
Inhibition of XPO-1 Mediated Nuclear Export through the Michael-Acceptor Character of Chalcones

 , , , , ,
, , , , , 
Abstract
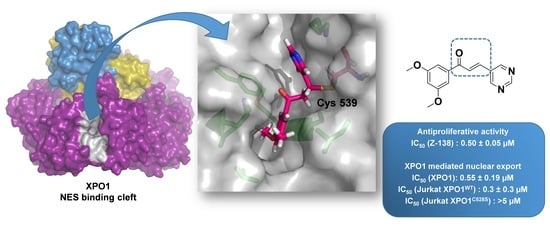
:
1. Introduction
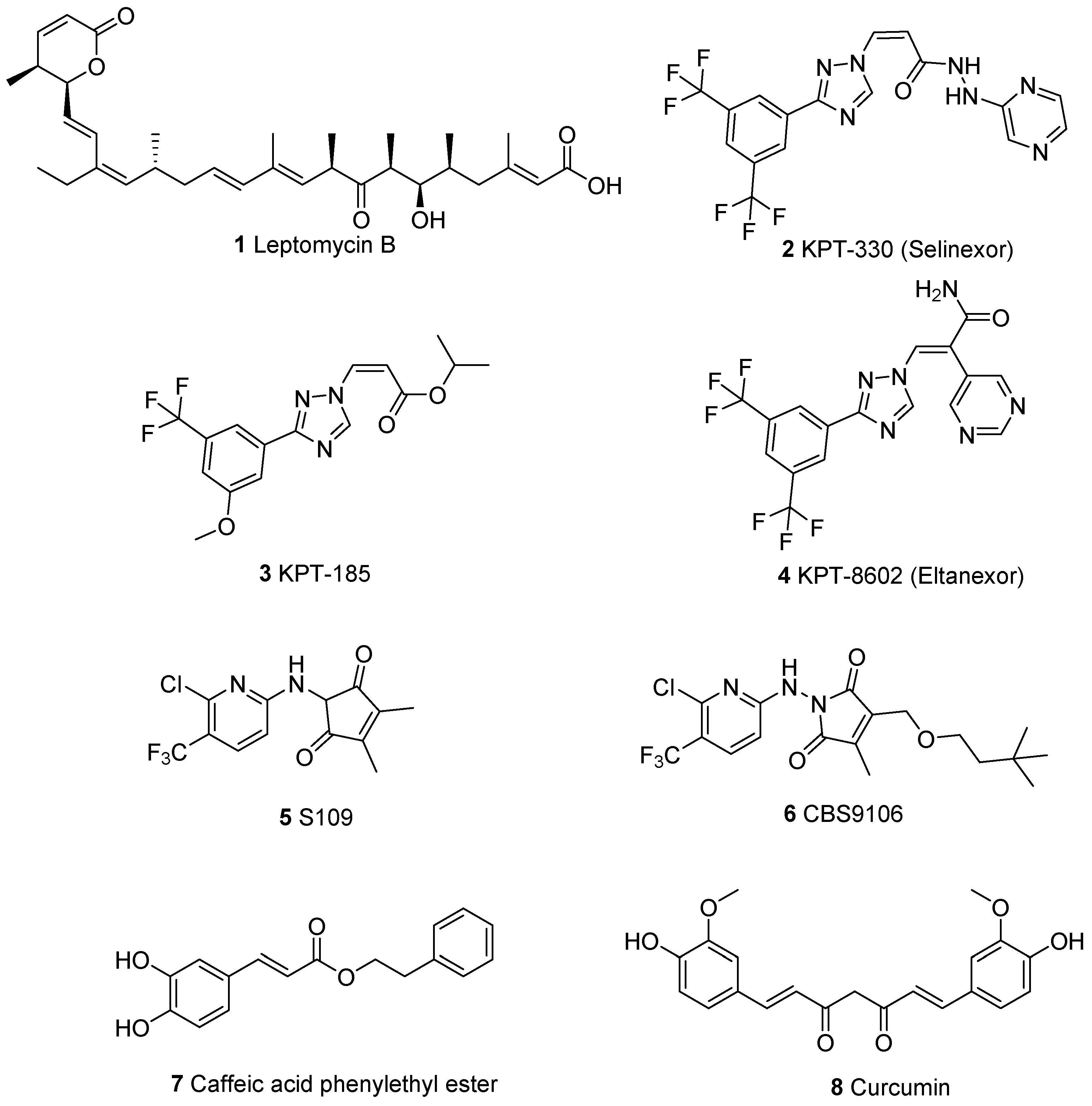
2. Results and Discussion
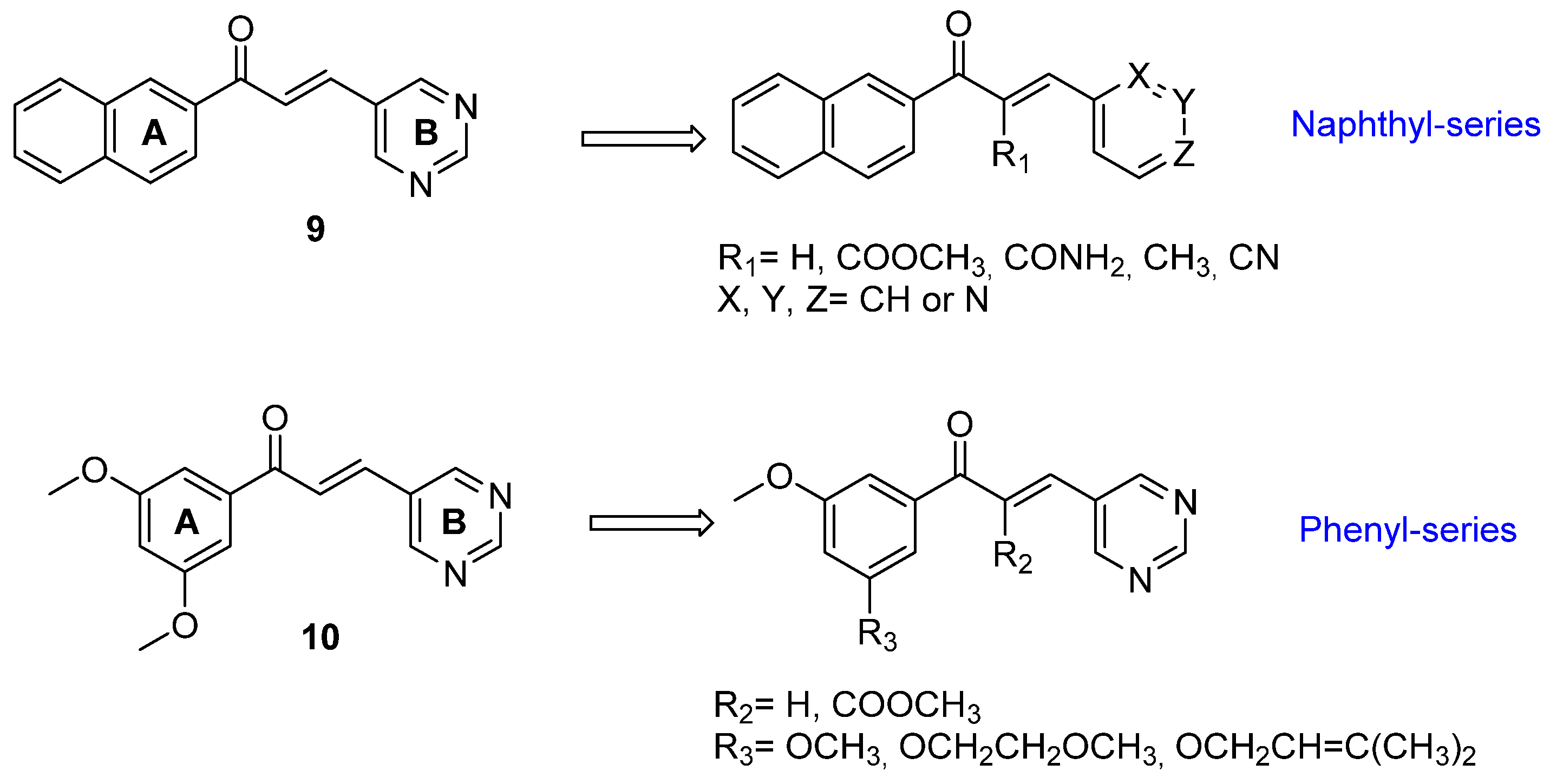
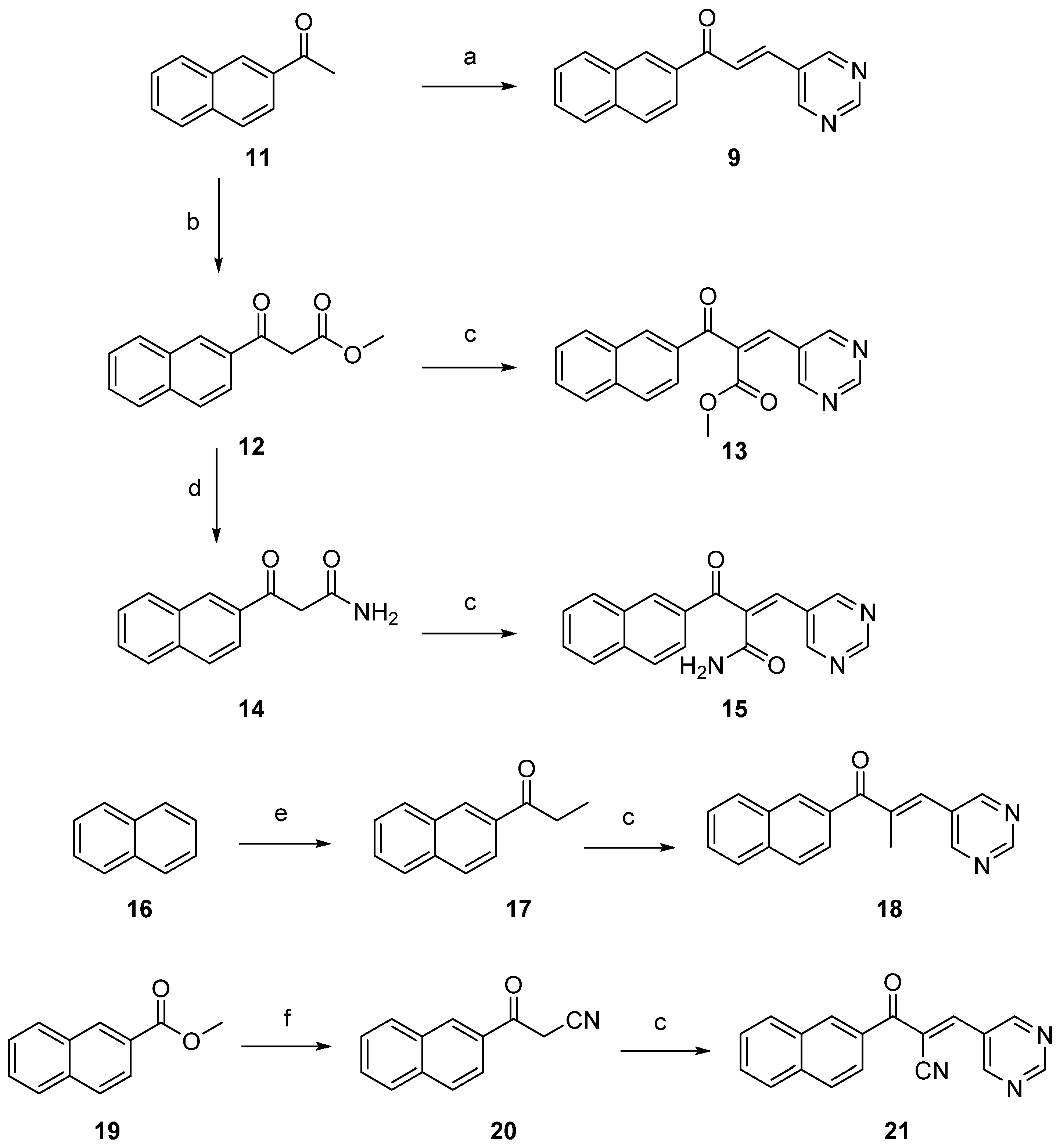
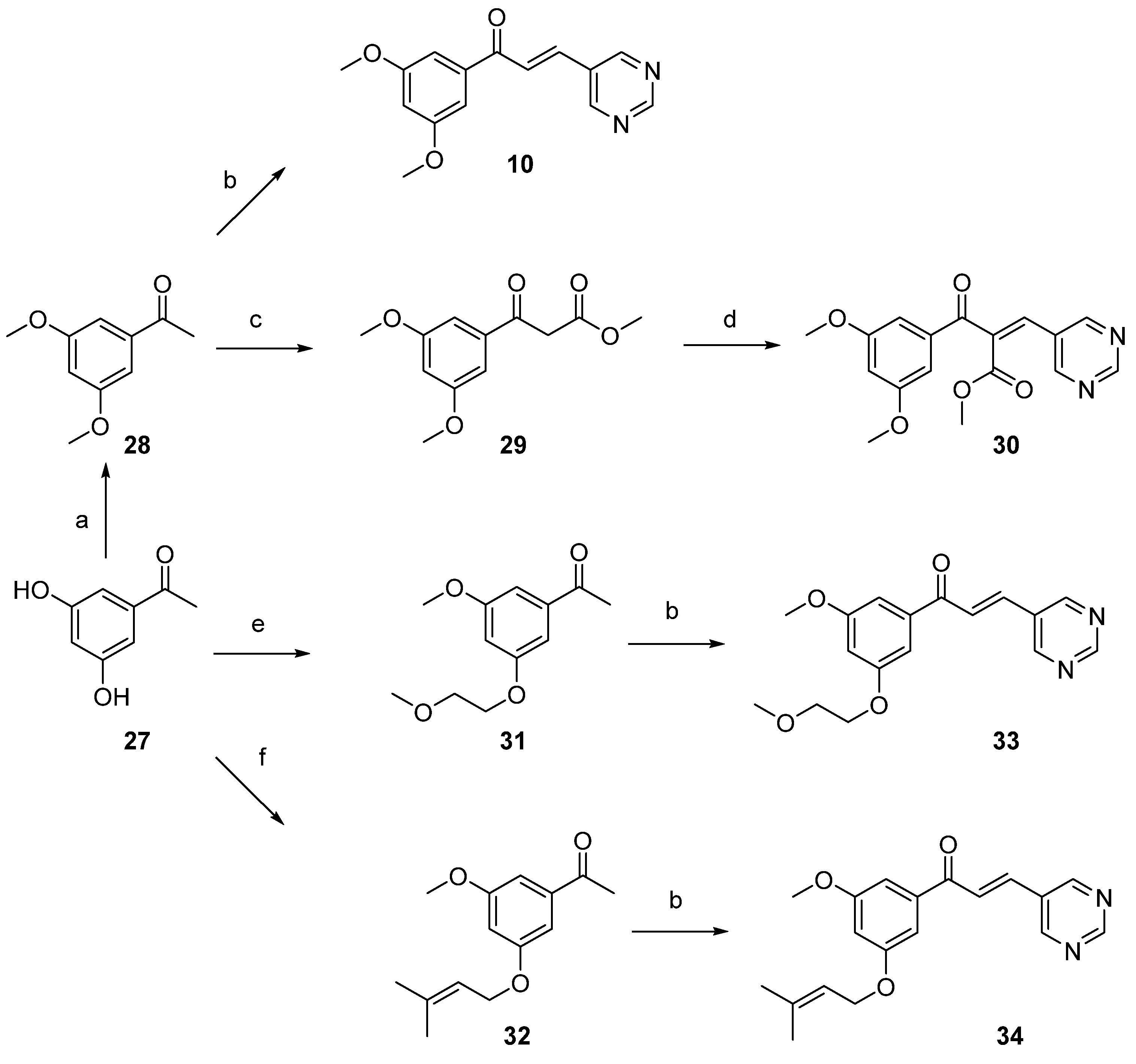
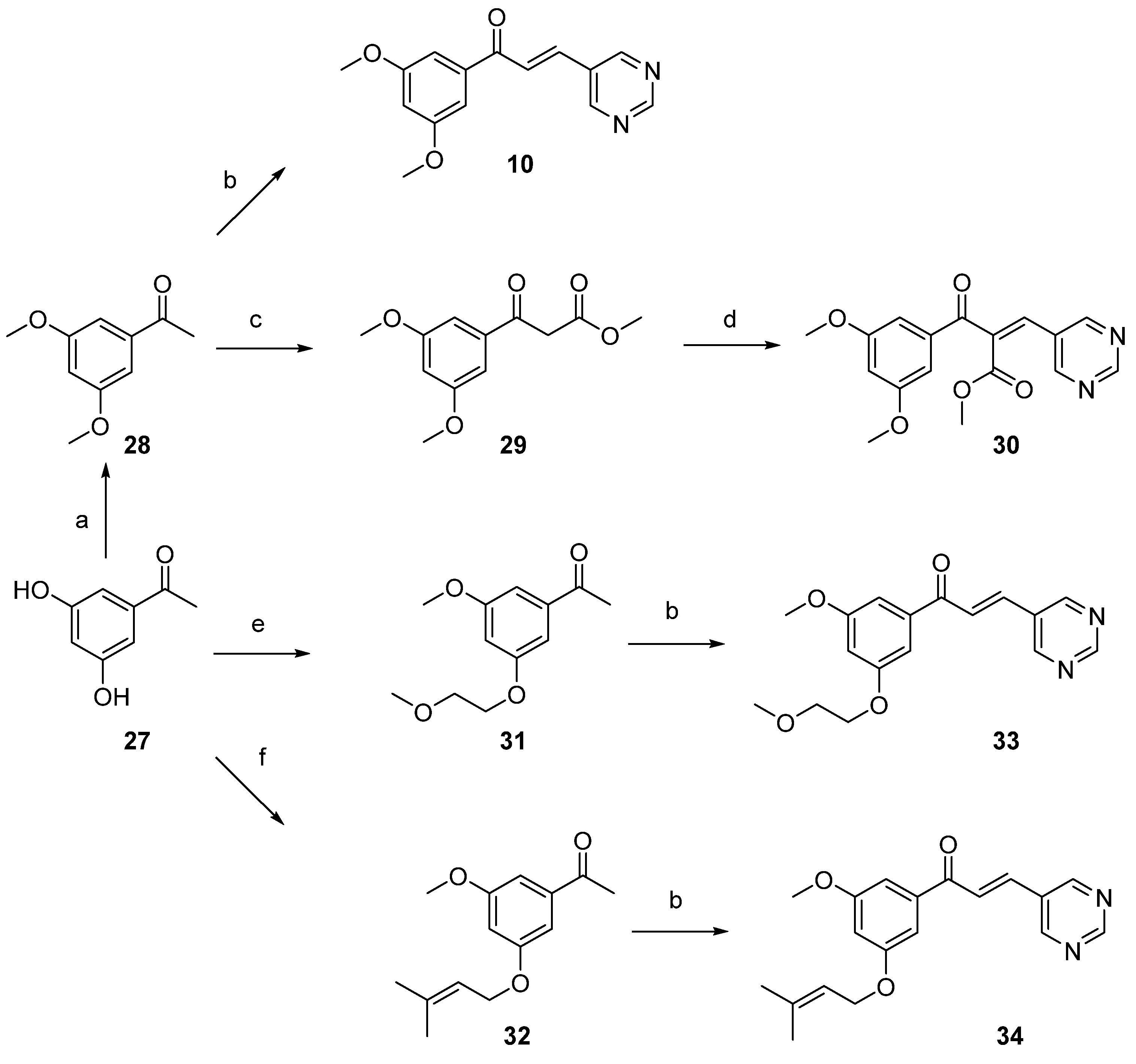
2.1. Synthesis
2.2. Incubation with GSH
2.3. Biological Results
2.3.1. Antiproliferative Activity
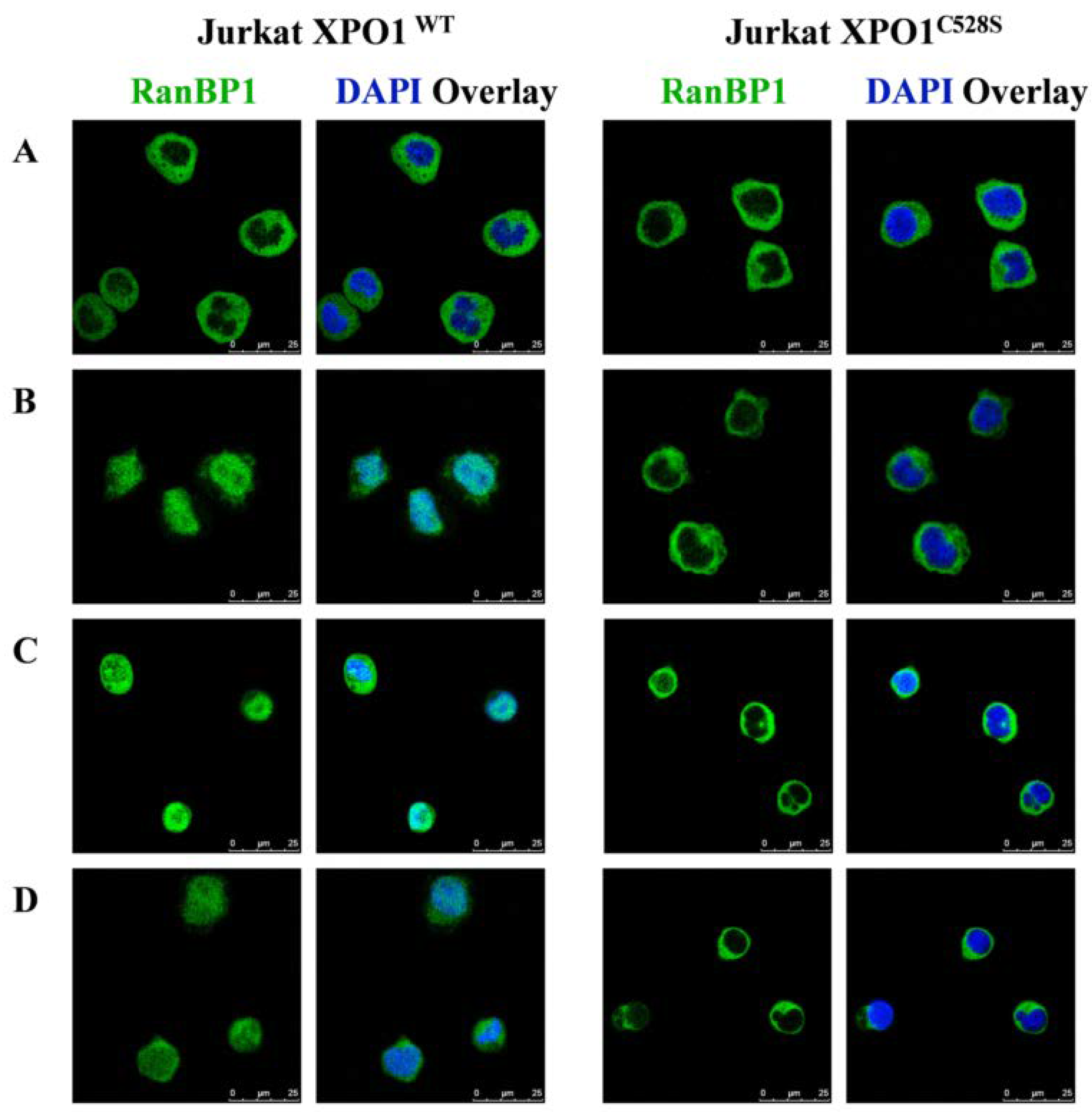
2.3.2. XPO1 Inhibition Studies
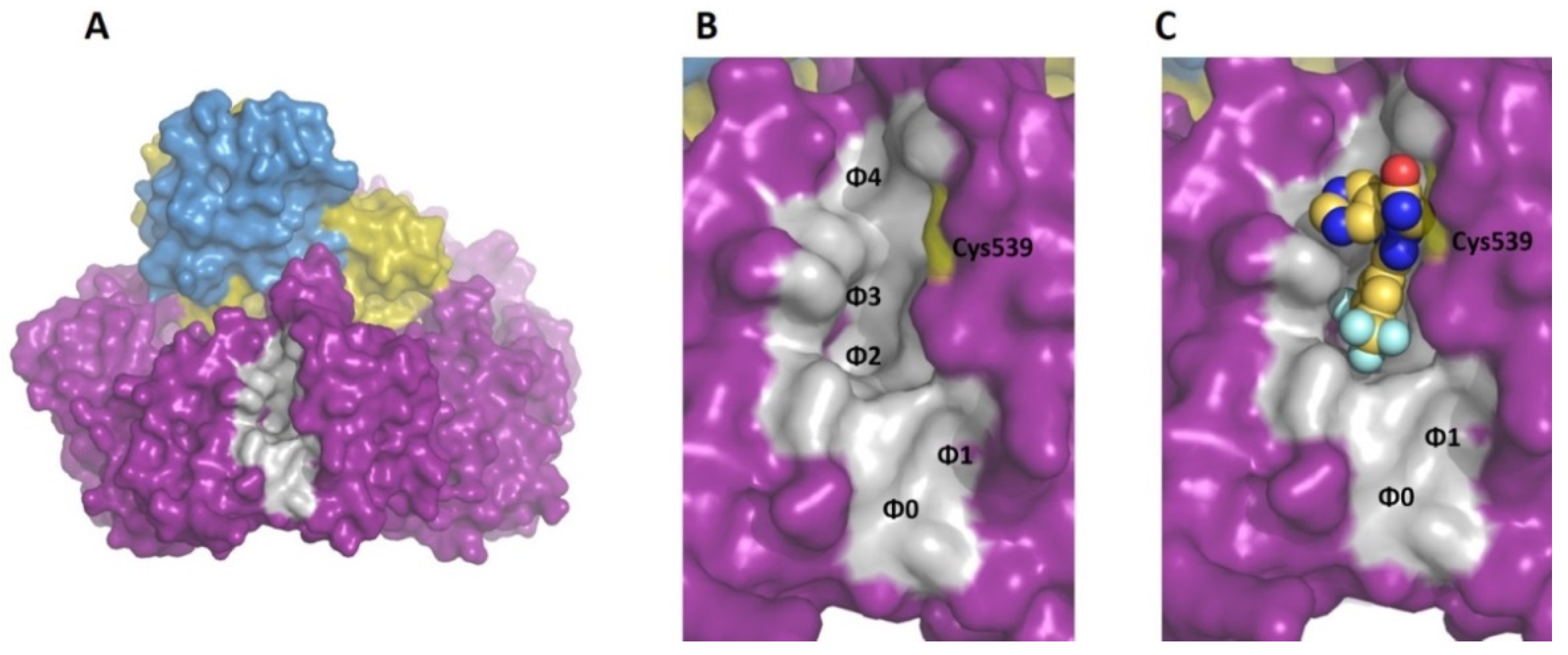
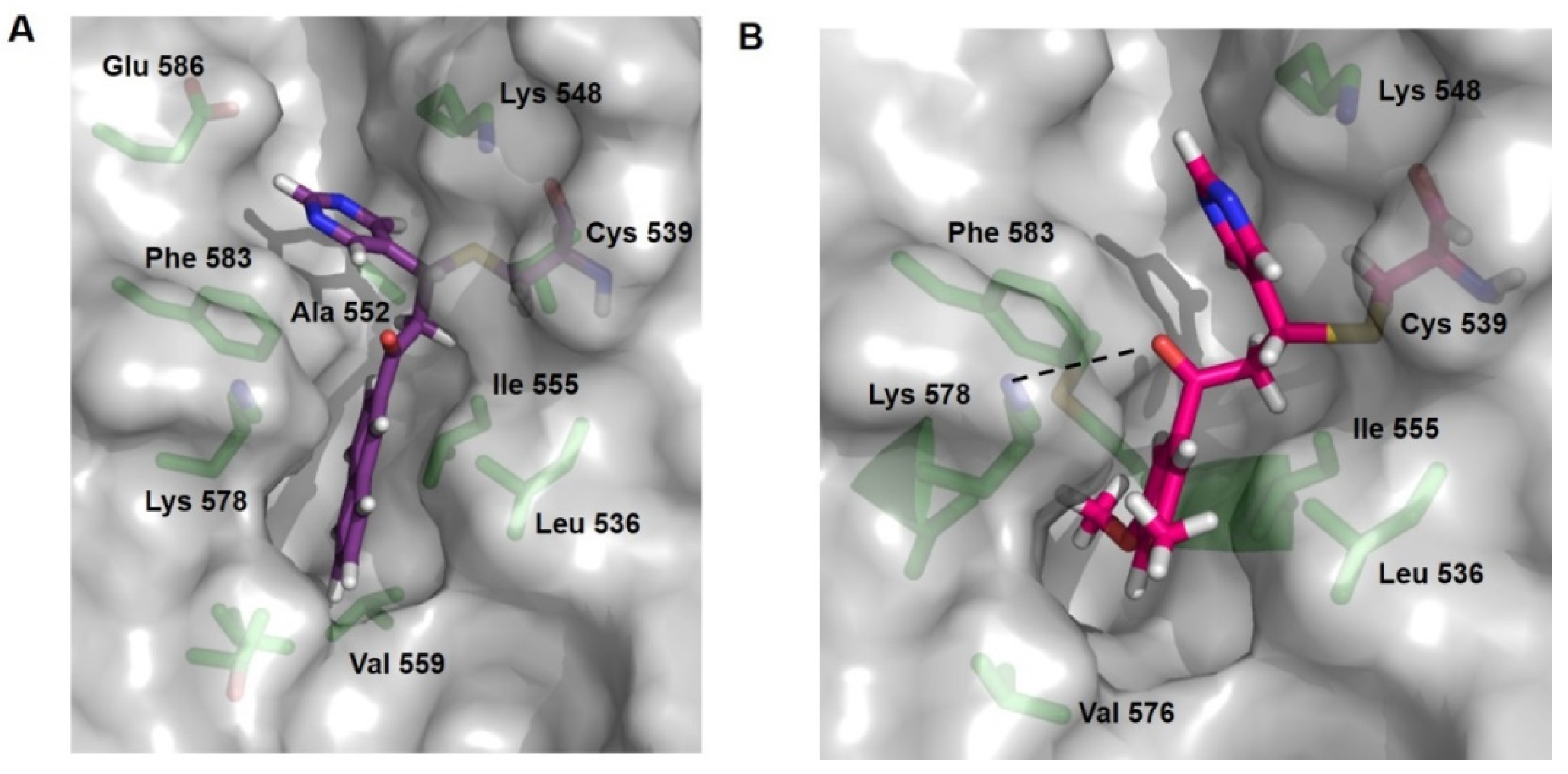
2.4. Docking Studies
3. Materials and Methods
3.1. Chemistry Procedures
3.2. GSH Reactivity Assay
3.3. Biological Assays
3.3.1. Cell Culture and Reference Compounds
3.3.2. Cell Proliferation Assays
3.3.3. XPO1 Phenotypic Reporter Assay
3.3.4. Immunofluorescence Staining of RanBP1
3.4. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, Y.H.; Han, M.-E.; Oh, S.-O. The molecular mechanism for nuclear transport and its application. Anat. Cell Biol. 2017, 50, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Kosyna, F.K.; Depping, R. Controlling the Gatekeeper: Therapeutic Targeting of Nuclear Transport. Cells 2018, 7, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickens, J.A.; Tripp, R.A. Verdinexor Targeting of CRM1 is a Promising Therapeutic Approach against RSV and Influenza Viruses. Viruses 2018, 10, 48. [Google Scholar] [CrossRef] [Green Version]
- Çağatay, T.; Chook, Y.M. Karyopherins in cancer. Curr. Opin. Cell Biol. 2018, 52, 30–42. [Google Scholar] [CrossRef]
- Mathew, C.; Ghildyal, R. CRM1 Inhibitors for Antiviral Therapy. Front. Microbiol. 2017, 8, 1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camus, V.; Miloudi, H.; Taly, A.; Sola, B.; Jardin, F. XPO1 in B cell hematological malignancies: From recurrent somatic mutations to targeted therapy. J. Hematol. Oncol. 2017, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Chen, X.; Zhou, Q.; Burstein, E.; Yang, S.; Jia, D. Inhibiting cancer cell hallmark features through nuclear export inhibition. Signal Transduct. Target. Ther. 2016, 1, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Dickmanns, A.; Monecke, T.; Ficner, R. Structural Basis of Targeting the Exportin CRM1 in Cancer. Cells 2015, 4, 538–568. [Google Scholar] [CrossRef] [Green Version]
- Muqbil, I.; Azmi, A.S.; Mohammad, R.M. Nuclear Export Inhibition for Pancreatic Cancer Therapy. Cancers 2018, 10, 138. [Google Scholar] [CrossRef] [Green Version]
- A Jans, D.; Martin, A.J.; Wagstaff, K.M. Inhibitors of nuclear transport. Curr. Opin. Cell Biol. 2019, 58, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, M.; Ishizawa, J.; Ruvolo, V.; Dilip, A.; Quintás-Cardama, A.; McDonnell, T.J.; Neelapu, S.S.; Kwak, L.; Shacham, S.; Kauffman, M.; et al. Induction of p53-mediated transcription and apoptosis by exportin-1 (XPO 1) inhibition in mantle cell lymphoma. Cancer Sci. 2014, 105, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Daelemans, D.; Afonina, E.; Nilsson, J.; Werner, G.; Kjems, J.; De Clercq, E.; Pavlakis, G.N.; Vandamme, A.-M. A synthetic HIV-1 Rev inhibitor interfering with the CRM1-mediated nuclear export. Proc. Natl. Acad. Sci. USA 2002, 99, 14440–14445. [Google Scholar] [CrossRef] [Green Version]
- Van Neck, T.; Pannecouque, C.; Vanstreels, E.; Stevens, M.; Dehaen, W.; Daelemans, D. Inhibition of the CRM1-mediated nucleocytoplasmic transport by N-azolylacrylates: Structure–activity relationship and mechanism of action. Bioorg. Med. Chem. 2008, 16, 9487–9497. [Google Scholar] [CrossRef]
- Lapalombella, R.; Sun, Q.; Williams, K.; Tangeman, L.; Jha, S.; Zhong, Y.; Goettl, V.; Mahoney, E.; Berglund, C.; Gupta, S.; et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood 2012, 120, 4621–4634. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; An, Q.; Shen, X.-F.; Sui, M.; Li, C.; Jia, D.; Luo, Y.; Sun, Q. Structure-Guided Design of the First Noncovalent Small-Molecule Inhibitor of CRM1. J. Med. Chem. 2021, 64, 6596–6607. [Google Scholar] [CrossRef] [PubMed]
- Shaikhqasem, A.; Dickmanns, A.; Neumann, P.; Ficner, R. Characterization of Inhibition Reveals Distinctive Properties for Human and Saccharomyces cerevisiae CRM1. J. Med. Chem. 2020, 63, 7545–7558. [Google Scholar] [CrossRef]
- Sun, Q.; Carrasco, Y.P.; Hu, Y.; Guo, X.; Mirzaei, H.; MacMillan, J.; Chook, Y.M. Nuclear export inhibition through covalent conjugation and hydrolysis of Leptomycin B by CRM1. Proc. Natl. Acad. Sci. USA 2013, 110, 1303–1308. [Google Scholar] [CrossRef] [Green Version]
- Van Der Watt, P.J.; Chi, R.-P.A.; Stelma, T.; Stowell, C.; Strydom, E.; Carden, S.; Angus, L.; Hadley, K.; Lang, D.; Wei, W.; et al. Targeting the Nuclear Import Receptor Kpnβ1 as an Anticancer Therapeutic. Mol. Cancer Ther. 2016, 15, 560–573. [Google Scholar] [CrossRef] [Green Version]
- Vercruysse, T.; De Bie, J.; Neggers, J.; Jacquemyn, M.; Vanstreels, E.; Schmid-Burgk, J.; Hornung, V.; Baloglu, E.; Landesman, Y.; Senapedis, W.; et al. The Second-Generation Exportin-1 Inhibitor KPT-8602 Demonstrates Potent Activity against Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2017, 23, 2528–2541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hing, Z.A.; Fung, H.Y.J.; Ranganathan, P.; Mitchell, S.; El-Gamal, D.; Woyach, J.A.; Williams, K.; Goettl, V.M.; Smith, J.; Yu, X.; et al. Next-generation XPO1 inhibitor shows improved efficacy and in vivo tolerability in hematological malignancies. Leukemia 2016, 30, 2364–2372. [Google Scholar] [CrossRef] [Green Version]
- FDA Grants Accelerated Approval to Selinexor for Multiple Myeloma|FDA. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-selinexor-multiple-myeloma (accessed on 24 September 2021).
- Sakakibara, K.; Saito, N.; Sato, T.; Suzuki, A.; Hasegawa, Y.; Friedman, J.; Kufe, D.W.; Vonhoff, D.D.; Iwami, T.; Kawabe, T. CBS9106 is a novel reversible oral CRM1 inhibitor with CRM1 degrading activity. Blood 2011, 118, 3922–3931. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chong, Y.; Liu, H.; Han, Y.; Niu, M. Novel reversible selective inhibitor of CRM1 for targeted therapy in ovarian cancer. J. Ovarian Res. 2015, 8, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Phase 1 Trial of a Novel XPO1 Inhibitor in Patients With Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT02667873 (accessed on 4 October 2021).
- Etchin, J.; Sun, Q.; Kentsis, A.; Farmer, A.; Zhang, Z.C.; Sanda, T.; Mansour, M.; Barceló, C.; McCauley, D.; Kauffman, M.; et al. Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia 2012, 27, 66–74. [Google Scholar] [CrossRef]
- Bradshaw, J.M.; McFarland, J.M.; Paavilainen, V.O.; Bisconte, A.; Tam, D.; Phan, V.T.; Romanov, S.; Finkle, D.; Shu, J.; Patel, V.; et al. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 2015, 11, 525–531. [Google Scholar] [CrossRef] [Green Version]
- Al-Rifai, N.; Rücker, H.; Amslinger, S. Opening or Closing the Lock? When Reactivity Is the Key to Biological Activity. Chem. A Eur. J. 2013, 19, 15384–15395. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.K.; Tao, J.; Tuck, T.N. Geminal Dichlorination of Phenyliodonium Ylides of β-Dicarbonyl Compounds through Double Ligand Transfer from (Dichloroiodo)benzene. Synthesis 2016, 48, 772–782. [Google Scholar] [CrossRef]
- Zheng, X.; Kerr, M.A. Synthesis and Cross-Coupling Reactions of 7-Azaindoles via a New Donor−Acceptor Cyclopropane. Org. Lett. 2006, 8, 3777–3779. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Takayama, J.; Aubin, Y.; Ratia, K.; Chaudhuri, R.; Baez, Y.; Sleeman, K.; Coughlin, M.; Nichols, D.B.; Mulhearn, D.C.; et al. Structure-Based Design, Synthesis, and Biological Evaluation of a Series of Novel and Reversible Inhibitors for the Severe Acute Respiratory Syndrome−Coronavirus Papain-Like Protease. J. Med. Chem. 2009, 52, 5228–5240. [Google Scholar] [CrossRef] [Green Version]
- Malmedy, F.; Wirth, T. Stereoselective Ketone Rearrangements with Hypervalent Iodine Reagents. Chem. A Eur. J. 2016, 22, 16072–16077. [Google Scholar] [CrossRef] [Green Version]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cara, C.L.; Salvador, M.K.; Preti, D.; Tabrizi, M.A.; Moorman, A.R.; Vincenzi, F.; Borea, P.A.; et al. Synthesis and biological effects of novel 2-amino-3-(4-chlorobenzoyl)-4-substituted thiophenes as allosteric enhancers of the A1 adenosine receptor. Eur. J. Med. Chem. 2013, 67, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, L.R.; Tolentino, D.R.; Gallon, B.J.; Schrodi, Y. Development of a Method for the Preparation of Ruthenium Indenylidene-Ether Olefin Metathesis Catalysts. Molecules 2012, 17, 5675–5689. [Google Scholar] [CrossRef] [Green Version]
- Parsons, D.E.; Frontier, A.J. Noncanonical Cation−π Cyclizations of Alkylidene β-Ketoesters: Synthesis of Spiro-fused and Bridged Bicyclic Ring Systems. Org. Lett. 2019, 21, 2008–2012. [Google Scholar] [CrossRef] [PubMed]
- Heinelt, U.; Wehner, V.; Herrmann, M.; Schoenafinger, K.; Steinhagen, H. Triazolium salts as PAR1 inhibitors, production thereof, and use as medicaments. PCT Int. Appl. WO200909 7971A1, 2009.
- Ducki, S.; Forrest, R.; Hadfield, J.A.; Kendall, A.; Lawrence, N.J.; McGown, A.T.; Rennison, D. Potent antimitotic and cell growth inhibitory properties of substituted chalcones. Bioorg. Med. Chem. Lett. 1998, 8, 1051–1056. [Google Scholar] [CrossRef]
- Lawrence, N.J.; Patterson, R.P.; Ooi, L.-L.; Cook, D.; Ducki, S. Effects of α-substitutions on structure and biological activity of anticancer chalcones. Bioorg. Med. Chem. Lett. 2006, 16, 5844–5848. [Google Scholar] [CrossRef] [PubMed]
- Biddle, M.M.; Lin, M.; Scheidt, K.A. Catalytic Enantioselective Synthesis of Flavanones and Chromanones. J. Am. Chem. Soc. 2007, 129, 3830–3831. [Google Scholar] [CrossRef] [PubMed]
- Böhme, A.; Thaens, D.; Paschke, A.; Schüürmann, G. Kinetic Glutathione Chemoassay To Quantify Thiol Reactivity of Organic Electrophiles—Application to α,β-Unsaturated Ketones, Acrylates, and Propiolates. Chem. Res. Toxicol. 2009, 22, 742–750. [Google Scholar] [CrossRef]
- Chan, K.; Poon, R.; O’Brien, P.J. Application of structure–activity relationships to investigate the molecular mechanisms of hepatocyte toxicity and electrophilic reactivity ofα,β-unsaturated aldehydes. J. Appl. Toxicol. 2008, 28, 1027–1039. [Google Scholar] [CrossRef]
- Cee, V.J.; Volak, L.P.; Chen, Y.; Bartberger, M.D.; Tegley, C.; Arvedson, T.; McCarter, J.; Tasker, A.S.; Fotsch, C. Systematic Study of the Glutathione (GSH) Reactivity of N-Arylacrylamides: 1. Effects of Aryl Substitution. J. Med. Chem. 2015, 58, 9171–9178. [Google Scholar] [CrossRef] [PubMed]
- Slawik, C.; Rickmeyer, C.; Brehm, M.; Böhme, A.; Schüürmann, G. Glutathione Adduct Patterns of Michael-Acceptor Carbonyls. Environ. Sci. Technol. 2017, 51, 4018–4026. [Google Scholar] [CrossRef]
- Neggers, J.; Vercruysse, T.; Jacquemyn, M.; Vanstreels, E.; Baloglu, E.; Shacham, S.; Crochiere, M.; Landesman, Y.; Daelemans, D. Identifying Drug-Target Selectivity of Small-Molecule CRM1/XPO1 Inhibitors by CRISPR/Cas9 Genome Editing. Chem. Biol. 2015, 22, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Warshaviak, D.T.; Golan, G.; Borrelli, K.W.; Zhu, K.; Kalid, O. Structure-Based Virtual Screening Approach for Discovery of Covalently Bound Ligands. J. Chem. Inf. Model. 2014, 54, 1941–1950. [Google Scholar] [CrossRef]
- Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking Covalent Inhibitors: A Parameter Free Approach To Pose Prediction and Scoring. J. Chem. Inf. Model. 2014, 54, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Devraj, R.; Kumaravel, G.; Lecci, C.; Loke, P.; Meniconi, M.; Monck, N.; North, C.; Ridgill, M.; Tye, H. Preparation of heteroaryl inhibitors of peptidylarginine deiminase 4. PCT Int. Appl. WO2018049296A1, 2018. [Google Scholar]
- Wang, K.-K.; Wang, P.; Ouyang, Q.; Du, W.; Chen, Y.-C. Substrate-controlled switchable asymmetric annulations to access polyheterocyclic skeletons. Chem. Commun. 2016, 52, 11104–11107. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, Y.-W.; Hu, S.-J.; Niu, W.-P.; Zhang, G.-N.; Zhu, M.; Wang, M.-H.; Zhang, F.; Li, X.-M.; Wang, J.-X. Design, synthesis, and antibacterial evaluation of PFK-158 derivatives as potent agents against drug-resistant bacteria. Bioorg. Med. Chem. Lett. 2021, 41, 127980. [Google Scholar] [CrossRef]
- Parnell, K.M.; McCall, J.; Romero, D. Preparation of functionalized pyrazoles and other nitrogen-containing heterocycles as inhibitors of MCT4 for the treatment of MCT4-mediated diseases. U.S. Pat. Appl. Publ. US20180162822A1, 2018. [Google Scholar]
- Amslinger, S.; Al-Rifai, N.; Winter, K.; Wörmann, K.; Scholz, R.; Baumeister, P.; Wild, M. Reactivity assessment of chalcones by a kinetic thiol assay. Org. Biomol. Chem. 2013, 11, 549–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Release 2018-3; Schrödinger, LLC: New York, NY, USA, 2018; Available online: https://www.schrodinger.com/citations (accessed on 13 October 2021).
- The PyMOL Molecular Graphics System; Version 2.0; Schrödinger, LLC: New York, NY, USA, 2017.
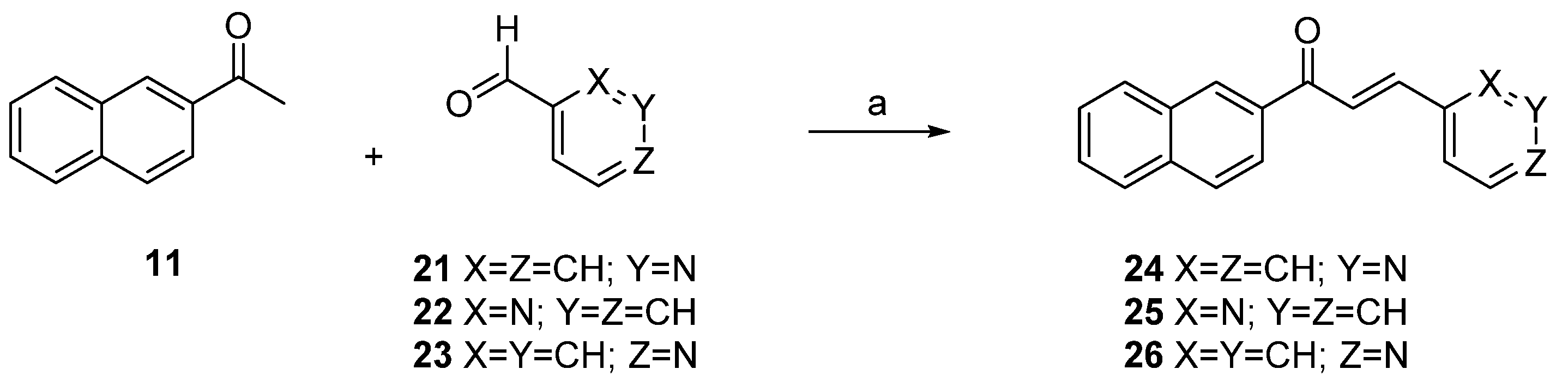

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (µM) a | |||||||
|---|---|---|---|---|---|---|---|
| Comp | Hap-1 b | HCT-116 b | NCI-H460 b | DND-41 b | HL-60 b | K-562 b | Z-138 b |
| 9 | 2.3 ± 0.2 | 2.4 ± 0.2 | 2.1 ± 0.1 | 2.1 ± 0.5 | 2.0 ± 0.5 | 1.8 ± 0.08 | 1.8 ± 0.05 |
| 10 | 2.1 ± 0.2 | 4.7 ± 1.7 | 3.5 ± 1.5 | 1.5 ± 0.2 | 2.2 ± 0.3 | 7.2 ± 1.9 | 0.5 ± 0.05 |
| 13 | ≥54.9 | ≥15.5 | ≥47.1 | ≥30.6 | >100 | >100 | >100 |
| 15 | 2.1 ± 0.7 | 5.3 ± 3.1 | 2.6 ± 0.7 | 8.9 ± 0.9 | 5.5 ± 3.6 | 2.1 ± 0.1 | 5.1 ± 2.7 |
| 18 | >100 | >100 | >100 | 39.1 ± 5.3 | ≥40.9 | 48.5 ± 2.4 | >100 |
| 24 | 1.6 ± 0.1 | 3.1 ± 0.9 | 4.3 ± 2.2 | 1.8 ± 0.5 | 3.5 ± 2.1 | 2.2 ± 0.03 | 1.6 ± 0.1 |
| 25 | 3.4 ± 0.5 | 3.9 ± 1.5 | 51.6 ± 11.7 | 2.6 ± 0.4 | 1.8 ± 0.05 | 4.8 ± 2.8 | 2.3 ± 0.5 |
| 26 | 2.3 ± 0.3 | 1.9 ± 0.4 | 7.7 ± 1.1 | 1.9 ± 0.4 | 0.9 ± 0.2 | 4.0 ± 0.2 | 1.1 ± 0.4 |
| 30 | ≥32.0 | ≥22.2 | 17.3 ± 4.6 | 43.2 ± 6.4 | 23.4 ± 1.9 | 42.9 ± 0.1 | 47.6 ± 1.1 |
| 33 | 4.3 ± 2.5 | 34.2 ± 6.5 | 16.9 ± 4.5 | 3.7 ± 1.6 | 10.0 ± 0.9 | 37.5 ± 0.9 | 1.5 ± 0.2 |
| 34 | 10.0 ± 2.0 | 52.9 ± 3.6 | 34.5 ± 5.2 | 10.1 ± 0.0 | 40.2 ± 10.1 | 4.1 ± 1.9 | 4.6 ± 2.7 |
| KPT-330 | 0.07 ± 0.03 | 0.10 ± 0.1 | 0.12 ± 0.08 | 0.05 ± 0.0 | 0.11 ± 0.1 | 0.09 ± 0.0 | 0.40 ± 0.4 |
| Comp | IC50 (µM) a |
|---|---|
| 9 | 2.46 ± 0.16 |
| 10 | 0.55 ± 0.19 |
| 13 | >100 |
| 5 | 9.18 ± 3.28 |
| 18 | >100 |
| 24 | 1.27 ± 0.29 |
| 25 | 7.28 ± 1.34 |
| 26 | 10.74 ± 3.24 |
| 30 | 87.34 ± 4.91 |
| 33 | 12.35 ± 1.28 |
| 34 | 1.59 ± 0.36 |
| KPT-330 | 0.05 |
| Comp | IC50 (μM) a | |
|---|---|---|
| Jurkat XPO1WT | Jurkat XPO1C528S | |
| KPT-330 | 0.07 ± 0.01 | >5 |
| 9 | 2.2 ± 0.03 | >5 |
| 10 | 0.3 ± 0.3 | >5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gargantilla, M.; López-Fernández, J.; Camarasa, M.-J.; Persoons, L.; Daelemans, D.; Priego, E.-M.; Pérez-Pérez, M.-J. Inhibition of XPO-1 Mediated Nuclear Export through the Michael-Acceptor Character of Chalcones. Pharmaceuticals 2021, 14, 1131. https://doi.org/10.3390/ph14111131
Gargantilla M, López-Fernández J, Camarasa M-J, Persoons L, Daelemans D, Priego E-M, Pérez-Pérez M-J. Inhibition of XPO-1 Mediated Nuclear Export through the Michael-Acceptor Character of Chalcones. Pharmaceuticals. 2021; 14(11):1131. https://doi.org/10.3390/ph14111131
Chicago/Turabian StyleGargantilla, Marta, José López-Fernández, Maria-Jose Camarasa, Leentje Persoons, Dirk Daelemans, Eva-Maria Priego, and María-Jesús Pérez-Pérez. 2021. "Inhibition of XPO-1 Mediated Nuclear Export through the Michael-Acceptor Character of Chalcones" Pharmaceuticals 14, no. 11: 1131. https://doi.org/10.3390/ph14111131
APA StyleGargantilla, M., López-Fernández, J., Camarasa, M.-J., Persoons, L., Daelemans, D., Priego, E.-M., & Pérez-Pérez, M.-J. (2021). Inhibition of XPO-1 Mediated Nuclear Export through the Michael-Acceptor Character of Chalcones. Pharmaceuticals, 14(11), 1131. https://doi.org/10.3390/ph14111131