
New Multi-Targeted Antiproliferative Agents: Design and Synthesis of IC261-Based Oxindoles as Potential Tubulin, CK1 and EGFR Inhibitors

 , ,
, ,  , ,
, , 
Abstract
1. Introduction
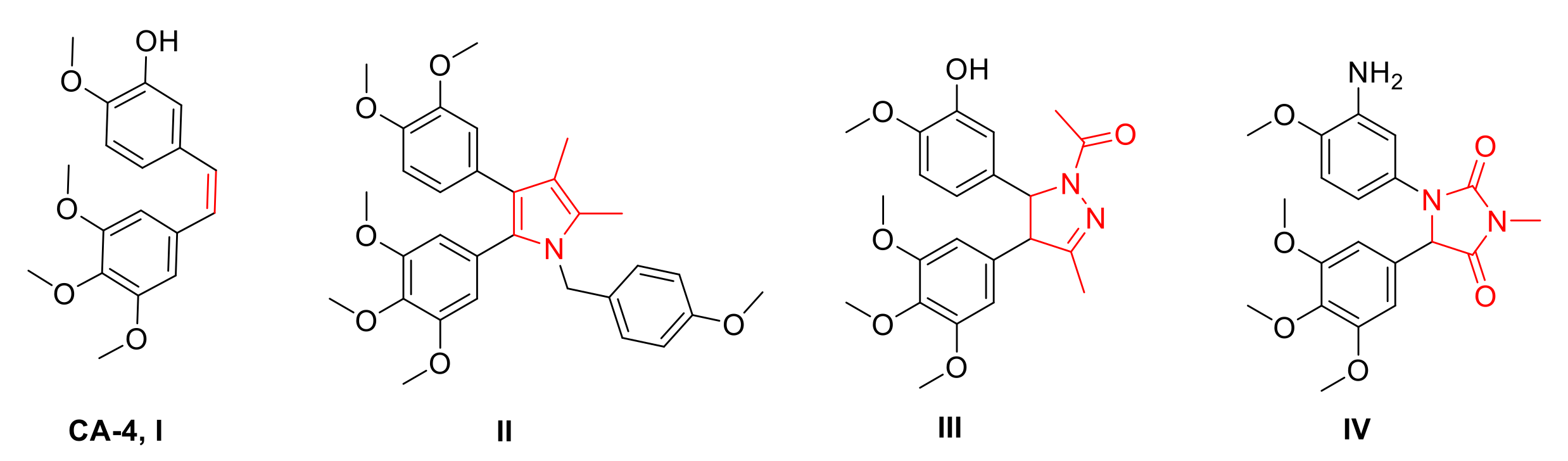
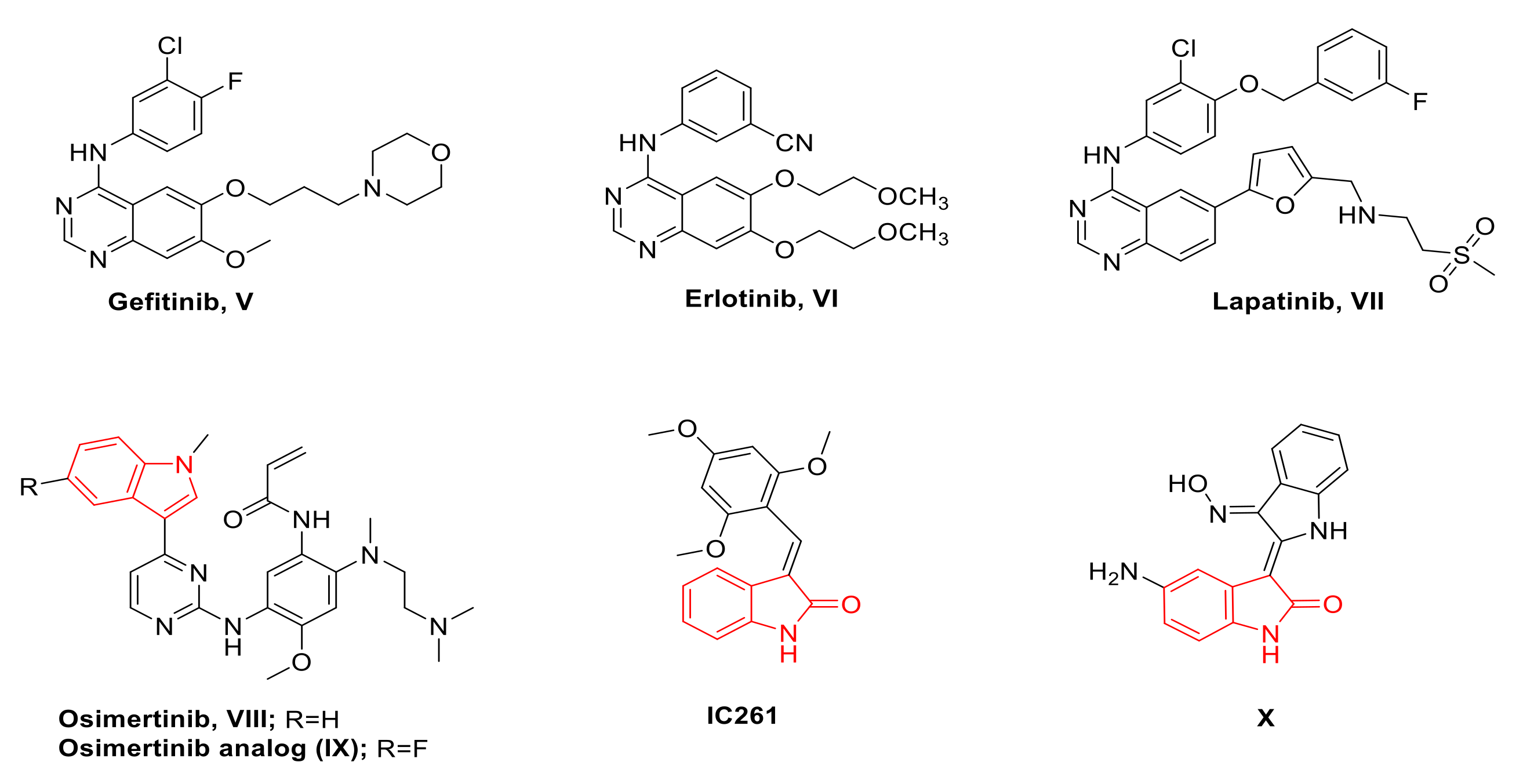
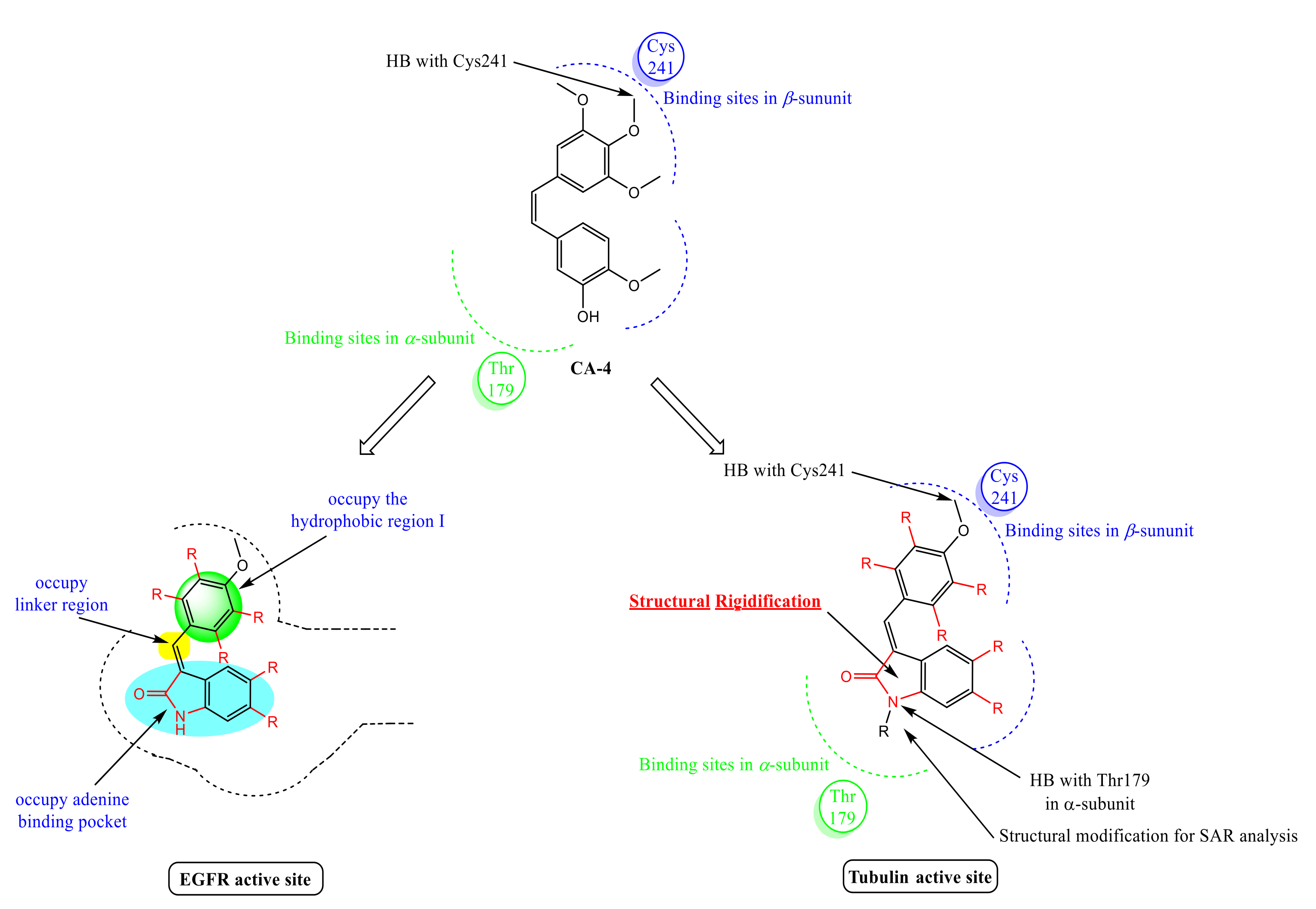
Rationale of Design
2. Results
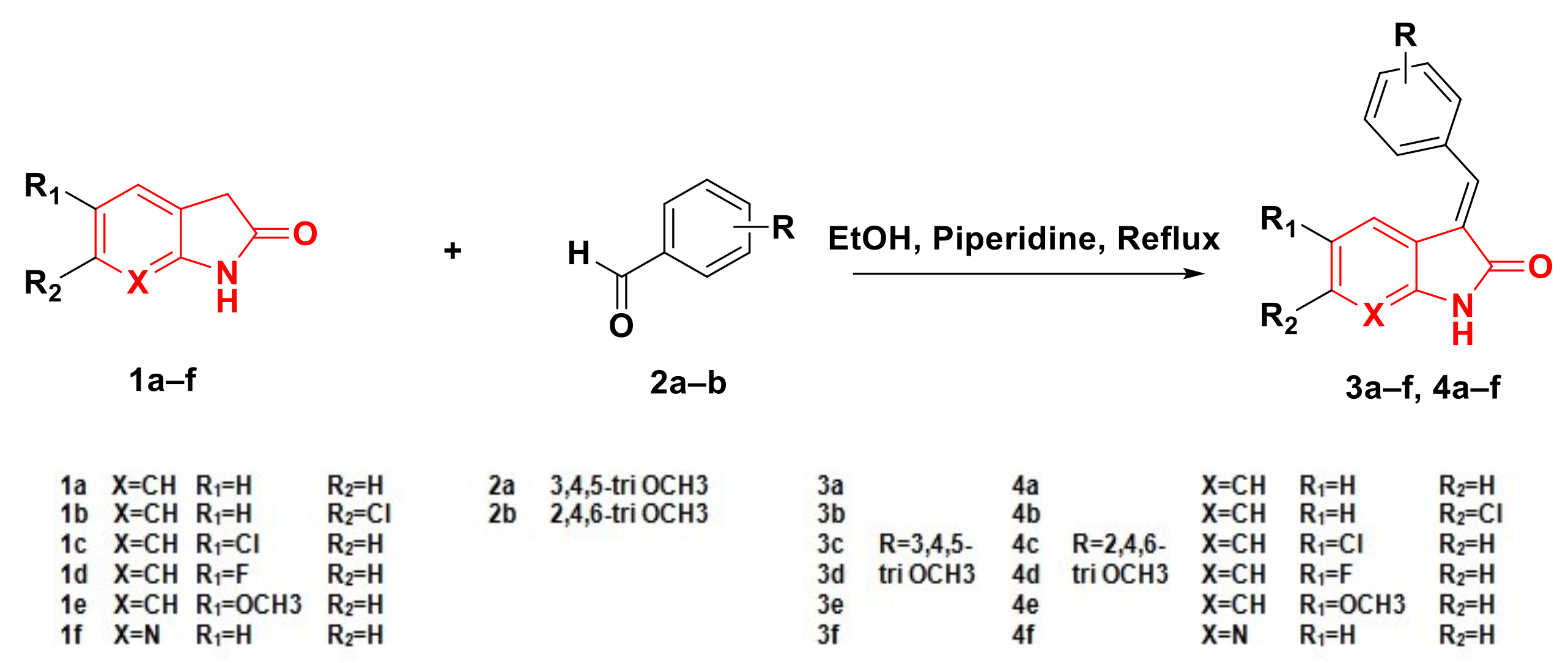
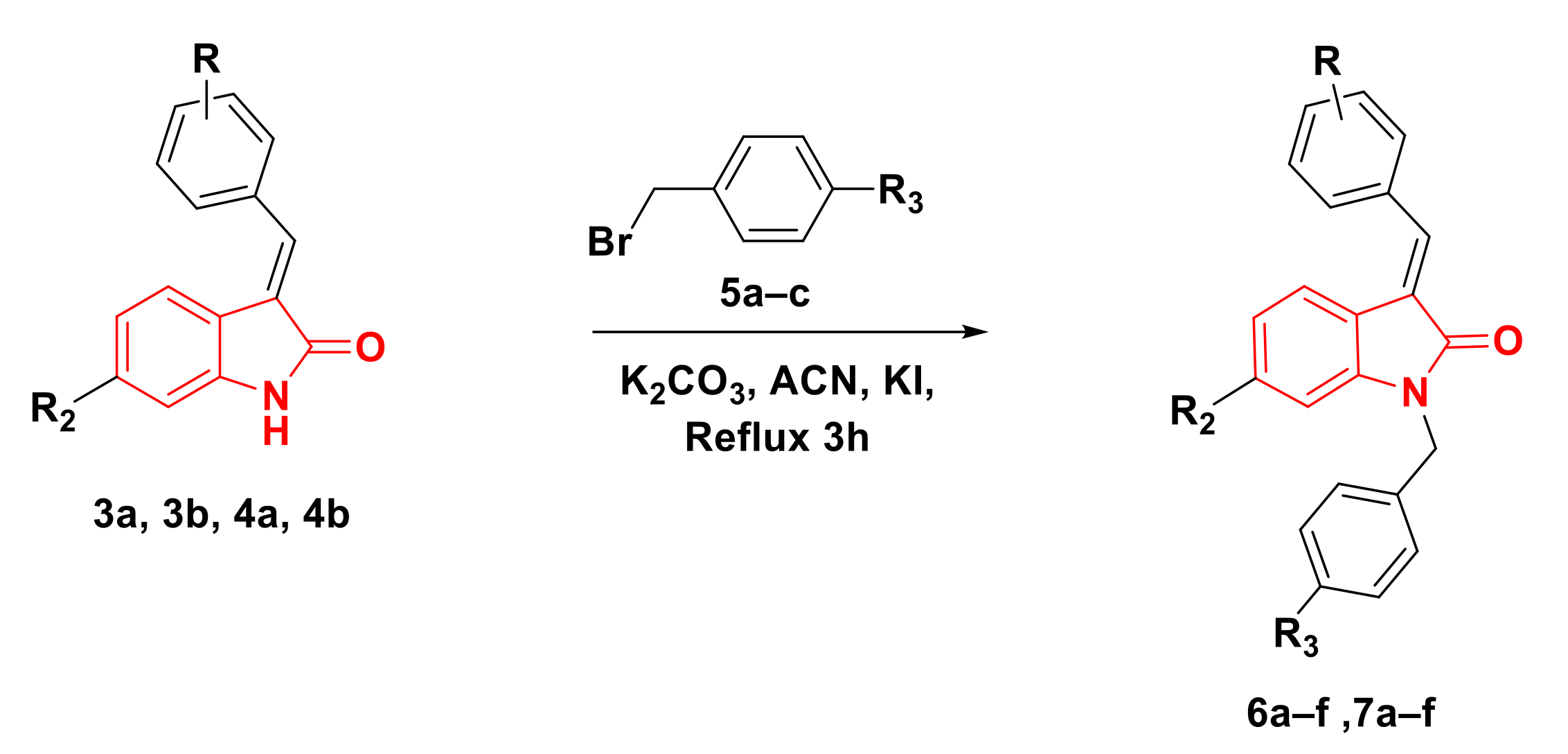
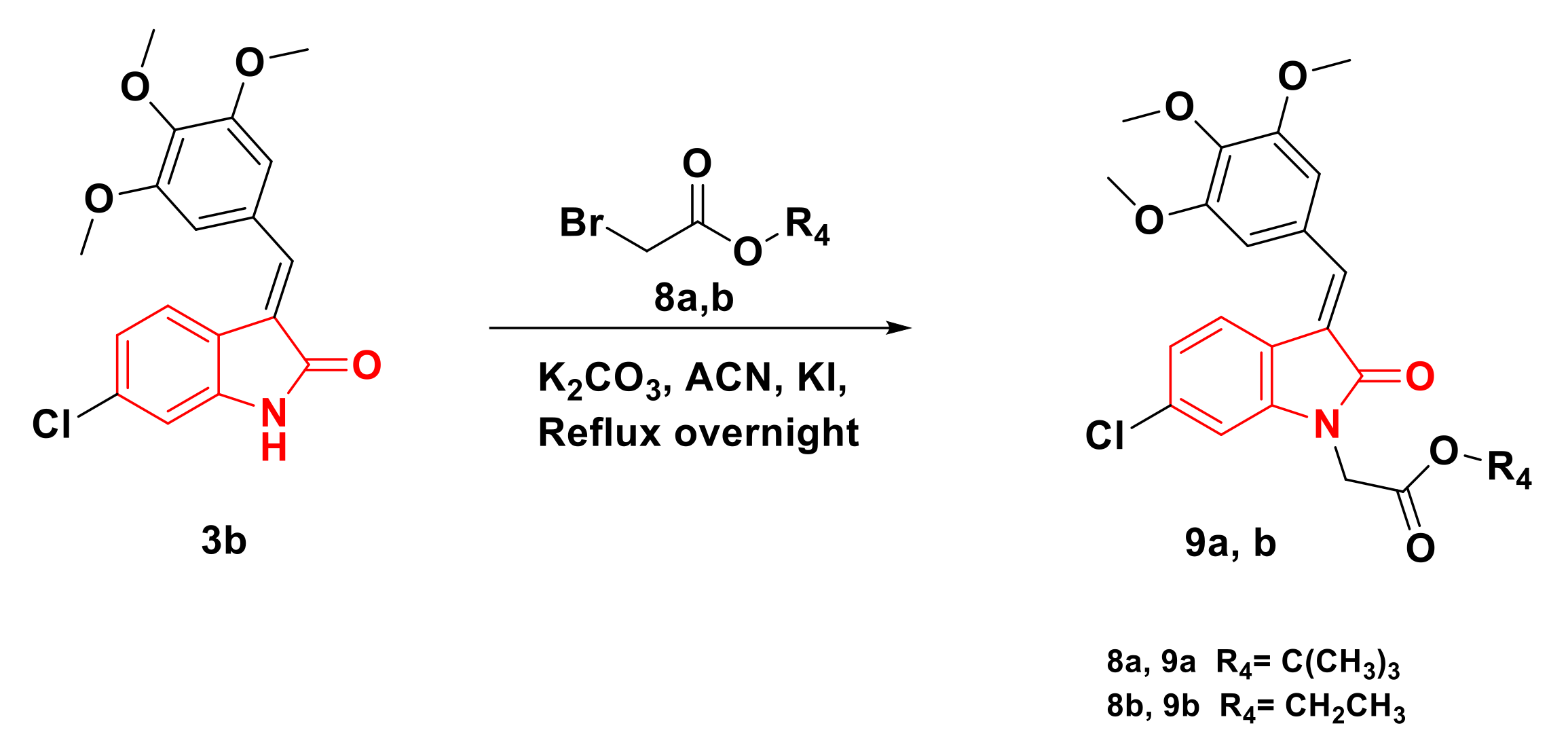
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. In Vitro Screening against COLO 205, MCF-7 and PC-3 Cell Lines
2.2.2. Antiproliferative Activity against COLO-205 and A459 Cell Lines
2.2.3. Assay of Tubulin Polymerization Inhibitory Activity
2.2.4. Inhibition of EGFR Activity
2.2.5. Inhibition of Casein Kinase (CK) Activity
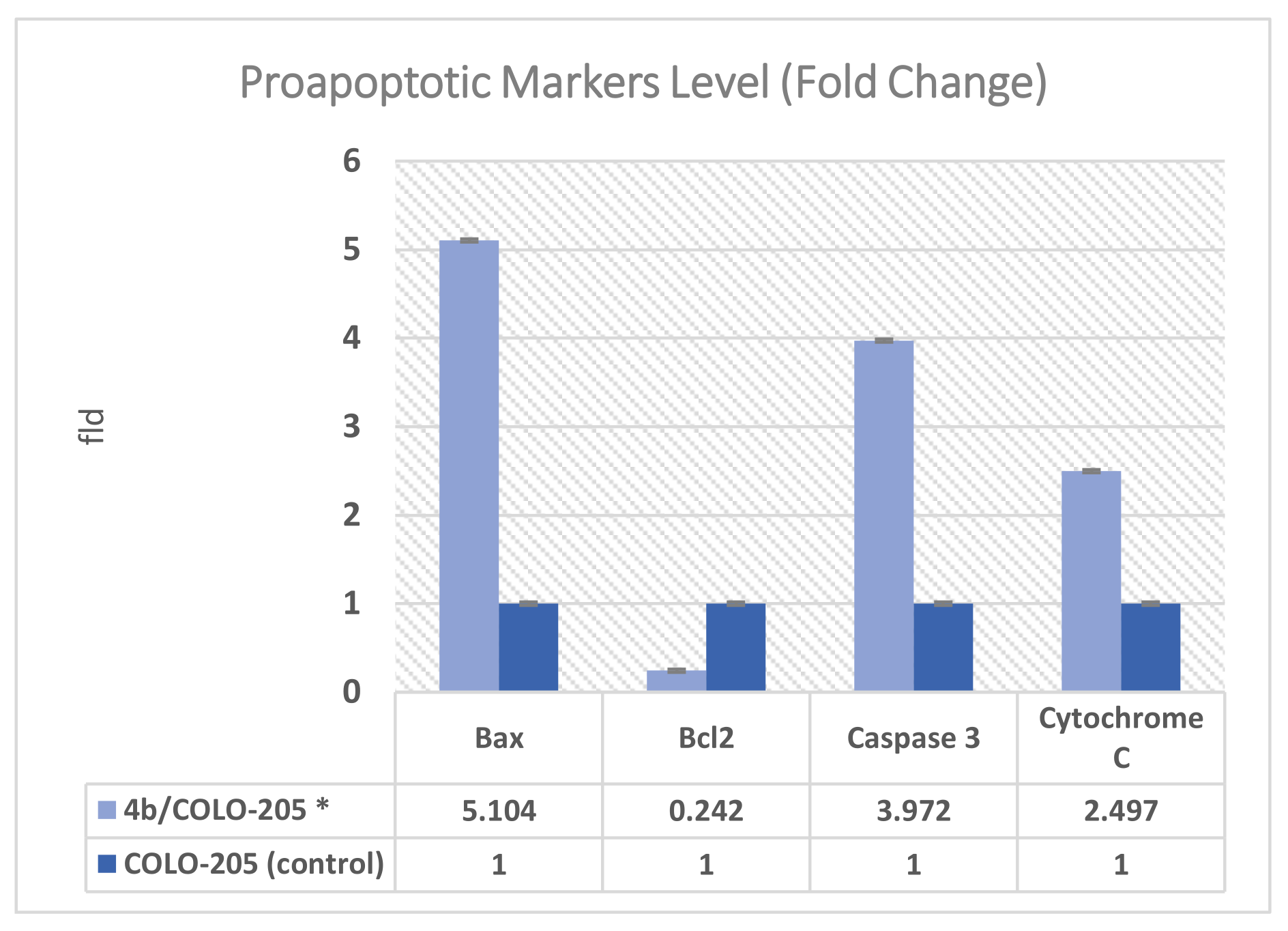
2.2.6. Effect on Bax, Bcl-2, Caspase 3 and Cytochrome C Expression Levels
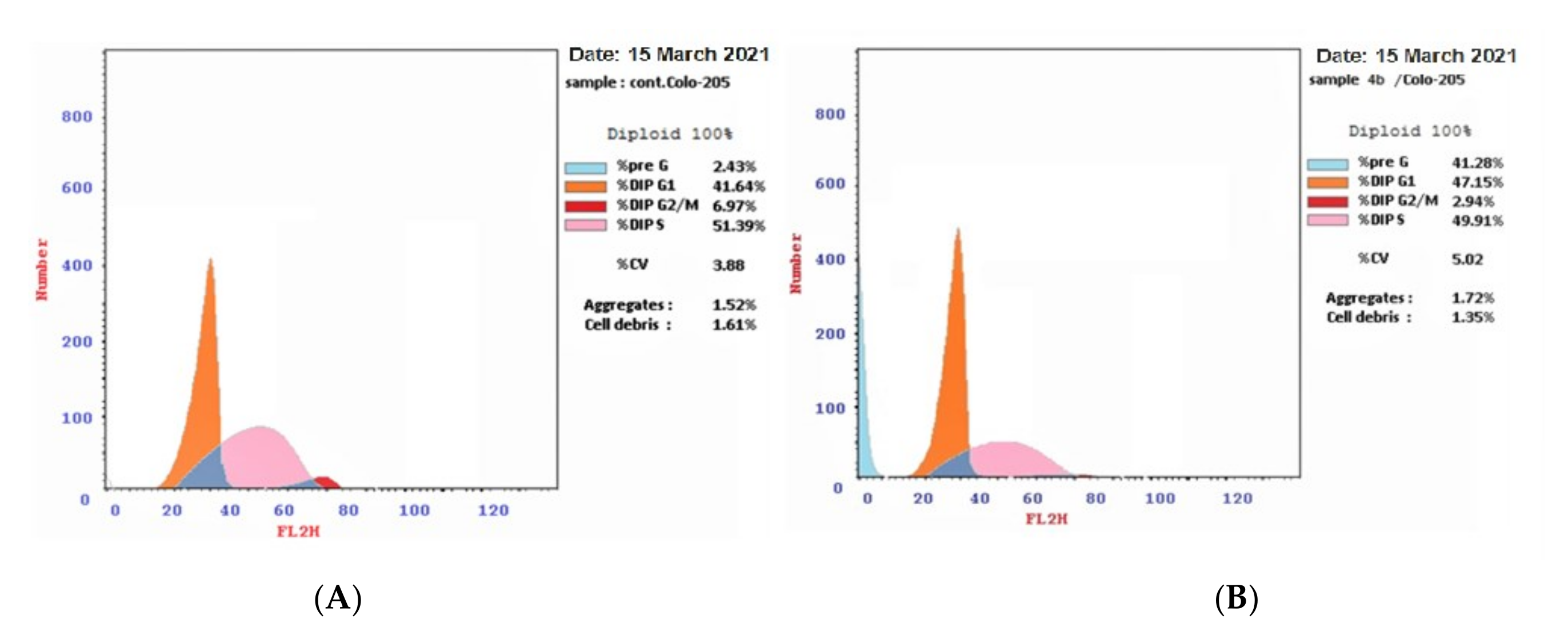
2.2.7. Cell Cycle Analysis
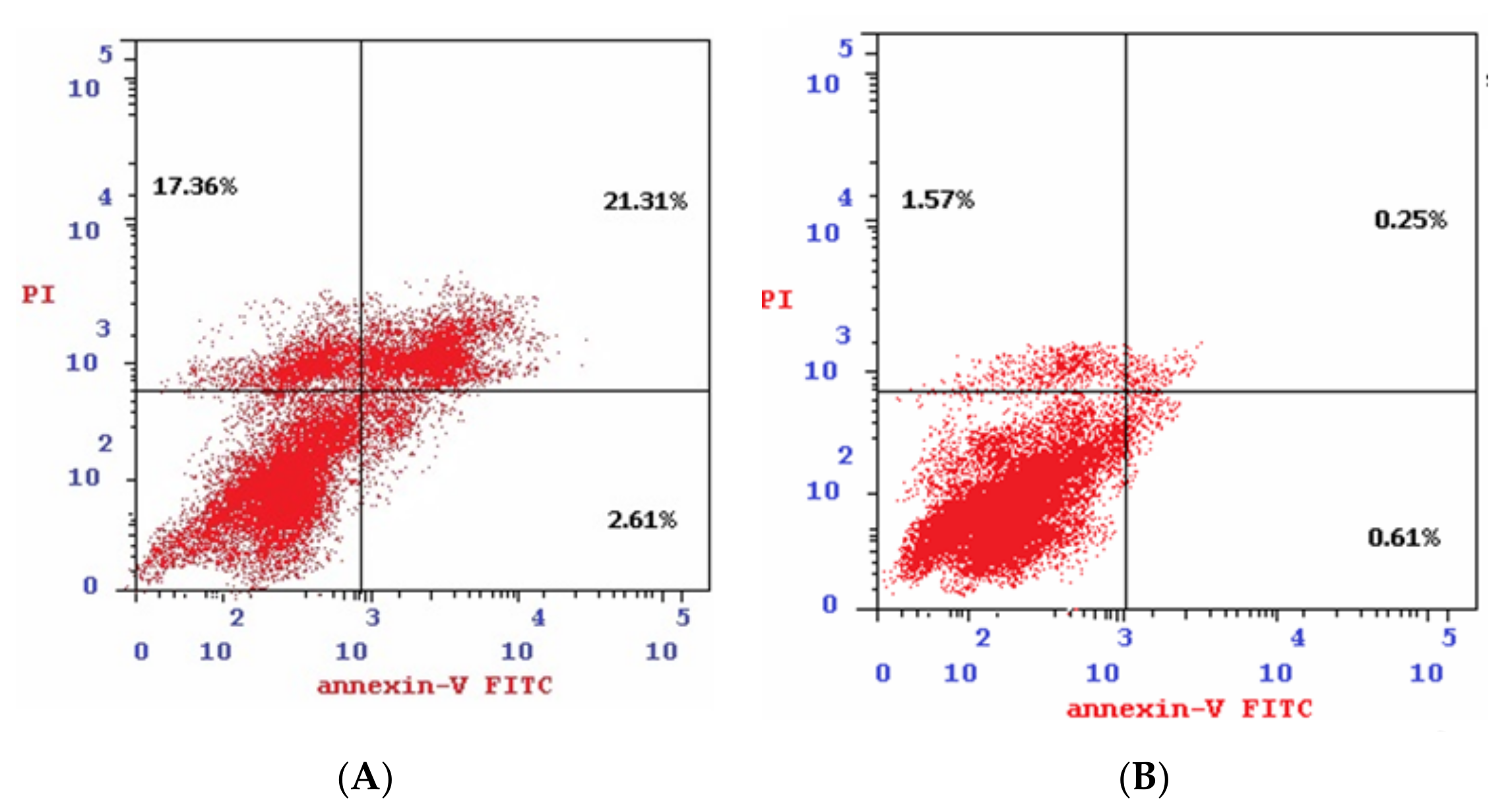
2.2.8. Annexin V-FITC Apoptosis Determination
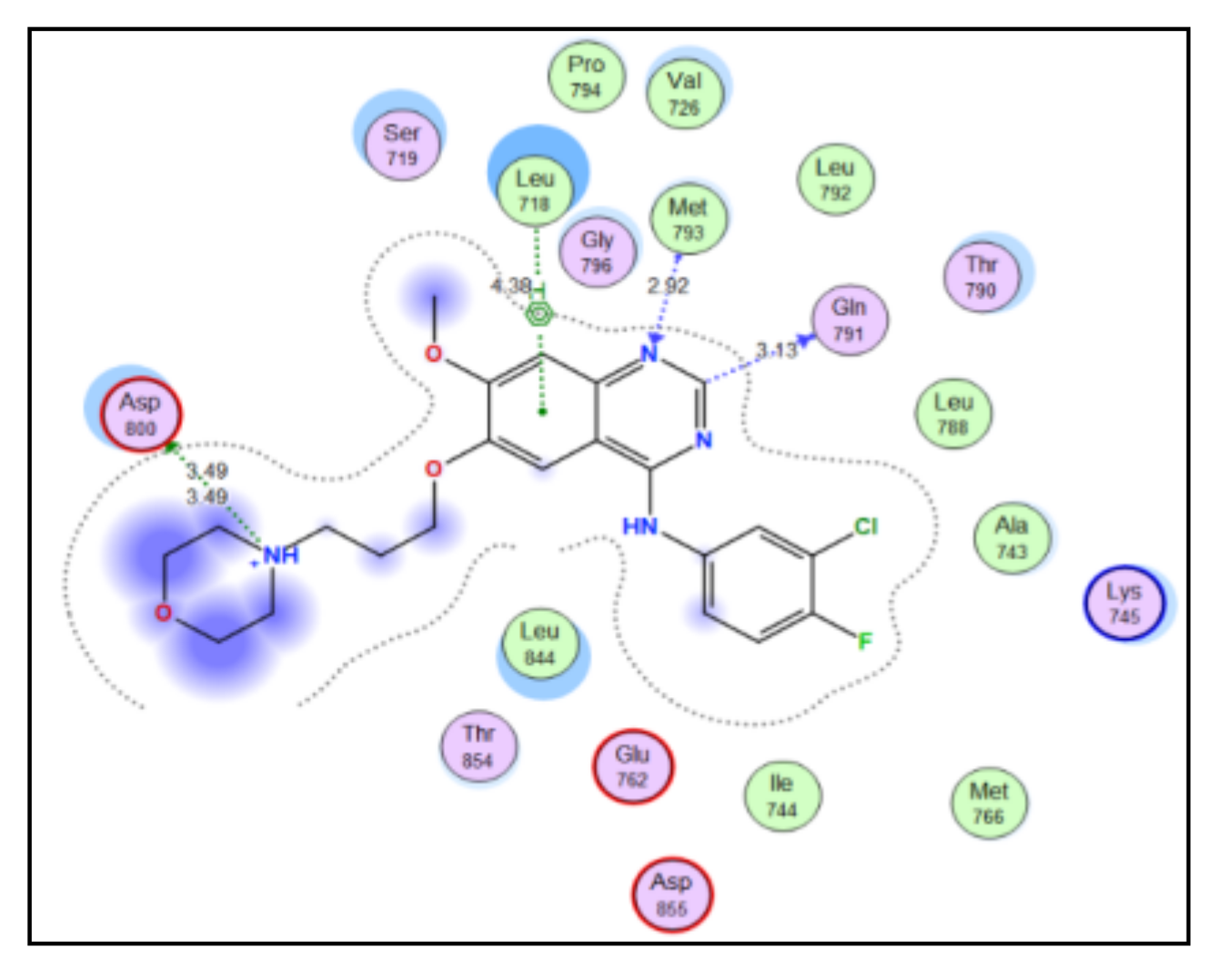
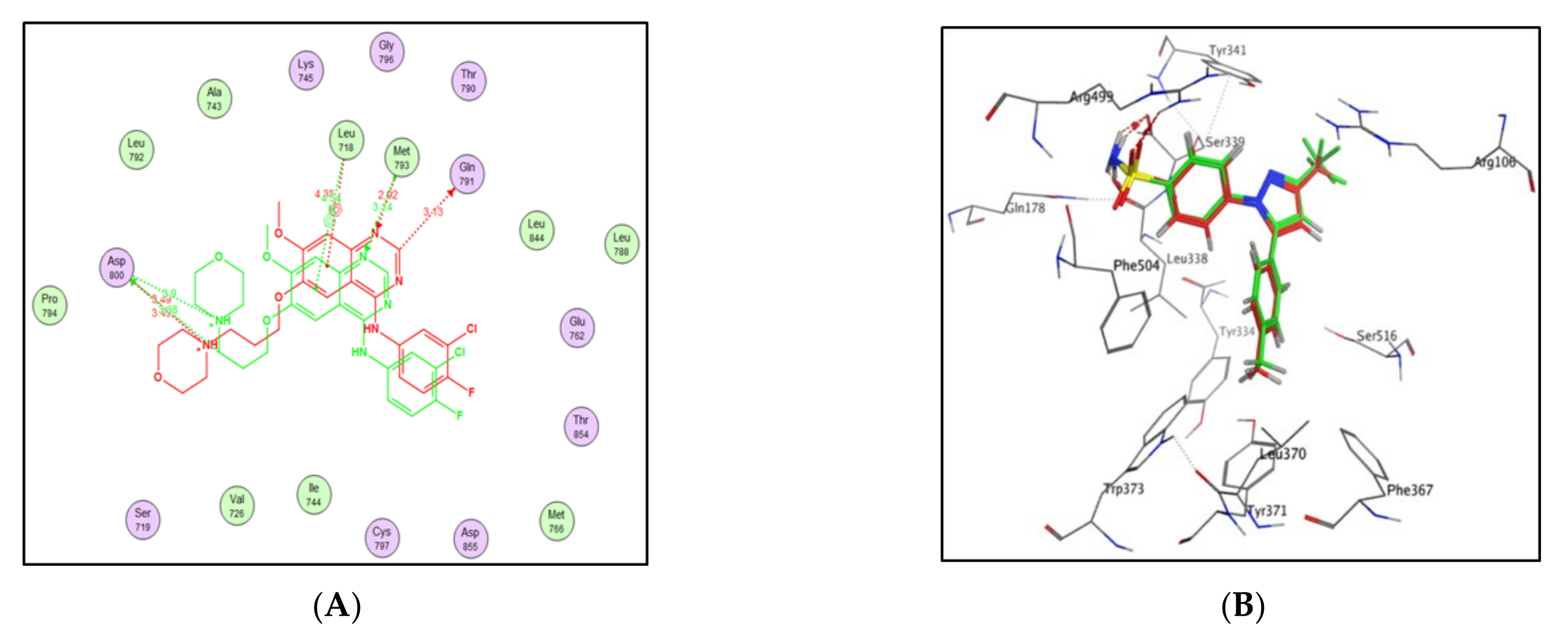
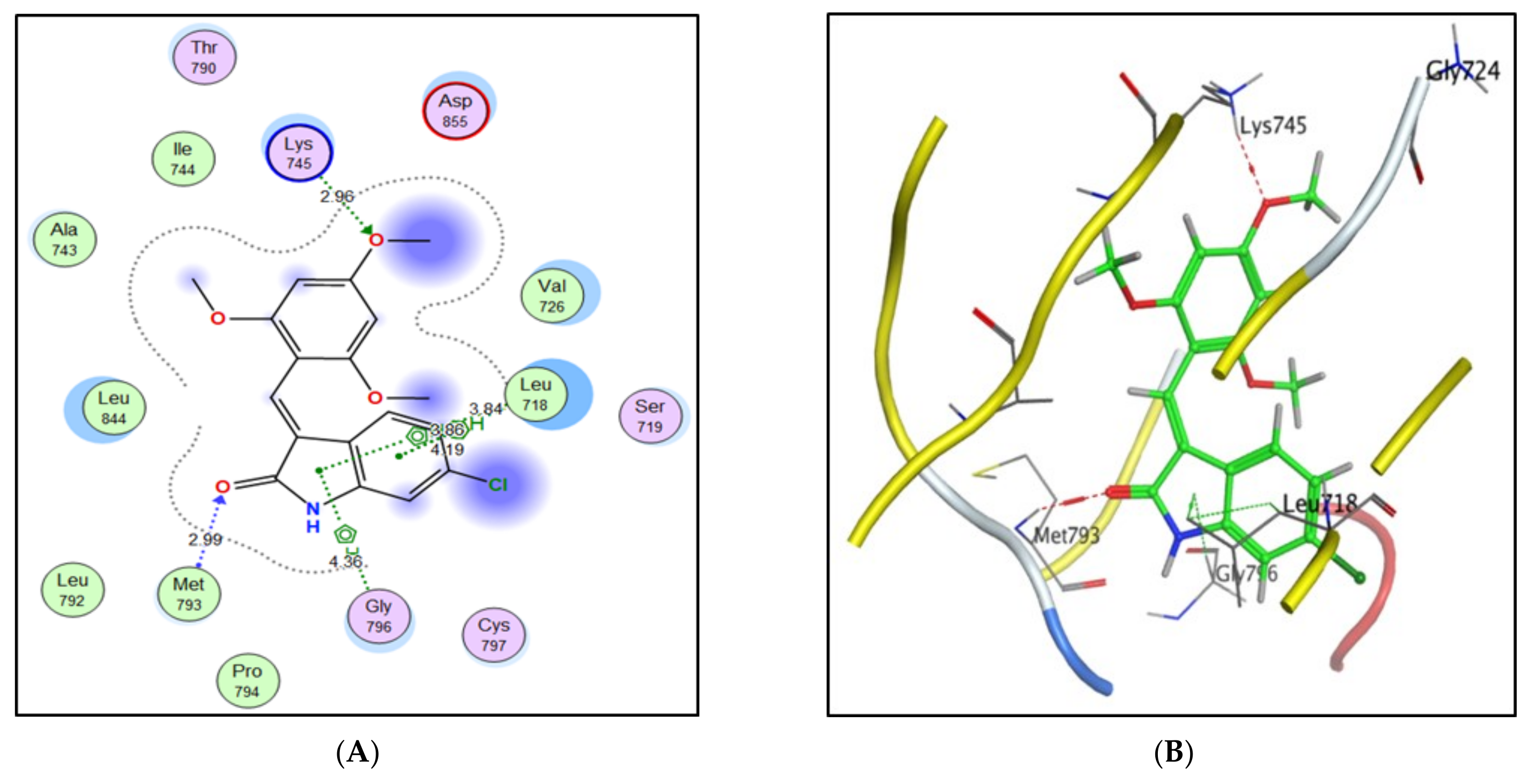
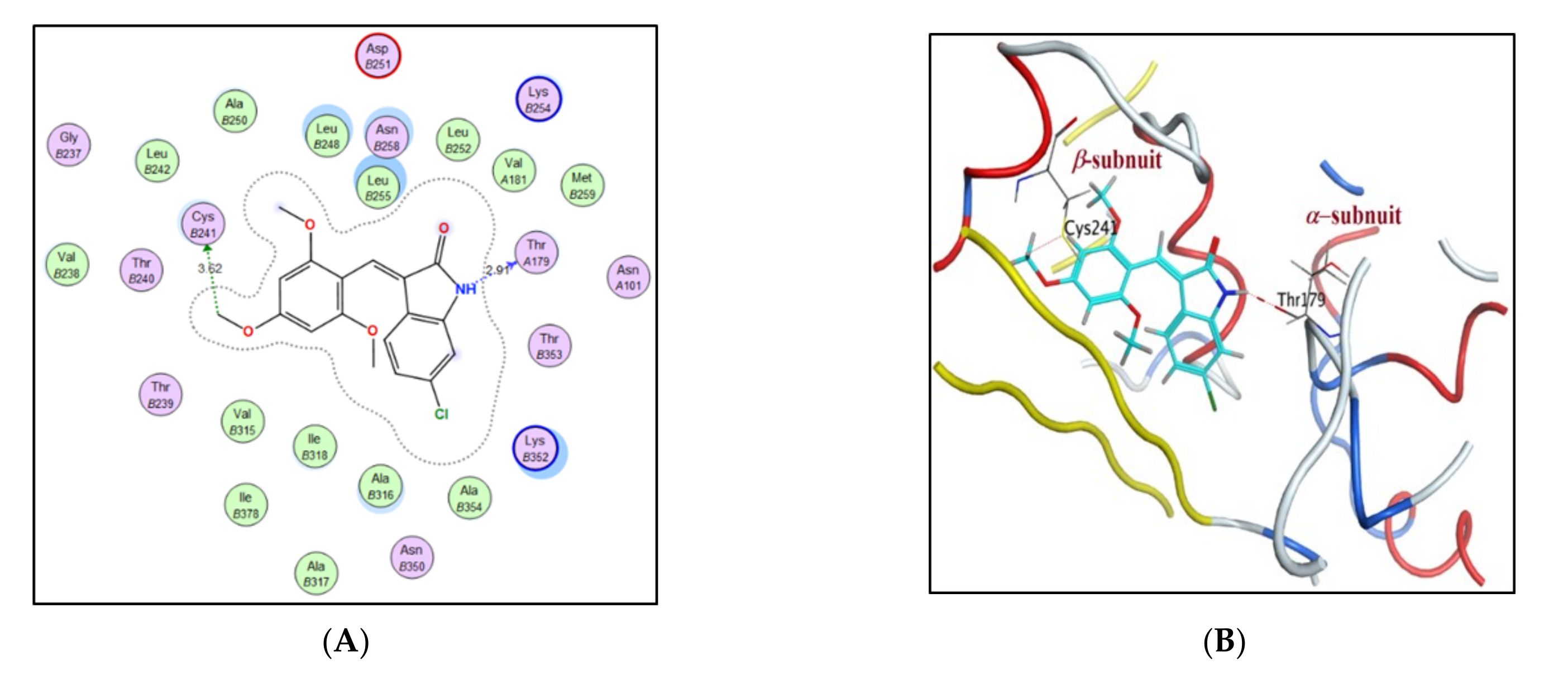
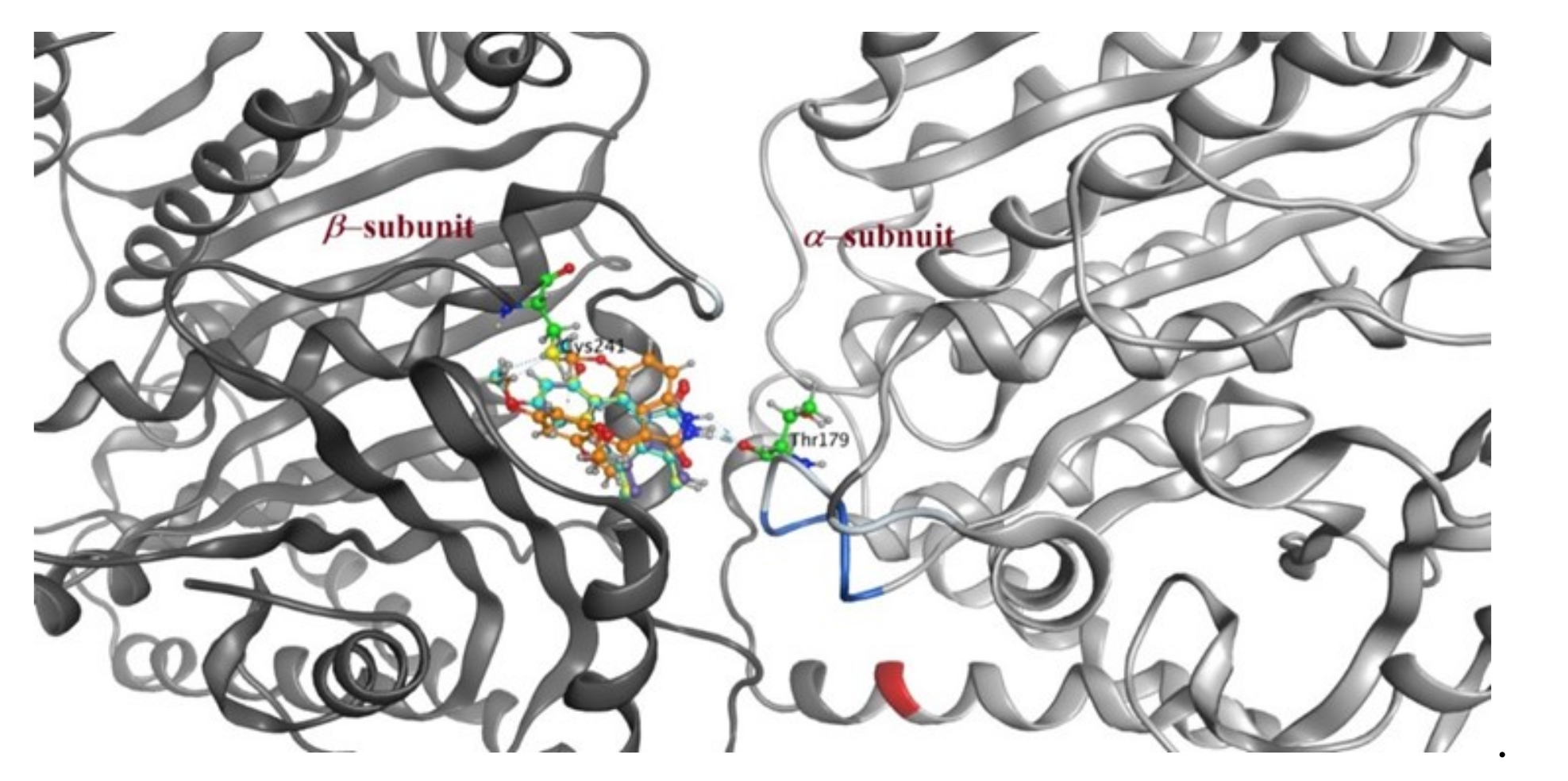
2.3. Molecular Docking Study
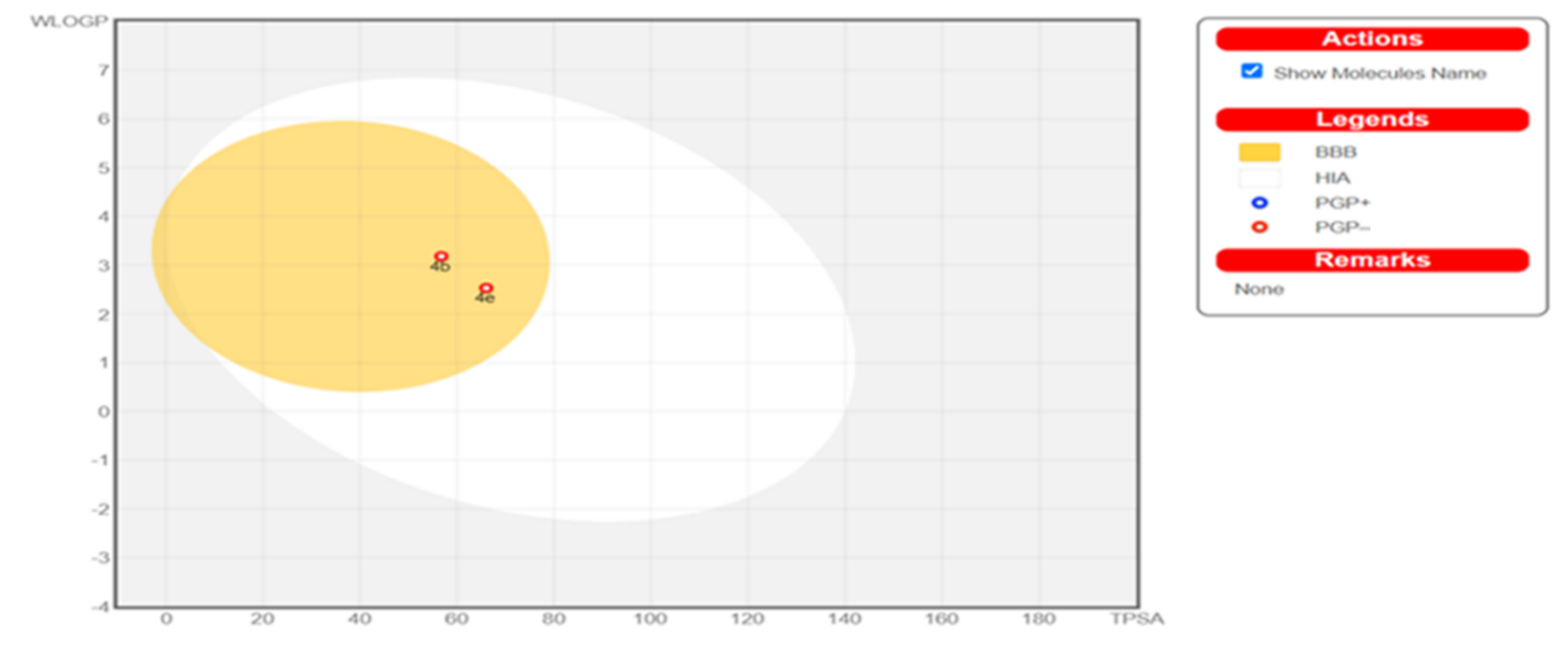
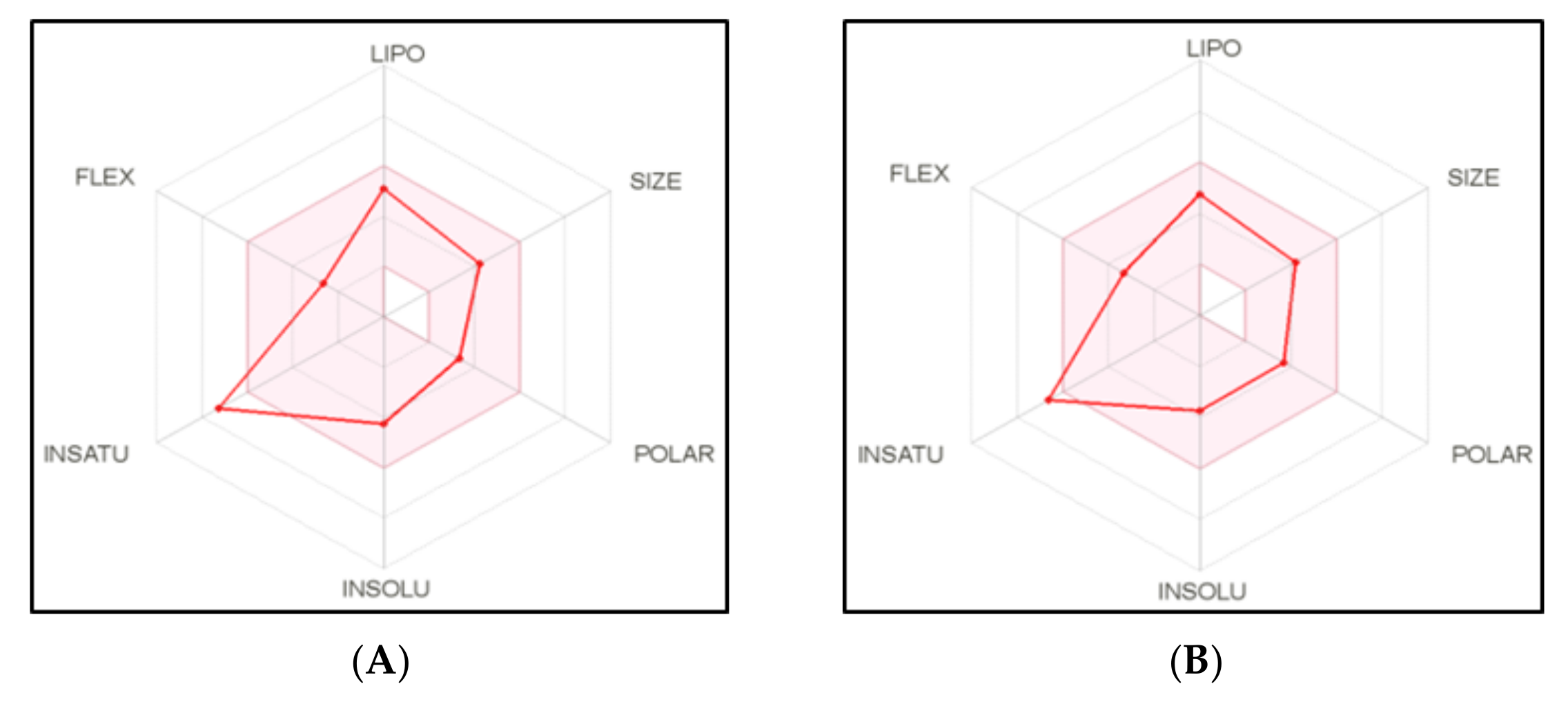
2.4. Physicochemical, ADME and Pharmacokinetic Properties Prediction
2.5. Pgp Permeability Assay
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Procedure for the Synthesis of Compounds 3a–f and 4a–f
- Synthesis of (E)-3-(3,4,5-trimethoxybenzylidene)indolin-2-one (3a) [54].
- Synthesis of (E)-6-chloro-3-(3,4,5-trimethoxybenzylidene)indolin-2-one (3b) [42].
- Synthesis of (E)-5-chloro-3-(3,4,5-trimethoxybenzylidene)indolin-2-one (3c) [54].
- Synthesis of (E)-5-fluoro-3-(3,4,5-trimethoxybenzylidene)indolin-2-one (3d) [54].
- Synthesis of (E)-5-methoxy-3-(3,4,5-trimethoxybenzylidene)indolin-2-one (3e) [54].
- Synthesis of (E)-3-(3,4,5-trimethoxybenzylidene)-1,3-dihydro-2H-pyrrolo[2,3-b] pyridin-2-one (3f) [54].
- Synthesis of (E)-3-(2,4,6-trimethoxybenzylidene)indolin-2-one (4a, IC261).
- Synthesis of (E)-6-chloro-3-(2,4,6-trimethoxybenzylidene)indolin-2-one (4b).
- Synthesis of (E)-5-chloro-3-(2,4,6-trimethoxybenzylidene)indolin-2-one (4c).
- Synthesis of (E)-5-fluoro-3-(2,4,6-trimethoxybenzylidene)indolin-2-one (4d).
- Synthesis of (E)-5-methoxy-3-(2,4,6-trimethoxybenzylidene)indolin-2-one (4e).
- Synthesis of (E)-3-(2,4,6-trimethoxybenzylidene)-1,3-dihydro-2H-pyrrolo[2,3-b] pyridin-2-one (4f) [77].
4.1.2. General Procedures for the Synthesis of Compounds 6a–f, 7a–f
- Synthesis of (E)-1-Benzyl-3-(3,4,5-trimethoxybenzylidene)indolin-2-one (6a).
- Synthesis of (E)-1-(4-Chlorobenzyl)-3-(3,4,5-trimethoxybenzylidene)indolin-2-one (6b) [78].
- Synthesis of (E)-1-(4-Fluorobenzyl)-3-(3,4,5-trimethoxybenzylidene)indolin-2-one (6c).
- Synthesis of (E)-1-benzyl-6-chloro-3-(3,4,5-trimethoxybenzylidene)indolin-2-one (6d).
- Synthesis of (E)-6-chloro-1-(4-chlorobenzyl)-3-(3,4,5-trimethoxybenzylidene) indolin-2-one (6e).
- Synthesis of (E)-6-chloro-1-(4-fluorobenzyl)-3-(3,4,5-trimethoxybenzylidene) indolin-2-one (6f).
- Synthesis of (E)-1-benzyl-3-(2,4,6-trimethoxybenzylidene)indolin-2-one (7a).
- Synthesis of (E)-1-(4-chlorobenzyl)-3-(2,4,6-trimethoxybenzylidene)indolin-2-one (7b).
- Synthesis of (E)-1-(4-fluorobenzyl)-3-(2,4,6-trimethoxybenzylidene)indolin-2-one (7c).
- Synthesis of (E)-1-benzyl-6-chloro-3-(2,4,6-trimethoxybenzylidene)indolin-2-one (7d).
- Synthesis of (E)-6-chloro-1-(4-chlorobenzyl)-3-(2,4,6-trimethoxybenzylidene) indolin-2-one (7e).
- Synthesis of (E)-6-chloro-1-(4-fluorobenzyl)-3-(2,4,6-trimethoxybenzylidene) indolin-2-one (7f).
4.1.3. General Procedures for Synthesizing Compounds 9a,b
- Synthesis of t-Butyl (E)-2-(6-chloro-2-oxo-3-(3,4,5-trimethoxybenzylidene) indolin-1-yl)acetate (9a).
- Synthesis of Ethyl (E)-2-(6-chloro-2-oxo-3-(3,4,5-trimethoxybenzylidene) indolin-1-yl)acetate (9b).
4.2. Biological Evaluation
4.2.1. Two Dose Antiproliferative Activity Screening
- a.
- Cell culture [79]
- b.
- Cytotoxicity assay [79]
4.2.2. Antiproliferative Activity against COLO-205 and A549 Cell Lines
4.2.3. Tubulin Polymerization Inhibitory Activity
4.2.4. Inhibition of EGFR Activity
4.2.5. Inhibition of Casein Kinase (CK) Activity
4.2.6. Cell Cycle Analysis
4.2.7. Annexin V-FITC Assay
4.2.8. Bax, Bcl-2, Caspase 3 and Cytochrome C level Assay
- a.
- Caspase 3 activation assay
- b.
- Bax and Bcl-2 assay
- c.
- Cytochrome c assay
4.3. Docking Study
4.4. Physicochemical, ADME and Pharmacokinetic Properties Prediction
4.5. PgP Permeability Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Ferlay, J. Global Cancer Statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Kassab, A.E.; Gedawy, E.M.; Hamed, M.I.A.; Doghish, A.S.; Hassan, R.A. Design, synthesis, anticancer evaluation, and molecular modelling studies of novel tolmetin derivatives as potential VEGFR-2 inhibitors and apoptosis inducers. J. Enzyme Inhib. Med. Chem. 2021, 36, 922–939. [Google Scholar] [CrossRef]
- Nemr, M.T.M.; AboulMagd, A.M.; Hassan, H.M.; Hamed, A.A.; Hamed, M.I.A.; Elsaadi, M.T. Design, synthesis and mechanistic study of new benzenesulfonamide derivatives as anticancer and antimicrobial agentsviacarbonic anhydrase IX inhibition. RSC Adv. 2021, 11, 26241–26257. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, X.; Wang, T.; Xiao, J. Quinolone hybrids and their anti-cancer activities: An overview. Eur. J. Med. Chem. 2019, 165, 59–79. [Google Scholar] [CrossRef]
- Rashid, H.U.; Xu, Y.; Muhammad, Y.; Wang, L.; Jiang, J. Research advances on anticancer activities of matrine and its derivatives: An updated overview. Eur. J. Med. Chem. 2019, 161, 205–238. [Google Scholar] [CrossRef]
- Akhtar, J.; Khan, A.A.; Ali, Z.; Haider, R.; Shahar Yar, M. Structure-Activity relationship (SAR) study and design strategies of nitrogen-containing heterocyclic moieties for their anticancer activities. Eur. J. Med. Chem. 2017, 125, 143–189. [Google Scholar] [CrossRef]
- Fadeyi, O.O.; Adamson, S.T.; Myles, E.L.; Okoro, C.O. Novel fluorinated acridone derivatives. Part 1: Synthesis and evaluation as potential anticancer agents. Bioorg. Med. Chem. Lett. 2008, 18, 4172–4176. [Google Scholar] [CrossRef]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting Microtubules by Natural Agents for Cancer Therapy. Mol. Cancer Ther. 2014, 13, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Dhuguru, J.; Skouta, R. Role of indole scaffolds as pharmacophores in the development of anti-lung cancer agents. Molecules 2020, 25, 1615. [Google Scholar] [CrossRef]
- Föhr, K.J.; Knippschild, U.; Herkommer, A.; Fauler, M.; Peifer, C.; Georgieff, M.; Adolph, O. State-Dependent block of voltage-gated sodium channels by the casein-kinase 1 inhibitor IC261. Investig. New Drugs 2017, 35, 277–289. [Google Scholar] [CrossRef]
- Zidar, N.; Secci, D.; Tomašič, T.; Mašič, L.P.; Kikelj, D.; Passarella, D.; Argaez, A.N.G.; Hyeraci, M.; Dalla Via, L. Synthesis, Antiproliferative Effect, and Topoisomerase II Inhibitory Activity of 3-Methyl-2-phenyl-1 H-indoles. ACS Med. Chem. Lett. 2020, 11, 691–697. [Google Scholar] [CrossRef]
- Zhao, B.; He, T. Chidamide, a histone deacetylase inhibitor, functions as a tumor inhibitor by modulating the ratio of Bax/Bcl-2 and P21 in pancreatic cancer. Oncol. Rep. 2015, 33, 304–310. [Google Scholar] [CrossRef]
- Walczak, C.E. Microtubule dynamics and tubulin interacting proteins. Curr. Opin. Cell Biol. 2000, 12, 52–56. [Google Scholar] [CrossRef]
- Vindya, N.G.; Sharma, N.; Yadav, M.; Ethiraj, K.R. Tubulins-The Target for Anticancer Therapy. Curr. Top. Med. Chem. 2015, 15, 73–82. [Google Scholar] [CrossRef]
- Kanthou, C.; Greco, O.; Stratford, A.; Cook, I.; Knight, R.; Benzakour, O.; Tozer, G. The Tubulin-Binding Agent Combretastatin A-4-Phosphate Arrests Endothelial Cells in Mitosis and Induces Mitotic Cell Death. Am. J. Pathol. 2004, 165, 1401–1411. [Google Scholar] [CrossRef]
- Pettit, G.R.; Singh, S.B.; Boyd, M.R.; Hamel, E.; Pettit, R.K.; Schmidt, J.M. Antineoplastic Agents 291. Isolation and Synthesis of Combretastatins A-4. J. Med. Chem. 1995, 38, 1666–1672. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Sham, H.L. Discovery and development of antimitotic agents that inhibit tubulin polymerisation for the treatment of cancer. Expert Opin. Ther. Pat. 2002, 12, 1663–1702. [Google Scholar] [CrossRef]
- Tzogani, K.; van Hennik, P.; Walsh, I.; de Graeff, P.; Folin, A.; Sjöberg, J.; Salmonson, T.; Bergh, J.; Laane, E.; Ludwig, H.; et al. EMA Review of Panobinostat (Farydak) for the Treatment of Adult Patients with Relapsed and/or Refractory Multiple Myeloma. Oncologist 2018, 23, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Metzler, M.; Neumann, H.G. Epoxidation of the stilbene double bond, a major pathway in aminostilbene metabolism. Xenobiotica 1977, 7, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; Yang, F.; Guo, D.; Xu, J.; Jiang, M.; Liu, C.; Bao, K.; Wu, Y.; Zhang, W. Synthesis and biological evaluation of novel 3,4-diaryl-1,2,5-selenadiazol analogues of combretastatin A-4. Eur. J. Med. Chem. 2014, 87, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mahal, K.; Biersack, B.; Schruefer, S.; Resch, M.; Ficner, R.; Schobert, R.; Mueller, T. Combretastatin A-4 derived 5-(1-methyl-4-phenyl-imidazol-5-yl)indoles with superior cytotoxic and anti-vascular effects on chemoresistant cancer cells and tumors. Eur. J. Med. Chem. 2016, 118, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Brockway, O.; Dandavati, A.; Tzou, S.; Sjoholm, R.; Satam, V.; Westbrook, C.; Lee, M. A novel class of trans-methylpyrazoline analogs of combretastatins: Synthesis and in-vitro biological testing. Eur. J. Med. Chem. 2011, 46, 3099–3104. [Google Scholar] [CrossRef]
- Kumar, D.; Patel, G.; Chavers, A.K.; Chang, K.H.; Shah, K. Synthesis of novel 1,2,4-oxadiazoles and analogues as potential anticancer agents. Eur. J. Med. Chem. 2011, 46, 3085–3092. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Cruz-Lopez, O.; Lopez Cara, C.; Carrion, M.D.; Brancale, A.; Hamel, E.; Chen, L.; Bortolozzi, R.; Basso, G.; et al. Synthesis and antitumor activity of 1,5-disubstituted 1,2,4-triazoles as cis-restricted combretastatin analogues. J. Med. Chem. 2010, 53, 4248–4258. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, M.; Shaik, T.B.; Malik, M.S.; Syed, R.; Mallipeddi, P.L.; Vardhan, M.V.P.S.V.; Kamal, A. Design and synthesis of cis-restricted benzimidazole and benzothiazole mimics of combretastatin A-4 as antimitotic agents with apoptosis inducing ability. Bioorg. Med. Chem. Lett. 2016, 26, 4527–4535. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Davis, R.; Vanderham, J.; Hills, P.; Mackay, H.; Brown, T.; Mooberry, S.L.; Lee, M. 1,2,3,4-Tetrahydro-2-thioxopyrimidine analogs of combretastatin-A4. Eur. J. Med. Chem. 2008, 43, 2011–2015. [Google Scholar] [CrossRef]
- Zhou, P.; Liang, Y.; Zhang, H.; Jiang, H.; Feng, K.; Xu, P.; Wang, J.; Wang, X.; Ding, K.; Luo, C.; et al. Design, synthesis, biological evaluation and cocrystal structures with tubulin of chiral β-lactam bridged combretastatin A-4 analogues as potent antitumor agents. Eur. J. Med. Chem. 2018, 144, 817–842. [Google Scholar] [CrossRef]
- Zhang, M.; Liang, Y.R.; Li, H.; Liu, M.M.; Wang, Y. Design, synthesis, and biological evaluation of hydantoin bridged analogues of combretastatin A-4 as potential anticancer agents. Bioorg. Med. Chem. 2017, 25, 6623–6634. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S. Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, S21–S26. [Google Scholar] [CrossRef]
- Singh, D.; Attri, B.K.; Gill, R.K.; Bariwal, J. Review on EGFR Inhibitors: Critical Updates. Mini Rev. Med. Chem. 2016, 16, 1134–1166. [Google Scholar] [CrossRef]
- Greig, S.L. Osimertinib: First Global Approval. Drugs 2016, 76, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Xiao, Z.; Qi, J.; Luo, R.; Lan, Z.; Zhang, Y.; Hu, X.; Tang, Q.; Zheng, P.; Xu, S.; et al. Design, synthesis and biological evaluation of AZD9291 derivatives as selective and potent EGFRL858R/T790M inhibitors. Eur. J. Med. Chem. 2019, 163, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.K.; Virshup, D.M. Casein kinase 1: Complexity In the family. Int. J. Biochem. Cell Biol. 2011, 43, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Schittek, B.; Sinnberg, T. Biological functions of casein kinase 1 isoforms and putative roles in tumorigenesis. Mol. Cancer 2014, 13, 231. [Google Scholar] [CrossRef] [PubMed]
- Narasimamurthy, R.; Hunt, S.R.; Lu, Y.; Fustin, J.; Okamura, H.; Partch, C.L. CK1 δ/e protein kinase primes the PER2 circadian phosphoswitch. Proc. Natl. Acad. Sci. USA 2018, 115, 5986–5991. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jiang, S.; Li, X.; Liu, Y.; Su, J.; Chen, J. Recent advances in trimethoxyphenyl (TMP) based tubulin inhibitors targeting the colchicine binding site. Eur. J. Med. Chem. 2018, 151, 482–494. [Google Scholar] [CrossRef]
- Brockschmidt, C.; Hirner, H.; Huber, N.; Eismann, H.; Hillenbrand, A.; Giamas, G.; Radunsky, B.; Ammerpohl, O.; Bohm, B.; Henne-Bruns, D.; et al. Anti-Apoptotic and growth-stimulatory functions of CK1 delta and epsilon in ductal adenocarcinoma of the pancreas are inhibited by IC261 in vitro and in vivo. Gut 2008, 57, 799–806. [Google Scholar] [CrossRef]
- Cheong, J.K.; Hung, N.T.; Wang, H.; Tan, P.; Voorhoeve, P.M.; Lee, S.H.; Virshup, D.M. IC261 induces cell cycle arrest and apoptosis of human cancer cells via CK1 δ/ε and Wnt/β-catenin independent inhibition of mitotic spindle formation. Oncogene 2011, 30, 2558–2596. [Google Scholar] [CrossRef]
- Herbst, R.S.; Oh, Y.; Wagle, A.; Lahn, M. Enzastaurin, a protein kinase Cβ-selective inhibitor, and its potential application as an anticancer agent in lung cancer. Clin. Cancer Res. 2007, 13, 4641–4646. [Google Scholar] [CrossRef]
- Espinoza-Fonseca, L.M. The benefits of the multi-target approach in drug design and discovery. Artif. Intell. Life Sci. 2006, 14, 896–897. [Google Scholar] [CrossRef]
- Nasir, S.; Bukhari, A.; Bharath, G.; Mohan, H.; Qin, H. Bioorganic Chemistry Development of combretastatins as potent tubulin polymerization inhibitors. Bioorg. Chem. 2017, 72, 130–147. [Google Scholar] [CrossRef]
- Andreani, A.; Burnelli, S.; Granaiola, M.; Leoni, A.; Locatelli, A.; Morigi, R.; Rambaldi, M.; Varoli, L.; Kunkel, M.W. Antitumor Activity of Substituted E-3-(3,4,5-Trimethoxybenzylidene)-1,3-dihydroindol-2-ones. J. Med. Chem. 2006, 49, 6922–6924. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Li, D.; Guo, M.; Fang, T.; Sun, J.; Qi, F.; Rao, Q.; Zhao, Z.; Huang, P.; Yang, B.; et al. IC261 suppresses progression of hepatocellular carcinoma in a casein kinase 1 δ/ε independent manner. Biochem. Biophys. Res. Commun. 2020, 523, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Utsuro, M. Neutron spin interference visibility in tunneling transmission through magnetic resonators. Phys. B Condens. Matter 2005, 358, 232–246. [Google Scholar] [CrossRef]
- Andree, H.A.M.; Reutelingsperger, C.P.M.; Hauptmann, R.; Hemker, H.C.; Hermens, W.T.; Willems, G.M. Binding of vascular anticoagulant α (VACα) to planar phospholipid bilayers. J. Biol. Chem. 1990, 265, 4923–4928. [Google Scholar] [CrossRef]
- Demchenko, A.P. Beyond annexin V: Fluorescence response of cellular membranes to apoptosis. Cytotechnology 2013, 65, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery 1 A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Szeliga, M. Thiadiazole derivatives as anticancer agents. Pharmacol. Rep. 2020, 72, 1079–1100. [Google Scholar] [CrossRef] [PubMed]
- Zdioruk, M.; Want, A.; Mietelska-Porowska, A.; Laskowska-Kaszub, K.; Wojsiat, J.; Klejman, A.; Użarowska, E.; Koza, P.; Olejniczak, S.; Pikul, S.; et al. A new inhibitor of tubulin polymerization kills multiple cancer cell types and reveals p21-mediated mechanism determining cell death after mitotic catastrophe. Cancers 2020, 12, 2161. [Google Scholar] [CrossRef]
- Andreani, A.; Granaiola, M.; Locatelli, A.; Morigi, R.; Rambaldi, M.; Varoli, L.; Vieceli Dalla Sega, F.; Prata, C.; Nguyen, T.L.; Bai, R.; et al. Cytotoxic activities of substituted 3-(3,4,5-trimethoxybenzylidene)-1,3-dihydroindol-2-ones and studies on their mechanisms of action. Eur. J. Med. Chem. 2013, 64, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Arnst, K.E.; Banerjee, S.; Chen, H.; Deng, S.; Hwang, D.-J.; Li, W.; Miller, D.D. Current advances of tubulin inhibitors as dual acting small molecules for cancer therapy. Med. Res. Rev. 2020, 39, 1398–1426. [Google Scholar] [CrossRef]
- Behrend, L.; Milne, D.; Stöter, M.; Deppert, W.; Campbell, L.E.; Meek, D.W.; Knippschild, U. IC261, a specific inhibitor of the protein kinases casein kinase 1-delta and -epsilon, triggers the mitotic checkpoint and induces p53-dependent postmitotic effects. Oncogene. 2000, 19, 5303–5313. [Google Scholar] [CrossRef] [PubMed]
- Stöter, M.; Krüger, M.; Banting, G.; Henne-Bruns, D.; Knippschild, U. Microtubules depolymerization caused by the CK1 inhibitor IC261 may be not mediated by CK1 blockage. PLoS ONE 2014, 9, e100090. [Google Scholar] [CrossRef]
- Xian, J.; Bu, F.; Wang, Y.; Long, F.; Zhang, Z.; Wu, C.; Tao, Y.; Wang, T.; Wang, G. A Rationale for Drug Design Provided by Co-Crystal Structure of IC261 in Complex with Tubulin. Molecules 2021, 26, 946. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, A.; Katayama, R.; Oh-Hara, T.; Sato, S.; Okuno, Y.; Fujita, N. Tivantinib (ARQ 197) exhibits antitumor activity by directly interacting with tubulin and overcomes ABC transporter-mediated drug resistance. Mol. Cancer Ther. 2014, 13, 2978–2990. [Google Scholar] [CrossRef]
- Smolinski, M.P.; Bu, Y.; Clements, J.; Gelman, I.H.; Hegab, T.; Cutler, D.L.; Fang, J.W.S.; Fetterly, G.; Kwan, R.; Barnett, A.; et al. Discovery of Novel Dual Mechanism of Action Src Signaling and Tubulin Polymerization Inhibitors (KX2-391 and KX2-361). J. Med. Chem. 2018, 61, 4704–4719. [Google Scholar] [CrossRef] [PubMed]
- Soltan, O.M.; Shoman, M.E.; Abdel-Aziz, S.A.; Narumi, A.; Konno, H.; Abdel-Aziz, M. Molecular hybrids: A five-year survey on structures of multiple targeted hybrids of protein kinase inhibitors for cancer therapy. Eur. J. Med. Chem. 2021, 225, 113768. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K. Microtubule Depolymerization by Kinase Inhibitors: Unexpected Findings of Dual Inhibitors. Int. J. Mol. Sci. 2017, 18, 2508. [Google Scholar] [CrossRef]
- Wu, X.; Sooman, L.; Lennartsson, J.; Bergström, S.; Bergqvist, M.; Gullbo, J.; Ekman, S. Microtubule inhibition causes epidermal growth factor receptor inactivation in oesophageal cancer cells. Int. J. Oncol. 2013, 6, 297–304. [Google Scholar] [CrossRef]
- Mphahlele, M.J.; Maluleka, M.M.; Parbhoo, N.; Malindisa, S.T. Synthesis, Evaluation for Cytotoxicity and Molecular Docking Studies of Benzo[c]furan-Chalcones for Potential to Inhibit Tubulin Polymerization and/or EGFR-Tyrosine Kinase Phosphorylation. Int. J. Mol. Sci. 2018, 19, 2552. [Google Scholar] [CrossRef]
- Zayed, M.F.; Ahmed, S.; Ihmaid, S.; Ahmed, H.E.A.; Rateb, H.S.; Ibrahim, S.R.M. Design, Synthesis, Cytotoxic Evaluation and Molecular Docking of New Fluoroquinazolinones as Potent Anticancer Agents with Dual EGFR Kinase and Tubulin Polymerization Inhibitory Effects. Int. J. Mol. Sci. 2018, 19, 1731. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Prencipe, F.; Oliva, P.; Baraldi, S.; Baraldi, P.G.; Ortega, S.S.; Chayah, M.; Salvador, M.K.; Lopez-Cara, L.C.; Brancale, A.; et al. Design, Synthesis, and Biological Evaluation of 6-Substituted Thieno[3,2-d ]pyrimidine Analogues as Dual Epidermal Growth Factor Receptor Kinase and Microtubule Inhibitors. J. Med. Chem. 2019, 62, 1274–1290. [Google Scholar] [CrossRef]
- Aouad, M.R.; Al-Mohammadi, H.M.; Al-blewi, F.F.; Ihmaid, S.; Elbadawy, H.M.; Althagfan, S.S.; Rezki, N. Anticancer agents with Dual Epidermal Growth Factor Receptor Kinase and Microtubule Inhibitors. Bioorg. Chem. 2019, 94, 103446. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.P.; Deng, X.; May, W.S. Phosphorylation of Bcl2 and regulation of apoptosis. Leukemia 2001, 15, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.W.; Botchan, M.R.; Berger, J.M. Mechanisms and regulation of DNA replication initiation in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 107–144. [Google Scholar] [CrossRef]
- Pathan, N.; Aime-Sempe, C.; Kitada, S.; Haldar, S.; Reed, J.C. Microtubule-Targeting drugs induce Bcl-2 phosphorylation and association with Pin1. Neoplasia 2001, 3, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Haldar, S. Microtubule-Damaging drugs triggered Bcl2 phosphorylation-requirement of phosphorylation on both serine-70 and serine-87 residues of Bcl2 protein. Int. J. Oncol. 1998, 13, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Naseri, M.H.; Mahdavi, M.; Davoodi, J.; Tackallou, H.S.; Goudarzvand, M.; Neishabouri, S.H. Up regulation of Bax and down regulation of Bcl2 during 3-NC mediated apoptosis in human cancer cells. Cancer Cell Int. 2015, 15, 55. [Google Scholar] [CrossRef]
- Nguyen, T.; Li, J.-X.; Thomas, B.F.; Wiley, J.L.; Kenakin, T.P.; Zhang, Y. Allosteric Modulation: An Alternate Approach Targeting the Cannabinoid CB1 Receptor Thuy. Physiol. Behav. 2018, 176, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J. Caspases: Executioners of Apoptosis. Pathobiol. Hum. Dis. A Dyn. Encycl. Dis. Mech. 2014, 16, 145–152. [Google Scholar] [CrossRef]
- Sokolowski, J.D.; Gamage, K.K.; Heffron, D.S.; Leblanc, A.C.; Deppmann, C.D.; Mandell, J.W. Caspase-mediated cleavage of actin and tubulin is a common feature and sensitive marker of axonal degeneration in neural development and injury. Acta Neuropathol. Commun. 2014, 2, 16. [Google Scholar] [CrossRef]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Weng, Q.; Fu, L.; Wang, Z.; Yu, P.; Liu, Z.; Li, X.; Zhang, H.; Liang, G. Synthesis and biological evaluation of novel oxindole-based RTK inhibitors as anti-cancer agents. Bioorg. Med. Chem. 2014, 22, 6953–6960. [Google Scholar] [CrossRef] [PubMed]
- Biehl, E.A.; Ankati, H.; Kamila, S. Compounds and derivatives of 2H-pyrido (3.2-b)(1,4) oxazin3)4H)-ones as raf kinase and lrrk2 inhibitors. U.S. Patent US 2012/0245347 A1, 27 September 2012. [Google Scholar]
- Rédei, G.P. Chemosensitivity. Encycl. Genet. Genom. Proteom. Inform. 2008, 110, 326. [Google Scholar] [CrossRef]
- Promega Corporation. Measuring Luminescence of the Kinase-Glo® Luminescent Kinase Assay using the GloMax® Discover System. Available online: https://www.promega.com/-/media/files/resources/protocols/technical-bulletins/101/kinase-glo-luminescent-kinase-assay-platform-protocol.pdf?la=en (accessed on 26 October 2021).
- Turky, A.; Bayoumi, A.H.; Ghiaty, A.; El-Azab, A.S.; Abdel-Aziz, A.A.-M.; Abulkhair, H.S. Design, synthesis, and antitumor activity of novel compounds based on 1,2,4-triazolophthalazine scaffold: Apoptosis-inductive and PCAF-inhibitory effects. Bioorg. Chem. 2020, 101, 104019. [Google Scholar] [CrossRef]
- Shum, J.; Leung, P.K.; Lo, K.K. Luminescent Ruthenium(II) Polypyridine Complexes for a Wide Variety of Biomolecular and Cellular Applications. Inorg. Chem. 2019, 58, 2231–2247. [Google Scholar] [CrossRef] [PubMed]
- Westermaier, M.; Mayr, H. Electrophilic Allylations and Benzylations of Indoles in Neutral Aqueous or Alcoholic Solutions. Org. Lett. 2006, 8, 4791–4794. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Number | Growth Inhibition (%) a | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MCF-7 | PC-3 | COLO-205 | ||||||||||
| 1 μM | 10 μM | 1 μM | 10 μM | 1 μM | 10 μM | |||||||
| Average | SEM | Average | SEM | Average | SEM | Average | SEM | Average | SEM | Average | SEM | |
| 3a | 1.17 | 0.17 | 3.44 | 0.18 | 1.92 | 0.92 | 20.44 | 1.16 | 1.18 | 0.38 | 4.71 | 1.35 |
| 3b | 1.14 | 0.3 | 41.17 | 0.75 | 7.69 | 1.33 | 38.99 | 0.44 | 5.70 | 0.80 | 32.58 | 1.32 |
| 3c | 1.06 | 0.46 | 8.56 | 0.81 | 1.42 | 0.24 | 14.31 | 1.39 | 2.86 | 0.28 | 6.41 | 0.79 |
| 3d | 0.98 | 0.31 | 3.60 | 1.00 | 2.02 | 0.30 | 1.91 | 0.07 | 7.01 | 1.00 | 11.85 | 0.18 |
| 3e | 20.98 | 0.76 | 65.3 b | 0.67 | 32.94 | 0.07 | 61.38 | 0.69 | 2.34 | 0.33 | 18.61 | 1.52 |
| 3f | 0.36 | 0.13 | 4.53 | 0.42 | 1.29 | 0.31 | 3.57 | 0.41 | 1.52 | 0.38 | 9.13 | 0.40 |
| 4a | 43.49 c | 1.11 | 58.93 | 0.99 | 59.19 | 0.35 | 67.87 | 0.52 | 63.60 | 0.80 | 77.5 | 0.15 |
| 4b | 45.3 | 1.23 | 59.15 | 0.95 | 64.05 | 1.19 | 71.66 | 1.54 | 73.32 | 1.07 | 82.5 | 1.23 |
| 4c | 17.88 | 0.31 | 65.1 | 1.65 | 28.93 | 0.24 | 62.25 | 0.61 | 2.67 | 0.38 | 58.44 | 1.00 |
| 4d | 10.02 | 0.60 | 66.11 | 0.33 | 23.81 | 0.14 | 57.98 | 0.89 | 9.69 | 0.51 | 64.01 | 1.07 |
| 4e | 34.48 | 0.19 | 68.91 | 0.57 | 42.36 | 0.71 | 63.27 | 0.16 | 8.05 | 0.86 | 31.89 | 1.44 |
| 4f | 11.49 | 0.13 | 65.85 | 1.13 | 23.02 | 0.25 | 69.12 | 1.14 | 7.64 | 0.66 | 63.79 | 0.66 |
| 6a | 1.74 | 0.23 | 48.54 | 0.57 | 4.30 | 1.40 | 5.03 | 1.72 | 2.46 | 1.33 | 4.97 | 0.77 |
| 6b | 1.13 | 0.89 | 32.21 | 0.47 | 1.04 | 0.14 | 28.88 | 0.67 | 3.90 | 0.90 | 45.84 | 0.87 |
| 6c | 5.68 | 0.58 | 25.03 | 1.71 | 0.69 | 0.22 | 8.22 | 0.57 | 4.66 | 0.57 | 13.63 | 0.57 |
| 6d | 0.68 | 0.29 | 48.38 | 1.08 | 4.24 | 0.30 | 7.34 | 0.34 | 2.38 | 0.92 | 8.68 | 0.21 |
| 6e | 3.69 | 1.01 | 6.00 | 0.20 | 3.22 | 0.33 | 5.65 | 0.13 | 6.49 | 0.66 | 72.57 | 1.37 |
| 6f | 3.56 | 0.88 | 29.87 | 0.61 | 0.40 | 0.17 | 23.37 | 0.99 | 3.67 | 1.14 | 63.77 | 0.87 |
| 7a | 0.60 | 0.38 | 15.31 | 0.48 | 0.65 | 0.26 | 8.59 | 0.58 | 11.91 | 0.26 | 20.02 | 1.03 |
| 7b | 3.81 | 0.98 | 5.48 | 0.48 | 1.05 | 0.11 | 3.15 | 1.21 | 3.00 | 1.19 | 3.33 | 0.33 |
| 7c | 4.08 | 0.11 | 25.95 | 0.88 | 0.89 | 0.30 | 13.82 | 0.45 | 4.66 | 0.57 | 18.28 | 1.19 |
| 7d | 0.49 | 0.23 | 40.55 | 0.90 | 1.47 | 0.25 | 19.9 | 0.99 | 2.38 | 1.06 | 15.56 | 1.19 |
| 7e | 1.59 | 0.30 | 46.25 | 0.32 | 1.77 | 0.21 | 51.68 | 1.17 | 3.82 | 1.06 | 22.15 | 1.37 |
| 7f | 10.27 | 0.58 | 15.96 | 0.71 | 0.75 | 0.05 | 7.08 | 0.17 | 3.33 | 0.33 | 3.97 | 0.65 |
| 9a | 2.11 | 1.07 | 7.11 | 0.64 | 1.19 | 0.15 | 5.56 | 0.75 | 3.05 | 1.32 | 12.98 | 1.66 |
| 9b | 2.30 | 1.11 | 6.82 | 0.18 | 1.66 | 0.41 | 13.20 | 0.49 | 35.14 | 1.75 | 40.87 | 0.66 |
| Compound | Cell Line | 4a (IC261) | 4b | 4d | 4e | 6e | 6f |
|---|---|---|---|---|---|---|---|
| IC50 (μM) | COLO-205 | 0.20 | 0.20 | 1.00 | 0.30 | 15.30 | 10.60 |
| A549 | 28.4 | 9.5 | - | - | - | - |
| Compound Number | Tubulin Polymerization Inhibition (IC50, μM) | Casein Kinase I Inhibition (IC50, μg/mL) | EGFR Inhibition (IC50, μg/mL) | |||
|---|---|---|---|---|---|---|
| Average | ±SD | Average | ±SD | Average | ±SD | |
| 4a | 5.045 | 0.31 | 2.39 | 0.12 | 0.106 | 0.002 |
| 4b | 1.66 | 0.08 | 1.92 | 0.09 | 0.19 | 0.004 |
| 4e | - | - | 13.08 | 0.66 | 0.401 | 0.008 |
| Standard | 0.42 (CA-4) | 0.02 | 2.39 (4a, IC261) | 0.12 | 0.057 (Gefitinib) | 0.001 |
| Sample | Results DNA Content | |||
|---|---|---|---|---|
| %G0-G1 | %S | %G2/M | %Pre-G1 | |
| 4b/Colo-205 | 47.15 | 49.91 | 2.94 * | 41.28 * |
| DMSO/Colo 205(control) | 41.64 | 51.39 | 6.97 | 2.43 |
| Sample | Apoptosis | Necrosis | ||
| Total | Early | Late | ||
| 4b/Colo-205 | 41.28 | 2.61 | 21.31 | 17.36 |
| Compound | S Score a Kcal/mol | Amino Acid/Bond | Distance (Å) | RMSD_Refine b |
|---|---|---|---|---|
| 4a | −7.9046 | Cys241/H-donor | 3.62 | 1.6019 |
| Thr179/H-donor | 2.91 | |||
| 4b | −8.2658 | Cys241/H-donor | 3.62 | 1.22 |
| Thr179/H-donor | 2.91 | |||
| 4d | −7.9163 | Cys241/H-donor | 3.63 | 1.1067 |
| Thr179/H-donor | 2.88 | |||
| 4e | −8.0707 | Cys241/H-donor | 2.75 | 1.5655 |
| Thr179/H-donor | 3.56 | |||
| CA-4 | −8.8842 | Cys241/H-donor | 3.72 | 1.3880 |
| Thr179/H-donor | 3.00 | |||
| Met259/H-donor | 3.55 |
| Compound No | Concentration (ng/mL) | |
|---|---|---|
| Average | SEM | |
| Control | 387.1 | 5.87 |
| 4b | 242.0 | 3.08 |
| Ketokonazole | 189.8 | 2.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fareed, M.R.; Shoman, M.E.; Hamed, M.I.A.; Badr, M.; Bogari, H.A.; Elhady, S.S.; Ibrahim, T.S.; Abuo-Rahma, G.E.-D.A.; Ali, T.F.S. New Multi-Targeted Antiproliferative Agents: Design and Synthesis of IC261-Based Oxindoles as Potential Tubulin, CK1 and EGFR Inhibitors. Pharmaceuticals 2021, 14, 1114. https://doi.org/10.3390/ph14111114
Fareed MR, Shoman ME, Hamed MIA, Badr M, Bogari HA, Elhady SS, Ibrahim TS, Abuo-Rahma GE-DA, Ali TFS. New Multi-Targeted Antiproliferative Agents: Design and Synthesis of IC261-Based Oxindoles as Potential Tubulin, CK1 and EGFR Inhibitors. Pharmaceuticals. 2021; 14(11):1114. https://doi.org/10.3390/ph14111114
Chicago/Turabian StyleFareed, Momen R., Mai E. Shoman, Mohammed I. A. Hamed, Mohamed Badr, Hanin A. Bogari, Sameh S. Elhady, Tarek S. Ibrahim, Gamal El-Din A. Abuo-Rahma, and Taha F. S. Ali. 2021. "New Multi-Targeted Antiproliferative Agents: Design and Synthesis of IC261-Based Oxindoles as Potential Tubulin, CK1 and EGFR Inhibitors" Pharmaceuticals 14, no. 11: 1114. https://doi.org/10.3390/ph14111114
APA StyleFareed, M. R., Shoman, M. E., Hamed, M. I. A., Badr, M., Bogari, H. A., Elhady, S. S., Ibrahim, T. S., Abuo-Rahma, G. E.-D. A., & Ali, T. F. S. (2021). New Multi-Targeted Antiproliferative Agents: Design and Synthesis of IC261-Based Oxindoles as Potential Tubulin, CK1 and EGFR Inhibitors. Pharmaceuticals, 14(11), 1114. https://doi.org/10.3390/ph14111114