
A Deep-Learning Proteomic-Scale Approach for Drug Design




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
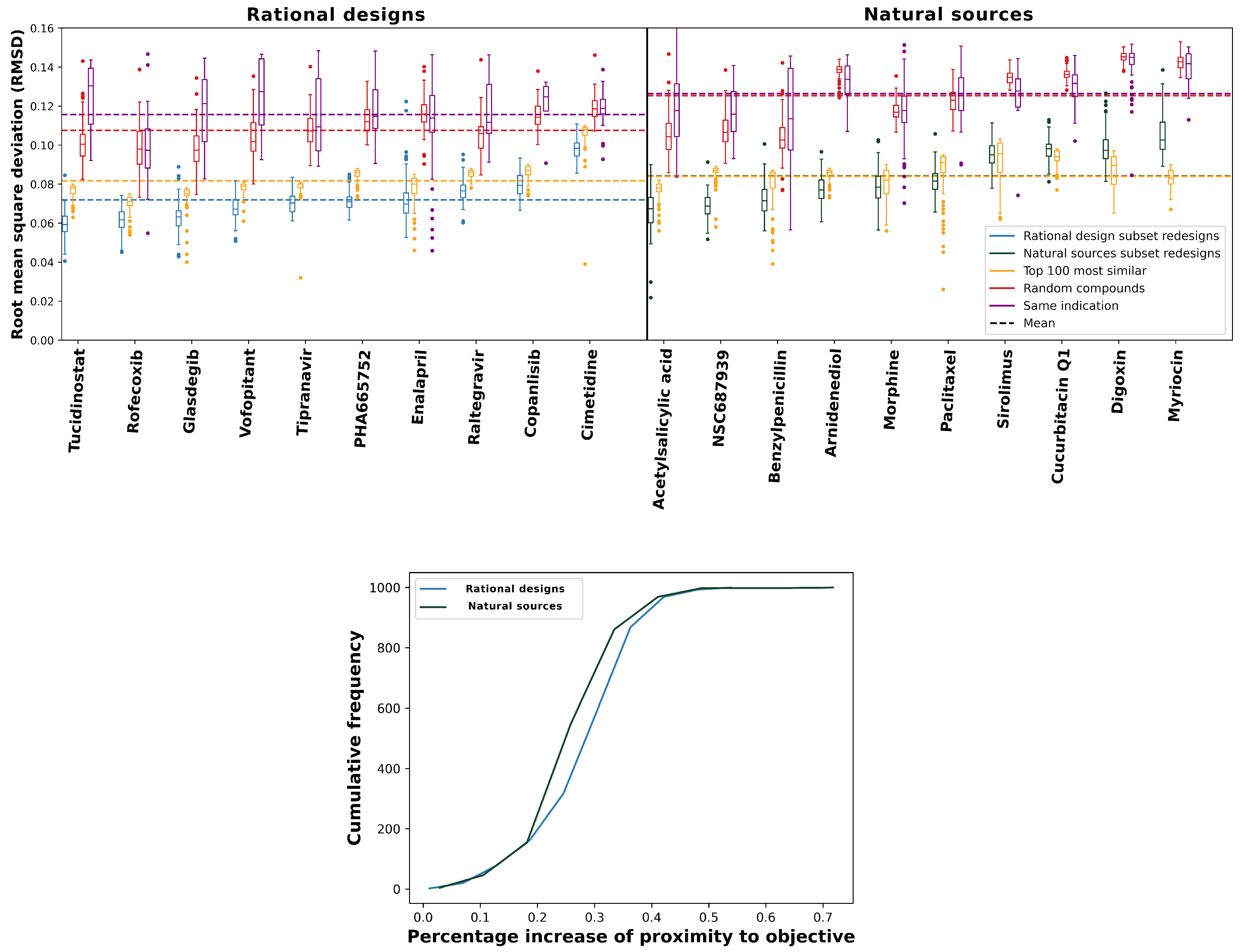
2.1. Behavioral Similarity of Designed Compounds to Their Objectives
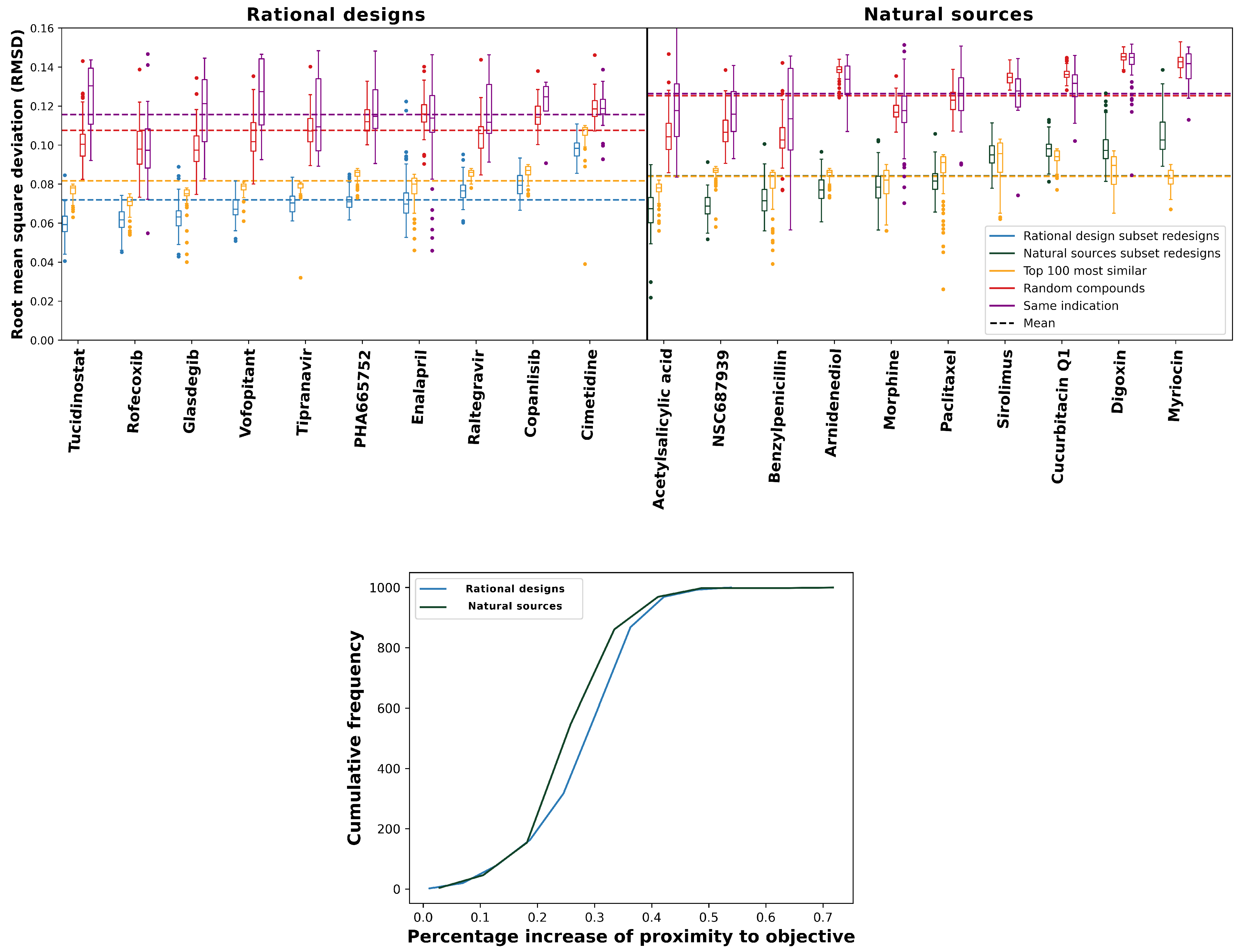
2.2. Relative Performance Gains of Designs Relative to Controls
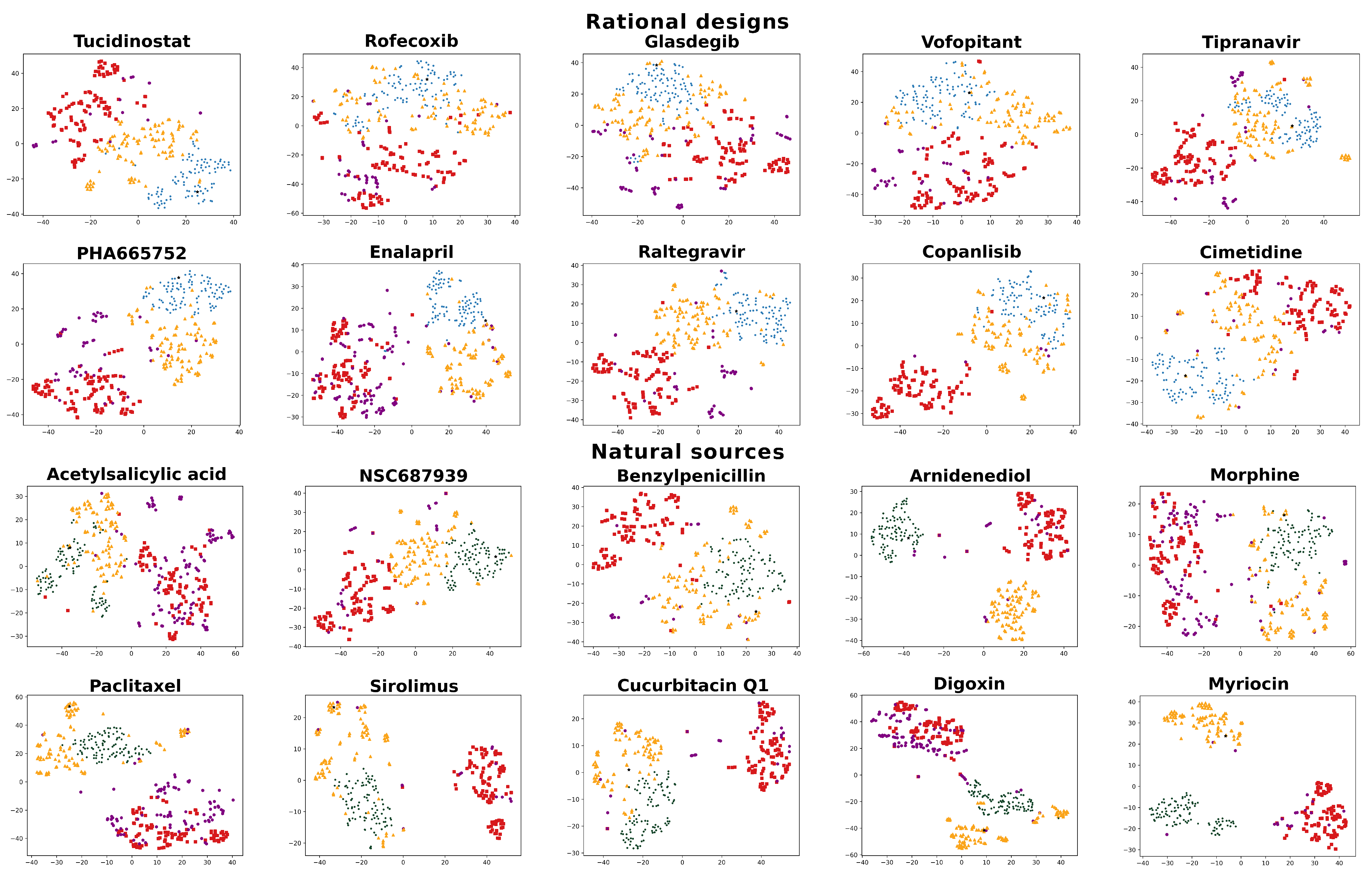
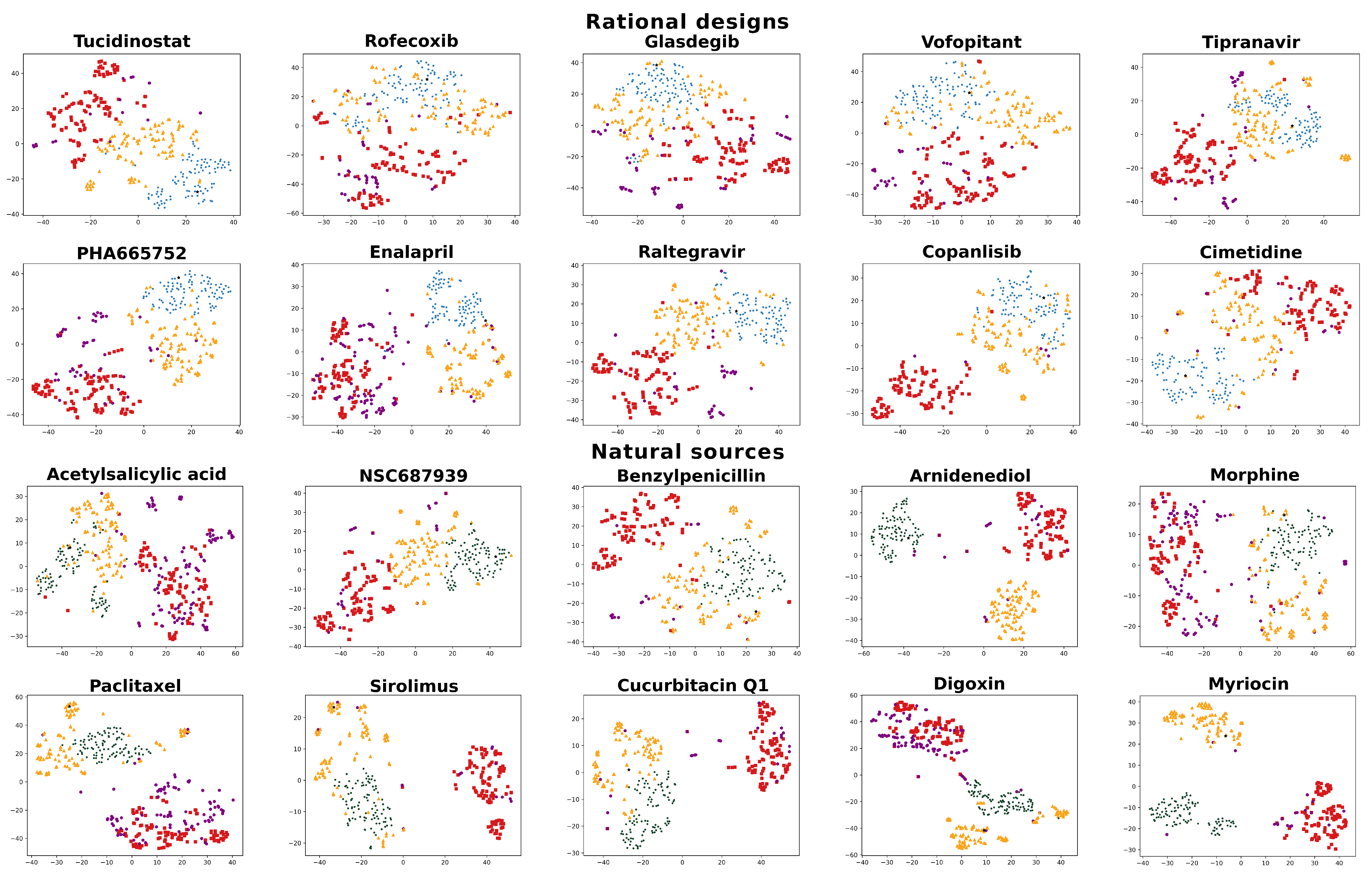
2.3. Visualizing and Filtering Using t-SNE Plots
2.4. Improving Cases with Sub-Optimal Performance
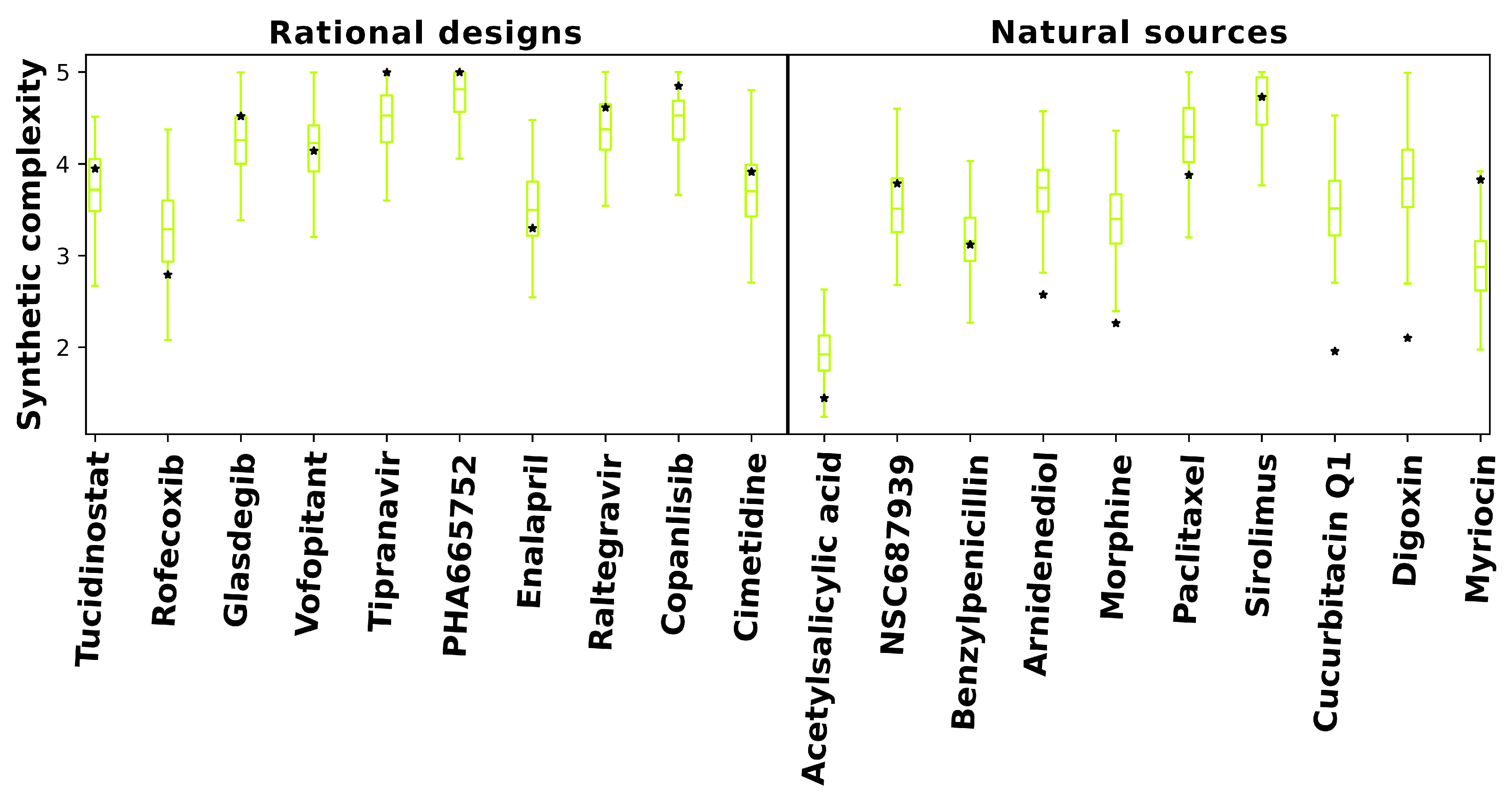
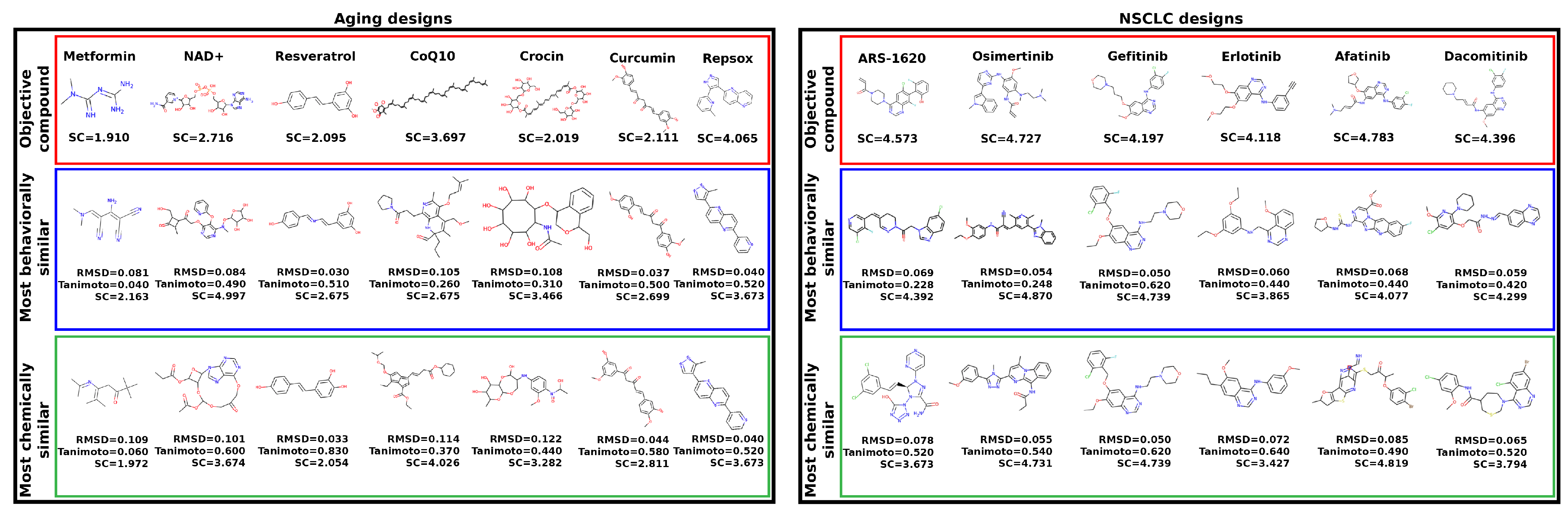
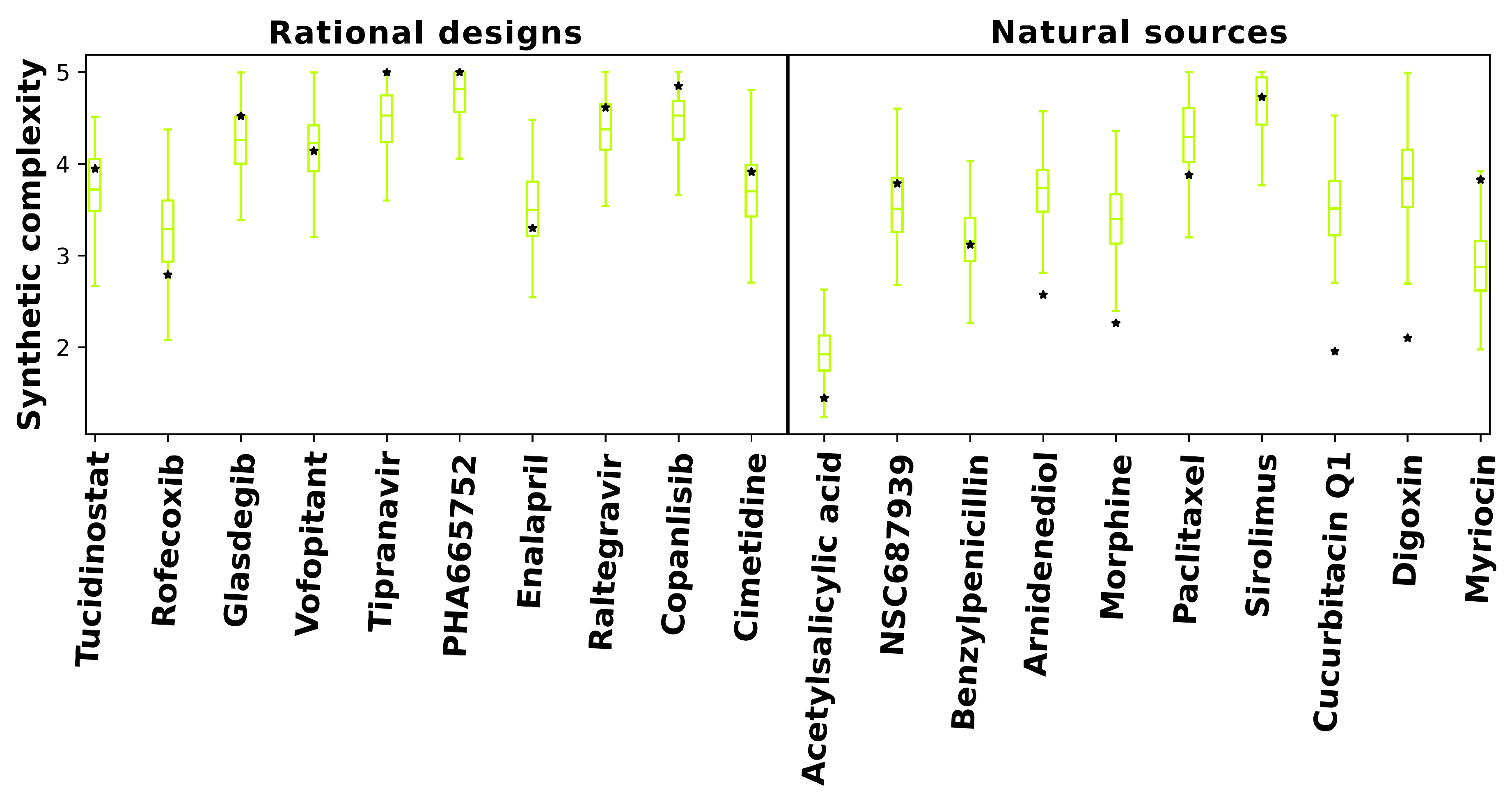
2.5. Synthetic Feasibility of Designed Compounds
2.6. Applications to Aging and Non-Small Cell Lung Cancer
2.7. Limitations and Future Work
3. Materials and Methods
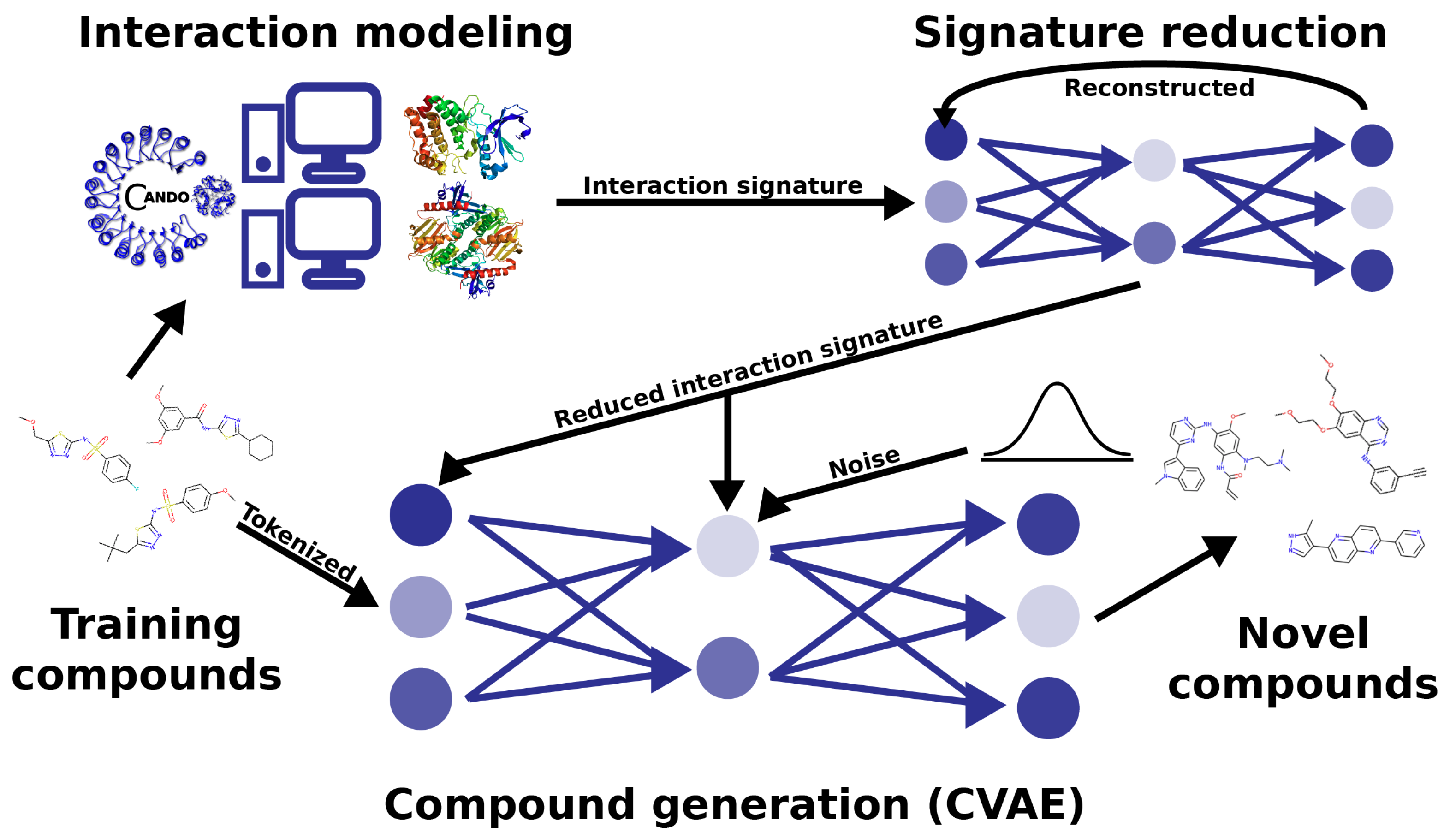
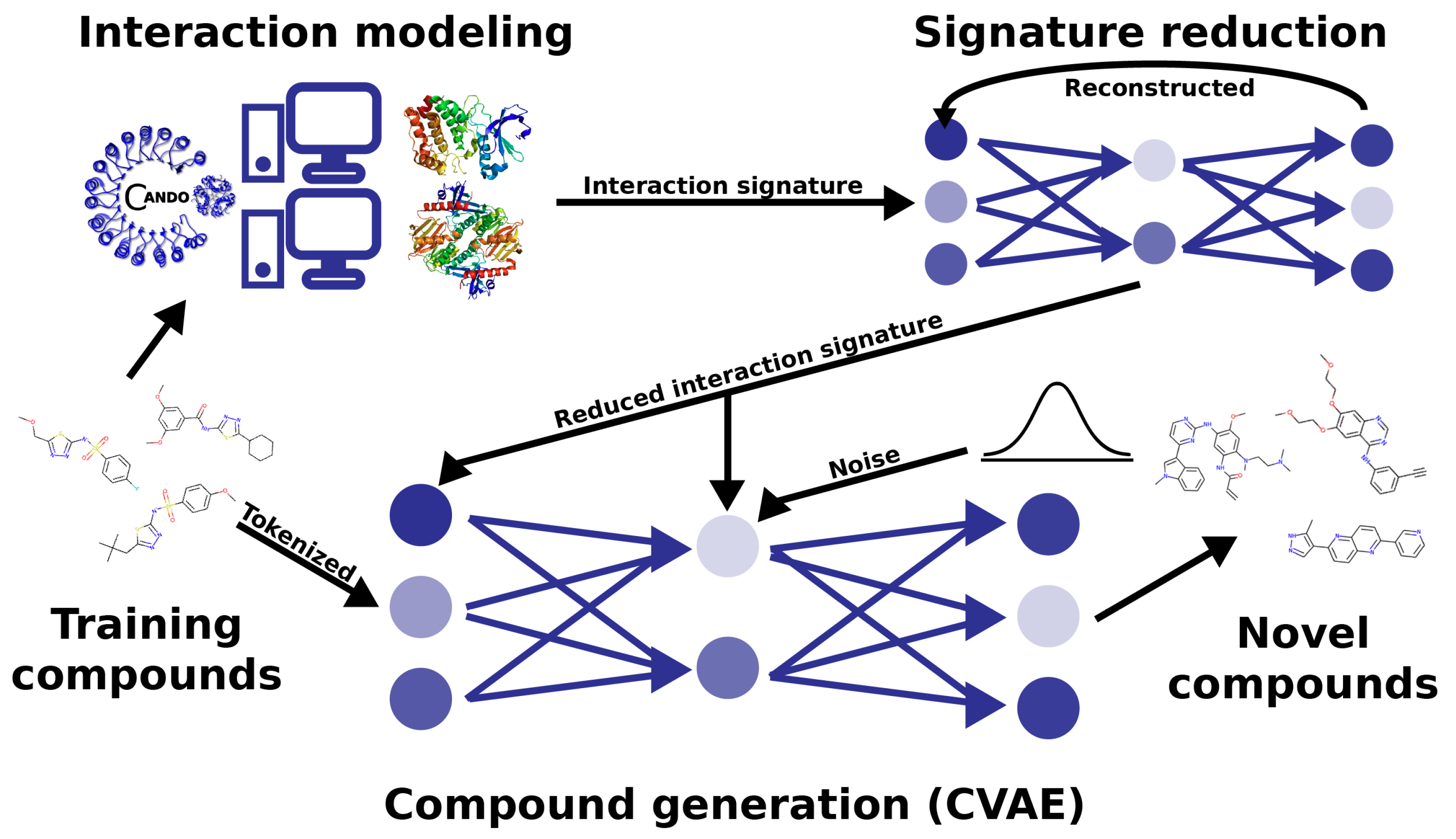
3.1. Compound–Proteome Interaction Signature Generation Using the CANDO Platform
3.2. Model Architecture and Data Generation
3.3. Benchmarking and Analysis of the RCVAE Design Pipeline Performance
3.4. Generating Novel Designs for Prospective Validation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schuhmacher, A.; Gassmann, O.; Hinder, M. Changing R&D models in research-based pharmaceutical companies. J. Transl. Med. 2016, 14, 105. [Google Scholar]
- Bajorath, J. Integration of virtual and high-throughput screening. Nat. Rev. Drug Discov. 2002, 1, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Dhasmana, A.; Raza, S.; Jahan, R.; Lohani, M.; Arif, J.M. Chapter 19-High-Throughput Virtual Screening (HTVS) of Natural Compounds and Exploration of Their Biomolecular Mechanisms: An In Silico Approach; Academic Press: Cambridge, MA, USA, 2019; pp. 523–548. [Google Scholar]
- Graff, D.E.; Shakhnovich, E.I.; Coley, C.W. Accelerating high-throughput virtual screening through molecular pool-based active learning. Chem. Sci. 2021, 12, 7866–7881. [Google Scholar] [CrossRef]
- Lim, J.; Ryu, S.; Park, K.; Choe, Y.J.; Ham, J.; Kim, W.Y. Predicting drug–target interaction using a novel graph neural network with 3D structure-embedded graph representation. J. Chem. Inf. Model. 2019, 59, 3981–3988. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Grosdidier, A.; Michielin, O. Docking, virtual high throughput screening and in silico fragment-based drug design. J. Cell. Mol. Med. 2009, 13, 238–248. [Google Scholar] [CrossRef] [Green Version]
- Ha, E.; Lwin, C.; Durrant, J. LigGrep: A tool for filtering docked poses to improve virtual-screening hit rates. J. Cheminform. 2020, 12, 69. [Google Scholar] [CrossRef]
- Lee, H.S.; Choi, J.; Kufareva, I.; Abagyan, R.; Filikov, A.; Yang, Y.; Yoon, S. Optimization of high throughput virtual screening by combining shape-matching and docking methods. J. Chem. Inf. Model. 2008, 48, 489–497. [Google Scholar] [CrossRef]
- Corbeil, C.R.; Moitessier, N. Docking ligands into flexible and solvated macromolecules. 3. Impact of input ligand conformation, protein flexibility, and water molecules on the accuracy of docking programs. J. Chem. Inf. Model. 2009, 49, 997–1009. [Google Scholar] [CrossRef]
- Feher, M.; Williams, C. Numerical errors and chaotic behavior in docking simulations. J. Chem. Inf. Model. 2012, 52, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Boran, A.; Iyengar, R. Systems approaches to polypharmacology and drug discovery. Curr. Opin. Drug Discov. Dev. 2010, 13, 297–309. [Google Scholar]
- Peyvandipour, A.; Saberian, N.; Shafi, A.; Donato, M.; Draghici, S. A novel computational approach for drug repurposing using systems biology. Bioinformatics 2018, 34, 2817–2825. [Google Scholar] [CrossRef] [Green Version]
- Shafi, A.; Nguyen, T.; Peyvandipour, A.; Nguyen, H.; Draghici, S. A multi-cohort and multi-omics meta-analysis framework to identify network-based Gene signatures. Front. Genet. 2019, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Tatonetti, N.P.; Liu, T.; Altman, R.B. Predicting drug side-effects by chemical systems biology. Genome Biol. 2009, 10, 238. [Google Scholar] [CrossRef]
- Liu, T.; Altman, R.B. Relating essential proteins to drug side-effects using canonical component analysis: A structure-based approach. J. Chem. Inf. Model. 2015, 55, 1483–1494. [Google Scholar] [CrossRef] [Green Version]
- Kirkpatrick, P.; Ellis, C. Chemical space. Nature 2004, 432, 832. [Google Scholar] [CrossRef]
- Shayakhmetov, R.; Kuznetsov, M.; Zhebrak, A.; Kadurin, A.; Nikolenko, S.; Aliper, A.; Polykovskiy, D. Molecular generation for desired transcriptome changes with adversarial autoencoders. Front. Pharmacol. 2020, 11, 269. [Google Scholar] [CrossRef]
- Minie, M.; Sethi, G.; Chopra, G.; Horst, J.; Roy, A.; White, G.; Samudrala, R. CANDO and the infinite drug discovery frontier. Drug Discov. Today 2014, 19, 1353–1363. [Google Scholar] [CrossRef] [Green Version]
- Sethi, G.; Chopra, G.; Samudrala, R. Multiscale modelling of relationships between protein classes and drug behavior across all diseases using the CANDO platform. Mini Rev. Med. Chem. 2015, 15, 705–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chopra, G.; Samudrala, R. Exploring polypharmacology in drug discovery and repurposing using the CANDO platform. Curr. Pharm. Des. 2016, 22, 3109–3123. [Google Scholar] [CrossRef] [Green Version]
- Chopra, G.; Kaushik, S.; Elkin, P.; Samudrala, R. Combating Ebola with repurposed therapeutics using the CANDO platform. Molecules 2016, 21, 1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangione, W.; Samudrala, R. Identifying protein features responsible for improved drug repurposing accuracies using the CANDO platform: Implications for drug design. Molecules 2019, 24, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falls, Z.; Mangione, W.; Schuler, J.; Samudrala, R. Exploration of interaction scoring criteria in the CANDO platform. BMC Bioinform. 2019, 12, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuler, J.; Samudrala, R. Fingerprinting CANDO: Increased accuracy with structure and ligand based shotgun drug repurposing. ACS Omega 2019, 4, 17393–17403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fine, J.; Lacker, R.; Samudrala, R.; Chopra, G. Computational chemoproteomics to understand the role of selected psychoactives in treating mental health disorders. Sci. Rep. 2019, 9, 13155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangione, W.; Falls, Z.; Melendy, T.; Chopra, G.; Samudrala, R. Shotgun drug repurposing biotechnology to tackle epidemics and pandemics. Drug Discov. Today 2020, 25, 1126–1129. [Google Scholar] [CrossRef]
- Mangione, W.; Falls, Z.; Chopra, G.; Samudrala, R. cando.py: Open source software for analyzing large scale drug-protein-disease data. J. Chem. Inf. Model. 2020, 60, 4131–4136. [Google Scholar] [CrossRef]
- Hudson, M.; Samudrala, R. Multiscale virtual screening optimization for shotgun drug repurposing using the CANDO platform. Molecules 2021, 26, 2581. [Google Scholar] [CrossRef] [PubMed]
- Schuler, J.; Falls, Z.; Mangione, W.; Hudson, M.; Bruggemann, L.; Samdurala, R. Evaluating performance of drug repurposing technologies. Drug Discov. Today 2021, in press. [Google Scholar] [CrossRef]
- Yang, L.; Wang, K.J.; Wang, L.S.; Jegga, A.G.; Qin, S.Y.; He, G.; Chen, J.; Xiao, Y.; He, L. Chemical-protein interactome and its application in off-target identification. Interdiscip. Sci. Comput. Life Sci. 2011, 3, 22–30. [Google Scholar] [CrossRef]
- Liu, T.; Tang, G.; Capriotti, E. Comparative modeling: The state of the art and protein drug target structure prediction. Comb. Chem. High Throughput Screen. 2011, 14, 532–547. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Gudivada, R.C.; Aronow, B.J.; Jegga, A.G. Computational drug repositioning through heterogeneous network clustering. BMC Syst. Biol. 2013, 7, S6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yella, J.; Yaddanapudi, S.; Wang, Y.; Jegga, A. Changing trends in computational drug repositioning. Pharmaceuticals 2018, 11, 57. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yella, J.; Jegga, A.G. Transcriptomic data mining and repurposing for computational drug discovery. In Methods in Molecular Biology; Springer: New York, NY, USA, 2018; pp. 73–95. [Google Scholar]
- Patel, L.; Shukla, T.; Huang, X.; Ussery, D.W.; Wang, S. Machine learning methods in drug discovery. Molecules 2020, 25, 5277. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Pei, J.; Lai, L. LigBuilder V3: A multi-target de novo drug design approach. Front. Chem. 2020, 8, 142. [Google Scholar] [CrossRef] [Green Version]
- Bai, Q.; Tan, S.; Xu, T.; Liu, H.; Huang, J.; Yao, X. MolAICal: A soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief. Bioinform. 2020, 22, bbaa161. [Google Scholar] [CrossRef]
- Chemistry 42. Available online: https://insilico.com/chemistry42 (accessed on 30 July 2021).
- Grechishnikova, D. Transformer neural network for protein-specific de novo drug generation as a machine translation problem. Sci. Rep. 2021, 11, 321. [Google Scholar] [CrossRef]
- Schmidhuber, J. Deep learning in neural networks: An overview. Neural Netw. 2015, 61, 85–117. [Google Scholar] [CrossRef] [Green Version]
- Kingma, D.P.; Welling, M. Auto-Encoding Variational Bayes. arXiv 2014, arXiv:1312.6114. [Google Scholar]
- Polykovskiy, D.; Vetrov, D. Deterministic Decoding for Discrete Data in Variational Autoencoders. In Proceedings of the Machine Learning Research, Proceedings of the Twenty Third International Conference on Artificial Intelligence and Statistics, Palermo, Italy, 3–5 June 2020; Chiappa, S., Calandra, R., Eds.; 2020; Volume 108, pp. 3046–3056. [Google Scholar]
- Sohn, K.; Lee, H.; Yan, X. Learning Structured Output Representation using Deep Conditional Generative Models. In Advances in Neural Information Processing Systems 28 (NIPS 2015); Curran Associates, Inc.: Red Hook, NY, USA, 2015; Volume 28. [Google Scholar]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Ryu, S.; Kim, J.; Kim, W. Molecular generative model based on conditional variational autoencoder for de novo molecular design. J. Cheminform. 2018, 10, 31. [Google Scholar] [CrossRef] [Green Version]
- Polykovskiy, D.; Zhebrak, A.; Sanchez-Lengeling, B.; Golovanov, S.; Tatanov, O.; Belyaev, S.; Kurbanov, R.; Artamonov, A.; Aladinskiy, V.; Veselov, M.; et al. Molecular Sets (MOSES): A benchmarking platform for molecular generation models. Front. Pharmacol. 2020, 11, 1931. [Google Scholar] [CrossRef]
- Jenwitheesuk, E.; Samudrala, R. Identification of potential multitarget antimalarial drugs. J. Am. Med. Assoc. 2005, 294, 1490–1491. [Google Scholar]
- Jenwitheesuk, E.; Samudrala, R. New paradigms for drug discovery: Computational multitarget screening. Trends Pharmacol. Sci. 2008, 29, 62–71. [Google Scholar] [CrossRef] [Green Version]
- Costin, J.; Jenwitheesuk, E.; Lok, S.; Hunsperger, E.; Conrads, K.; Fontaine, K.; Rees, C.; Rossmann, M.; Isern, S.; Samudrala, R.; et al. Structural optimization and de novo design of dengue virus entry inhibitory peptides. PLoS Negl. Trop. Dis. 2010, 4, e721. [Google Scholar] [CrossRef]
- Palanikumar, L.; Karpauskaite, L.; Al-Sayegh, M.; Chehade, I.; Alam, M.; Hassan, S.; Maity, D.; Ali, L.; Kalmouni, M.; Hunashal, Y.; et al. Protein mimetic amyloid inhibitor potently abrogates cancer-associated mutant p53 aggregation and restores tumor suppressor function. Nat. Commun. 2021, 12, 3962. [Google Scholar] [CrossRef] [PubMed]
- MedicineNet. Available online: https://www.medicinenet.com/me-too_drug/definition.htm (accessed on 30 July 2021).
- Cheng, G.; Qian, B.; Samudrala, R.; Baker, D. Improvement in protein functional site prediction by distinguishing structural and functional constraints on protein family evolution using computational design. Nucleic Acids Res. 2005, 33, 5861–5867. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Z.; Morris-Natschke, S.L.; Lee, K.H. Strategies for the optimization of natural leads to anticancer drugs or drug candidates. Med. Res. Rev. 2016, 36, 32–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterling, T.; Irwin, J. ZINC 15—Ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Coley, C.; Rogers, L.; Green, W.; Jensen, K. SCScore: Synthetic complexity Learned from a reaction corpus. J. Chem. Inf. Model. 2018, 58, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Genheden, S.; Thakkar, A.; Chadimová, V.; Reymond, J.L.; Engkvist, O.; Bjerrum, E. AiZynthFinder: A fast, robust and flexible open-source software for retrosynthetic planning. J. Cheminform. 2020, 12, 70. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.; Fiscato, M.; Segler, M.H.; Vaucher, A.C. GuacaMol: Benchmarking Models for de Novo Molecular Design. J. Chem. Inf. Model. 2019, 59, 1096–1108. [Google Scholar] [CrossRef]
- Li, J.; Zhang, C.X.; Liu, Y.M.; Chen, K.L.; Chen, G. A comparative study of anti-aging properties and mechanism: Resveratrol and caloric restriction. Oncotarget 2017, 8, 65717–65729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soukas, A.; Hao, H.; Wu, L. Metformin as anti-Aging therapy: Is it for everyone? Trends Endocrinol. Metab. 2019, 30, 745–755. [Google Scholar] [CrossRef]
- Aman, Y.; Qiu, Y.; Tao, J.; Fang, E. Therapeutic potential of boosting NAD+ in aging and age-related diseases. Transl. Med. Aging 2018, 2, 30–37. [Google Scholar] [CrossRef]
- Hernández-Camacho, J.; Bernier, M.; López-Lluch, G.; Navas, P. Coenzyme Q10 supplementation in aging and disease. Front. Physiol. 2018, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Fagot, D.; Pham, D.; Laboureau, J.; Planel, E.; Guerin, L.; Nègre, C.; Donovan, M.; Bernard, B. Crocin, a natural molecule with potentially beneficial effects against skin ageing. Int. J. Cosmet. Sci. 2018, 40, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Bielak-Zmijewska, A.; Grabowska, W.; Ciolko, A.; Bojko, A.; Mosieniak, G.; Bijoch, L.; Sikora, E. The role of curcumin in the modulation of ageing. Int. J. Mol. Sci. 2019, 20, 1239. [Google Scholar] [CrossRef] [Green Version]
- Ichida, J.K.; Blanchard, J.; Lam, K.; Son, E.Y.; Chung, J.E.; Egli, D.; Loh, K.; Carter, A.C.; Di Giorgio, F.P.; Koszka, K.; et al. A small-molecule inhibitor of TGF-β signaling replaces sox2 in reprogramming by inducing nanog. Cell Stem Cell 2009, 5, 491–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamoah, E.N.; Li, M.; Shah, A.; Elliott, K.L.; Cheah, K.; Xu, P.X.; Phillips, S.; Young, S.M., Jr.; Eberl, D.F.; Fritzsch, B. Using sox2 to alleviate the hallmarks of age-related hearing loss. Ageing Res. Rev. 2020, 59, 101042. [Google Scholar] [CrossRef]
- Tominaga, K.; Suzuki, H. TGF-β signaling in cellular senescence and aging-related pathology. Int. J. Mol. Sci. 2019, 20, 5002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, B.K.; Pennypacker, J.K. Drugs that modulate aging: The promising yet difficult path ahead. Transl. Res. 2014, 163, 456–465. [Google Scholar] [CrossRef] [Green Version]
- Jiao, D.; Yang, S. Overcoming resistance to drugs targeting KRASG12C mutation. Innovation 2020, 1, 100035. [Google Scholar]
- Shah, R.; Lester, J. Tyrosine Kinase inhibitors for the treatment of EGFR mutation-positive non-small-cell lung cancer: A clash of the generations. Clin. Lung Cancer 2020, 21, 216–228. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Tsuboi, M.; He, J.; John, T.; Grohe, C.; Majem, M.; Goldman, J.W.; Laktionov, K.; Kim, S.W.; Kato, T.; et al. Osimertinib in resected EGFR-mutated Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 1711–1723. [Google Scholar] [CrossRef]
- Molina-Arcas, M.; Moore, C.; Rana, S.; van Maldegem, F.; Mugarza, E.; Romero-Clavijo, P.; Herbert, E.; Horswell, S.; Li, L.S.; Janes, M.R.; et al. Development of combination therapies to maximize the impact of KRAS-G12C inhibitors in lung cancer. Sci. Transl. Med. 2019, 11, eaaw7999. [Google Scholar] [CrossRef]
- Moore, A.; Rosenberg, S.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Lau, S.C.M.; Batra, U.; Mok, T.S.K.; Loong, H.H. Dacomitinib in the management of advanced Non-Small-Cell Lung Cancer. Drugs 2019, 79, 823–831. [Google Scholar] [CrossRef]
- Keating, G.M. Afatinib: A review in advanced Non-Small Cell Lung Cancer. Target. Oncol. 2016, 11, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Piperdi, B.; Perez-Soler, R. Role of Erlotinib in the treatment of Non-Small Cell Lung Cancer. Drugs 2012, 72, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Cui, G.; Hu, S.; Zhang, Z.; Yang, C.; Liu, Z.; Wang, L.; Li, C.; Sun, M. Graph Neural Networks: A review of methods and applications. arXiv 2021, arXiv:1812.08434. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.; Peck, D.; Modell, J.; Blat, I.; Wrobel, M.; Lerner, J.; Brunet, J.; Subramanian, A.; Ross, K.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Narayan, R.; Corsello, S.; Peck, D.; Natoli, T.; Lu, X.; Gould, J.; Davis, J.; Tubelli, A.; Asiedu, J.; et al. A next generation Connectivity Map: L1000 platform and the first 1,000,000 profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef] [PubMed]
- Wleklinski, M.; Loren, B.; Ferreira, C.; Jaman, Z.; Avramova, L.; Sobreira, T.; Thompson, D.; Cooks, R. High throughput reaction screening using desorption electrospray ionization mass spectrometry. Sci. Rep. 2018, 9, 1647–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morato, N.; Holden, D.; Cooks, R. High-throughput label-free enzymatic assays using desorption electrospray-ionization mass spectrometry. Angew. Chem. Int. Ed. Engl. 2020, 59, 20459–20464. [Google Scholar] [CrossRef]
- Logsdon, D.; Li, Y.; Sobreira, T.; Ferreira, C.; Thompson, D.; Cooks, R. High-throughput screening of reductive amination reactions using desorption electrospray ionization mass spectrometry. Org. Process Res. Dev. 2020, 24, 1647–1657. [Google Scholar] [CrossRef]
- Sobreira, T.; Avramova, L.; Szilagyi, B.; Logsdon, D.; Loren, B.; Jaman, Z.; Hilger, R.; Hosler, R.; Ferreira, C.; Koswara, A.; et al. High-throughput screening of organic reactions in microdroplets using desorption electrospray ionization mass spectrometry (DESI-MS): Hardware and software implementation. Methods 2020, 12, 3654–3669. [Google Scholar] [CrossRef]
- Le, M.; Morato, N.; Kaerner, A.; Welch, C.; Cooks, R. Fragmentation of polyfunctional compounds recorded using automated high-throughput desorption electrospray ionization. J. Am. Soc. Mass Spectrom. 2021, 32, 2261–2273. [Google Scholar] [CrossRef] [PubMed]
- Morato, N.; Le, M.; Holden, D.; Cooks, R. Automated high-throughput system combining small-scale synthesis with bioassays and reaction screening. SLAS Technol. 2021, 26, 555–571. [Google Scholar] [CrossRef]
- Biyani, S.; Qi, Q.; Wu, J.; Moriuchi, Y.; Larocque, E.A.; Sintim, H.; Thompson, D. Use of high-throughput tools for telescoped continuous flow synthesis of an alkynylnaphthyridine anticancer agent, HSN608. Org. Process Res. Dev. 2020, 24, 2240–2251. [Google Scholar] [CrossRef]
- Jaman, Z.; Mufti, A.; Sah, S.; Avramova, L.; Thompson, D. High Throughput Experimentation and Continuous Flow Validation of Suzuki-Miyaura Cross-Coupling Reactions. Chem. Eur. J. 2018, 24, 9546–9554. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Xie, Z.; Kuvelkar, R.; Shah, V.; Bateman, K.; McLaren, D.; Cooks, R. high-throughput bioassays using “dip-and-go” multiplexed electrospray mass spectrometry. Angew. Chem. Int. Ed. Engl. 2019, 58, 17594–17598. [Google Scholar] [CrossRef]
- Berman, H.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.; Weissig, H.; Shindyalov, I.; Bourne, P. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Mortuza, S.; He, B.; Wang, Y.; Zhang, Y. Template-based and free modeling of I-TASSER and QUARK pipelines using predicted contact maps in CASP12. Proteins Struct. Funct. Genet. 2018, 86 (Suppl. S1), 136–151. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, J.; He, B.; Walker, S.; Zhang, H.; Govindarajoo, B.; Virtanen, J.; Xue, Z.; Shen, H.; Zhang, Y. Integration of QUARK and I-TASSER for ab initio protein structure prediction in CASP11. Proteins Struct. Funct. Genet. 2016, 84 (Suppl. S1), 76–86. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, W.; He, B.; Walker, S.; Zhang, H.; Govindarajoo, B.; Virtanen, J.; Xue, Z.; Shen, H.; Zhang, Y. Template-based protein structure prediction in CASP11 and retrospect of I-TASSER in the last decade. Proteins Struct. Funct. Genet. 2016, 84 (Suppl. S1), 233–246. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Zhang, J.; Roy, A.; Zhang, Y. Automated protein structure modeling in CASP9 by I-TASSER pipeline combined with QUARK-based ab initio folding and FG-MD-based structure refinement. Proteins Struct. Funct. Genet. 2011, 79 (Suppl. S10), 147–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Peng, Z.; Zhang, Y.; Yang, J. COACH-D: Improved protein-ligand binding sites prediction with refined ligand-binding poses through molecular docking. Nucleic Acids Res. 2018, 46, W430–W438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inform. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Kramer, M. Nonlinear principal component analysis using autoassociative neural networks. AIChE J. 1991, 37, 233–243. [Google Scholar] [CrossRef]
- Santhanam, S. Context based text-generation using LSTM networks. arXiv 2020, arXiv:2005.00048. [Google Scholar]
- Hancock, J.T.; Khoshgoftaar, T.M. Survey on categorical data for neural networks. J. Big Data 2020, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Hochreiter, S.; Schmidhuber, J. Long Short-Term Memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Yu, Y.; Si, X.; Hu, C.; Zhang, J. A Review of recurrent neural networks: LSTM cells and network architectures. Neural Comput. 2019, 31, 1235–1270. [Google Scholar] [CrossRef] [PubMed]
- Kullback, S.; Leibler, R. On information and sufficiency. Ann. Math. Statist. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Davis, A.; Grondin, C.; Johnson, R.; Sciaky, D.; Wiegers, J.; Wiegers, T.; Mattingly, C. Comparative Toxicogenomics Database (CTD): Update 2021. Nucleic Acids Res. 2021, 49, D1138. [Google Scholar] [CrossRef]
- Tatonetti, N.; Ye, P.; Daneshjou, R.; Altman, R. Data-driven prediction of drug effects and interactions. Sci. Transl. Med. 2012, 4, 125–129. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, M.; Letunic, I.; Jensen, L.; Bork, P. The SIDER database of drugs and side effects. Nucleic Acids Res. 2016, 44, D1075–D1079. [Google Scholar] [CrossRef]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feller, W. On the Kolmogorov-Smirnov Limit Theorems for Empirical Distributions. Ann. Math. Stat. 1948, 19, 177–189. [Google Scholar] [CrossRef]
- Karson, M. Handbook of Methods of Applied Statistics. Volume I: Techniques of Computation Descriptive Methods, and Statistical Inference. Volume II: Planning of Surveys and Experiments. I. M. Chakravarti, R. G. Laha, and J. Roy, New York, John Wiley; 1967, $9.00. J. Am. Stat. Assoc. 1968, 63, 1047–1049. [Google Scholar] [CrossRef]
- Cox, D.R. Statistical significance tests. Br. J. Clin. Pharmacol. 1982, 14, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.A.; Shokhirev, M.N.; Wyss-Coray, T.; Lehallier, B. Systematic review and analysis of human proteomics aging studies unveils a novel proteomic aging clock and identifies key processes that change with age. Ageing Res. Rev. 2020, 60, 101070. [Google Scholar] [CrossRef]
- Lorusso, J.S.; Sviderskiy, O.A.; Labunskyy, V.M. Emerging omics approaches in aging research. Antioxid. Redox Signal. 2018, 29, 985–1002. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.; Parry, A.; Santos, F.; Hernando-Herraez, I.; Stubbs, T.M.; Milagre, I.; Reik, W. Multi-omic rejuvenation of human cells by maturation phase transient reprogramming. bioRxiv 2021. [Google Scholar] [CrossRef]
- Natarajan, K.N.; Teichmann, S.A.; Kolodziejczyk, A.A. Single cell transcriptomics of pluripotent stem cells: Reprogramming and differentiation. Curr. Opin. Genet. Dev. 2017, 46, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Koomen, J.M.; Haura, E.B.; Bepler, G.; Sutphen, R.; Remily-Wood, E.R.; Benson, K.; Hussein, M.; Hazlehurst, L.A.; Yeatman, T.J.; Hildreth, L.T.; et al. Proteomic contributions to personalized cancer care. Mol. Cell. Proteom. 2008, 7, 1780–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yumura, M.; Nagano, T.; Nishimura, Y. Novel multitarget therapies for lung cancer and respiratory disease. Molecules 2020, 25, 3987. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Overhoff, B.; Falls, Z.; Mangione, W.; Samudrala, R. A Deep-Learning Proteomic-Scale Approach for Drug Design. Pharmaceuticals 2021, 14, 1277. https://doi.org/10.3390/ph14121277
Overhoff B, Falls Z, Mangione W, Samudrala R. A Deep-Learning Proteomic-Scale Approach for Drug Design. Pharmaceuticals. 2021; 14(12):1277. https://doi.org/10.3390/ph14121277
Chicago/Turabian StyleOverhoff, Brennan, Zackary Falls, William Mangione, and Ram Samudrala. 2021. "A Deep-Learning Proteomic-Scale Approach for Drug Design" Pharmaceuticals 14, no. 12: 1277. https://doi.org/10.3390/ph14121277
APA StyleOverhoff, B., Falls, Z., Mangione, W., & Samudrala, R. (2021). A Deep-Learning Proteomic-Scale Approach for Drug Design. Pharmaceuticals, 14(12), 1277. https://doi.org/10.3390/ph14121277