 2.2 | −19.6 ± 2.2 |
| 2182602 | −30.5 ± 1.8 | −14.2 ± 2.1 |
| 2182602 * | −31.5 ± 2.1 | −15.6 ± 2.7 |
| 2753201 | −24.9 ± 2.9 | −12.5 ± 2.2 |
| 2753202 | −18.9 ± 3.8 | −5.3 ± 2.6 |
| 2185851 | −32.4 ± 2.1 | −12.2 ± 2.3 |
| 2185852 | −32.3 ± 2.6 | −13.2 ± 2.8 |
| 2185853 | −34.4 ± 2.0 | −14.5 ± 2.4 |
| 2185854 | −34.3 ± 2.2 | −18.2 ± 2.4 |
| 2185855 | −35.1 ± 2.2 | −18.6 ± 2.6 |
| 2185856 | −35.0 ± 2.3 | −19.7 ± 2.5 |
| 320sadae | −35.7 ± 2.2 | −20.3 ± 2.5 |
| SAM | −45.7 ± 4.9 | −25.0 ± 4.1 |
| SAH | −40.2 ± 3.4 | −21.8 ± 3.2 |
| Sinefungin | −39.6 ± 3.8 | −24.8 ± 3.6 |
| SADAE | −67.7 ± 3.5 | −35.8 ± 3.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galy, R.; Ballereau, S.; Génisson, Y.; Mourey, L.; Plaquevent, J.-C.; Maveyraud, L. Fragment-Based Ligand Discovery Applied to the Mycolic Acid Methyltransferase Hma (MmaA4) from Mycobacterium tuberculosis: A Crystallographic and Molecular Modelling Study. Pharmaceuticals 2021, 14, 1282. https://doi.org/10.3390/ph14121282
Galy R, Ballereau S, Génisson Y, Mourey L, Plaquevent J-C, Maveyraud L. Fragment-Based Ligand Discovery Applied to the Mycolic Acid Methyltransferase Hma (MmaA4) from Mycobacterium tuberculosis: A Crystallographic and Molecular Modelling Study. Pharmaceuticals. 2021; 14(12):1282. https://doi.org/10.3390/ph14121282
Chicago/Turabian StyleGaly, Romain, Stéphanie Ballereau, Yves Génisson, Lionel Mourey, Jean-Christophe Plaquevent, and Laurent Maveyraud. 2021. "Fragment-Based Ligand Discovery Applied to the Mycolic Acid Methyltransferase Hma (MmaA4) from Mycobacterium tuberculosis: A Crystallographic and Molecular Modelling Study" Pharmaceuticals 14, no. 12: 1282. https://doi.org/10.3390/ph14121282
APA StyleGaly, R., Ballereau, S., Génisson, Y., Mourey, L., Plaquevent, J.-C., & Maveyraud, L. (2021). Fragment-Based Ligand Discovery Applied to the Mycolic Acid Methyltransferase Hma (MmaA4) from Mycobacterium tuberculosis: A Crystallographic and Molecular Modelling Study. Pharmaceuticals, 14(12), 1282. https://doi.org/10.3390/ph14121282