
Hydroquinone 5-O-Cinnamoyl Ester of Renieramycin M Suppresses Lung Cancer Stem Cells by Targeting Akt and Destabilizes c-Myc


 and
and 
Abstract
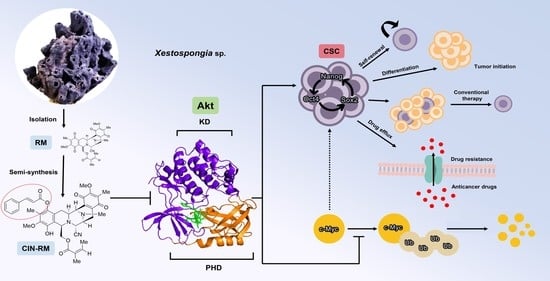
:
1. Introduction
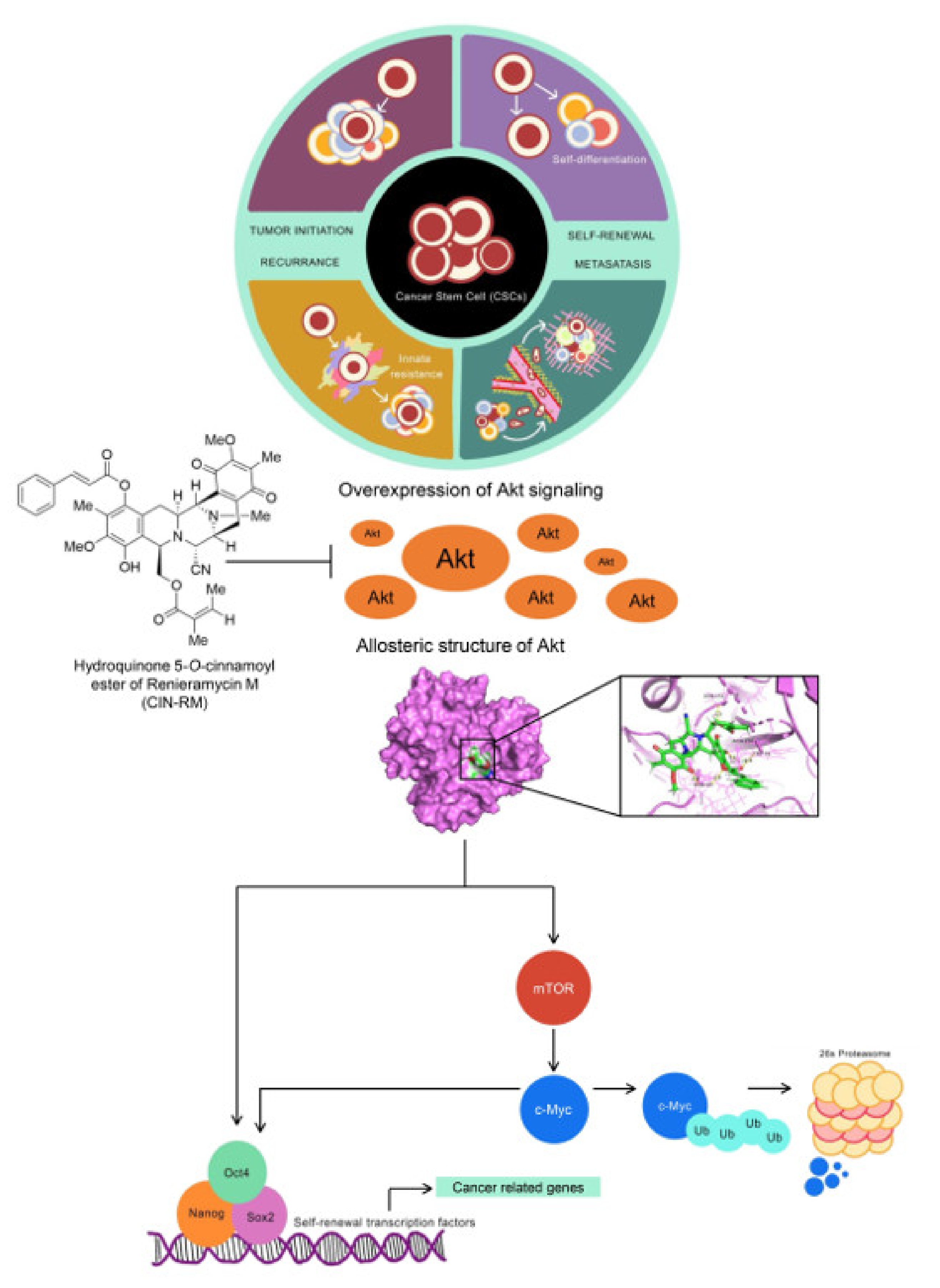
2. Results
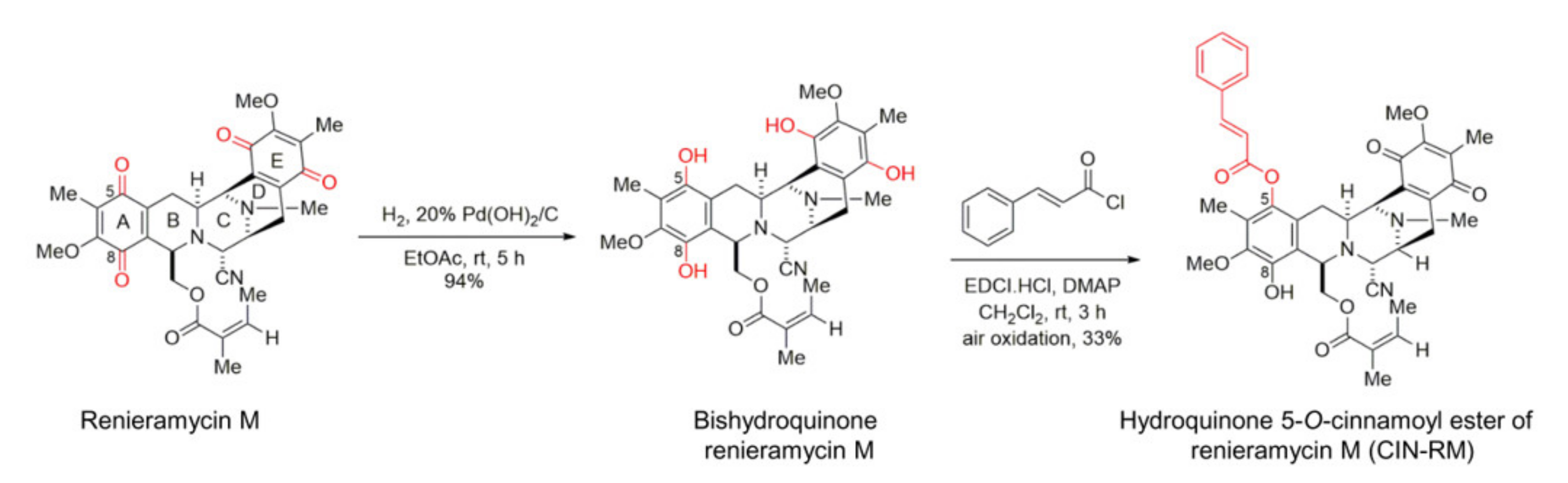
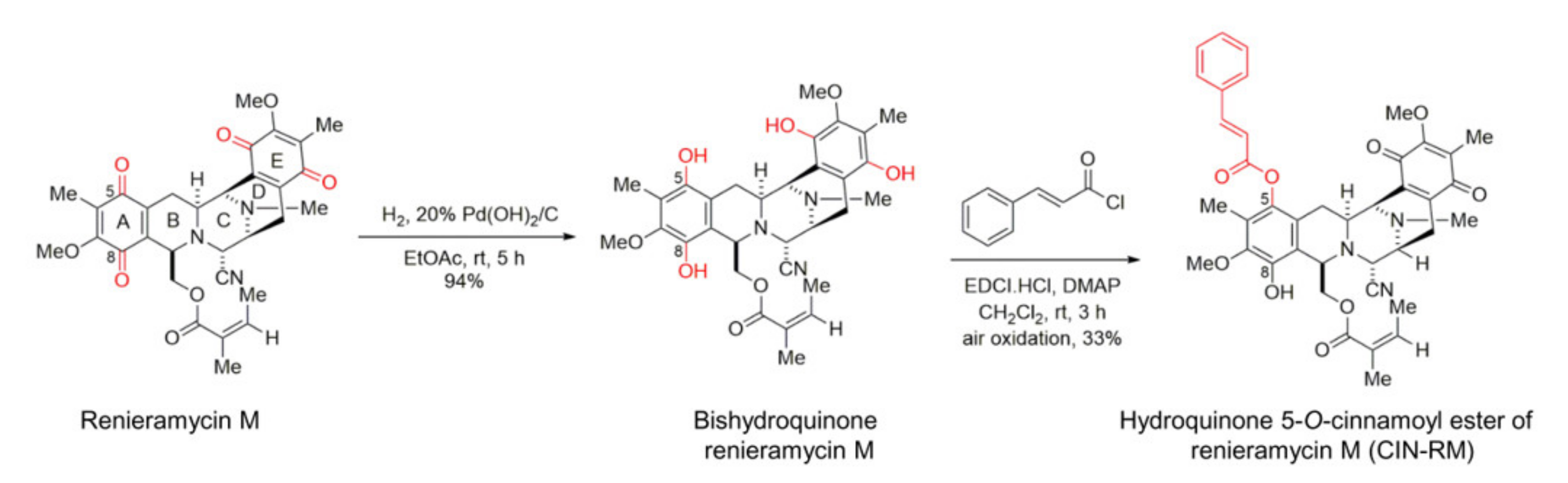
2.1. Semi-Synthesis of Hydroquinone 5-O-Cinnamoyl Ester of Renieramycin M (CIN-RM)
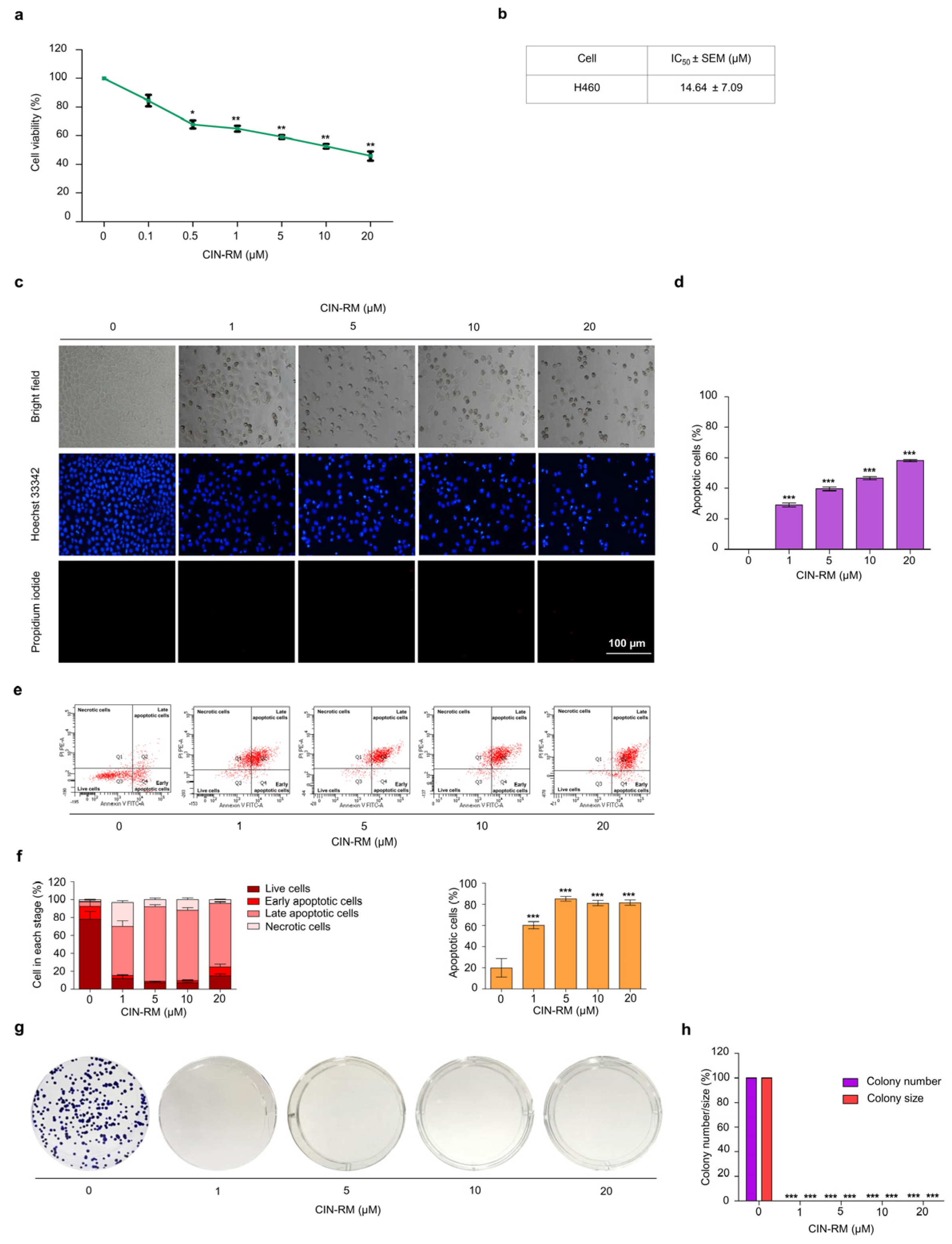
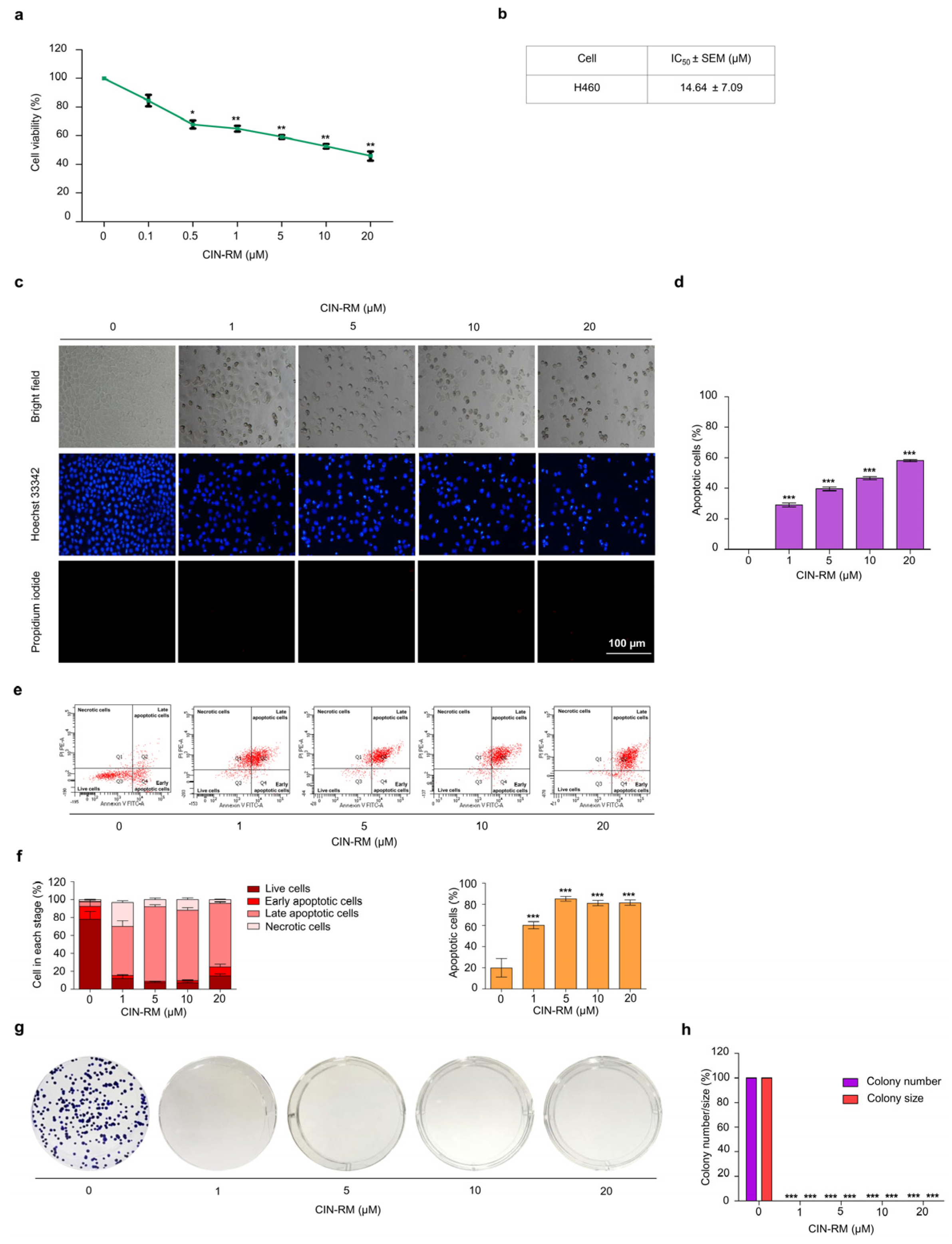
2.2. Selective Cytotoxicity of CIN-RM in Human Lung Cancer Cells
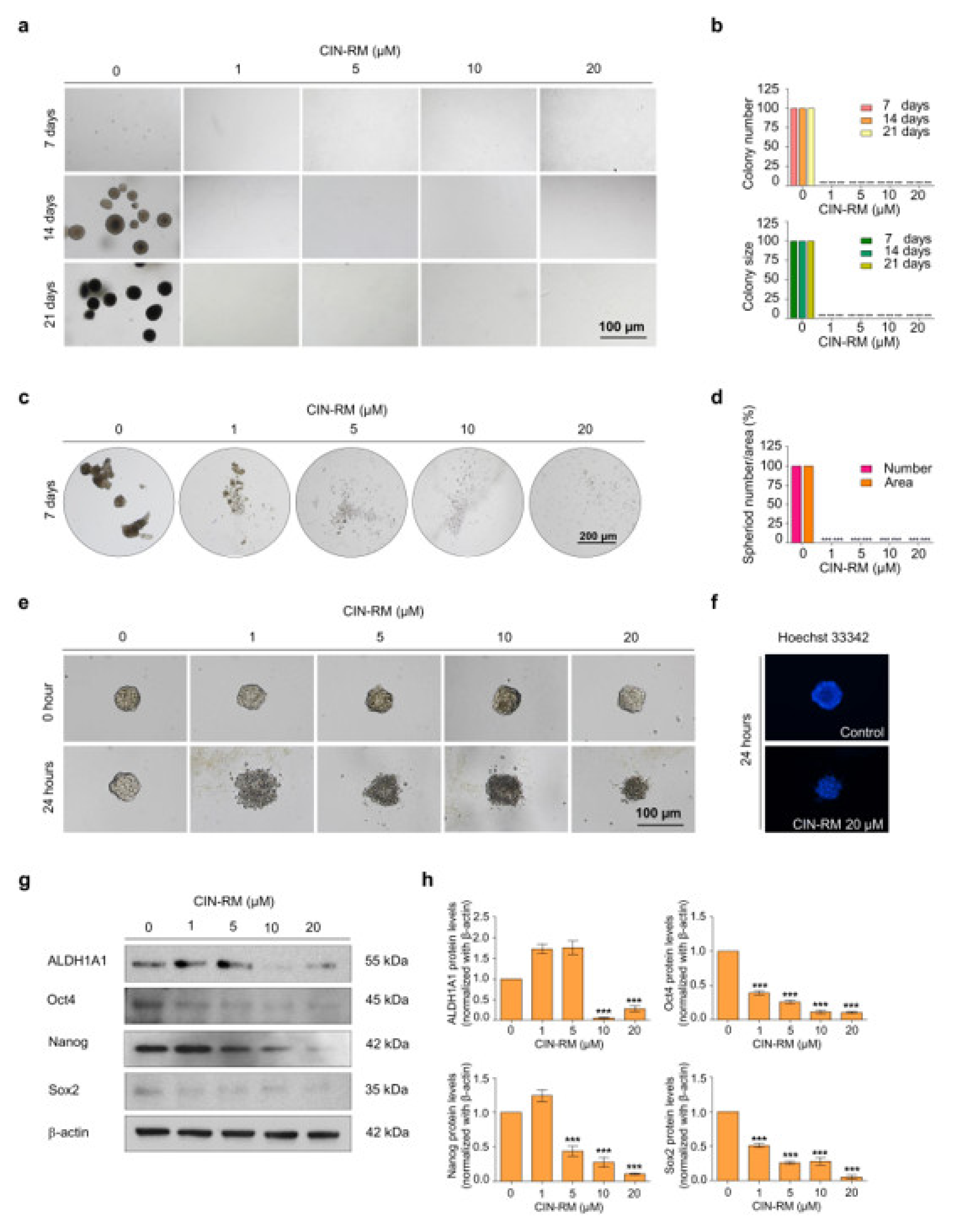
2.3. CIN-RM Attenuates Anchorage-Independent Growth and Suppresses CSC Spheroid Formation
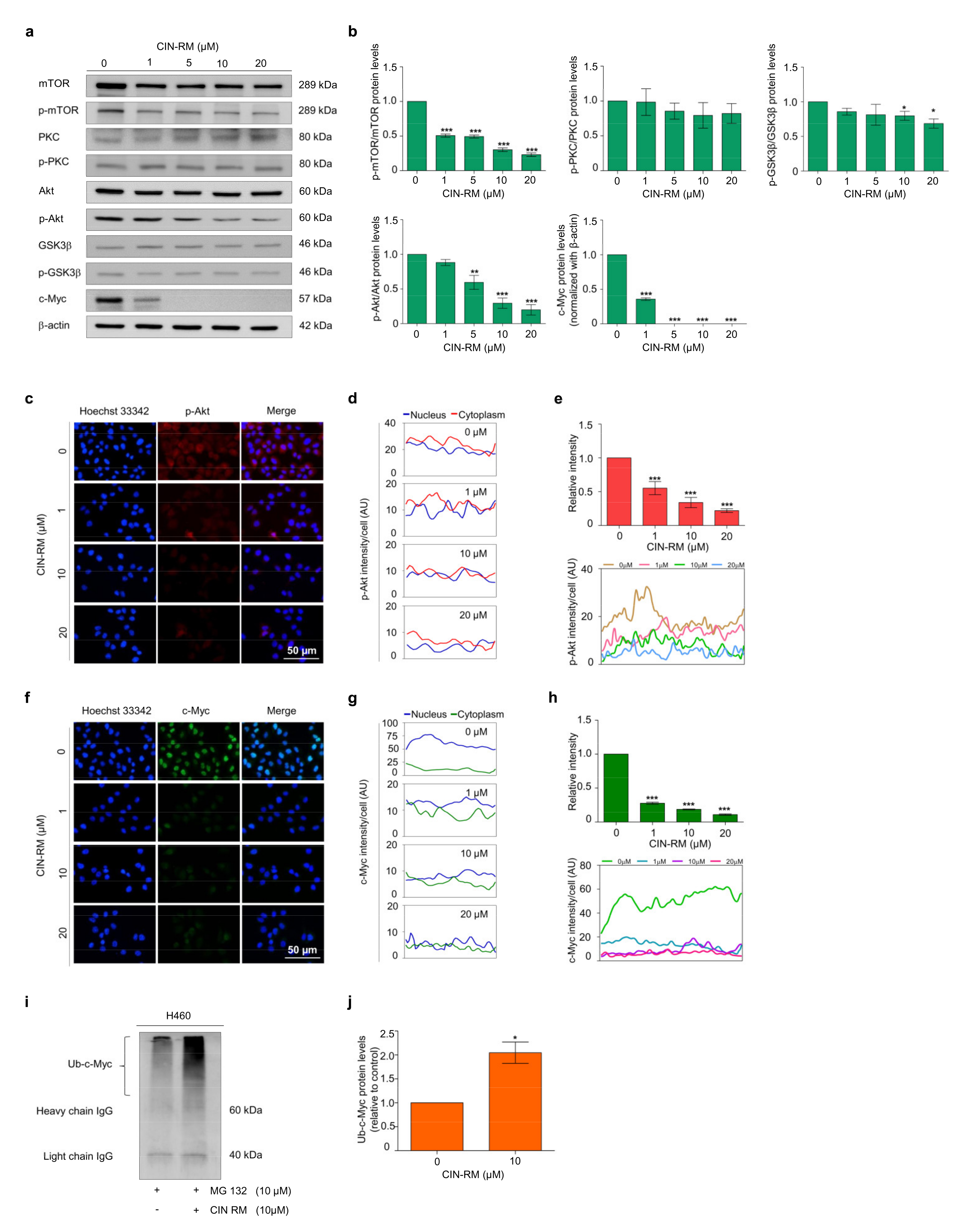
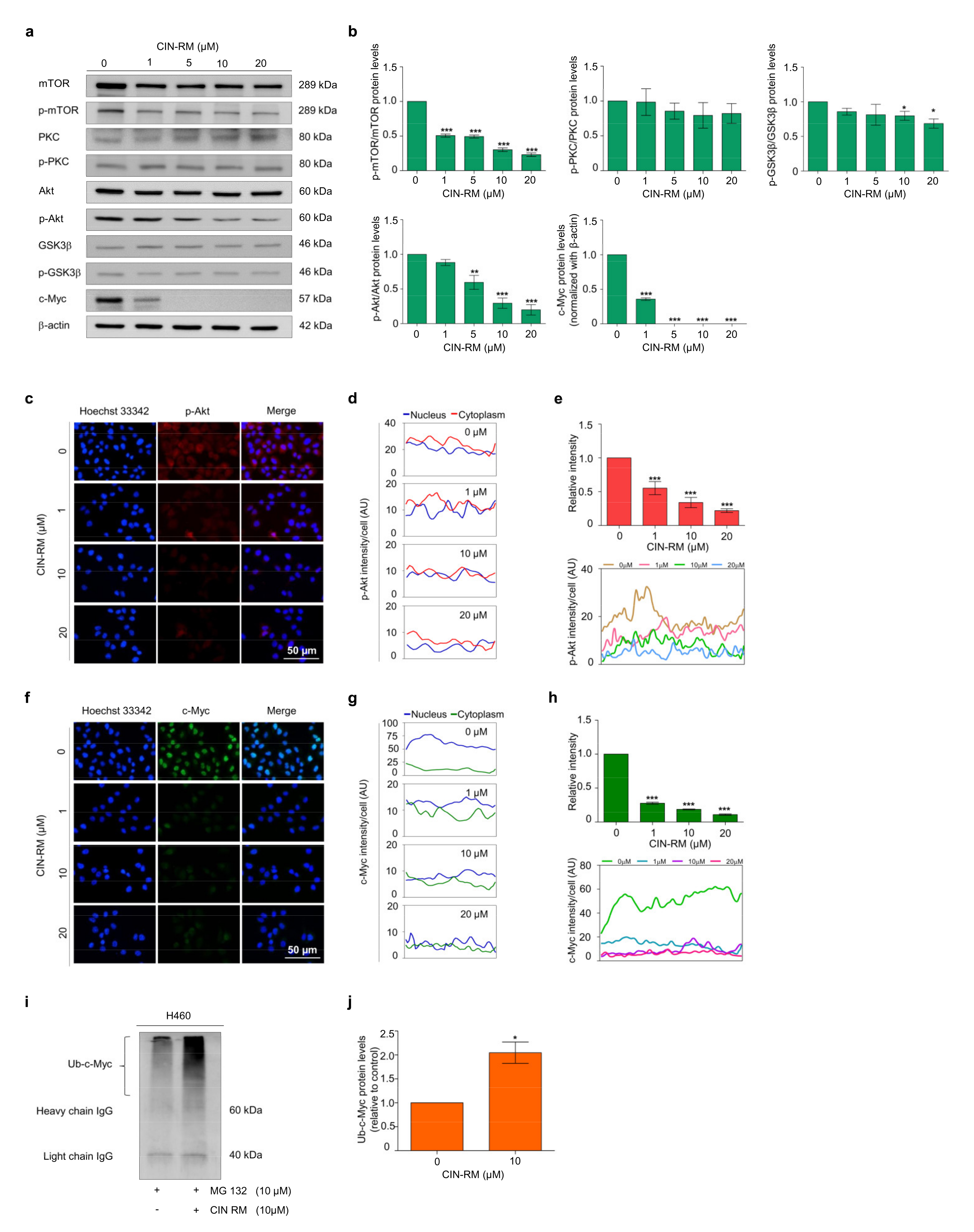
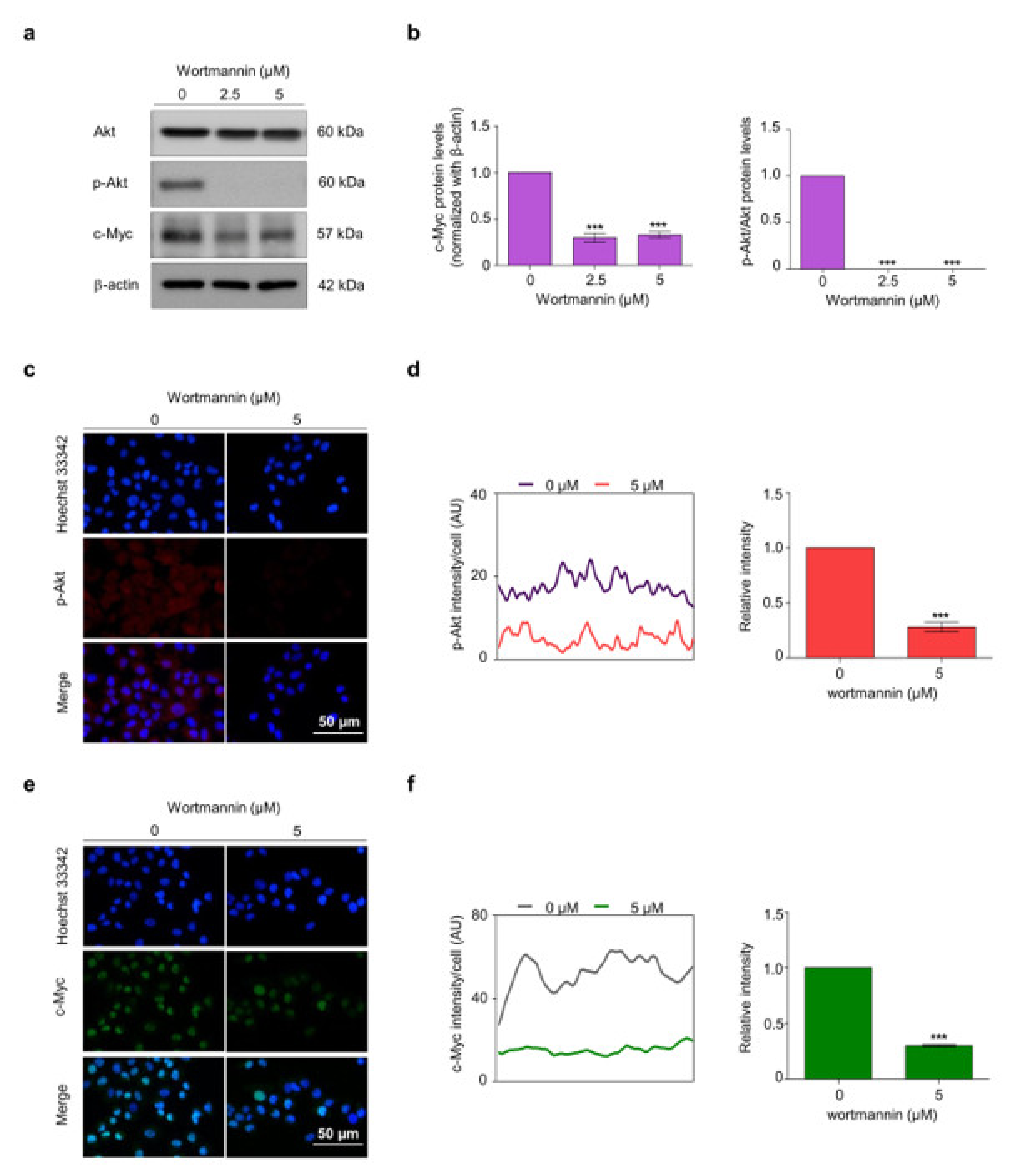
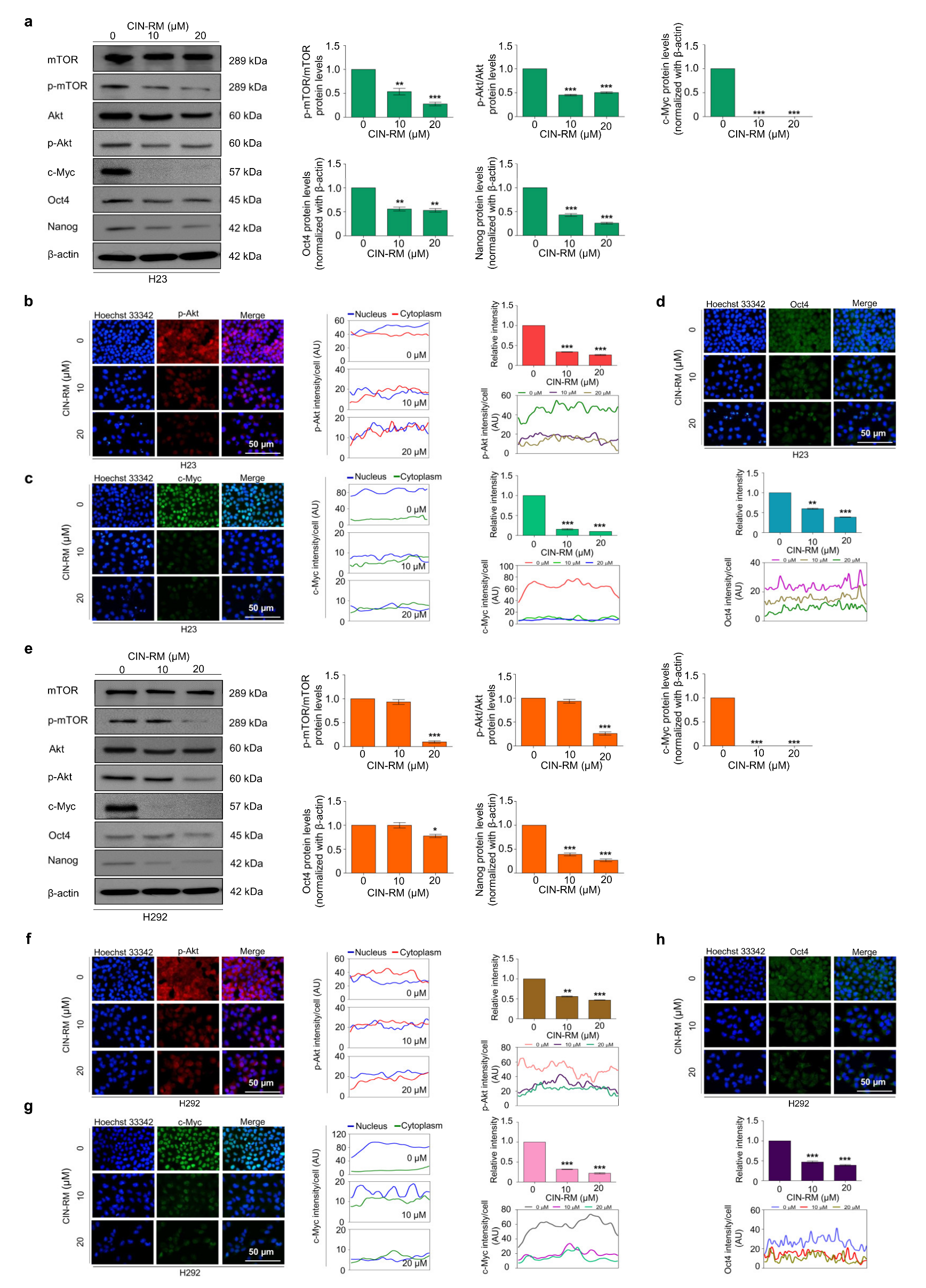
2.4. CIN-RM Suppression of CSC Is Mediated via Akt Inhibition
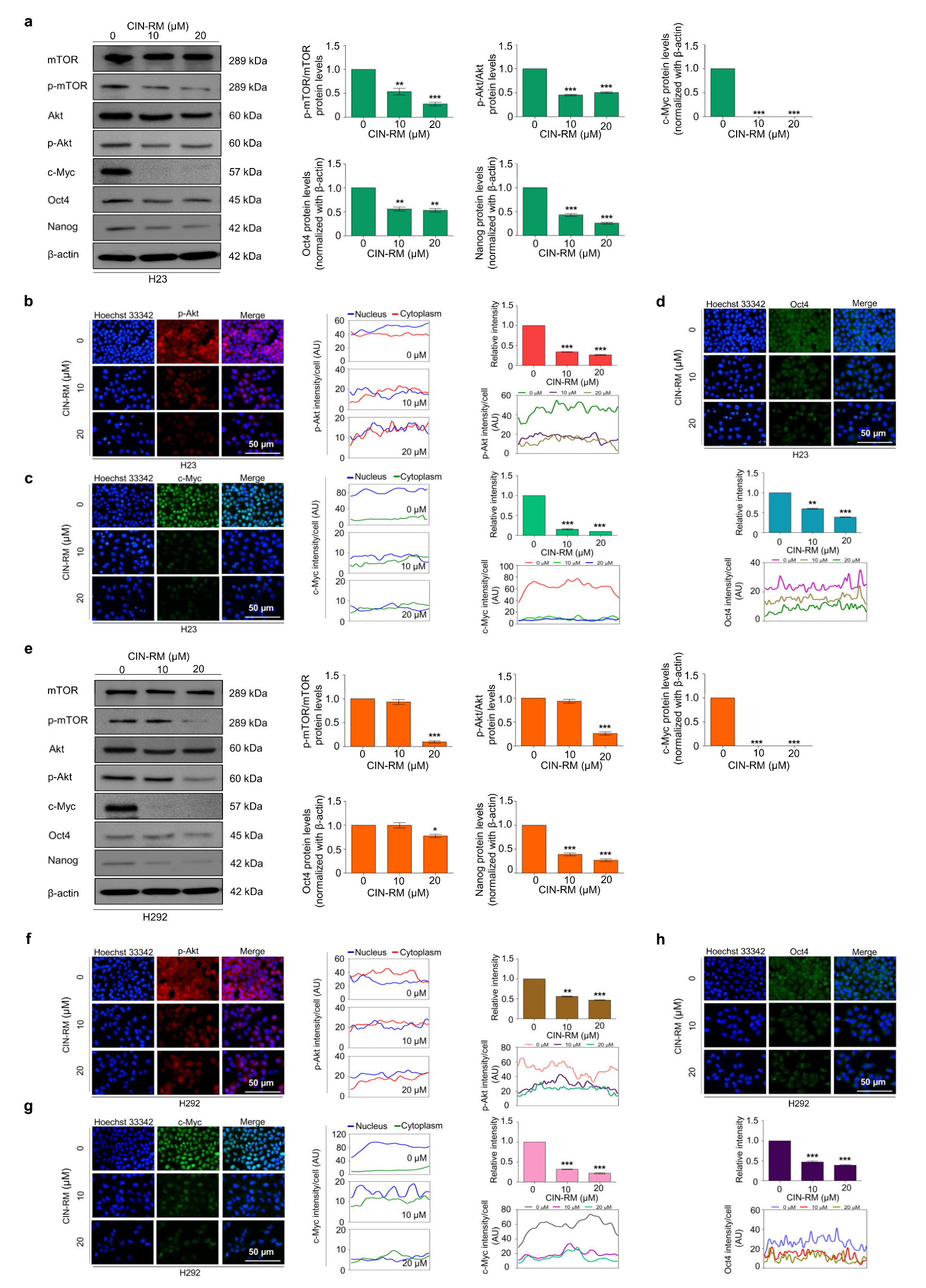
2.5. CIN-RN Suppression of CSC on Other Lung Cancer Cells
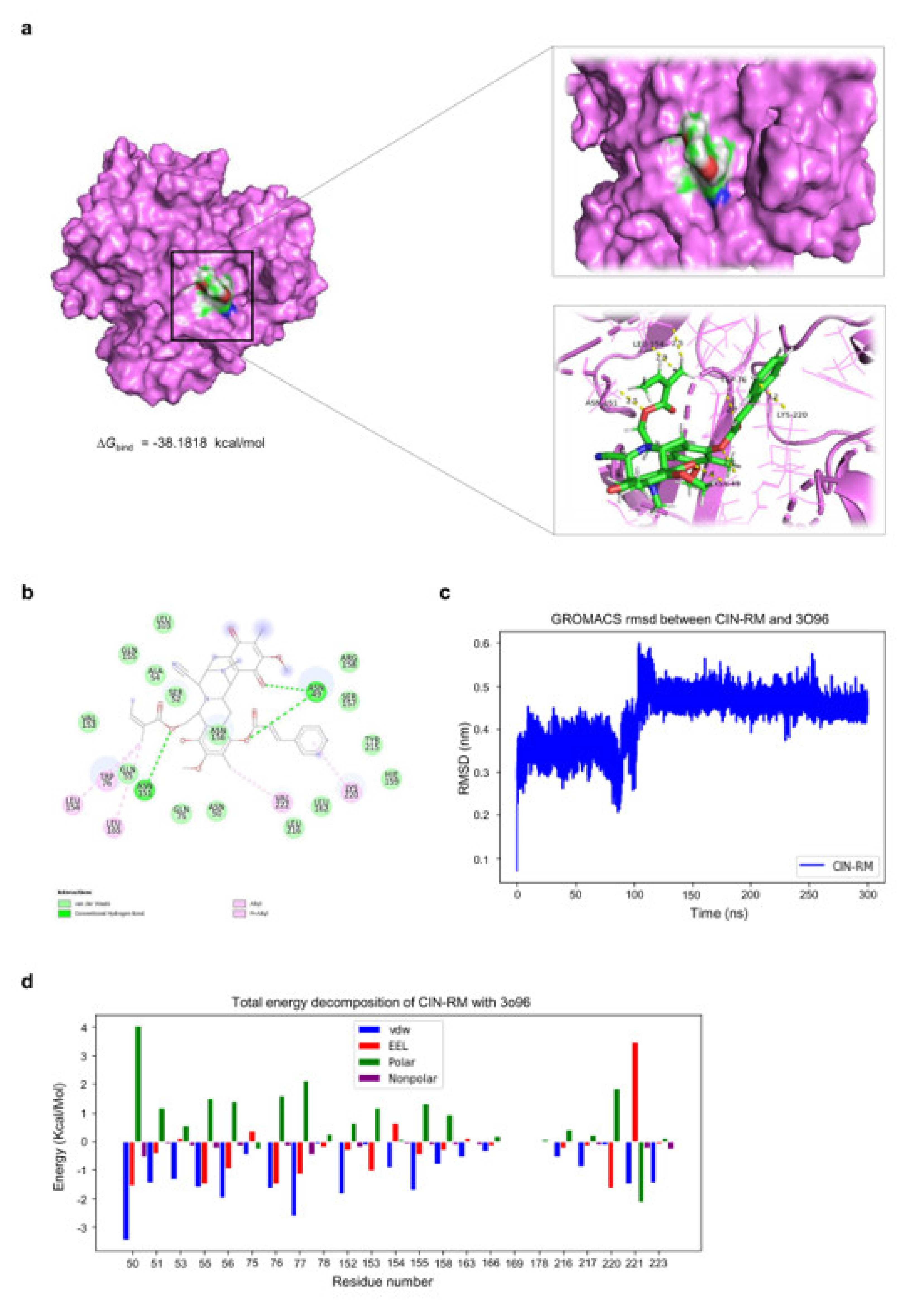
2.6. Molecular Docking Simulation Demonstrates the Interaction of CIN-RM with the Akt Protein
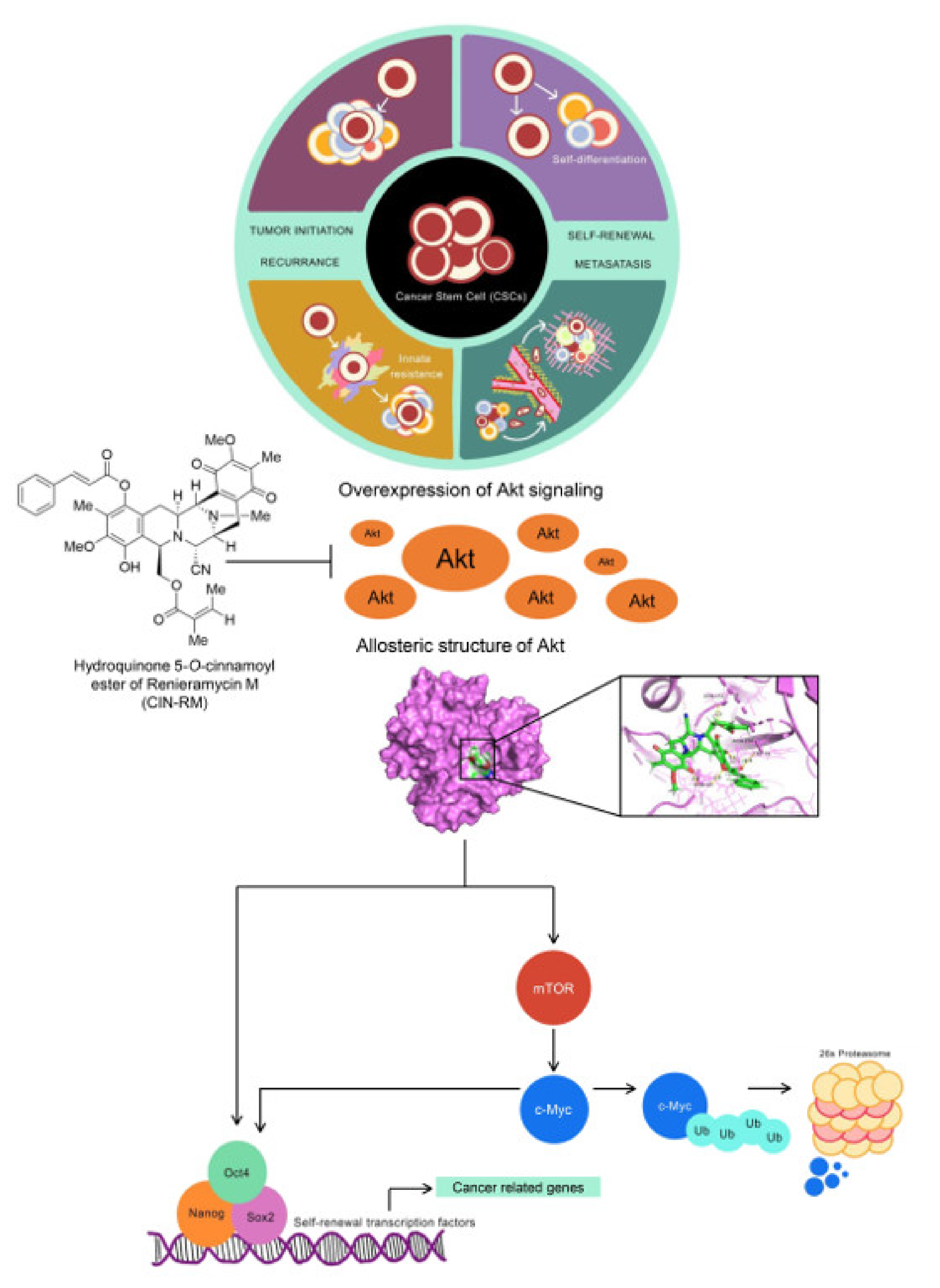
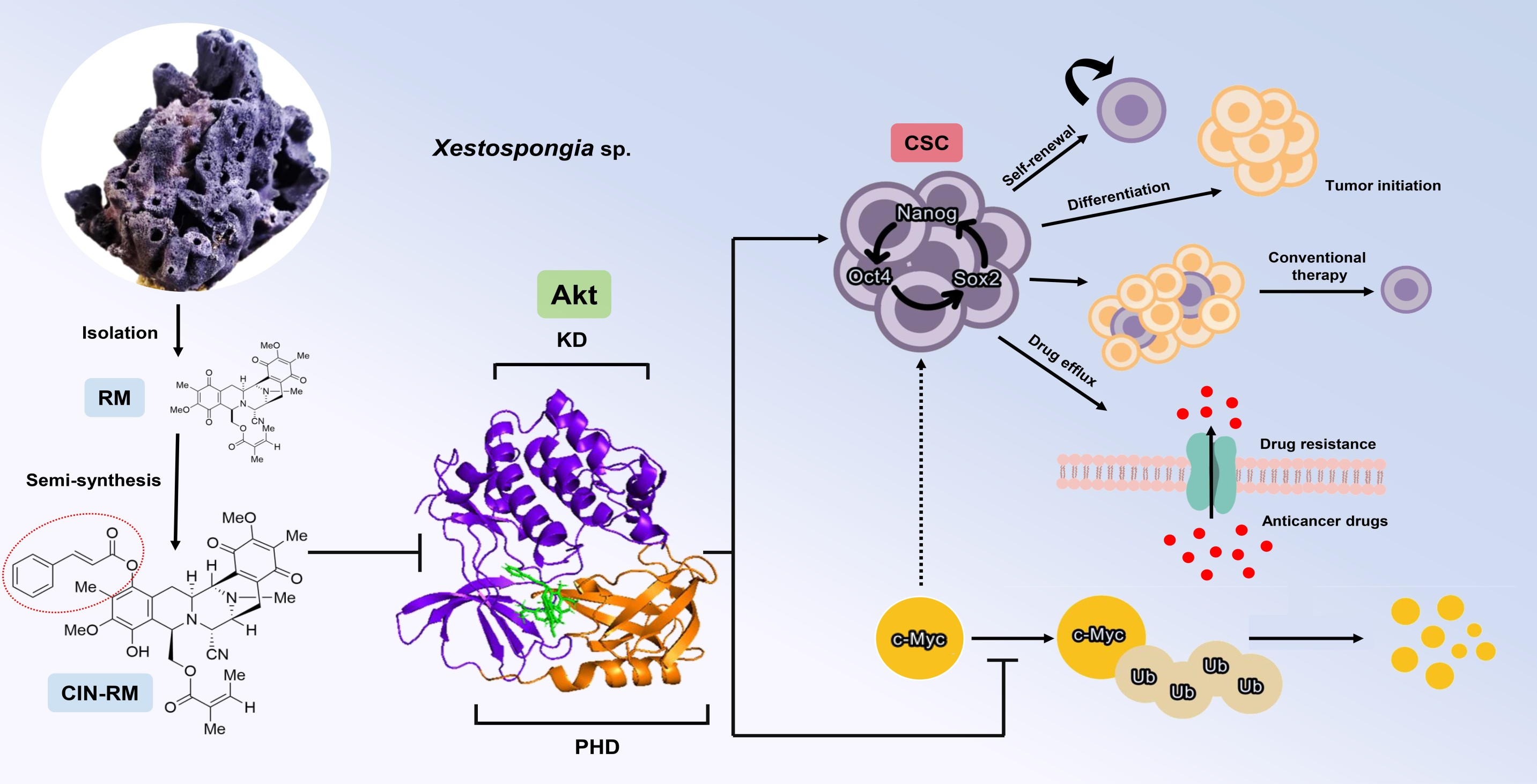
3. Discussion
4. Materials and Methods
4.1. Non-Small Cell Lung Cancer Cell Lines and Cultures
4.2. Semi-Synthesis of Hydroquinone 5-O-Cinnamoyl Ester of Renieramycin M (CIN-RM)
4.3. Reagents and Antibodies
4.4. Cytotoxicity Assay
4.5. Apoptosis Assay
4.6. Nuclear Staining Assay
4.7. Colony Formation Assay
4.8. Anchorage-Independent Growth Assay
4.9. Spheroid Formation Assay
4.10. Immunofluorescence
4.11. Immunoprecipitation Assay
4.12. Western Blot Analysis
4.13. Computational Akt Modelling and Molecular Docking
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, T.; Song, X.; Xu, D.; Tiek, D.; Goenka, A.; Wu, B.; Sastry, N.; Hu, B.; Cheng, S.-Y. Stem cell programs in cancer initiation, progression, and therapy resistance. Theranostics 2020, 10, 8721–8743. [Google Scholar] [CrossRef] [PubMed]
- Sowa, T.; Menju, T.; Sonobe, M.; Nakanishi, T.; Shikuma, K.; Imamura, N.; Motoyama, H.; Hijiya, K.; Aoyama, A.; Chen, F. Association between epithelial-mesenchymal transition and cancer stemness and their effect on the prognosis of lung adenocarcinoma. Cancer Med. 2015, 4, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 1–35. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Yu, X.; Liu, S. Pluripotency transcription factors and cancer stem cells: Small genes make a big difference. Chin. J. Cancer 2013, 32, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.F.; Wu, C.; Alshareef, A.; Gupta, N.; Zhao, Q.; Xu, X.E.; Jiao, J.W.; Li, E.M.; Xu, L.Y.; Lai, R. The PI3K/AKT/c-MYC axis promotes the acquisition of cancer stem-like features in esophageal squamous cell carcinoma. Stem Cells 2016, 34, 2040–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, P.; Xu, X.-Y. PI3K/Akt/mTOR signaling pathway in cancer stem cells: From basic research to clinical application. Am. J. Cancer Res. 2015, 5, 1602–1609. [Google Scholar] [PubMed]
- Murphy, M.J.; Wilson, A.; Trumpp, A. More than just proliferation: Myc function in stem cells. Trends Cell Biol. 2005, 15, 128–137. [Google Scholar] [CrossRef]
- Elbadawy, M.; Usui, T.; Yamawaki, H.; Sasaki, K. Emerging roles of C-Myc in cancer stem cell-related signaling and resistance to cancer chemotherapy: A potential therapeutic target against colorectal cancer. Int. J. Mol. Sci. 2019, 20, 2340. [Google Scholar] [CrossRef] [Green Version]
- Deb, T.B.; Coticchia, C.M.; Dickson, R.B. Calmodulin-mediated activation of Akt regulates survival of c-Myc-overexpressing mouse mammary carcinoma cells. J. Biol. Chem. 2004, 279, 38903–38911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanvorachote, P.; Sriratanasak, N.; Nonpanya, N. C-myc contributes to malignancy of lung Cancer: A potential anticancer drug target. Anticancer Res. 2020, 40, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Swords, R.T.; Schenk, T.; Stengel, S.; Gil, V.S.; Petrie, K.R.; Perez, A.; Ana, R.; Watts, J.M.; Vargas, F.; Elias, R.; et al. Inhibition of the PI3K/AKT/mTOR pathway leads to down-regulation of c-Myc and overcomes resistance to ATRA in acute myeloid leukemia. Blood 2015, 126, 1363. [Google Scholar] [CrossRef]
- Zhang, X.; Ai, Z.; Chen, J.; Yi, J.; Liu, Z.; Zhao, H.; Wei, H. Glycometabolic adaptation mediates the insensitivity of drug-resistant K562/ADM leukaemia cells to adriamycin via the AKT-mTOR/c-Myc signalling pathway. Mol. Med. Rep. 2017, 15, 1869–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagnocchi, L.; Zippo, A. Multiple roles of MYC in integrating regulatory networks of pluripotent stem cells. Front. Cell Dev. Biol. 2017, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Oron, E.; Nelson, B.; Razis, S.; Ivanova, N. Distinct lineage specification roles for NANOG, OCT4, and SOX2 in human embryonic stem cells. Cell Stem Cell 2012, 10, 440–454. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Chen, J.; Hu, G.; Yu, J.; Zhu, X.; Lin, Y.; Chen, S.; Yuan, J. Statistical research on the bioactivity of new marine natural products discovered during the 28 years from 1985 to 2012. Mar. Drugs 2015, 13, 202–221. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, S.A.; Elias, N.; Farag, M.A.; Chen, L.; Saeed, A.; Hegazy, M.-E.F.; Moustafa, M.S.; El-Wahed, A.; Al-Mousawi, S.M.; Musharraf, S.G.; et al. Marine natural products: A source of novel anticancer drugs. Mar. Drugs 2019, 17, 491. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Marine-sourced anti-cancer and cancer pain control agents in clinical and late preclinical development. Mar. Drugs 2014, 12, 255–278. [Google Scholar] [CrossRef] [Green Version]
- Scott, J.D.; Williams, R.M. Chemistry and biology of the tetrahydroisoquinoline antitumor antibiotics. Chem. Rev. 2002, 102, 1669–1730. [Google Scholar] [CrossRef]
- Chamni, S.; Sirimangkalakitti, N.; Chanvorachote, P.; Suwanborirux, K.; Saito, N. Chemistry of Renieramycins. Part 19: Semi-Syntheses of 22-O-Amino Ester and Hydroquinone 5-O-Amino Ester Derivatives of Renieramycin M and Their Cytotoxicity against Non-Small-Cell Lung Cancer Cell Lines. Mar. Drugs 2020, 18, 418. [Google Scholar] [CrossRef] [PubMed]
- Sirimangkalakitti, N.; Chamni, S.; Suwanborirux, K.; Chanvorachote, P. Renieramycin m attenuates cancer stem cell-like phenotypes in h460 lung cancer cells. Anticancer Res. 2017, 37, 615–621. [Google Scholar] [CrossRef] [Green Version]
- Maiuthed, A.; Pinkhien, T.; Chamni, S.; Suwanborirux, K.; Saito, N.; Petpiroon, N.; Chanvorachote, P. Apoptosis-inducing effect of hydroquinone 5-o-cinnamoyl ester analog of renieramycin m on non-small cell lung cancer cells. Anticancer Res. 2017, 37, 6259–6267. [Google Scholar] [PubMed]
- Chamni, S.; Sirimangkalakitti, N.; Chanvorachote, P.; Saito, N.; Suwanborirux, K. Chemistry of renieramycins. 17. A new generation of renieramycins: Hydroquinone 5-O-monoester analogues of renieramycin M as potential cytotoxic agents against non-small-cell lung cancer cells. J. Nat. Prod. 2017, 80, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Chang, J.T.; Andrechek, E.R.; Matsumura, N.; Baba, T.; Yao, G.; Kim, J.W.; Gatza, M.; Murphy, S.; Nevins, J.R. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene 2009, 28, 2796–2805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, A.S.; Sears, R.C. MYC degradation. Cold Spring Harb. Perspect. Med. 2014, 4, a014365. [Google Scholar] [CrossRef]
- Mossahebi-Mohammadi, M.; Quan, M.; Zhang, J.-S.; Li, X. FGF signaling pathway: A key regulator of stem cell pluripotency. Front. Cell Dev. Biol. 2020, 8, 79. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [Green Version]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Würth, R.; Barbieri, F.; Florio, T. New molecules and old drugs as emerging approaches to selectively target human glioblastoma cancer stem cells. BioMed Res. Int. 2014, 2014, 126586. [Google Scholar] [CrossRef]
- Radke, J.; Bortolussi, G.; Pagenstecher, A. Akt and c-Myc induce stem-cell markers in mature primary p53−/− astrocytes and render these cells gliomagenic in the brain of immunocompetent mice. PLoS ONE 2013, 8, e56691. [Google Scholar] [CrossRef]
- Quan, Y.; Wang, N.; Chen, Q.; Xu, J.; Cheng, W.; Di, M.; Xia, W.; Gao, W.-Q. SIRT3 inhibits prostate cancer by destabilizing oncoprotein c-MYC through regulation of the PI3K/Akt pathway. Oncotarget 2015, 6, 26494–26507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chantarawong, W.; Chamni, S.; Suwanborirux, K.; Saito, N.; Chanvorachote, P. 5-O-Acetyl-Renieramycin T from Blue Sponge Xestospongia sp. Induces Lung Cancer Stem Cell Apoptosis. Mar. Drugs 2019, 17, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottinger, S.; Klöppel, A.; Rausch, V.; Liu, L.; Kallifatidis, G.; Gross, W.; Gebhard, M.M.; Brümmer, F.; Herr, I. Targeting of pancreatic and prostate cancer stem cell characteristics by Crambe crambe marine sponge extract. Int. J. Cancer 2012, 130, 1671–1681. [Google Scholar] [CrossRef] [PubMed]
- Suresh, R.; Ali, S.; Ahmad, A.; Philip, P.A.; Sarkar, F.H. The role of cancer stem cells in recurrent and drug-resistant lung cancer. In Lung Cancer and Personalized Medicine: Novel Therapies and Clinical Management; Springer: Cham, Switzerland, 2016; pp. 57–74. [Google Scholar]
- Sullivan, J.P.; Spinola, M.; Dodge, M.; Raso, M.G.; Behrens, C.; Gao, B.; Schuster, K.; Shao, C.; Larsen, J.E.; Sullivan, L.A. Aldehyde dehydrogenase activity selects for lung adenocarcinoma stem cells dependent on notch signaling. Cancer Res. 2010, 70, 9937–9948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.-P.; Tsai, M.-F.; Chang, T.-H.; Tang, W.-C.; Chen, S.-Y.; Lai, H.-H.; Lin, T.-Y.; Yang, J.C.-H.; Yang, P.-C.; Shih, J.-Y.; et al. ALDH-positive lung cancer stem cells confer resistance to epidermal growth factor receptor tyrosine kinase inhibitors. Cancer Lett. 2013, 328, 144–151. [Google Scholar] [CrossRef]
- Schaefer, T.; Steiner, R.; Lengerke, C. SOX2 and p53 expression control converges in PI3K/AKT signaling with versatile implications for stemness and cancer. Int. J. Mol. Sci. 2020, 21, 4902. [Google Scholar] [CrossRef]
- Srinual, S.; Chanvorachote, P.; Pongrakhananon, V. Suppression of cancer stem-like phenotypes in NCI-H460 lung cancer cells by vanillin through an Akt-dependent pathway. Int. J. Oncol. 2017, 50, 1341–1351. [Google Scholar] [CrossRef]
- Su, T.; Dan, S.; Wang, Y. Akt–Oct4 regulatory circuit in pluripotent stem cells. Chin. Sci. Bull. 2014, 59, 936–943. [Google Scholar] [CrossRef]
- Zayed, H.; Petersen, I. Stem cell transcription factor SOX2 in synovial sarcoma and other soft tissue tumors. Pathol.-Res. Pract. 2018, 214, 1000–1007. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Yang, Y.; Li, W.; Chen, Q.; Li, J.; Pan, X.; Zhou, L.; Liu, C.; Chen, C.; He, J.; et al. Reciprocal regulation of Akt and Oct4 promotes the self-renewal and survival of embryonal carcinoma cells. Mol. Cell 2012, 48, 627–640. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafson, W.; Weiss, W. Myc proteins as therapeutic targets. Oncogene 2010, 29, 1249–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Shi, A.; Wang, N.; Li, M.; He, X.; Yin, C.; Tu, Q.; Shen, X.; Tao, Y.; Wang, Q. Polyphenolic Proanthocyanidin-B2 suppresses proliferation of liver cancer cells and hepatocellular carcinogenesis through directly binding and inhibiting AKT activity. Redox Biol. 2020, 37, 101701. [Google Scholar] [CrossRef] [PubMed]
- Elghazi, L.; Balcazar, N.; Bernal-Mizrachi, E. Emerging role of protein kinase B/Akt signaling in pancreatic β-cell mass and function. Int. J. Biochem. Cell Biol. 2006, 38, 689–695. [Google Scholar] [CrossRef]
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.A.; Libra, M.; Tsatsakis, A.M. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use. Int. J. Oncol. 2016, 48, 869–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suwanborirux, K.; Amnuoypol, S.; Plubrukarn, A.; Pummangura, S.; Kubo, A.; Tanaka, C.; Saito, N. Chemistry of renieramycins. Part 3. Isolation and structure of stabilized renieramycin type derivatives possessing antitumor activity from Thai sponge Xestospongia species, pretreated with potassium cyanide. J. Nat. Prod. 2003, 66, 1441–1446. [Google Scholar] [CrossRef]
- Kaur, K.; Kaur, P.; Mittal, A.; Nayak, S.K.; Khatik, G.L. Design and molecular docking studies of novel antimicrobial peptides using autodock molecular docking software. Asian J. Pharm. Clin. Res. 2017, 10, 28–31. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.-I.; Voegtli, W.C.; Sturgis, H.L.; Dizon, F.P.; Vigers, G.P.; Brandhuber, B.J. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS ONE 2010, 5, e12913. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Guex, N. Swiss-PdbViewer: A fast and easy-to-use PDB viewer for Macintosh and PC. Protein Data Bank Q. Newsl. 1996, 77, 7. [Google Scholar]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, A.W.S.; Vranken, W.F. ACPYPE-Antechamber python parser interface. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Pawar, S.S.; Rohane, S.H. Review on Discovery Studio: An important Tool for Molecular Docking. Asian J. Res. Chem. 2021, 14, 86–88. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CIN-RM/Akt (∆Gbind) | |
|---|---|
| ∆Eele | −18.4679 ± 4.0324 |
| ∆EvdW | −63.3923 ± 2.7583 |
| ∆EMM | −81.8602 ± 6.7907 |
| ∆Gsolv,non-polar | −8.3395 ± 0.2798 |
| ∆Gsolv,polar | 41.3801 ± 4.1900 |
| ∆Gtotal | −48.8170 ± 3.3050 |
| −TDS | 10.6352 |
| ∆Gbind | −38.1818 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hongwiangchan, N.; Sriratanasak, N.; Wichadakul, D.; Aksorn, N.; Chamni, S.; Chanvorachote, P. Hydroquinone 5-O-Cinnamoyl Ester of Renieramycin M Suppresses Lung Cancer Stem Cells by Targeting Akt and Destabilizes c-Myc. Pharmaceuticals 2021, 14, 1112. https://doi.org/10.3390/ph14111112
Hongwiangchan N, Sriratanasak N, Wichadakul D, Aksorn N, Chamni S, Chanvorachote P. Hydroquinone 5-O-Cinnamoyl Ester of Renieramycin M Suppresses Lung Cancer Stem Cells by Targeting Akt and Destabilizes c-Myc. Pharmaceuticals. 2021; 14(11):1112. https://doi.org/10.3390/ph14111112
Chicago/Turabian StyleHongwiangchan, Nattamon, Nicharat Sriratanasak, Duangdao Wichadakul, Nithikoon Aksorn, Supakarn Chamni, and Pithi Chanvorachote. 2021. "Hydroquinone 5-O-Cinnamoyl Ester of Renieramycin M Suppresses Lung Cancer Stem Cells by Targeting Akt and Destabilizes c-Myc" Pharmaceuticals 14, no. 11: 1112. https://doi.org/10.3390/ph14111112
APA StyleHongwiangchan, N., Sriratanasak, N., Wichadakul, D., Aksorn, N., Chamni, S., & Chanvorachote, P. (2021). Hydroquinone 5-O-Cinnamoyl Ester of Renieramycin M Suppresses Lung Cancer Stem Cells by Targeting Akt and Destabilizes c-Myc. Pharmaceuticals, 14(11), 1112. https://doi.org/10.3390/ph14111112