
Design, Synthesis and Biological Evaluation of Aromatase Inhibitors Based on Sulfonates and Sulfonamides of Resveratrol

 ,
,  ,
,  ,
,  ,
, 
 ,
,  ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
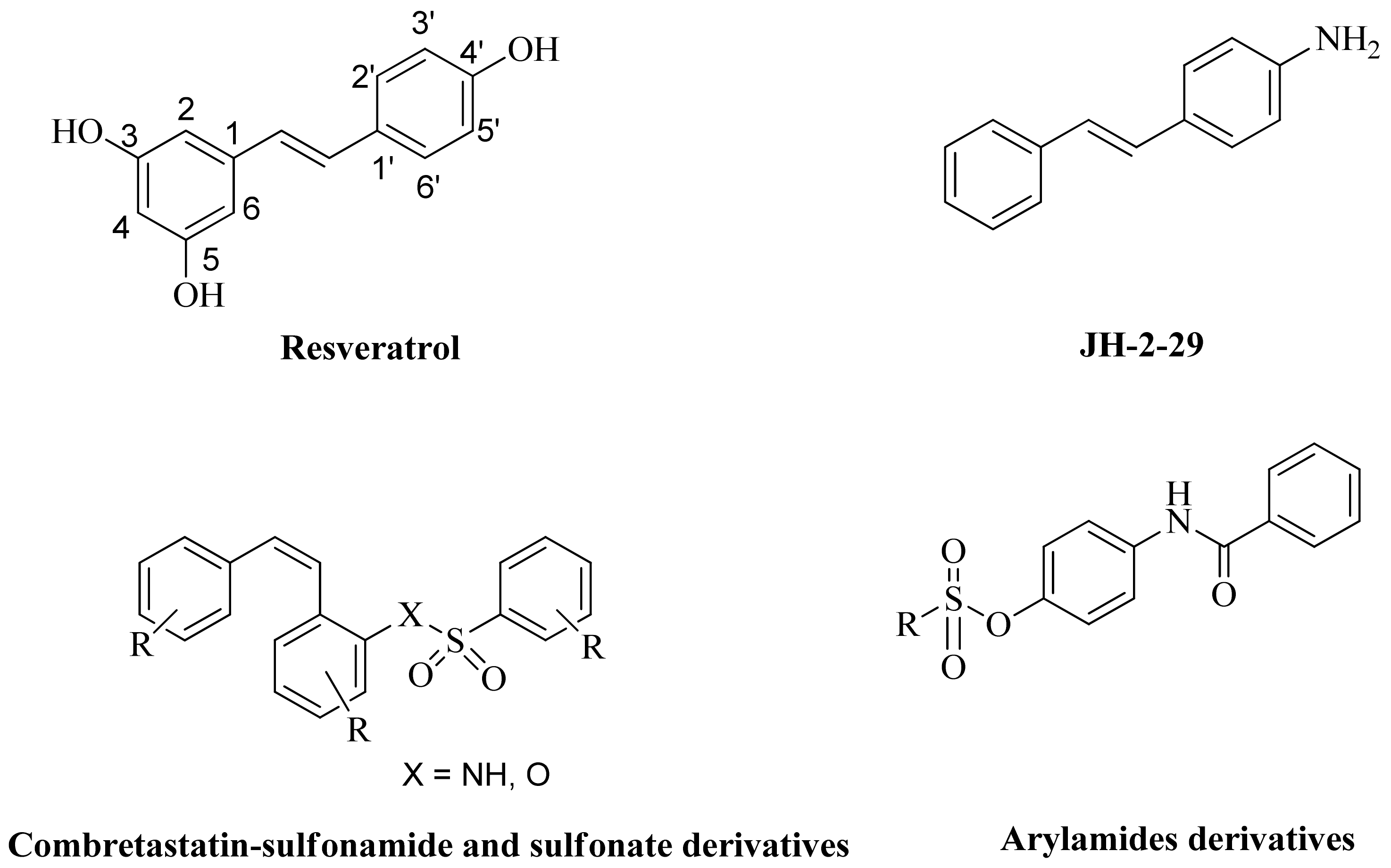
1. Introduction
2. Results and Discussions
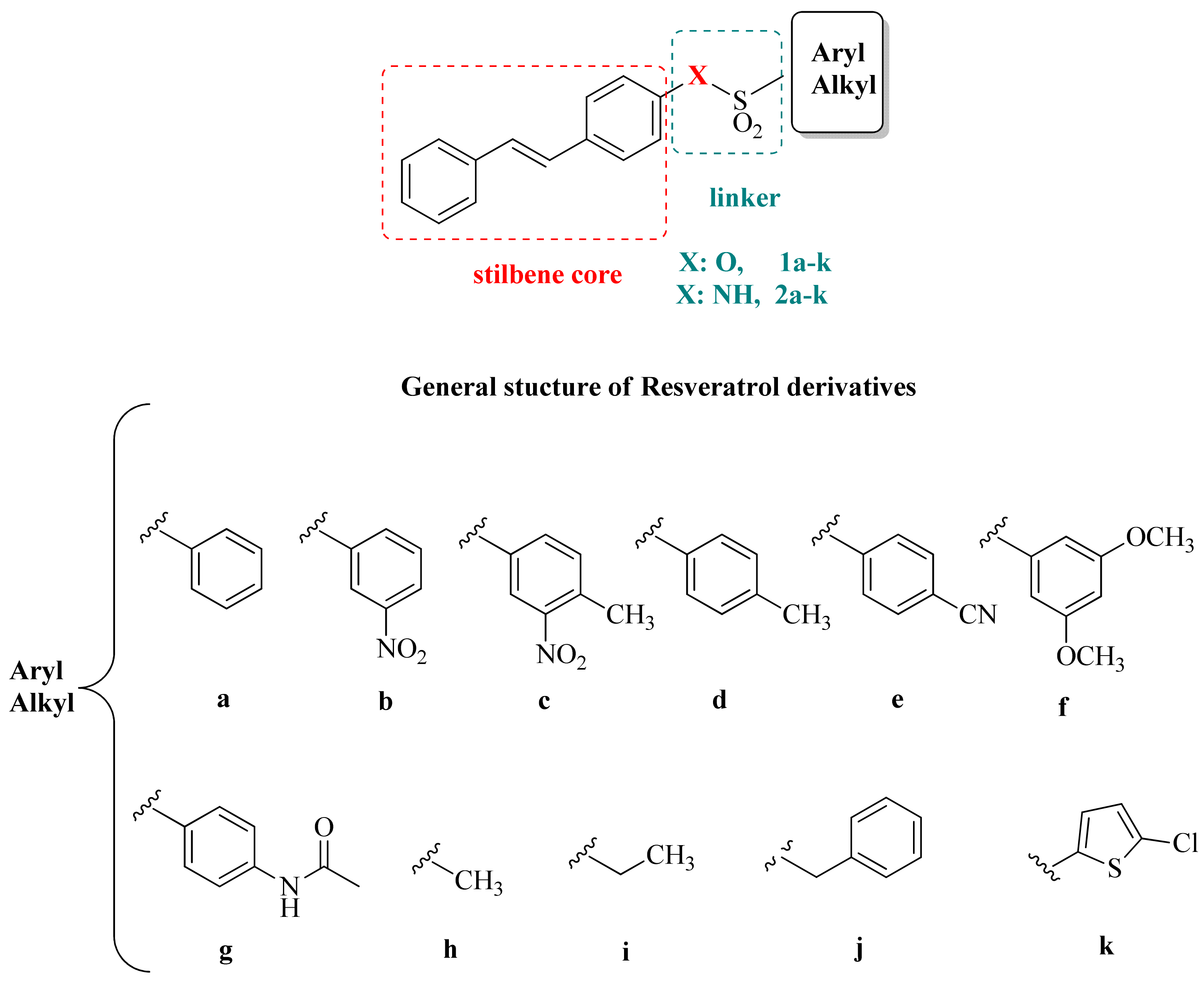
2.1. Chemistry
2.2. Aromatase Inhibition Studies
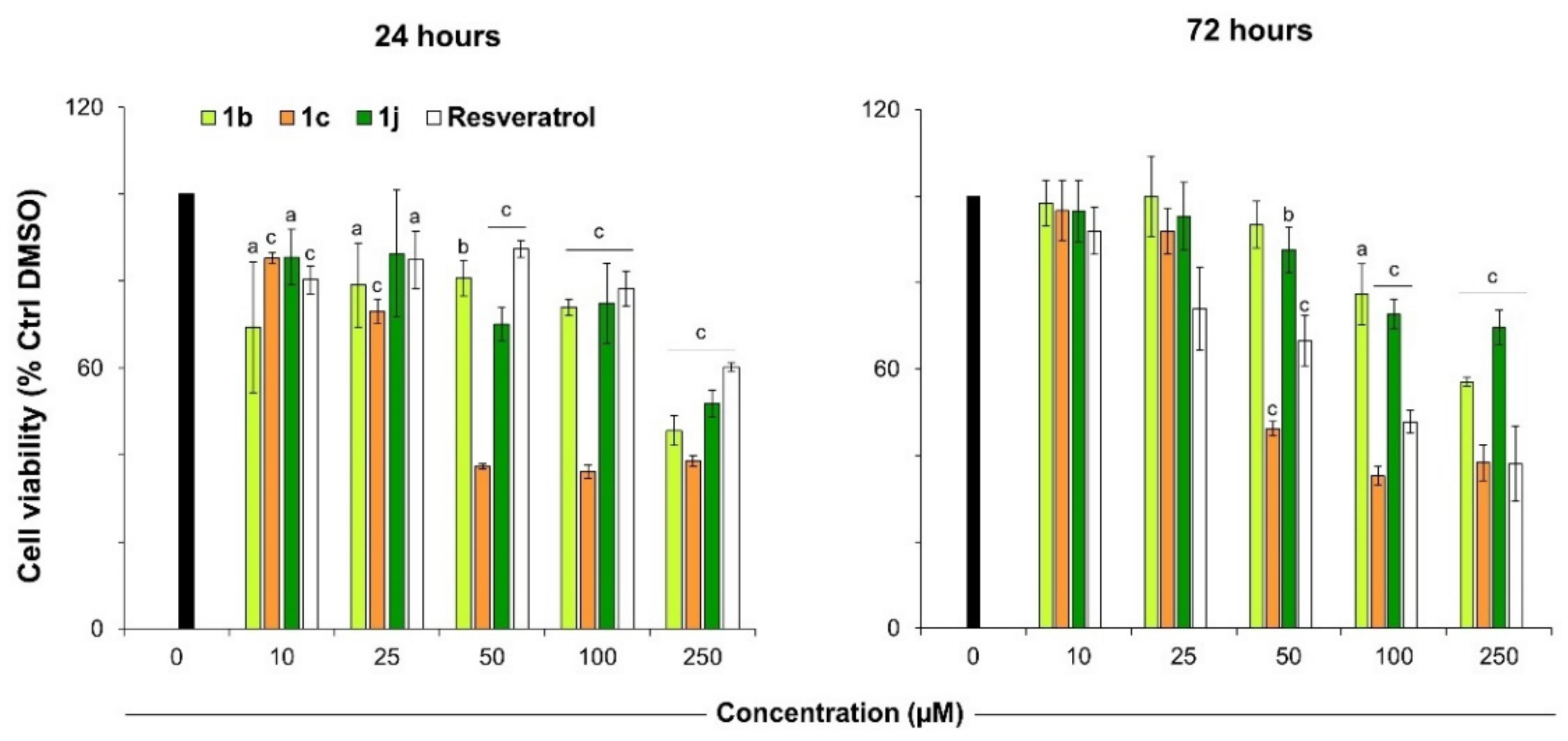
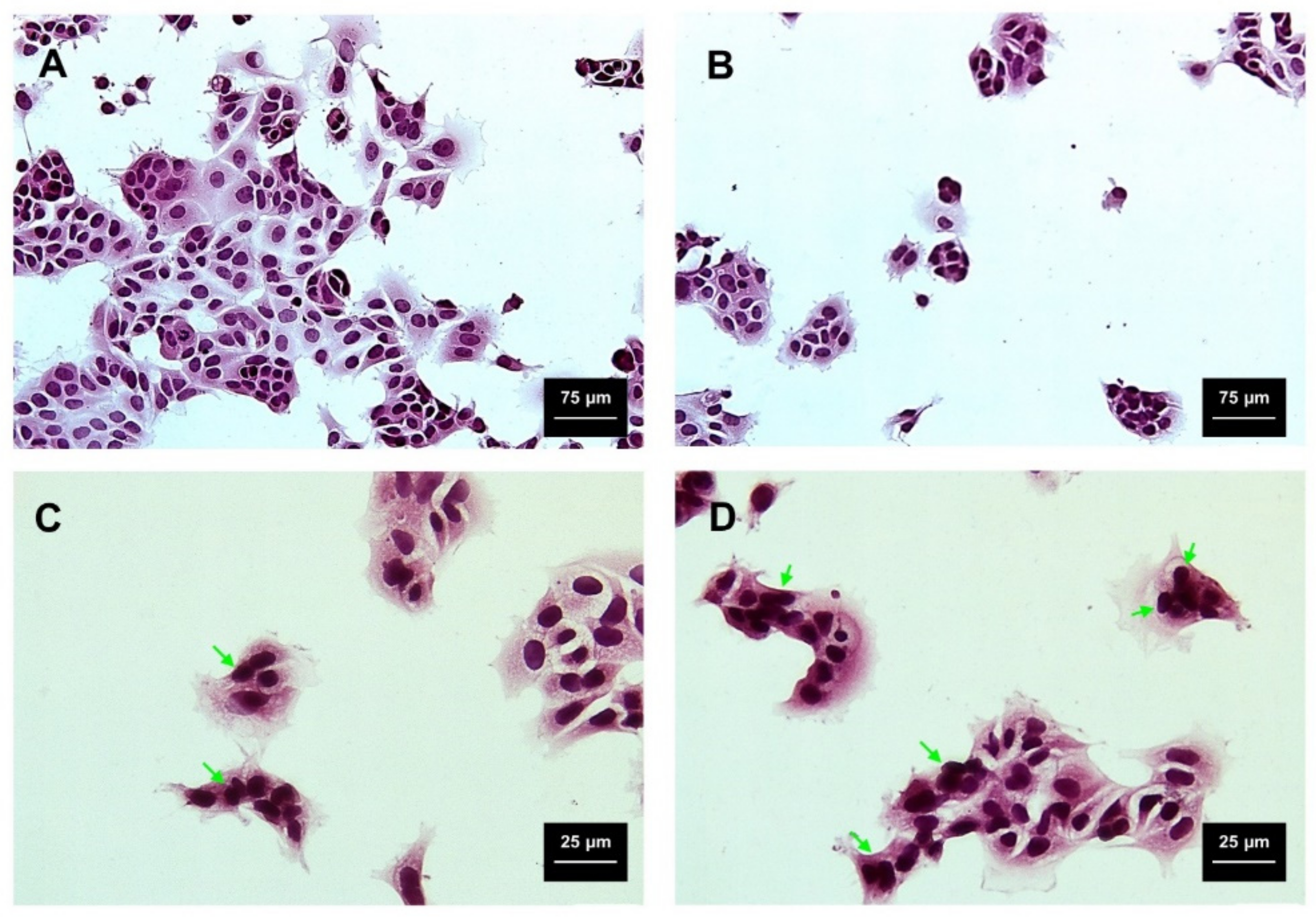
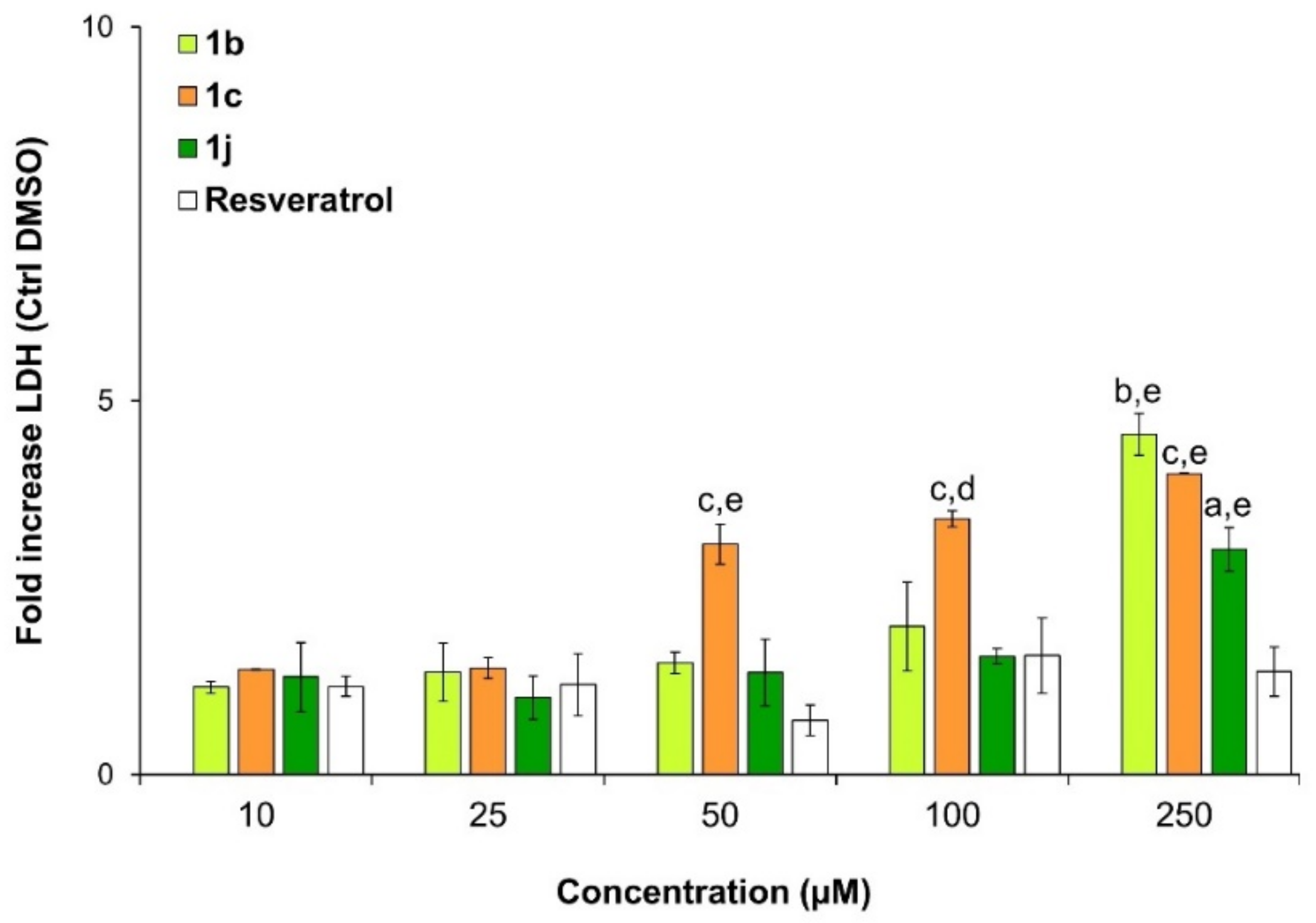
2.3. Viability and Cytotoxicity Assay
2.4. Molecular Docking Studies
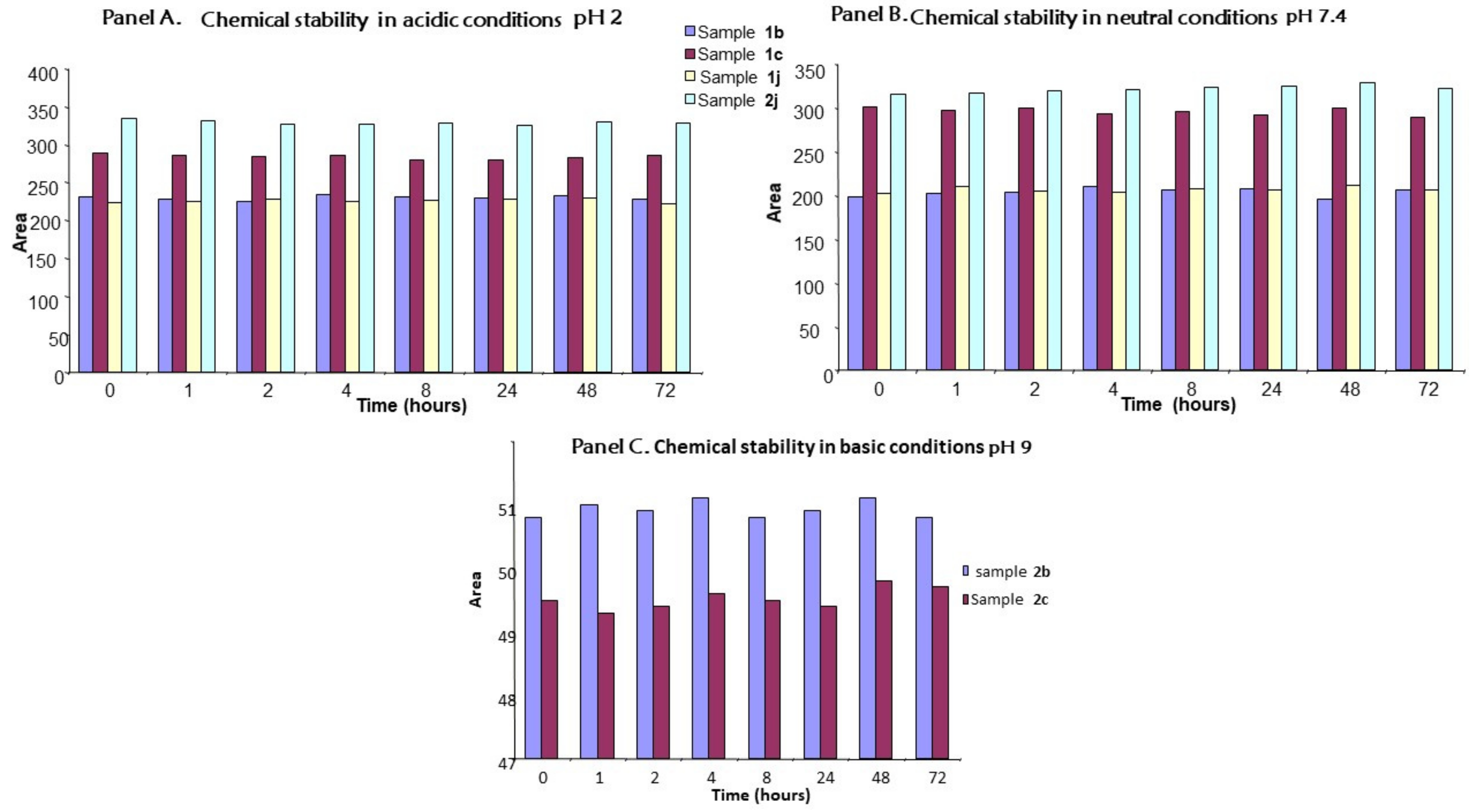
2.5. Chemical Stability
3. Materials and Methods
3.1. Chemistry
3.2. General Method for the Synthesis of Sulfonates 1a–k
3.2.1. 4-[(E)-2-Phenylvinyl]phenylbenzenesulfonate 1a
3.2.2. 4-[(E)-2-Phenylvinyl]phenyl 3-nitrobenzenesulfonate 1b
3.2.3. 4-[(E)-2-Phenylvinyl]phenyl 4-methyl-3-nitrobenzenesulfonate 1c
3.2.4. 4-[(E)-2-Phenylvinyl]phenyl 4-methylbenzenesulfonate 1d
3.2.5. 4-[(E)-2-Phenylvinyl]phenyl 4-cyanobenzenesulfonate 1e
3.2.6. 4-[(E)-2-Phenylvinyl]phenyl 4-(acetylamino)benzenesulfonate 1f
3.2.7. 4-[(E)-2-Phenylvinyl]phenyl 2,4-dimethoxybenzenesulfonate 1g
3.2.8. 4-[(E)-2-Phenylvinyl]phenylmethanesulfonate 1h
3.2.9. 4-[(E)-2-Phenylvinyl]phenyl ethanesulfonate 1i
3.2.10. 4-[(E)-2-Phenylvinyl]phenyl phenylmethanesulfonate 1j
3.2.11. 4-[(E)-2-Phenylvinyl]phenyl 5-chlorothiophene-2-sulfonate 1k
3.3. General Method for the Synthesis of Sulfonamides 2a–k
3.3.1. N-{4-[(E)-2-Phenylvinyl]phenyl}benzenesulfonamide 2a
3.3.2. 3-Nitro-N-{4-[(E)-2-phenylvinyl]phenyl}benzenesulfonamide 2b
3.3.3. 4-Methyl-3-nitro-N-{4-[(E)-2-phenylvinyl]phenyl}benzenesulfonamide 2c
3.3.4. 4-Methyl-N-{4-[(E)-2-phenylvinyl]phenyl}benzenesulfonamide 2d
3.3.5. 4-Cyano-N-{4-[(E)-2-phenylvinyl]phenyl}benzenesulfonamide 2e
3.3.6. N-{4-[({4-[(E)-2-Phenylvinyl]phenyl}amino)sulfonyl]phenyl}acetamide 2f
3.3.7. 2,4-Dimethoxy-N-{4-[(E)-2-phenylvinyl]phenyl}benzenesulfonamide 2g
3.3.8. N-{4-[(E)-2-Phenylvinyl]phenyl}methanesulfonamide 2h
3.3.9. N-{4-[(E)-2-Phenylvinyl]phenyl}ethanesulfonamide 2i
3.3.10. 1-Phenyl-N-{4-[(E)-2-phenylvinyl]phenyl}methanesulfonamide 2j
3.3.11. 5-Chloro-N-{4-[(E)-2-phenylvinyl]phenyl}thiophene-2-sulfonamide 2k
3.4. Biological Assays
3.4.1. Aromatase Activity Inhibition Assay and Calculation of the IC50
3.4.2. Cell Viability Evaluation
Cell Viability Assay (MTT)
Cytotoxicity Test (Lactate Dehydrogenase Release)
Hematoxylin/Eosin Staining
Statistics
3.5. Molecular Modeling
3.6. Chemical Stability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sainsbury, R. The development of endocrine therapy for women with breast cancer. Cancer Treat. Rev. 2013, 39, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhou, L.; Shangguan, A.J.; Bulun, S.E. Aromatase expression and regulation in breast and endometrial cancer. J. Mol. Endocrinol. 2016, 57, R19–R33. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I. Shagufta Recent developments in steroidal and nonsteroidal aromatase inhibitors for the chemoprevention of estrogen-dependent breast cancer. Eur. J. Med. Chem. 2015, 102, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.R.; Barmade, M.; Tamboli, R.S.; Murumkar, P. Developing steroidal aromatase inhibitors-an effective armament to win the battle against breast cancer. Eur. J. Med. Chem. 2015, 105, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Xiao, X.; Huang, C.; Yuan, Y.; Tang, D.; Dai, X.; Zeng, X. Potent aromatase inhibitors and molecular mechanism of inhibitory action. Eur. J. Med. Chem. 2018, 143, 426–437. [Google Scholar] [CrossRef]
- Favia, A.D.; Nicolotti, O.; Stefanachi, A.; Leonetti, F.; Carotti, A. Computational methods for the design of potent aromatase inhibitors. Expert Opin. Drug Discov. 2013, 8, 395–409. [Google Scholar] [CrossRef]
- Adhikari, N.; Amin, S.A.; Saha, A.; Jha, T. Combating breast cancer with non-steroidal aromatase inhibitors (NSAIs): Understanding the chemico-biological interactions through comparative SAR/QSAR study. Eur. J. Med. Chem. 2017, 137, 365–438. [Google Scholar] [CrossRef]
- Xie, H.; Qiu, K.; Xie, X. Pharmacophore modeling, virtual screening, and 3D-QSAR studies on a series of non-steroidal aromatase inhibitors. Med. Chem. Res. 2015, 24, 1901–1915. [Google Scholar] [CrossRef]
- De Araújo, F.F.; de Paulo Farias, D.; Neri-Numa, I.A.; Pastore, G.M. Polyphenols and their applications: An approach in food chemistry and innovation potential. Food Chem. 2020, 338, 127535. [Google Scholar] [CrossRef]
- Ratre, P.; Mishra, K.; Dubey, A.; Vyas, A.; Jain, A.; Thareja, S. Aromatase Inhibitors for the Treatment of Breast Cancer: A Journey from the Scratch. Anti-Cancer Agents Med. Chem. 2020, 20, 1994–2004. [Google Scholar] [CrossRef]
- Jayaprakash, J.S.; Gowda, D.V.; Kulkarni, P.K. Therapeutic application of Resveratrol in human diseases. Int. J. Res. Pharm. Sci. 2020, 11, 1447–1456. [Google Scholar] [CrossRef]
- Ogas, T.; Kondratyuk, T.P.; Pezzuto, J.M. Resveratrol analogs: Promising chemopreventive agents. Ann. N. Y. Acad. Sci. 2013, 1290, 21–29. [Google Scholar] [CrossRef]
- Singh, C.K.; Ndiaye, M.A.; Ahmad, N. Resveratrol and cancer: Challenges for clinical translation. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 1178–1185. [Google Scholar] [CrossRef]
- Xu, Q.-H.; Xiao, Y.; Li, X.-Q.; Fan, L.; Zhou, C.-C.; Cheng, L.; Jiang, Z.-D.; Wang, G.-H. Resveratrol Counteracts Hypoxia-Induced Gastric Cancer Invasion and EMT through Hedgehog Pathway Suppression. Anti-Cancer Agents Med. Chem. 2020, 20, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Delmas, D.; Aires, V.; Limagne, E.; Dutartre, P.; Mazué, F.; Ghiringhelli, F.; Latruffe, N. Transport, stability, and biological activity of resveratrol. Ann. N. Y. Acad. Sci. 2011, 1215, 48–59. [Google Scholar] [CrossRef]
- Neves, A.R.; Lúcio, M.; Lima, J.L.C.; Reis, S. Resveratrol in Medicinal Chemistry: A Critical Review of its Pharmacokinetics, Drug-Delivery, and Membrane Interactions. Curr. Med. Chem. 2012, 19, 1663–1681. [Google Scholar] [CrossRef] [PubMed]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E., Jr.; Walle, U.K. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab. Dispos. 2004, 32, 1377–1382. [Google Scholar] [CrossRef]
- De Filippis, B.; Ammazzalorso, A.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Amoroso, R. Anticancer activity of stilbene-based derivatives. ChemMedChem 2017, 12, 558–570. [Google Scholar] [CrossRef]
- Kondratyuk, T.P.; Park, E.-J.; Marler, L.E.; Ahn, S.; Yuan, Y.; Choi, Y.; Yu, R.; van Breemen, R.; Sun, B.; Hoshino, J.; et al. Resveratrol derivatives as promising chemopreventive agents with improved potency and selectivity. Mol. Nutr. Food Res. 2011, 55, 1249–1265. [Google Scholar] [CrossRef] [PubMed]
- Orsini, F.; Verotta, L.; Klimo, K.; Gerhäuser, C. Synthesis of Resveratrol Derivatives andIn VitroScreening for Potential Cancer Chemopreventive Activities. Arch. Pharm. 2016, 349, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Mayhoub, A.S.; Marler, L.; Kondratyuk, T.P.; Park, E.-J.; Pezzuto, J.M.; Cushman, M. Optimization of the aromatase inhibitory activities of pyridylthiazole analogues of resveratrol. Bioorg. Med. Chem. 2012, 20, 2427–2434. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, E.; Rupiani, S.; Guidotti, L.; Recanatini, M.; Roberti, M. The Use of Stilbene Scaffold in Medicinal Chemistry and Multi- Target Drug Design. Curr. Med. Chem. 2016, 23, 2439–2489. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Sarkar, N.; Biswas, J.; Bishayee, A. Resveratrol for breast cancer prevention and therapy: Preclinical evidence and molecular mechanisms. Semin. Cancer Biol. 2016, 40–41, 209–232. [Google Scholar] [CrossRef] [PubMed]
- Chottanapund, S.; van Duursen, M.; Navasumrit, P.; Hunsonti, P.; Timtavorn, S.; Ruchirawat, M.; Berg, M.V.D. Anti-aromatase effect of resveratrol and melatonin on hormonal positive breast cancer cells co-cultured with breast adipose fibroblasts. Toxicol. In Vitro 2014, 28, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Hoshino, J.; Jermihov, K.; Marler, L.; Pezzuto, J.M.; Mesecar, A.; Cushman, M. Design, synthesis, and biological evaluation of resveratrol analogues as aromatase and quinone reductase 2 inhibitors for chemoprevention of cancer. Bioorg. Med. Chem. 2010, 18, 5352–5366. [Google Scholar] [CrossRef]
- Supuran, C.T. Special Issue: Sulfonamides. Molecules 2017, 22, 1642. [Google Scholar] [CrossRef] [PubMed]
- Gul, H.I.; Yamali, C.; Sakagami, H.; Angeli, A.; Leitans, J.; Kazaks, A.; Tars, K.; Ozgun, D.O.; Supuran, C.T. New anticancer drug candidates sulfonamides as selective hCA IX or hCA XII inhibitors. Bioorg. Chem. 2018, 77, 411–419. [Google Scholar] [CrossRef]
- Riaz, S.; Khan, I.U.; Bajda, M.; Ashraf, M.; Ain, Q.-U.; Shaukat, A.; Rehman, T.U.; Mutahir, S.; Hussain, S.; Mustafa, G.; et al. Pyridine sulfonamide as a small key organic molecule for the potential treatment of type-II diabetes mellitus and Alzheimer’s disease: In Vitro studies against yeast α-glucosidase, acetylcholinesterase and butyrylcholinesterase. Bioorg. Chem. 2015, 63, 64–71. [Google Scholar] [CrossRef]
- Zajdel, P.; Partyka, A.; Marciniec, K.; Bojarski, A.J.; Pawlowski, M.; Wesolowska, A. Quinoline- and isoquinoline-sulfonamide analogs of aripiprazole: Novel antipsychotic agents? Future Med. Chem. 2014, 6, 57–75. [Google Scholar] [CrossRef]
- Mutahir, S.; Jończyk, J.; Bajda, M.; Khan, I.U.; Khan, M.A.; Ullah, N.; Ashraf, M.; Ain, Q.U.; Riaz, S.; Hussain, S.; et al. Novel biphenyl bis -sulfonamides as acetyl and butyrylcholinesterase inhibitors: Synthesis, biological evaluation and molecular modeling studies. Bioorg. Chem. 2016, 64, 13–20. [Google Scholar] [CrossRef]
- Khan, F.A.; Mushtaq, S.; Naz, S.; Farooq, U.; Zaidi, A.; Bukhari, S.M.; Rauf, A.; Mubarak, M.S. Sulfonamides as Potential Bioactive Scaffolds. Curr. Org. Chem. 2018, 22, 818–830. [Google Scholar] [CrossRef]
- Nocentini, A.; Supuran, C.T. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin. Drug Discov. 2019, 14, 1175–1197. [Google Scholar] [CrossRef]
- Mustafa, M.; El Kardocy, A.; Mostafa, Y.A. Development of new hetero-steroid hybrids with antiproliferative activity against MCF-7 breast cancer cells. Mon. Chem.-Chem. Mon. 2021, 152, 137–149. [Google Scholar] [CrossRef]
- Ghorab, M.M.; Alsaid, M.S.; Samir, N.; Abdel-Latif, G.A.; Soliman, A.M.; Ragab, F.A.; El Ella, D.A.A. Aromatase inhibitors and apoptotic inducers: Design, synthesis, anticancer activity and molecular modeling studies of novel phenothiazine derivatives carrying sulfonamide moiety as hybrid molecules. Eur. J. Med. Chem. 2017, 134, 304–315. [Google Scholar] [CrossRef]
- Fantacuzzi, M.; De Filippis, B.; Gallorini, M.; Ammazzalorso, A.; Giampietro, L.; Maccallini, C.; Aturki, Z.; Donati, E.; Ibrahim, R.S.; Shawky, E.; et al. Synthesis, biological evaluation, and docking study of indole aryl sulfonamides as aromatase inhibitors. Eur. J. Med. Chem. 2020, 185, 111815. [Google Scholar] [CrossRef] [PubMed]
- Ammazzalorso, A.; Gallorini, M.; Fantacuzzi, M.; Gambacorta, N.; De Filippis, B.; Giampietro, L.; Maccallini, C.; Nicolotti, O.; Cataldi, A.; Amoroso, R. Design, synthesis and biological evaluation of imidazole and triazole-based carbamates as novel aromatase inhibitors. Eur. J. Med. Chem. 2021, 211, 113115. [Google Scholar] [CrossRef] [PubMed]
- Leechaisit, R.; Pingaew, R.; Prachayasittikul, V.; Worachartcheewan, A.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Synthesis, molecular docking, and QSAR study of bis-sulfonamide derivatives as potential aromatase inhibitors. Bioorg. Med. Chem. 2019, 27, 115040. [Google Scholar] [CrossRef]
- Pingaew, R.; Mandi, P.; Prachayasittikul, V.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Synthesis, molecular docking, and QSAR study of sulfonamide-based indoles as aromatase inhibitors. Eur. J. Med. Chem. 2018, 143, 1604–1615. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.M.; Barreiro, L.M.L.A.E.J. Bioisosterism: A Useful Strategy for Molecular Modification and Drug Design. Curr. Med. Chem. 2005, 12, 23–49. [Google Scholar] [CrossRef]
- El-Gamal, M.I.; Semreen, M.H.; Foster, P.A.; Potter, B.V. Design, synthesis, and biological evaluation of new arylamide derivatives possessing sulfonate or sulfamate moieties as steroid sulfatase enzyme inhibitors. Bioorg. Med. Chem. 2016, 24, 2762–2767. [Google Scholar] [CrossRef][Green Version]
- Foster, P.A.; Newman, S.P.; Chander, S.K.; Stengel, C.; Jhalli, R.; Woo, L.L.W.; Potter, B.V.L.; Reed, M.J.; Purohit, A. In vivo Efficacy of STX213, A Second-Generation Steroid Sulfatase Inhibitor, for Hormone-Dependent Breast Cancer Therapy. Clin. Cancer Res. 2006, 12, 5543–5549. [Google Scholar] [CrossRef]
- Hoshino, J.; Park, E.-J.; Kondratyuk, T.P.; Marler, L.; Pezzuto, J.M.; van Breemen, R.B.; Mo, S.; Li, Y.; Cushman, M. Selective Synthesis and Biological Evaluation of Sulfate-Conjugated Resveratrol Metabolites. J. Med. Chem. 2010, 53, 5033–5043. [Google Scholar] [CrossRef]
- Mahesh, R.; Nayak, V.L.; Babu, K.S.; Riyaz, S.; Shaik, T.B.; Kumar, G.B.; Mallipeddi, P.L.; Reddy, C.R.; Shekar, K.C.; Jose, J.; et al. Design, Synthesis, and in vitro and in vivo Evaluations of (Z)-3,4,5-Trimethoxystyrylbenzenesulfonamides/sulfonates as Highly Potent Tubulin Polymerization Inhibitors. ChemMedChem 2017, 12, 678–700. [Google Scholar] [CrossRef][Green Version]
- Leporini, L.; Giampietro, L.; Amororso, R.; Ammazzalorso, A.; Fantacuzzi, M.; Menghini, L.; Maccallini, C.; Ferrante, C.; Orlando, G.; De Filippis, B. In vitro protective effects of resveratrol and stilbene alkanoic derivatives on induced oxidative stress on C2C12 and MCF7 cells. J. Boil. Regul. Homeost. Agents 2017, 31, 589–601. [Google Scholar]
- Amoroso, R.; Leporini, L.; Cacciatore, I.; Marinelli, L.; Ammazzalorso, A.; Bruno, I.; De Filippis, B.; Fantacuzzi, M.; Maccallini, C.; Menghini, L.; et al. Synthesis, Characterization and Evaluation of Gemfibrozil-Stilbene Hybrid as Antioxidant Agent. Lett. Drug Des. Discov. 2018, 15, 1230–1238. [Google Scholar] [CrossRef]
- Giampietro, L.; Laghezza, A.; Cerchia, C.; Florio, R.; Recinella, L.; Capone, F.; Ammazzalorso, A.; Bruno, I.; De Filippis, B.; Fantacuzzi, M.; et al. Novel Phenyldiazenyl Fibrate Analogues as PPAR α/γ/δ Pan-Agonists for the Amelioration of Metabolic Syndrome. ACS Med. Chem. Lett. 2019, 10, 545–551. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, B.; Agamennone, M.; Ammazzalorso, A.; Bruno, I.; D’Angelo, A.; Di Matteo, M.; Fantacuzzi, M.; Giampietro, L.; Giancristofaro, A.; Maccallini, C.; et al. PPARα agonists based on stilbene and its bioisosteres: Biological evaluation and docking studies. MedChemComm 2015, 6, 1513–1517. [Google Scholar] [CrossRef]
- Ammazzalorso, A.; D’Angelo, A.; Giancristofaro, A.; De Filippis, B.; Di Matteo, M.; Fantacuzzi, M.; Giampietro, L.; Linciano, P.; Maccallini, C.; Amoroso, R. Fibrate-derived N-(methylsulfonyl)amides with antagonistic properties on PPARα. Eur. J. Med. Chem. 2012, 58, 317–322. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, B.; De Lellis, L.; Florio, R.; Ammazzalorso, A.; Amoia, P.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Amoroso, R.; Veschi, S.; et al. Synthesis and cytotoxic effects on pancreatic cancer cells of resveratrol analogs. Med. Chem. Res. 2019, 28, 984–991. [Google Scholar] [CrossRef]
- Di Filippo, E.S.; Giampietro, L.; De Filippis, B.; Balaha, M.; Ferrone, V.; Locatelli, M.; Pietrangelo, T.; Tartaglia, A.; Amoroso, R.; Fulle, S. Synthesis and Biological Evaluation of Halogenated E-Stilbenols as Promising Antiaging Agents. Molecules 2020, 25, 5770. [Google Scholar] [CrossRef] [PubMed]
- Di Fermo, P.; Di Lodovico, S.; Amoroso, R.; De Filippis, B.; D’Ercole, S.; Di Campli, E.; Cellini, L.; Di Giulio, M. Searching for New Tools to Counteract the Helicobacter pylori Resistance: The Positive Action of Resveratrol Derivatives. Antibiotics 2020, 9, 891. [Google Scholar] [CrossRef]
- Di Matteo, M.; Ammazzalorso, A.; Andreoli, F.; Caffa, I.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Nencioni, A.; Parenti, M.D.; et al. Synthesis and biological characterization of 3-(imidazol-1-ylmethyl)piperidine sulfonamides as aromatase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 3192–3194. [Google Scholar] [CrossRef]
- Maccallini, C.; Gallorini, M.; Sisto, F.; Akdemir, A.; Ammazzalorso, A.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Carradori, S.; Cataldi, A.; et al. New azolyl-derivatives as multitargeting agents against breast cancer and fungal infections: Synthesis, biological evaluation and docking study. J. Enzym. Inhib. Med. Chem. 2021, 36, 1632–1645. [Google Scholar] [CrossRef] [PubMed]
- Giampietro, L.; Gallorini, M.; Gambacorta, N.; Ammazzalorso, A.; De Filippis, B.; Della Valle, A.; Fantacuzzi, M.; Maccallini, C.; Mollica, A.; Cataldi, A.; et al. Synthesis, structure-activity relationships and molecular docking studies of phenyldiazenyl sulfonamides as aromatase inhibitors. Eur. J. Med. Chem. 2021, 224, 113737. [Google Scholar] [CrossRef] [PubMed]
- Amaral, C.; Varela, C.; Azevedo, M.; da Silva, E.T.; Roleira, F.; Chen, S.; Correia-Da-Silva, G.; Teixeira, N. Effects of steroidal aromatase inhibitors on sensitive and resistant breast cancer cells: Aromatase inhibition and autophagy. J. Steroid Biochem. Mol. Biol. 2013, 135, 51–59. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A. Exploring QSAR: Fundamentals and Applications in Chemistry and Biology (ACS Professional Reference Book); American Chemical Society: Washington, DC, USA, 1995. [Google Scholar]
- Nicolotti, O.; Catto, M.; Giangreco, I.; Barletta, M.; Leonetti, F.; Stefanachi, A.; Pisani, L.; Cellamare, S.; Tortorella, P.; Loiodice, F.; et al. Design, synthesis and biological evaluation of 5-hydroxy, 5-substituted-pyrimidine-2,4,6-triones as potent inhibitors of gelatinases MMP-2 and MMP-9. Eur. J. Med. Chem. 2012, 58, 368–376. [Google Scholar] [CrossRef]
- Veschi, S.; Carradori, S.; De Lellis, L.; Florio, R.; Brocco, D.; Secci, D.; Guglielmi, P.; Spano, M.; Sobolev, A.P.; Cama, A. Synthesis and evaluation of a large library of nitroxoline derivatives as pancreatic cancer antiproliferative agents. J. Enzym. Inhib. Med. Chem. 2020, 35, 1331–1344. [Google Scholar] [CrossRef]
- He, D.-X.; Wu, X.-L.; Lu, C.-X.; Gu, X.-T.; Zhang, G.-Y.; Ma, X.; Liu, D.-Q. Genome-wide analysis of the three-way interplay among gene expression, estrogen receptor expression and chemotherapeutic sensitivity in breast cancer. Oncol. Rep. 2017, 38, 3392–3402. [Google Scholar] [CrossRef] [PubMed]
- Simstein, R.; Burow, M.; Parker, A.; Weldon, C.; Beckman, B. Apoptosis, Chemoresistance, and Breast Cancer: Insights From the MCF-7 Cell Model System. Exp. Biol. Med. 2003, 228, 995–1003. [Google Scholar] [CrossRef]
- Howgate, D.J.; Gamie, Z.; Panteliadis, P.; Bhalla, A.; Mantalaris, A.; Tsiridis, E. The potential adverse effects of aromatase inhibitors on wound healing: In Vitro and in vivo evidence. Expert Opin. Drug Saf. 2009, 8, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Metzler, M.; Neumann, H.-G. Zur bedeutung chemisch-biologischer wechselwirkungen für die toxische und krebserzeugende wirkung aromatischer amine—III: Synthese und analytik einiger stoffwechselprodukte von trans-dimethylaminostilben, cis-4-dimethyl-aminostilbenund 4-dimethylaminobibenzyl. Tetrahedron 1971, 27, 2225–2246. [Google Scholar]
- Giampietro, L.; Gallorini, M.; De Filippis, B.; Amoroso, R.; Cataldi, A.; di Giacomo, V. PPAR-γ agonist GL516 reduces oxidative stress and apoptosis occurrence in a rat astrocyte cell line. Neurochem. Int. 2019, 126, 239–245. [Google Scholar] [CrossRef]
- Maccallini, C.; Arias, F.; Gallorini, M.; Amoia, P.; Ammazzalorso, A.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Cataldi, A.; Camacho, M.E.; et al. Antiglioma Activity of Aryl and Amido-Aryl Acetamidine Derivatives Targeting iNOS: Synthesis and Biological Evaluation. ACS Med. Chem. Lett. 2020, 11, 1470–1475. [Google Scholar] [CrossRef]
- Alagöz, M.; Özdemir, Z.; Uysal, M.; Carradori, S.; Gallorini, M.; Ricci, A.; Zara, S.; Mathew, B. Synthesis, Cytotoxicity and Anti-Proliferative Activity against AGS Cells of New 3(2H)-Pyridazinone Derivatives Endowed with a Piperazinyl Linker. Pharmaceuticals 2021, 14, 183. [Google Scholar] [CrossRef]
- Ghosh, D.; Griswold, J.; Erman, M.; Pangborn, W. Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nature 2009, 457, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2020-4: Protein Preparation Wizard; Epik, Schrödinger, LLC: New York, NY, USA; Impact, Schrödinger, LLC: New York, NY, USA, 2016; Prime, Schrödinger, LLC: New York, NY, USA, 2020.
- Schrödinger Release 2020-4: Semiempirical NDDO Protocol; Jaguar, Schrödinger, LLC: New York, NY, USA; MOPAC, Schrödinger, LLC: New York, NY, USA, 2021.
- Cho, A.E.; Guallar, V.; Berne, B.J.; Friesner, R. Importance of accurate charges in molecular docking: Quantum mechanical/molecular mechanical (QM/MM) approach. J. Comput. Chem. 2005, 26, 915–931. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-4: LigPrep; Schrödinger, LLC: New York, NY, USA, 2020.
- Mateos-Vivas, M.; Fanali, S.; Rodríguez-Gonzalo, E.; Carabias-Martínez, R.; Aturki, Z. Rapid determination of nucleotides in infant formula by means of nano-liquid chromatography. Electrophoresis 2016, 37, 1873–1880. [Google Scholar] [CrossRef] [PubMed]
- Zátopková, R.; Aturki, Z.; Bednář, P. Stereoisomer separation of flavanones and flavanone-7-O-glycosides by means of nanoliquid chromatography employing derivatized β-cyclodextrins as mobile-phase additive. J. Sep. Sci. 2020, 43, 3382–3390. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd | Aromatase IC50 (μM) a,b | MCF7 Cells IC50 (μM) c | ||
|---|---|---|---|---|
| 24 h | 72 h | |||
| 1b |  | 2.51 | 159.3 ± 9.9 | 240.3 ± 6.7 |
| 1c |  | 3.17 | 29.2 ± 1.9 | 39.9 ± 2.1 |
| 1j |  | 2.21 | >250 | >250 |
| RSV | 80 d | >250 | 83.0 ± 3.8 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fantacuzzi, M.; Gallorini, M.; Gambacorta, N.; Ammazzalorso, A.; Aturki, Z.; Balaha, M.; Carradori, S.; Giampietro, L.; Maccallini, C.; Cataldi, A.; et al. Design, Synthesis and Biological Evaluation of Aromatase Inhibitors Based on Sulfonates and Sulfonamides of Resveratrol. Pharmaceuticals 2021, 14, 984. https://doi.org/10.3390/ph14100984
Fantacuzzi M, Gallorini M, Gambacorta N, Ammazzalorso A, Aturki Z, Balaha M, Carradori S, Giampietro L, Maccallini C, Cataldi A, et al. Design, Synthesis and Biological Evaluation of Aromatase Inhibitors Based on Sulfonates and Sulfonamides of Resveratrol. Pharmaceuticals. 2021; 14(10):984. https://doi.org/10.3390/ph14100984
Chicago/Turabian StyleFantacuzzi, Marialuigia, Marialucia Gallorini, Nicola Gambacorta, Alessandra Ammazzalorso, Zeineb Aturki, Marwa Balaha, Simone Carradori, Letizia Giampietro, Cristina Maccallini, Amelia Cataldi, and et al. 2021. "Design, Synthesis and Biological Evaluation of Aromatase Inhibitors Based on Sulfonates and Sulfonamides of Resveratrol" Pharmaceuticals 14, no. 10: 984. https://doi.org/10.3390/ph14100984
APA StyleFantacuzzi, M., Gallorini, M., Gambacorta, N., Ammazzalorso, A., Aturki, Z., Balaha, M., Carradori, S., Giampietro, L., Maccallini, C., Cataldi, A., Nicolotti, O., Amoroso, R., & De Filippis, B. (2021). Design, Synthesis and Biological Evaluation of Aromatase Inhibitors Based on Sulfonates and Sulfonamides of Resveratrol. Pharmaceuticals, 14(10), 984. https://doi.org/10.3390/ph14100984