
Recent Developments in the Synthesis of β-Diketones



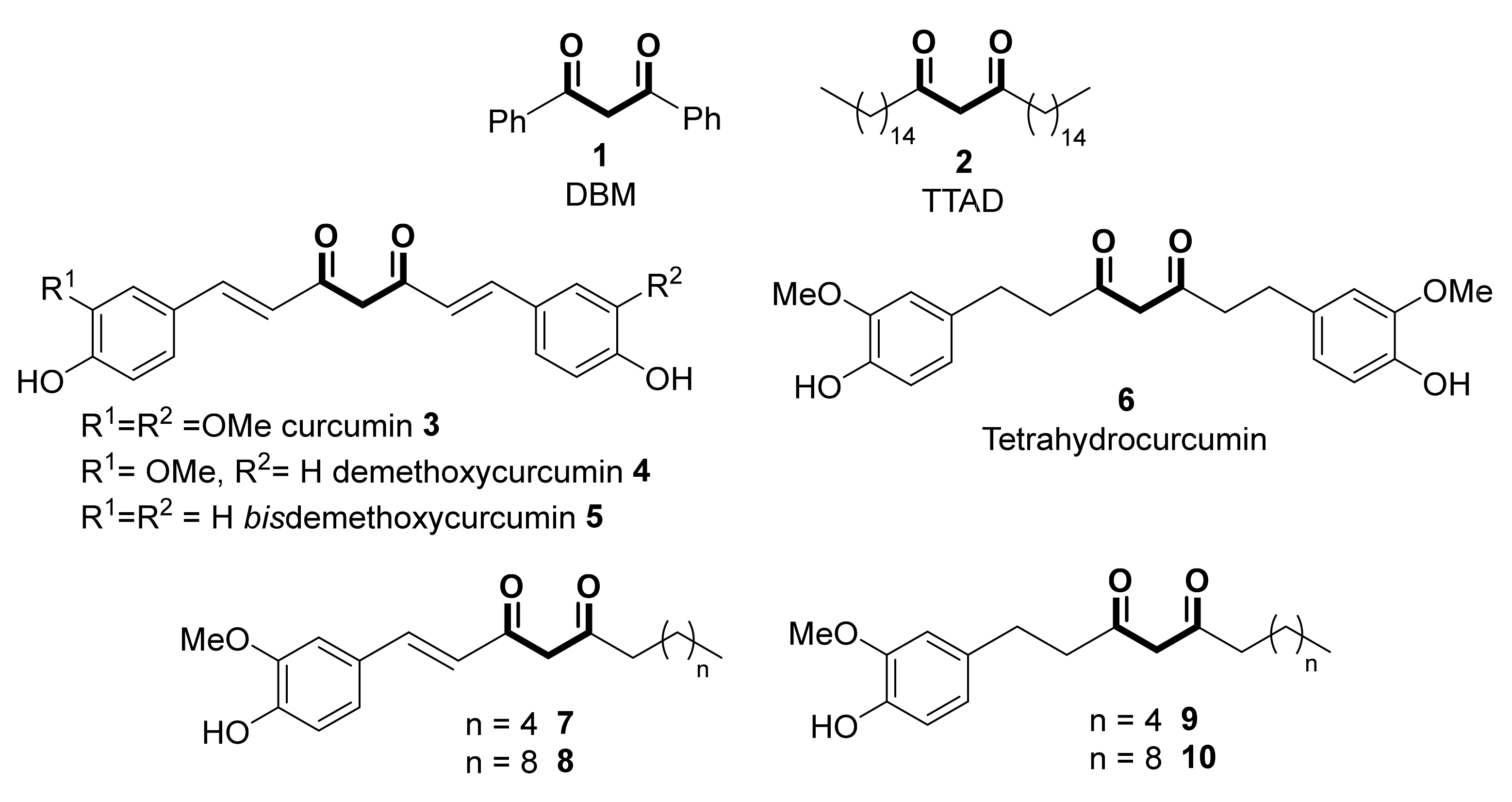{kind=link}
{kind=link}
{kind=link}
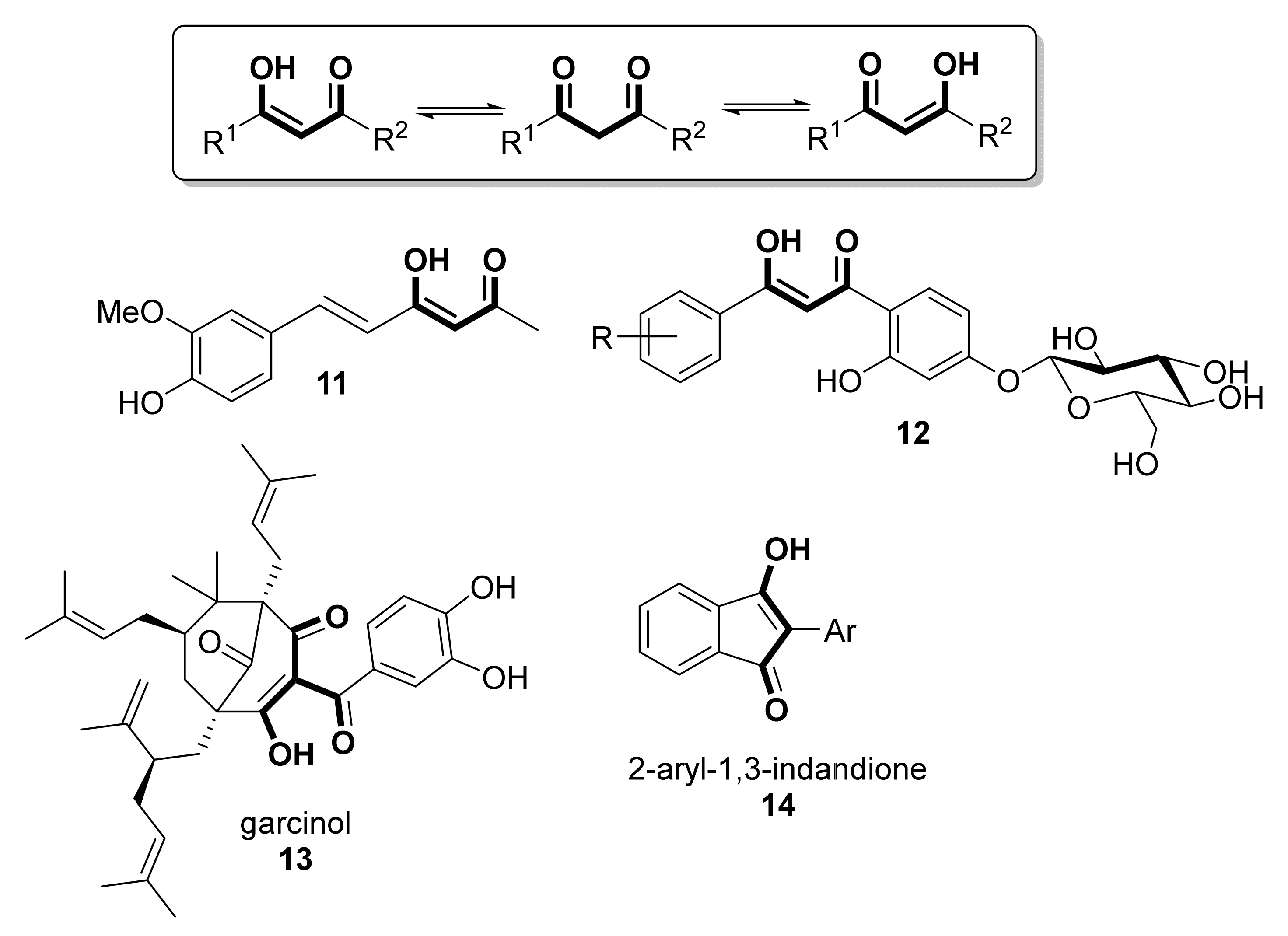
{kind=link}
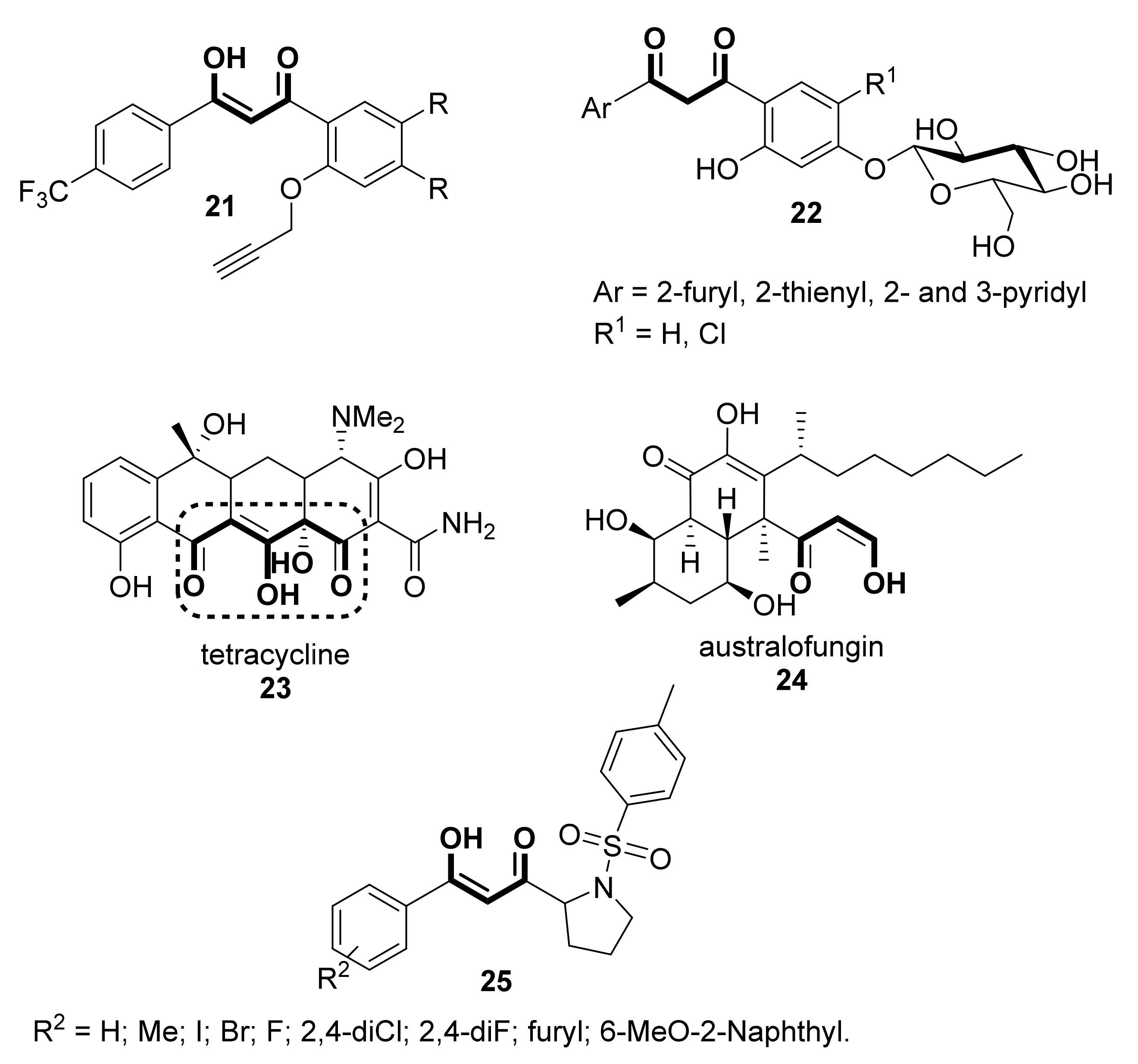
{kind=link}
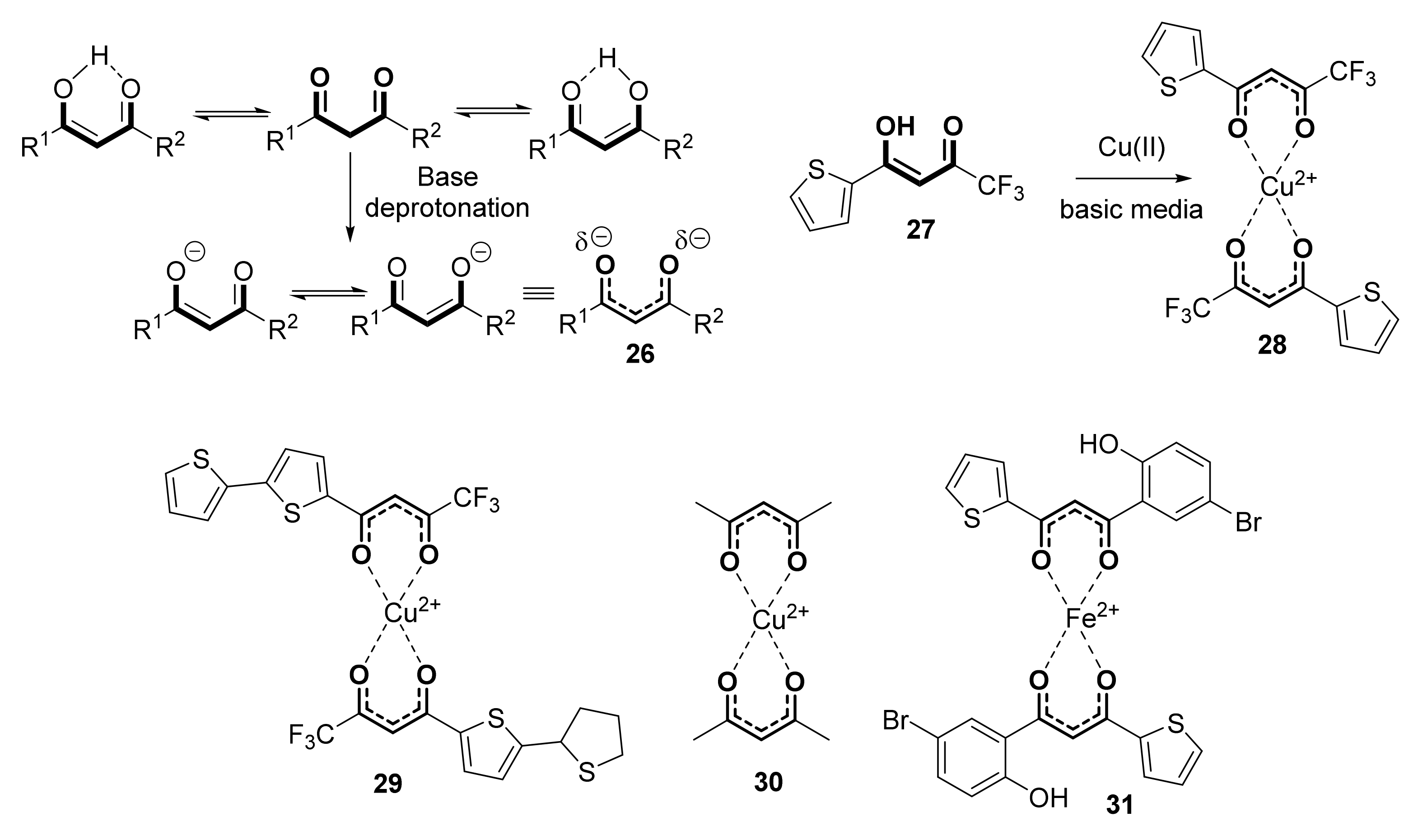
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Synthetic Methodologies for the Preparation of β-Diketones
2.1. Synthesis of β-Diketones by Claisen Approach
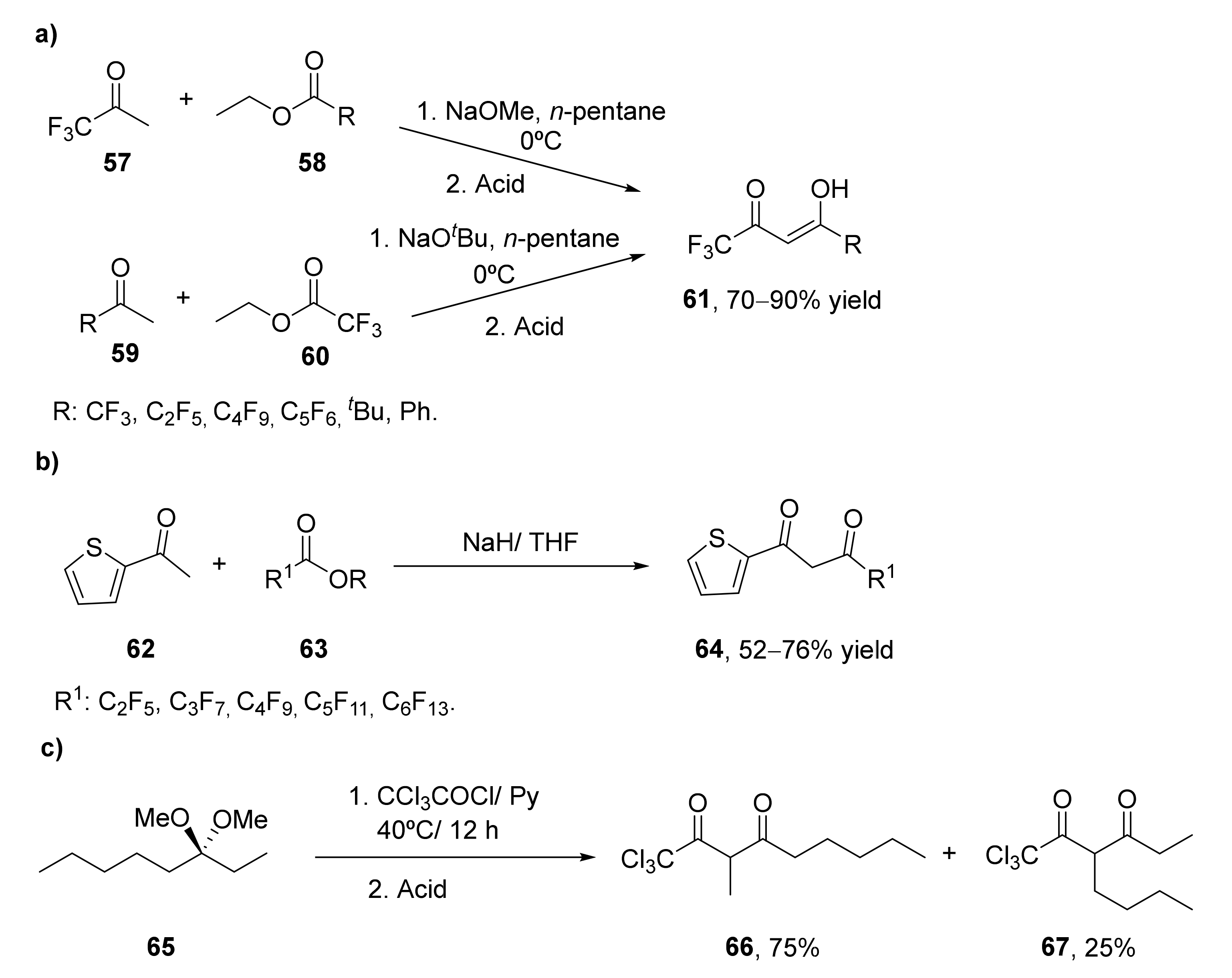
2.1.1. Preparation of Halogenated Diketones
2.1.2. Strategies for the Preparation of 1,3-Diketones Using Other Enolates
2.2. Synthesis of 1,3-Diketones by Hydration of Alkynones
2.3. Decarboxylative Coupling Reactions for the Synthesis of 1,3-Diketones
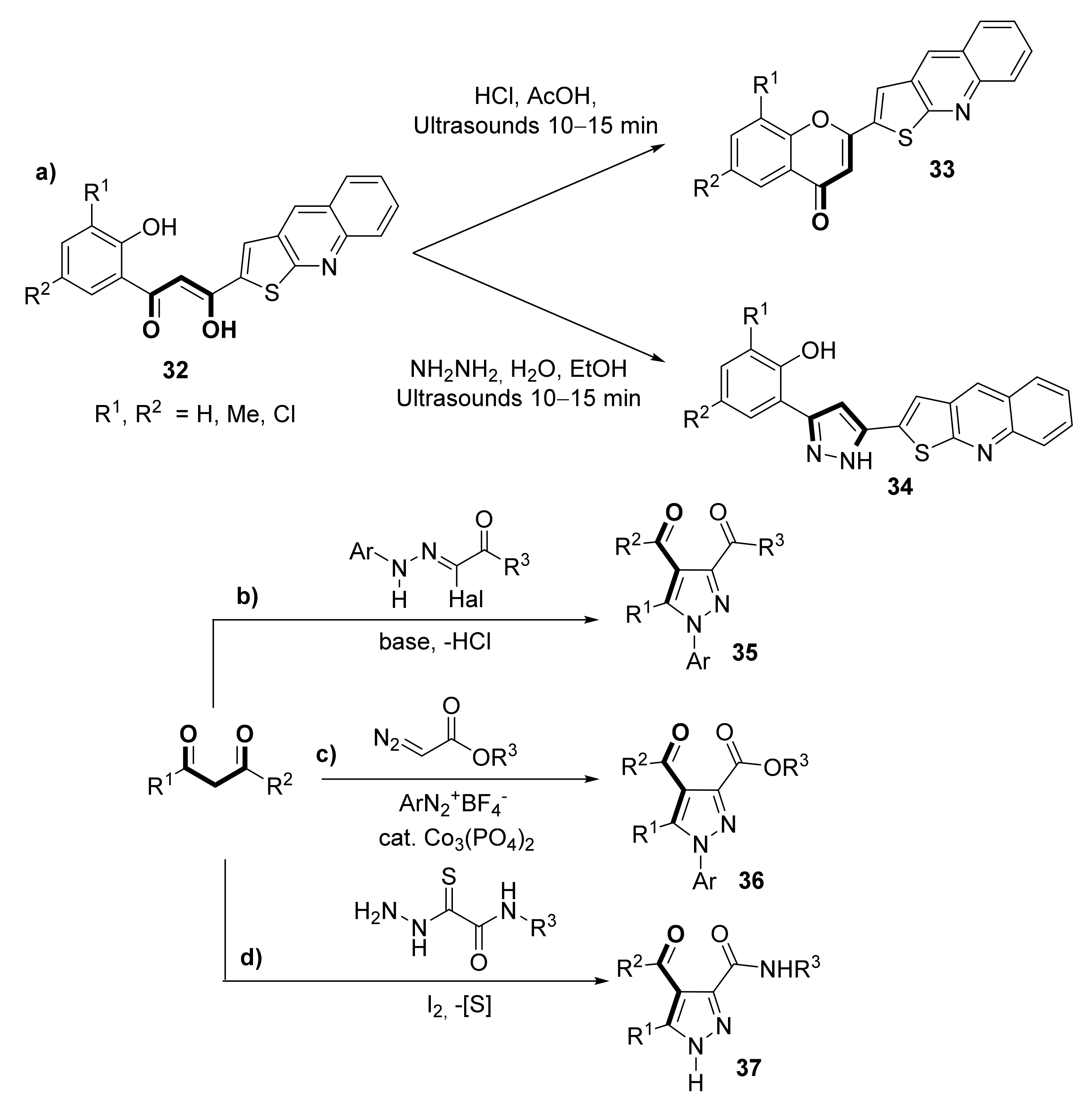
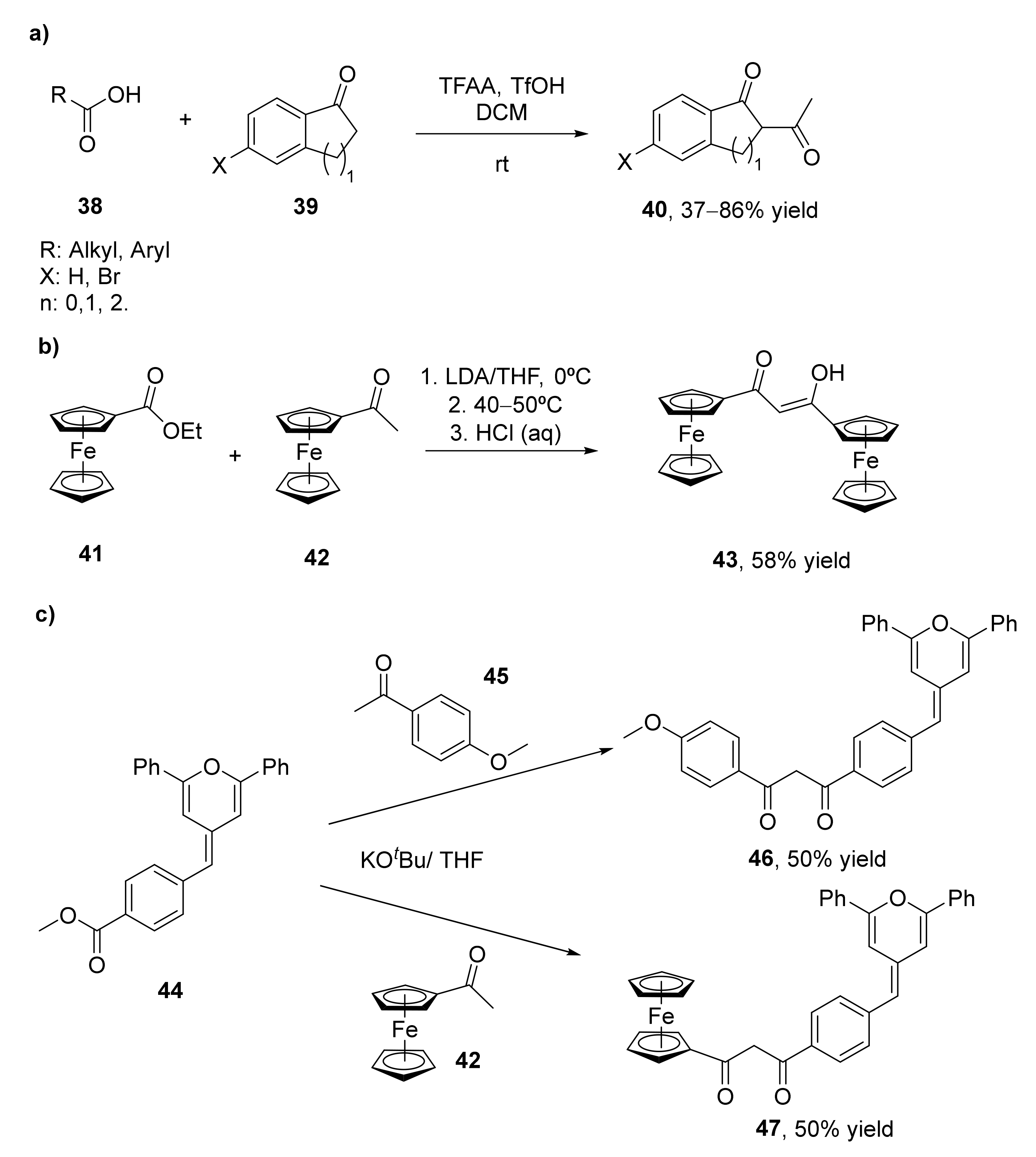
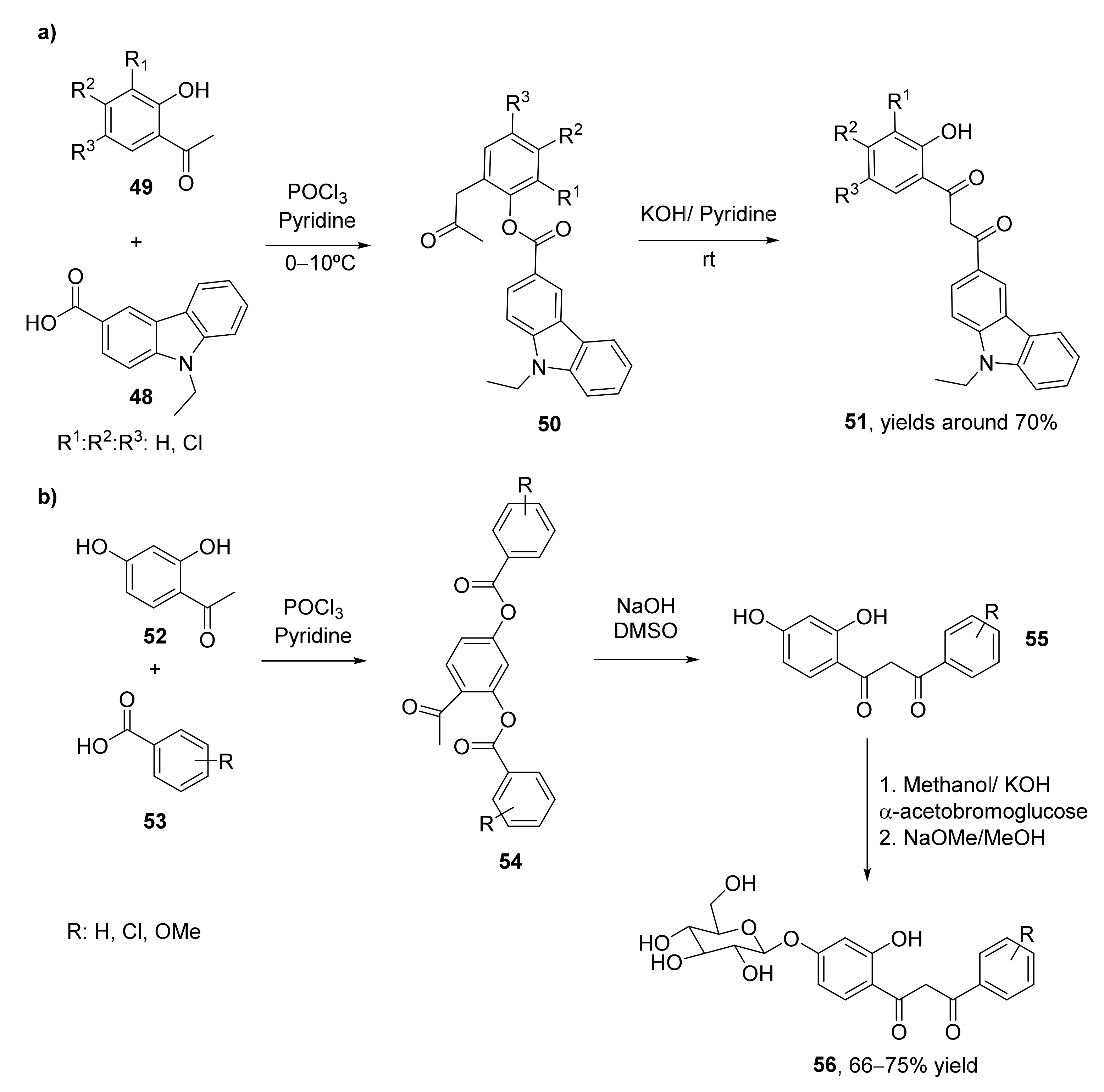
2.4. Miscellaneous Methodologies
3. Catalytic Methods for the Preparation of 1,3-Diketones
3.1. Biocatalytic Synthesis of 1,3-Diketones
3.2. Organocatalytic Methodologies for the Preparation 1,3-Diketones
3.3. Synthesis of 1,3-Diketones Using Metal-Based Catalysts
4. Synthesis by α-Functionalization of Other 1,3-Diketones
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brezani, V.; Šmejkal, K. Secondary metabolites isolated from the genus Eucalyptus. Curr. Trends Med. Chem. 2013, 7, 65–95. [Google Scholar]
- Gominho, J.; Lourenço, A.; Marques, A.V.; Pereira, H. An extensive study on the chemical diversity of lipophilic extractives from Eucalyptus globulus wood. Phytochemistry 2020, 180, 112520. [Google Scholar] [CrossRef]
- Mancia, M.D.; Reid, M.E.; DuBose, E.S.; Campbell, J.A.; Jackson, K.M. Qualitative identification of dibenzoylmethane in licorice root (Glycyrrhiza glabra) using gas chromatography-triple quadrupole mass spectrometry. Nat. Pro. Comm. 2014, 9, 91–94. [Google Scholar] [CrossRef]
- Brunschwig, C.; Collard, F.X.; Bianchini, J.P.; Raharivelomanana, P. Evaluation of chemical variability of cured vanilla beans (Vanilla tahitensis and Vanilla planifolia). Nat. Pro. Comm. 2009, 4, 1393–1400. [Google Scholar] [CrossRef] [Green Version]
- Schulz, S.; Arsene, C.; Tauber, M.; McNeil, J.N. Composition of lipids from sunflower pollen (Helianthus annuus). Phytochemistry 2000, 54, 325–336. [Google Scholar] [CrossRef]
- Jackson, K.M.; Frazier, M.C.; Mancia, M.D.; Shaw, R.N. Recent advances in the licorice root constituent dibenzoylmethane as a potential therapeutic option for cancer. In Studies in Natural Products Chemistry; Elsevier B.V.: Amsterdam, The Netherlands, 2019; Volume 63, pp. 1–19. [Google Scholar]
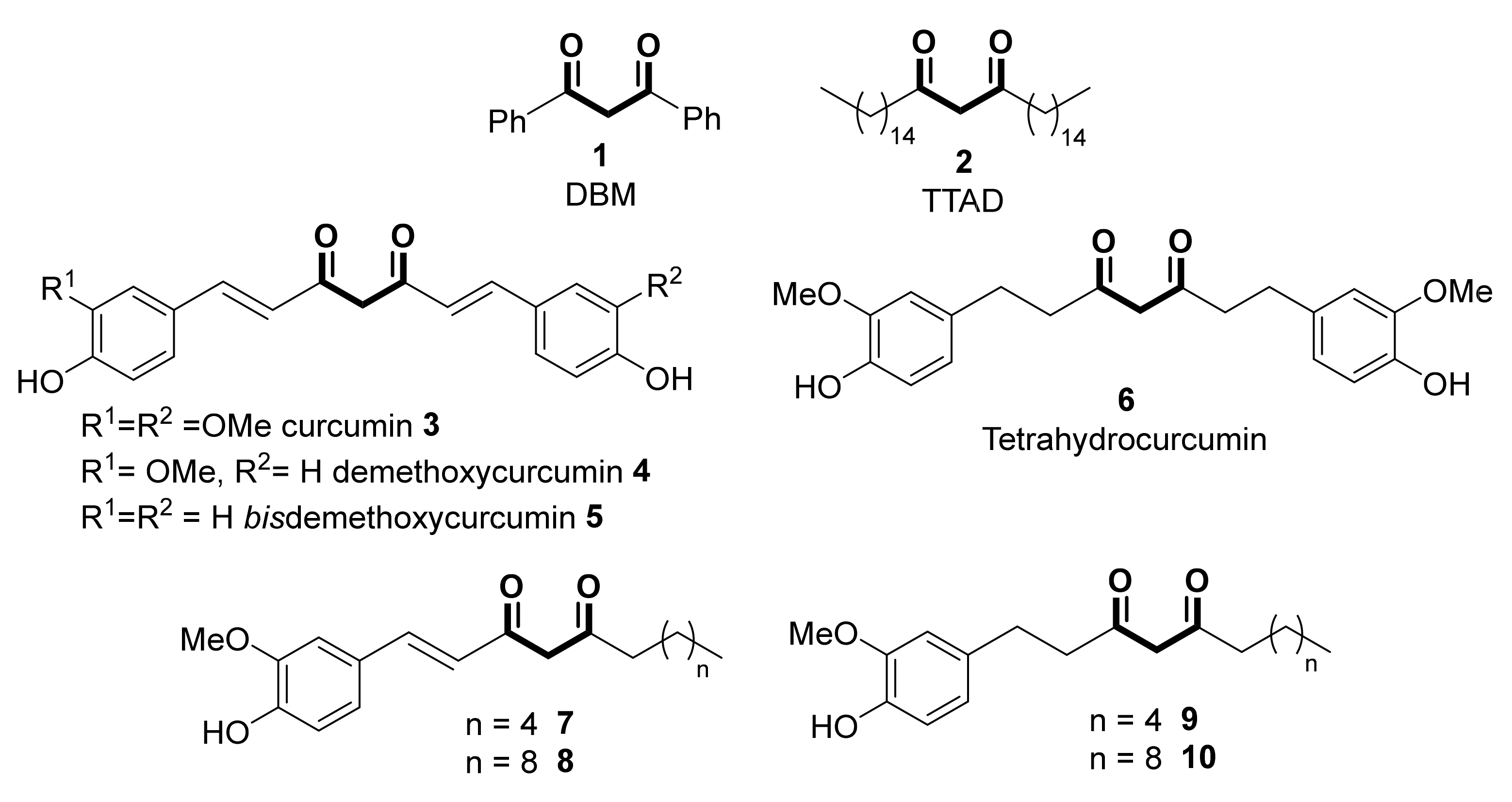
- Schraufstätter, E.; Bernt, H. Antibacterial action of curcumin and related compounds. Nature 1949, 164, 456–457. [Google Scholar] [CrossRef]
- Li, S.; Yuan, W.; Deng, G.; Wang, P.; Yang, P.; Aggarwal, B.B. Chemical composition and product quality control of turmeric (Curcuma longa L.). Pharm. Crops 2011, 2, 28–54. [Google Scholar] [CrossRef]
- Amalraj, A.; Pius, A.; Gopi, S.; Gopi, S. Biological activities of curcuminoids, other biomolecules from turmeric and their derivatives—A review. J. Tradit. Complement. Med. 2017, 7, 205–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotha, R.R.; Luthria, D.L. Curcumin: Biological, pharmaceutical, nutraceutical, and analytical aspects. Molecules 2019, 24, 2930. [Google Scholar] [CrossRef] [Green Version]
- Slika, L.; Patra, D. Traditional uses, therapeutic effects and recent advances of curcumin: A mini-review. Mini-Rev. Med. Chem. 2020, 20, 1072–1082. [Google Scholar] [CrossRef]
- Rahmani, A.H.; Alsahli, M.A.; Aly, S.M.; Khan, M.A.; Aldebasi, Y.H. Role of curcumin in disease prevention and treatment. Adv. Biomed. Res. 2018, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Hassan, F.U.; Rehman, M.S.U.; Khan, M.S.; Ali, M.A.; Javed, A.; Nawaz, A.; Yang, C. Curcumin as an alternative epigenetic modulator: Mechanism of action and potential effects. Front. Genet. 2019, 10, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.S.; Ho, C.T.; Pan, M.H. The cancer chemopreventive and therapeutic potential of tetrahydrocurcumin. Biomolecules 2020, 10, 831. [Google Scholar] [CrossRef] [PubMed]
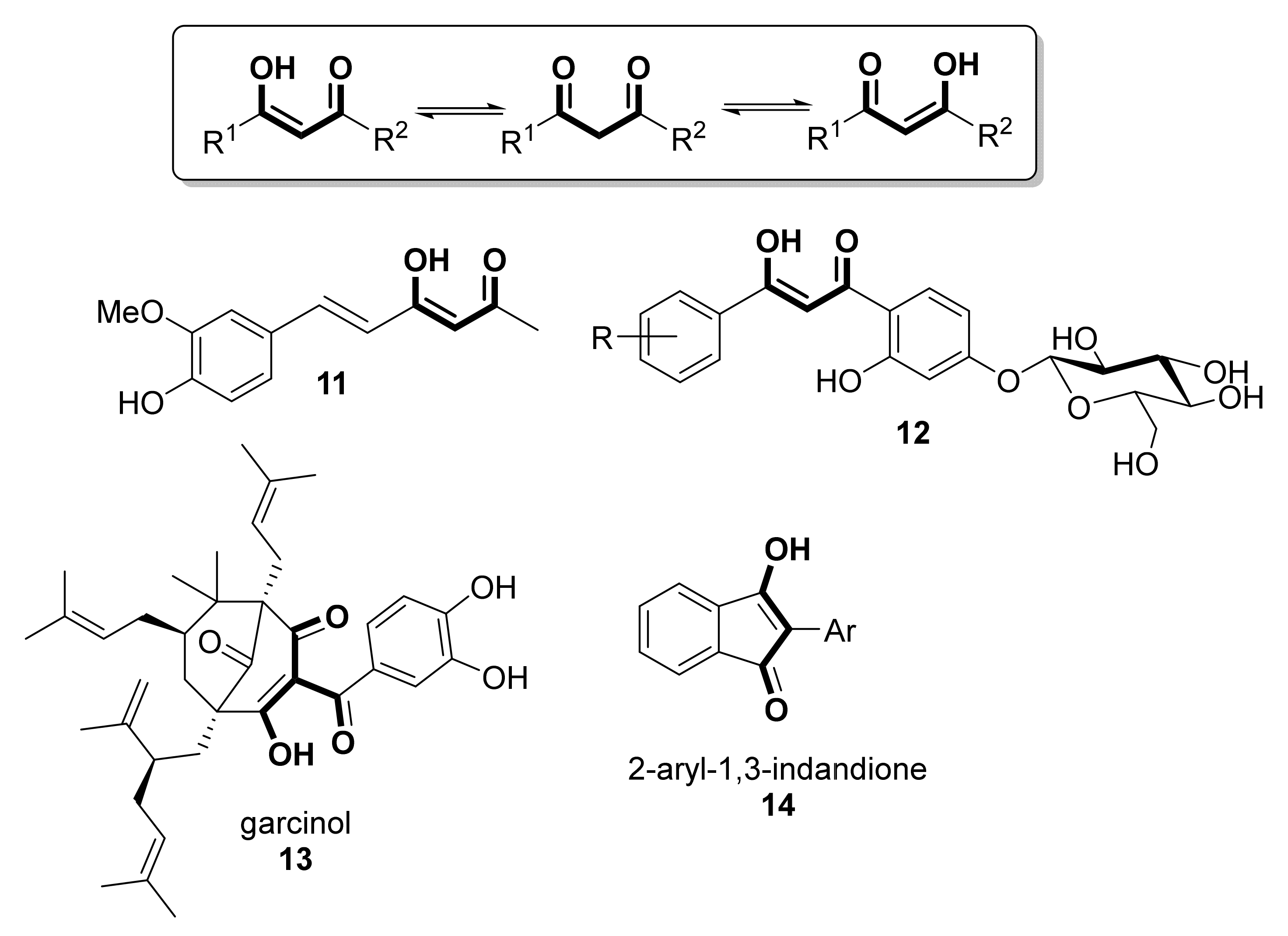
- Patro, B.S.; Rele, S.; Chintalwar, G.J.; Chattopadhyay, S.; Adhikari, S.; Mukherjee, T. Protective activities of some phenolic 1,3-diketones against lipid peroxidation: Possible involvement of the 1,3-diketone moiety. ChemBioChem 2002, 3, 364–370. [Google Scholar] [CrossRef]
- Feng, J.Y.; Liu, Z.Q. Feruloylacetone as the model compound of half-curcumin: Synthesis and antioxidant properties. Eur. J. Med. Chem. 2011, 46, 1198–1206. [Google Scholar] [CrossRef]
- Sheikh, J.; Parvez, A.; Juneja, H.; Ingle, V.; Chohan, Z.; Youssoufi, M.; Ben Hadda, T. Synthesis, biopharmaceutical characterization, antimicrobial and antioxidant activities of 1-(4′-O-β-d-glucopyranosyloxy-2′- hydroxyphenyl)-3-aryl-propane-1,3-diones. Eur. J. Med. Chem. 2011, 46, 1390–1399. [Google Scholar] [CrossRef]
- Padhye, S.; Ahmad, A.; Oswal, N.; Sarkar, F.H. Emerging role of Garcinol, the antioxidant chalcone from Garcinia indica Choisy and its synthetic analogs. J. Hematol. Oncol. 2009, 2, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Schobert, R.; Biersack, B. Chemical and Biological Aspects of Garcinol and Isogarcinol: Recent Developments. Chem. Biodivers. 2019, 16, e1900366. [Google Scholar] [CrossRef] [Green Version]
- Kopytko, P.; Piotrowska, K.; Janisiak, J.; Tarnowski, M. Garcinol—A natural histone acetyltransferase inhibitor and new anti-cancer epigenetic drug. Int. J. Mol. Sci. 2021, 22, 2828. [Google Scholar] [CrossRef]
- Nishiyama, T.; Shiotsu, S.; Tsujita, H. Antioxidative activity and active site of 1,3-indandiones with the β-diketone moiety. Polym. Degradation Stab. 2002, 76, 435–439. [Google Scholar] [CrossRef]
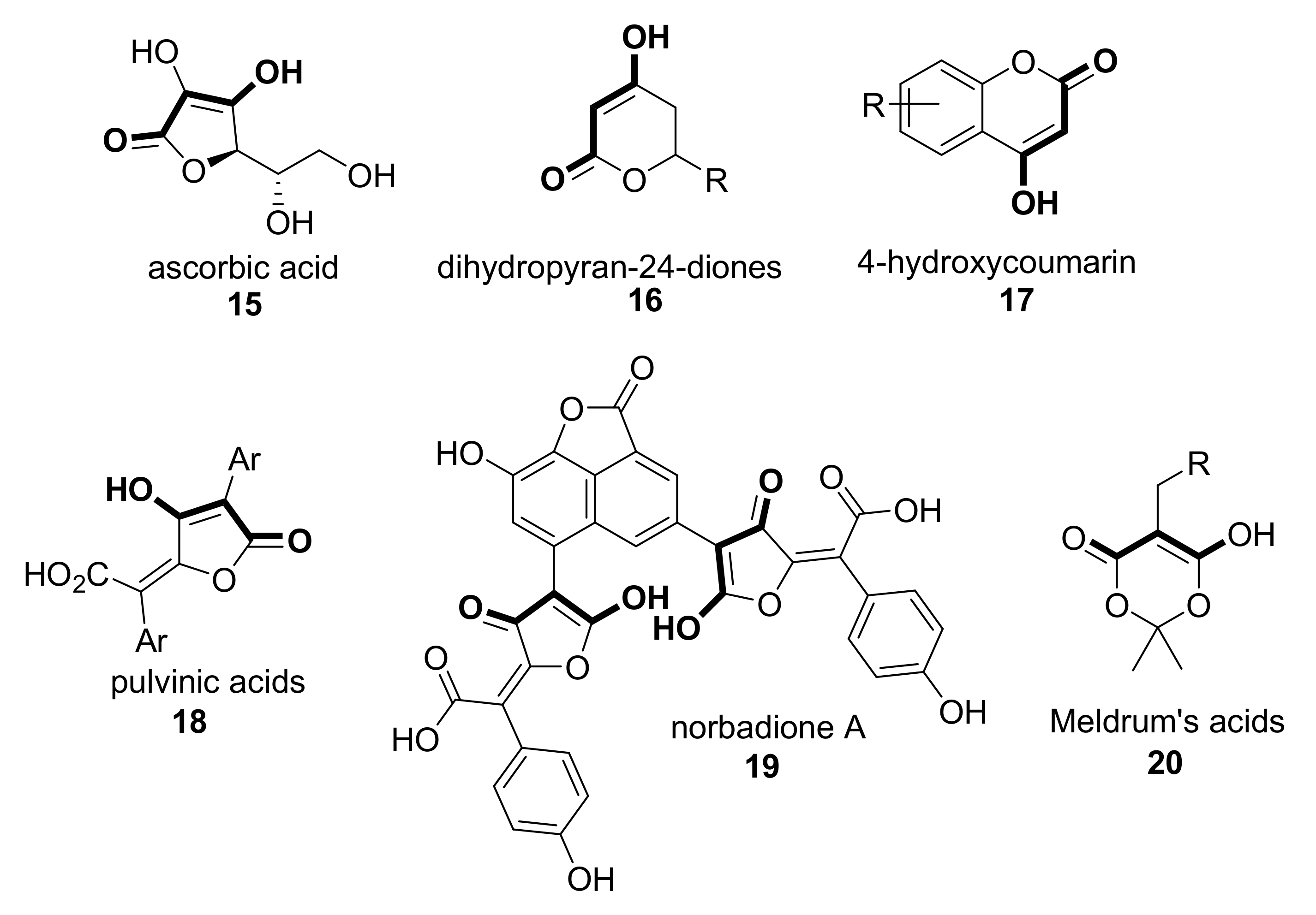
- Arrigoni, O.; De Tullio, M.C. Ascorbic acid: Much more than just an antioxidant. Biochim. Biophys. Acta Gen. Subj. 2002, 1569, 1–9. [Google Scholar] [CrossRef]
- Njus, D.; Kelley, P.M.; Tu, Y.J.; Schlegel, H.B. Ascorbic acid: The chemistry underlying its antioxidant properties. Free Radic. Biol. Med. 2020, 159, 37–43. [Google Scholar] [CrossRef]
- De Souza, L.C.; De Araújo, S.M.S.; De Oliveira Imbroisi, D. Determination of the free radical scavenging activity of dihydropyran-2,4-diones. Bioorg. Med. Chem. Lett. 2004, 14, 5859–5861. [Google Scholar] [CrossRef]
- Rouaiguia-Bouakkaz, S.; Benayahoum, A. The antioxidant activity of 4-hydroxycoumarin derivatives and some sulfured analogs. J. Phys. Org. Chem. 2015, 28, 714–722. [Google Scholar] [CrossRef]
- Nadal, B.; Thetiot-Laurent, S.A.L.; Pin, S.; Renault, J.P.; Cressier, D.; Rima, G.; Le Roux, A.; Meunier, S.; Wagner, A.; Lion, C.; et al. Synthesis and antioxidant properties of pulvinic acids analogues. Bioorg. Med. Chem. 2010, 18, 7931–7939. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, A.; Kuzmanovski, I.; Habrant, D.; Meunier, S.; Bischoff, P.; Nadal, B.; Thetiot-Laurent, S.A.L.; Le Gall, T.; Wagner, A.; Novič, M. Design and synthesis of new antioxidants predicted by the model developed on a set of pulvinic acid derivatives. J. Chem. Inf. Model. 2011, 51, 3050–3059. [Google Scholar] [CrossRef] [PubMed]
- Meunier, S.; Hanédanian, M.; Desage-El Murr, M.; Nowaczyk, S.; Le Gall, T.; Pin, S.; Renault, J.P.; Boquet, D.; Créminon, C.; Mioskowski, C.; et al. High-throughput evaluation of antioxidant and pro-oxidant activities of polyphenols with thymidine protection assays. ChemBioChem 2005, 6, 1234–1241. [Google Scholar] [CrossRef]
- Ivanov, A.S. Meldrum’s acid and related compounds in the synthesis of natural products and analogs. Chem. Soc. Rev. 2008, 37, 789–811. [Google Scholar] [CrossRef]
- Brosge, F.; Singh, P.; Almqvist, F.; Bolm, C. Selected applications of Meldrum’s acid—A tutorial. Org. Biomol. Chem. 2021, 19, 5014–5027. [Google Scholar] [CrossRef]
- Dalal, A.; Kumar, P.; Khanna, R.; Kumar, D.; Paliwal, D.; Kamboj, R.C. Hydroxyenone derivatives: In vitro anti-malarial and docking studies against P. Falciparum. Infect. Disord. Drug Targets 2020, 20, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, J.; Hadda, T.B. Antibacterial, antifungal and antioxidant activity of some new water-soluble β-diketones. Med. Chem. Res. 2013, 22, 964–975. [Google Scholar] [CrossRef]
- Pluskota, R.; Koba, M. Indandione and its derivatives - Chemical compounds with high biological potential. Mini-Rev. Med. Chem. 2018, 18, 1321–1330. [Google Scholar] [CrossRef]
- Chopra, I.; Roberts, M. Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, M.C. Tetracycline therapy: Update. Clin. Infect. Dis. 2003, 36, 462–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakeri, B.; Wright, G.D. Chemical biology of tetracycline antibiotics. Biochem. Cell Biol. 2008, 86, 124–136. [Google Scholar] [CrossRef]
- Nguyen, F.; Starosta, A.L.; Arenz, S.; Sohmen, D.; Dönhöfer, A.; Wilson, D.N. Tetracycline antibiotics and resistance mechanisms. Biol. Chem. 2014, 395, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Genilloud, O.; Vicente, F. Tetracycline antibiotics and novel analogs. In Antimicrobials: New and Old Molecules in the Fight Against Multi-Resistant Bacteria; Genilloud, O., Vicente, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 9783642399688, pp. 231–245. [Google Scholar]
- Liu, F.; Myers, A.G. Development of a platform for the discovery and practical synthesis of new tetracycline antibiotics. Curr. Opin. Chem. Biol. 2016, 32, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Kong, Q.; Yang, Y. Recent advances in antibacterial agents. Bioorg. Med. Chem. Lett. 2021, 35. [Google Scholar] [CrossRef]
- Williams, D.R.; Cullen Klein, J.; Kopel, L.C.; Nguyen, N.; Tantillo, D.J. Studies toward australifungin. A synthesis dilemma of regioselective keto-enol tautomerization. Org. Lett. 2016, 18, 424–427. [Google Scholar] [CrossRef]
- Porchezhiyan, V.; Kalaivani, D.; Shobana, J.; Noorjahan, S.E. Synthesis, docking and in vitro evaluation of L-proline derived 1,3-diketones possessing anti-cancer and anti-inflammatory activities. J. Mol. Struct. 2020, 1206, 127754. [Google Scholar] [CrossRef]
- Porchezhiyan, V.; Kalaivani, D.; Shobana, J.; Noorjahan, S.E. Synthesis, characterization, in silico and in vitro evaluations of symmetrical 1,3-Diketones. Asian J. Chem. 2020, 32, 853–864. [Google Scholar] [CrossRef]
- Conradie, J. Density functional theory calculations of Rh-β-diketonato complexes. Dalton Trans. 2015, 44, 1503–1515. [Google Scholar] [CrossRef] [PubMed]
- Freitag, R.; Conradie, J. Electrochemical and Computational Chemistry Study of Mn(β-diketonato)3 complexes. Electrochim. Acta 2015, 158, 418–426. [Google Scholar] [CrossRef]
- Conradie, M.M.; Conradie, J. Electrochemical behaviour of Tris(β-diketonato)iron(III) complexes: A DFT and experimental study. Electrochim. Acta 2015, 152, 512–519. [Google Scholar] [CrossRef]
- Liu, R.; Conradie, J. Tris(β-diketonato)chromium(III) complexes: Effect of the β-diketonate ligand on the redox properties. Electrochim. Acta 2015, 185, 288–296. [Google Scholar] [CrossRef]
- Chiyindiko, E.; Malan, F.P.; Langner, E.H.G.; Conradie, J. Conformational study of [Cu(CF3COCHCO(C4H3X))2] (X = O or S), a combined experimental and DFT study. J. Mol. Struct. 2019, 1198, 126916. [Google Scholar] [CrossRef]
- Chiyindiko, E.; Stuurman, N.F.; Langner, E.H.G.; Conradie, J. Electrochemical behaviour of bis(β-diketonato)copper(II) complexes containing γ-substituted β-diketones. J. Electroanal. Chem. 2020, 860, 113929. [Google Scholar] [CrossRef]
- Radzyminska-Lenarcik, E.; Witt, K. Solvent extraction of copper ions by 3-substituted derivatives of β-diketones. Sep. Sci. Technol. 2018, 53, 1223–1229. [Google Scholar] [CrossRef]
- Valdéz-Camacho, J.R.; Ramírez-Solís, A.; Escalante, J.; Ruiz-Azuara, L.; Hô, M. Theoretical determination of half-wave potentials for phenanthroline-, bipyridine-, acetylacetonate-, and glycinate-containing copper (II) complexes. J. Mol. Model. 2020, 26, 1–13. [Google Scholar] [CrossRef]
- Xu, D.F.; Shen, Z.H.; Shi, Y.; He, Q.; Xia, Q.C. Synthesis, characterization, crystal structure, and biological activity of the copper complex. Russ. J. Coord. Chem. 2010, 36, 458–462. [Google Scholar] [CrossRef]
- Mateyise, N.G.S.; Ghosh, S.; Gryzenhout, M.; Chiyindiko, E.; Conradie, M.M.; Langner, E.H.G.; Conradie, J. Synthesis, characterization, DFT and biological activity of oligothiophene β-diketone and Cu-complexes. Polyhedron 2021, 205, 115290. [Google Scholar] [CrossRef]
- Sampal, S.N.; Thakur, S.V.; Rajbhoj, A.S.; Gaikwad, S.T. Synthesis, characterization and antimicrobial screening of 1,3-dione with their metal complexes. Asian J. Chem. 2018, 30, 398–402. [Google Scholar] [CrossRef]
- Sheikh, J.; Juneja, H.; Ingle, V.; Ali, P.; Hadda, T.B. Synthesis and in vitro biology of Co(II), Ni(II), Cu(II) and Zinc(II) complexes of functionalized beta-diketone bearing energy buried potential antibacterial and antiviral O,O pharmacophore sites. J. Saudi Chem. Soc. 2013, 17, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Kel’in, A.V.; Maioli, A. Recent advances in the chemistry of 1,3-diketones: Structural modifications and synthetic applications. Curr. Org. Chem. 2003, 7, 1855–1886. [Google Scholar] [CrossRef]
- Kumar, V.; Kaur, K.; Gupta, G.K.; Sharma, A.K. Pyrazole containing natural products: Synthetic preview and biological significance. Eur. J. Med. Chem. 2013, 69, 735–753. [Google Scholar] [CrossRef]
- Ansari, A.; Ali, A.; Asif, M. Review: Biologically active pyrazole derivatives. New J. Chem. 2016, 41, 16–41. [Google Scholar] [CrossRef]
- Khan, M.F.; Alam, M.M.; Verma, G.; Akhtar, W.; Akhter, M.; Shaquiquzzaman, M. The therapeutic voyage of pyrazole and its analogs: A review. Eur. J. Med. Chem. 2016, 120, 170–201. [Google Scholar] [CrossRef]
- Faria, J.V.; Vegi, P.F.; Miguita, A.G.C.; dos Santos, M.S.; Boechat, N.; Bernardino, A.M.R. Recently reported biological activities of pyrazole compounds. Bioorg. Med. Chem. 2017, 25, 5891–5903. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Gao, C.; Ren, Q.C.; Song, X.F.; Feng, L.S.; Lv, Z.S. Recent advances of pyrazole-containing derivatives as anti-tubercular agents. Eur. J. Med. Chem. 2017, 139, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, P.; Nikam, M.; Asrondkar, A.; Bobade, A.; Gill, C. Synthesis, Antioxidant, and Anti-Inflammatory Evaluation of Novel Thiophene-Fused Quinoline Based β-Diketones and Derivatives. J. Heterocycl. Chem. 2017, 54, 1415–1422. [Google Scholar] [CrossRef]
- Mohamed, T.K.; Batran, R.Z.; Elseginy, S.A.; Ali, M.M.; Mahmoud, A.E. Synthesis, anticancer effect and molecular modeling of new thiazolylpyrazolyl coumarin derivatives targeting VEGFR-2 kinase and inducing cell cycle arrest and apoptosis. Bioorg. Chem. 2019, 85, 253–273. [Google Scholar] [CrossRef]
- Farag, A.M.; Mayhoub, A.S.; Barakat, S.E.; Bayomi, A.H. Regioselective synthesis and antitumor screening of some novel N-phenylpyrazole derivatives. Bioorg. Med. Chem. 2008, 16, 881–889. [Google Scholar] [CrossRef]
- Shao, Y.; Zheng, H.; Qian, J.; Wan, X. In Situ Generation of Nitrilimines from Aryldiazonium Salts and Diazo Esters: Synthesis of Fully Substituted Pyrazoles under Room Temperature. Org. Lett. 2018, 20, 2412–2415. [Google Scholar] [CrossRef] [PubMed]
- Komendantova, A.S.; Lyssenko, K.A.; Zavarzin, I.V.; Volkova, Y.A. Iodine-promoted synthesis of pyrazoles from 1,3-dicarbonyl compounds and oxamic acid thiohydrazides. Org. Chem. Front. 2020, 7, 1640–1646. [Google Scholar] [CrossRef]
- Shokova, E.A.; Kim, J.K.; Kovalev, V.V. 1,3-Diketones. Synthesis and properties. Russ. J. Org. Chem. 2015, 51, 755–830. [Google Scholar] [CrossRef]
- Kim, J.; Shokova, E.; Tafeenko, V.; Kovalev, V. (CF3CO)2O/CF3SO3H-mediated synthesis of 1,3-diketones from carboxylic acids and aromatic ketones. Beilstein J. Org. Chem. 2014, 10, 2270–2278. [Google Scholar] [CrossRef] [Green Version]
- Lehrich, S.W.; Mahrholdt, J.; Korb, M.; Hildebrandt, A.; Swarts, J.C.; Lang, H. Synthesis, characterization, and electrochemistry of diferrocenyl β-diketones, -diketonates, and pyrazoles. Molecules 2020, 25, 4476. [Google Scholar] [CrossRef]
- Du Plessis, W.C.; Davis, W.L.; Cronje, S.J.; Swarts, J.C. Structural, thermodynamic and kinetic consequences of a spectroscopic study of the equilibrium between isomeric forms of ferrocene-containing β-diketones. Inorg. Chim. Acta 2001, 314, 97–104. [Google Scholar] [CrossRef]
- Novoa, N.; Roisnel, T.; Dorcet, V.; Hamon, J.R.; Carrillo, D.; Manzur, C.; Guen, F.R.L.; Cabon, N. Anisyl and ferrocenyl adducts of methylenepyran-containing β-diketone: Synthesis, spectral, structural, and redox properties Dedicated to our distinguished colleague and friend Professor Bertrand Caro for his scientific achievements in the field of organometallic methylenepyran chemistry. J. Organomet. Chem. 2014, 762, 19–28. [Google Scholar] [CrossRef]
- Xue, Y.J.; Li, M.Y.; Jin, X.J.; Zheng, C.J.; Piao, H.R. Design, synthesis and evaluation of carbazole derivatives as potential antimicrobial agents. J. Enzyme Inhib. Med. Chem. 2021, 36, 295–306. [Google Scholar] [CrossRef]
- Caruso, A.; Ceramella, J.; Iacopetta, D.; Saturnino, C.; Mauro, M.V.; Bruno, R.; Aquaro, S.; Sinicropi, M.S. Carbazole derivatives as antiviral agents: An overview. Molecules 2019, 24, 1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vairavelu, L.; Zeller, M.; Rajendra Prasad, K.J. Solvent-free synthesis of heteroannulated carbazoles: A novel class of anti-tumor agents. Bioorg. Chem. 2014, 54, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Du, Y.; Gao, Z.; Shen, J. Molecular modeling studies on carbazole carboxamide based BTK inhibitors using docking and structure-based 3D-QSAR. Int. J. Mol. Sci. 2018, 19, 1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadnor, V.A.; Mhaske, G.R.; Shelke, S.N. Synthesis and Antimicrobial Evaluation of Novel Carbazole Based β-diketones and its Pyrazole Derivatives. Croat. Chem. Acta 2018, 91, 367–375. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Acenã, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II-III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef]
- Johnson, B.M.; Shu, Y.Z.; Zhuo, X.; Meanwell, N.A. Metabolic and Pharmaceutical Aspects of Fluorinated Compounds. J. Med. Chem. 2020, 63, 6315–6386. [Google Scholar] [CrossRef]
- Grünebaum, M.; Buchheit, A.; Günther, C.; Wiemhöfer, H.D. New efficient synthetic routes to trifluoromethyl substituted pyrazoles and corresponding β-diketones. Tetrahedron Lett. 2016, 57, 1555–1559. [Google Scholar] [CrossRef]
- Taydakov, I.V.; Kreshchenova, Y.M.; Dolotova, E.P. A convenient and practical synthesis of β-diketones bearing linear perfluorinated alkyl groups and a 2-thienyl moiety. Beilstein J. Org. Chem. 2018, 14, 3106–3111. [Google Scholar] [CrossRef]
- Flores, A.F.C.; Blanco, R.F.; Souto, A.A.; Malavolta, J.L.; Flores, D.C. Synthesis of fatty trichloromethyl-β-diketones and new 1H-pyrazoles as unusual FAMEs and FAEEs. J. Braz. Chem. Soc. 2013, 24, 2059–2065. [Google Scholar] [CrossRef]
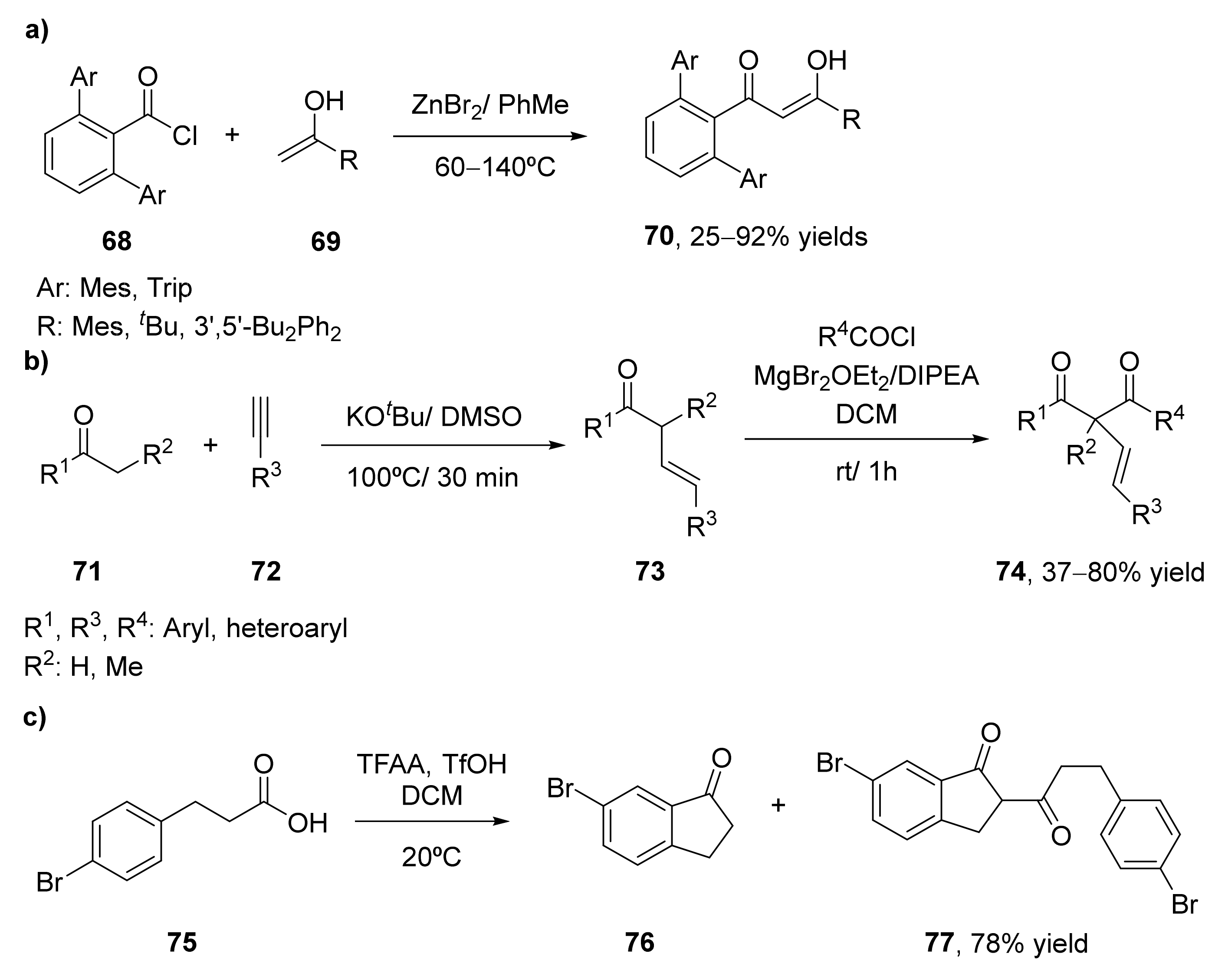
- Crossman, A.S.; Larson, A.T.; Shi, J.X.; Krajewski, S.M.; Akturk, E.S.; Marshak, M.P. Synthesis of Sterically Hindered β-Diketones via Condensation of Acid Chlorides with Enolates. J. Org. Chem. 2019, 84, 7434–7442. [Google Scholar] [CrossRef]
- Shabalin, D.A.; Ivanova, E.V.; Ushakov, I.A.; Schmidt, E.Y.; Trofimov, B.A. Retrosynthetic Analysis of α-Alkenyl-β-Diketones: Regio-and Stereoselective Two-Step Synthesis of Highly Arylated Representatives from Acetylenes, Ketones, and Acyl Chlorides. J. Org. Chem. 2020, 85, 8429–8436. [Google Scholar] [CrossRef]
- Bezzubov, S.I.; Zharinova, I.S.; Khusyainova, A.A.; Kiselev, Y.M.; Taydakov, I.V.; Varaksina, E.A.; Metlin, M.T.; Tobohova, A.S.; Korshunov, V.M.; Kozyukhin, S.A.; et al. Aromatic β-Diketone as a Novel Anchoring Ligand in Iridium(III) Complexes for Dye-Sensitized Solar Cells. Eur. J. Inorg. Chem. 2020, 2020, 3277–3286. [Google Scholar] [CrossRef]
- Kim, D.K.; Shokova, E.A.; Tafeenko, V.A.; Kovalev, V.V. Synthesis of 1,3-diketones from 3-(4-R-phenyl)propionic acids. Russ. J. Org. Chem. 2014, 50, 464–468. [Google Scholar] [CrossRef]
- Lima, S.R.; Coelho, F. Synthesis of 1,4,6-Tricarbonyl Compounds via Regioselective Gold(I)-Catalyzed Alkyne Hydration and Their Application in the Synthesis of α-Arylidene-butyrolactones. ACS Omega 2020, 5, 8032–8045. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, T.R.; Park, J.K. An Overview of Water-Mediated Alkyne Functionalization by Neighboring Group Participation of Carbonyl Groups. Adv. Synth. Catal. 2020, 362, 4833–4860. [Google Scholar] [CrossRef]
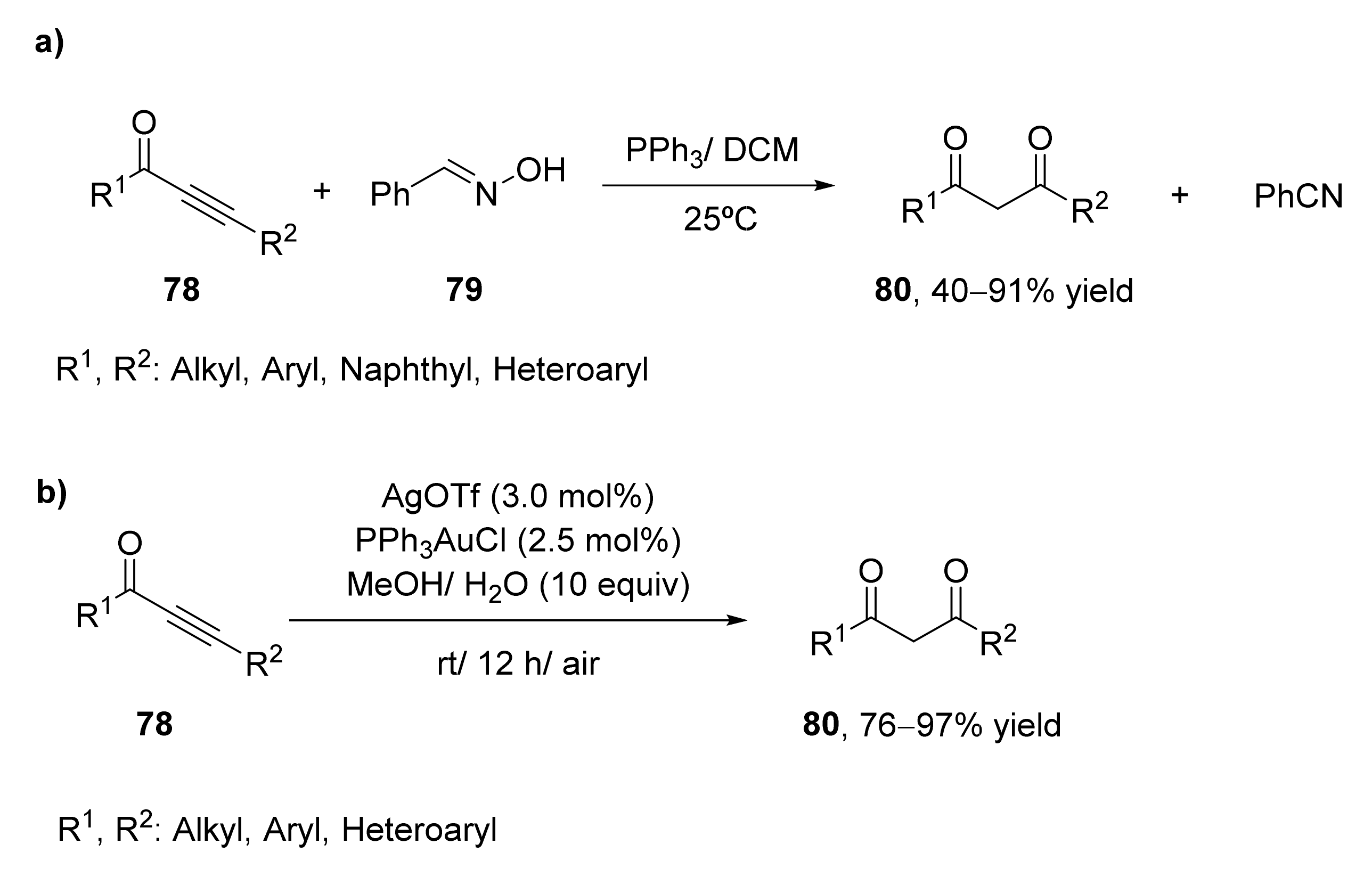
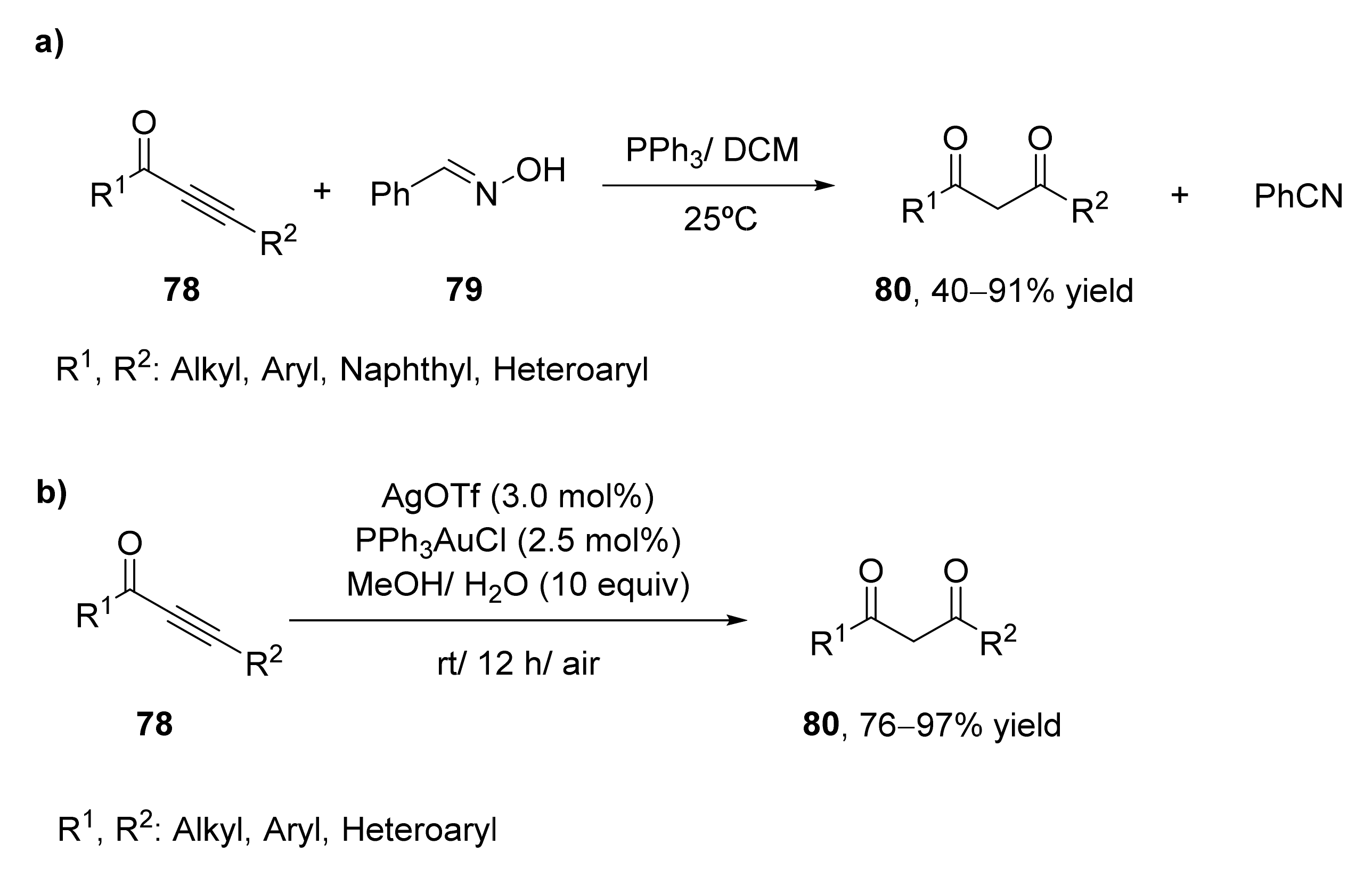
- Chen, P.; Zhang, Q.Q.; Guo, J.; Chen, L.L.; Wang, Y.B.; Zhang, X. An effective preparation of both 1,3-diketones and nitriles from alkynones with oximes as hydroxide sources. Org. Biomol. Chem. 2018, 16, 6958–6966. [Google Scholar] [CrossRef] [PubMed]
- Kuang, J.; Zhou, T.; You, T.; Chen, J.; Su, C.; Xia, Y. Facile access to 1,3-diketones by gold(I)-catalyzed regioselective hydration of ynones. Org. Biomol. Chem. 2019, 17, 3940–3944. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, N.; Goossen, L.J. Decarboxylative coupling reactions: A modern strategy for C–C-bond formation. Chem. Soc. Rev. 2011, 40, 5030–5048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Guo, L.N.; Duan, X.H. Decarboxylative acylation of cyclic enamides with α-oxocarboxylic acids by palladium-catalyzed C-H activation at room temperature. Org. Lett. 2012, 14, 4358–4361. [Google Scholar] [CrossRef] [PubMed]
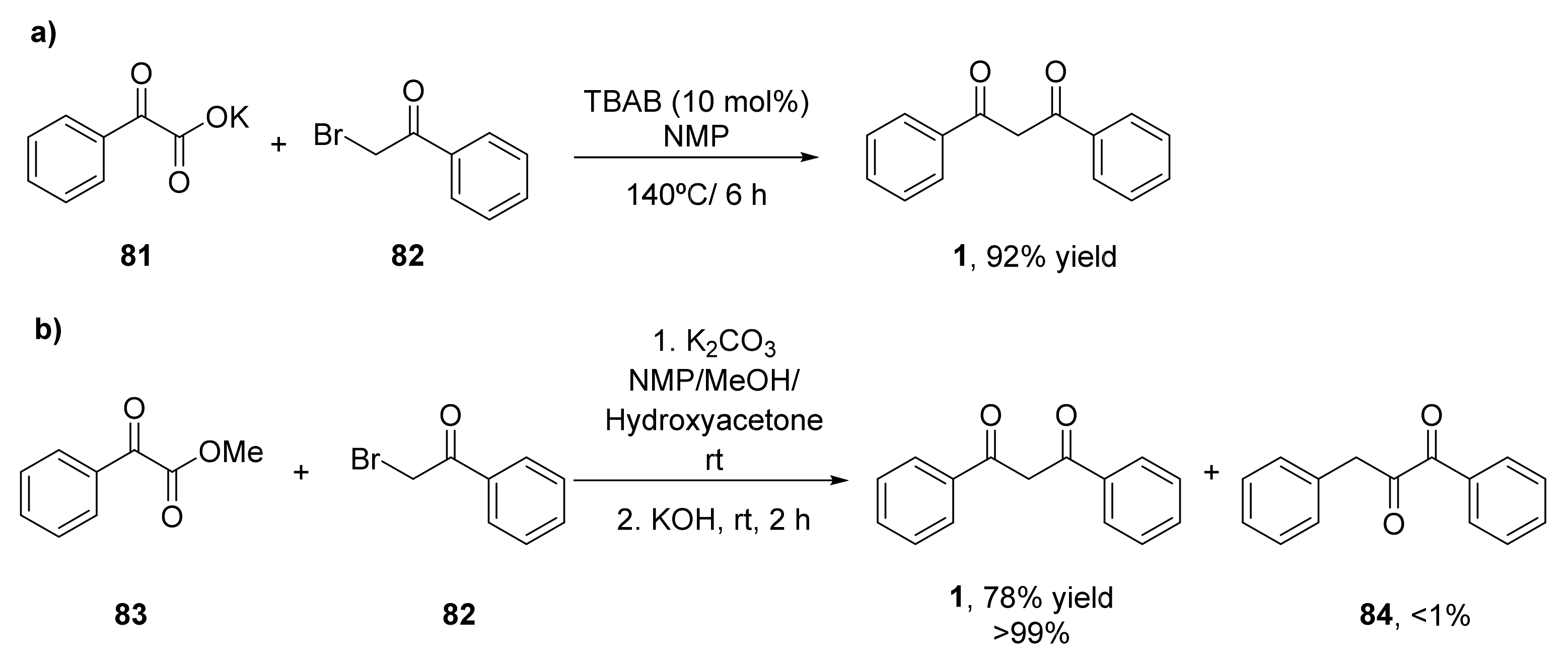
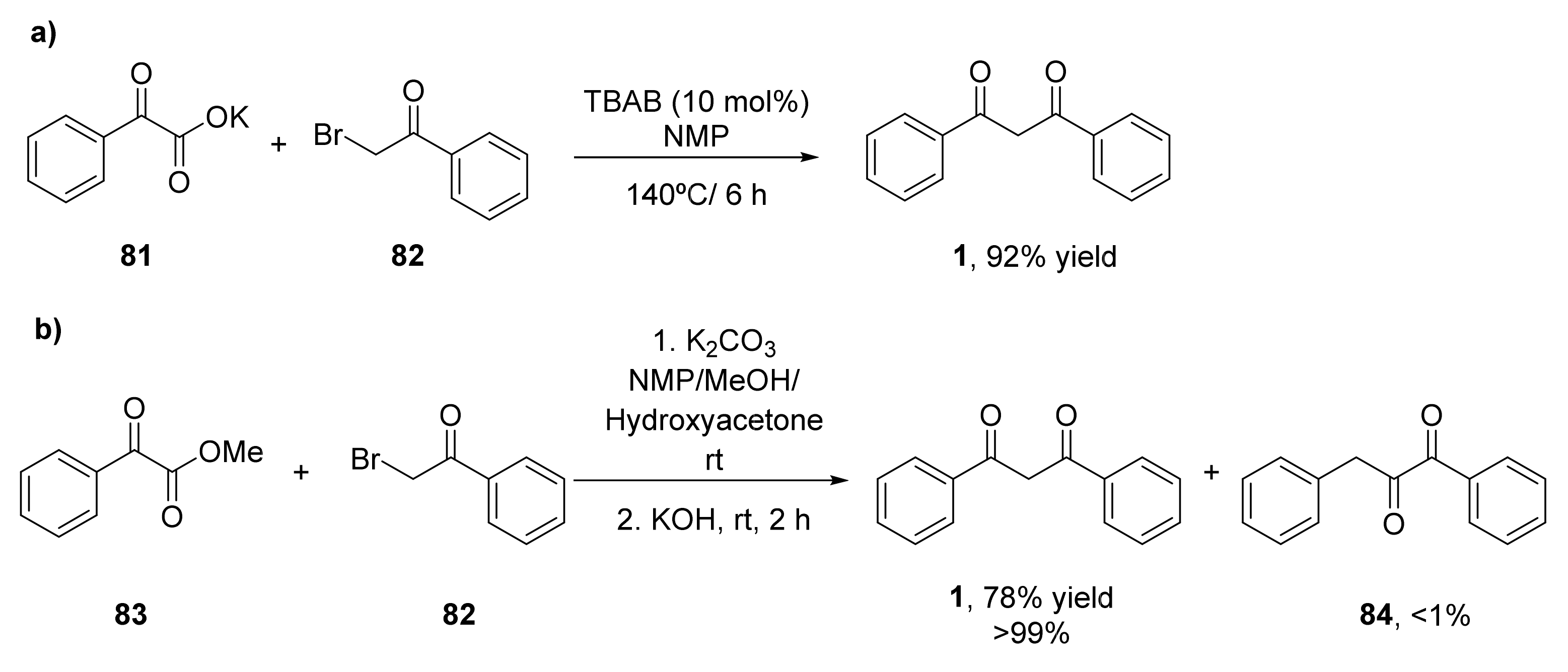
- He, Z.; Qi, X.; Li, S.; Zhao, Y.; Gao, G.; Lan, Y.; Wu, Y.; Lan, J.; You, J. Transition-metal-free formal decarboxylative coupling of α-oxocarboxylates with α-bromoketones under neutral conditions: A simple access to 1,3-diketones. Angew. Chem. Int. Ed. 2015, 54, 855–859. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Qi, X.; She, Z.; Zhao, Y.; Li, S.; Tang, J.; Gao, G.; Lan, Y.; You, J. Room-Temperature Coupling/Decarboxylation Reaction of α-Oxocarboxylates with α-Bromoketones: Solvent-Controlled Regioselectivity for 1,2- and 1,3-Diketones. J. Org. Chem. 2017, 82, 1403–1411. [Google Scholar] [CrossRef]
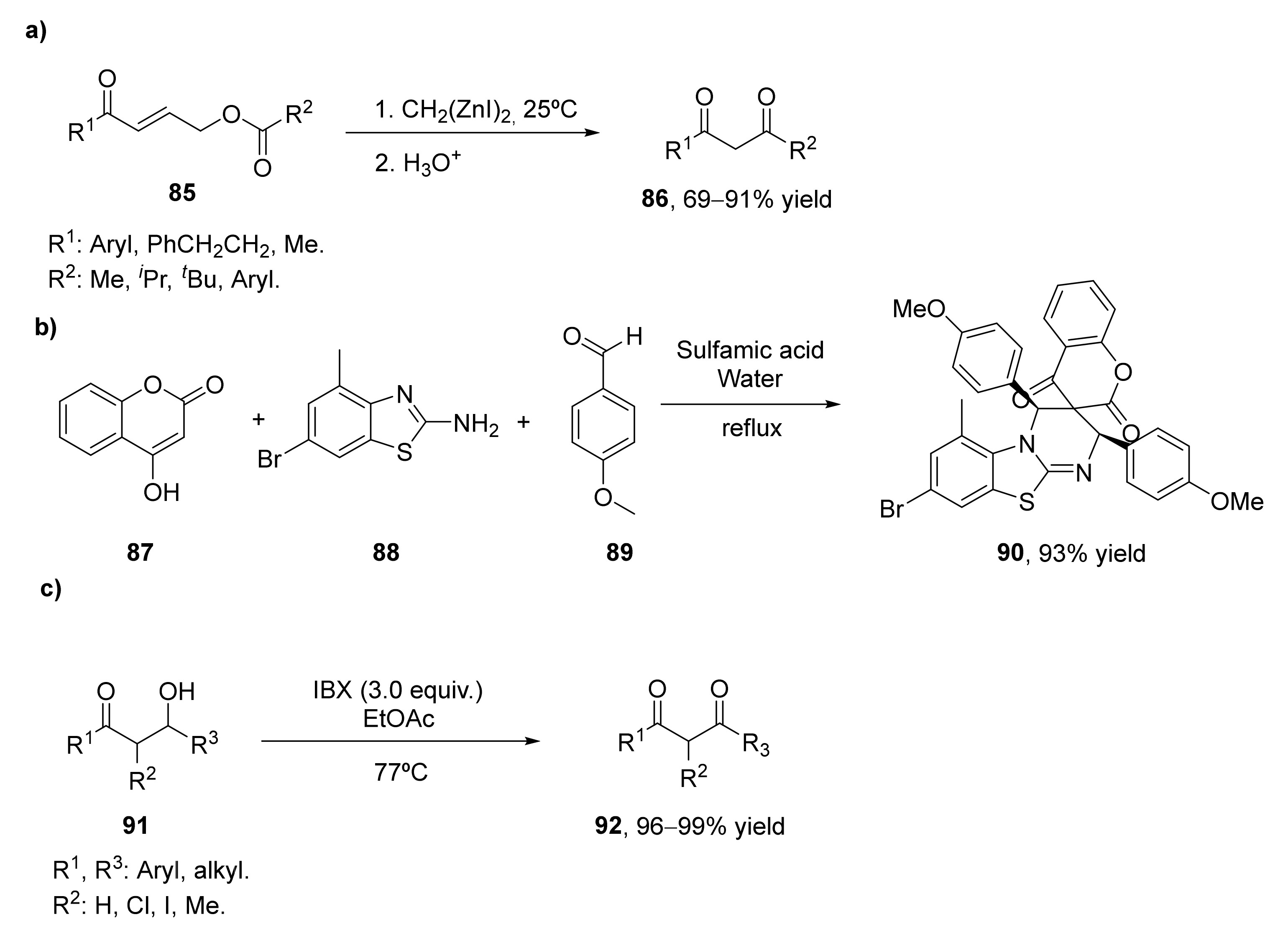
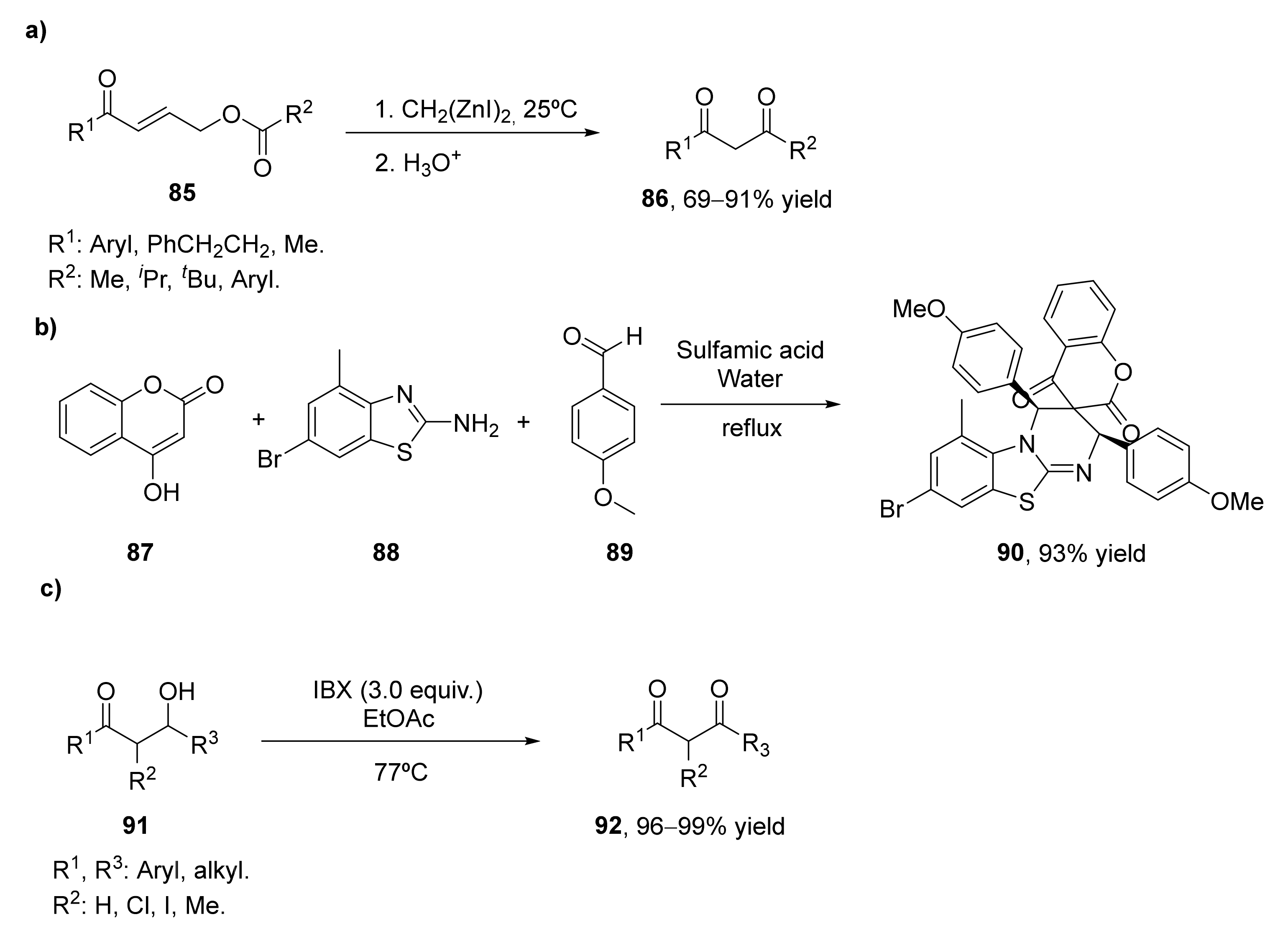
- Sada, M.; Matsubara, S. A tandem reaction initiated by 1,4-addition of bis(iodozincio)methane for 1,3-diketone formation. J. Am. Chem. Soc. 2010, 132, 432–433. [Google Scholar] [CrossRef]
- Kumar, M.; Arya, A.K.; George, J.; Arya, K.; Pardasani, R.T. DFT Studied Hetero-Diels–Alder Cycloaddition for the Domino Synthesis of Spiroheterocycles Fused to Benzothiazole and Chromene/Pyrimidine Rings in Aqueous Media. J. Heterocycl. Chem. 2017, 54, 3418–3426. [Google Scholar] [CrossRef]
- Bartlett, S.L.; Beaudry, C.M. High-yielding oxidation of β-hydroxyketones to β-diketones using o-iodoxybenzoic acid. J. Org. Chem. 2011, 76, 9852–9855. [Google Scholar] [CrossRef] [PubMed]
- Hoyos, P.; Pace, V.; Hernáiz, M.J.; Alcántara, A.R. Biocatalysis in the Pharmaceutical Industry. A greener future. Curr. Green Chem. 2014, 1, 155–181. [Google Scholar] [CrossRef]
- Hoyos, P.; Hernáiz, M.J.; Alcántara, A.R. Biocatalyzed Production of Fine Chemicals. In Reference Module in Life Sciences; Moo-Young, M., Ed.; Pergamon: Oxford, UK, 2017. [Google Scholar] [CrossRef]
- Hoyos, P.; Pace, V.; Alcántara, A.R. Chiral Building Blocks for Drugs Synthesis via Biotransformations. In Asymmetric Synthesis of Drugs and Natural Products; Nag, A., Ed.; CRC Press: Boca Raton, FL, USA, 2018; pp. 346–448. [Google Scholar]
- Domiínguez de Mariía, P.; de Gonzalo, G. Biocatalysis: An Industrial Perspective; Royal Society of Chemistry: London, UK, 2018. [Google Scholar] [CrossRef]
- Alcántara, A.R. Biocatalysis and Pharmaceuticals: A Smart Tool for Sustainable Development. Catalysts 2019, 9, 792. [Google Scholar] [CrossRef] [Green Version]
- Domínguez de María, P.; de Gonzalo, G.; Alcántara, A.R. Biocatalysis as useful tool in asymmetric synthesis: An assessment of recently granted patents (2014–2019). Catalysts 2019, 9, 802. [Google Scholar] [CrossRef] [Green Version]
- Bell, E.L.; Finnigan, W.; France, S.P.; Green, A.P.; Hayes, M.A.; Hepworth, L.J.; Lovelock, S.L.; Niikura, H.; Osuna, S.; Romero, E.; et al. Biocatalysis. Nat. Rev. Methods Primers 2021, 1, 46. [Google Scholar] [CrossRef]
- Hall, M. Enzymatic strategies for asymmetric synthesis. RSC. Chem. Biol. 2021, 2, 958–989. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Brady, D. Streamlining design, engineering, and applications of enzymes for sustainable biocatalysis. ACS Sustainable Chem. Eng. 2021, 9, 8032–8052. [Google Scholar] [CrossRef]
- Wohlgemuth, R. Biocatalysis-Key enabling tools from biocatalytic one-step and multi-step reactions to biocatalytic total synthesis. New Biotech. 2021, 60, 113–123. [Google Scholar] [CrossRef]
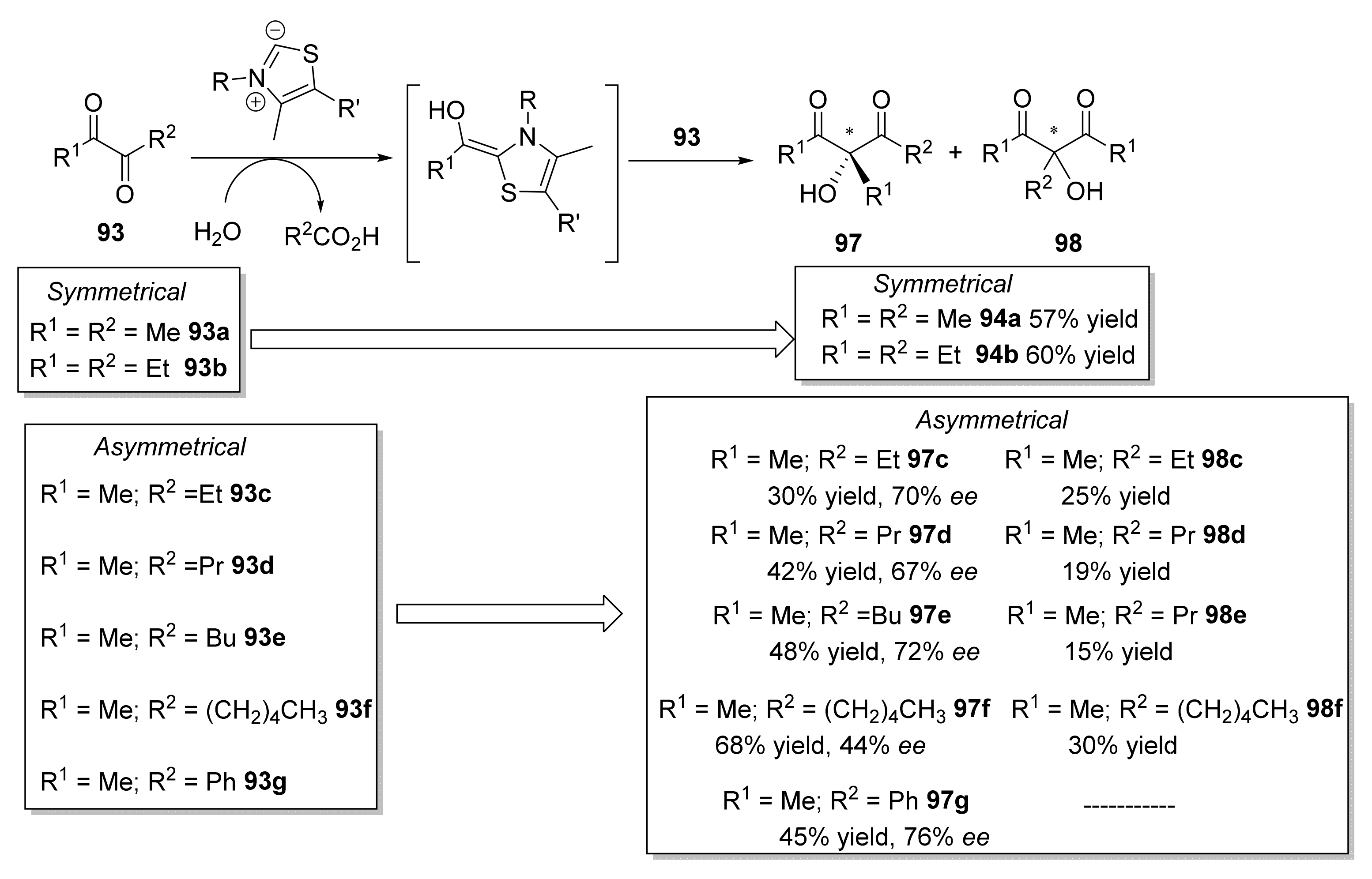
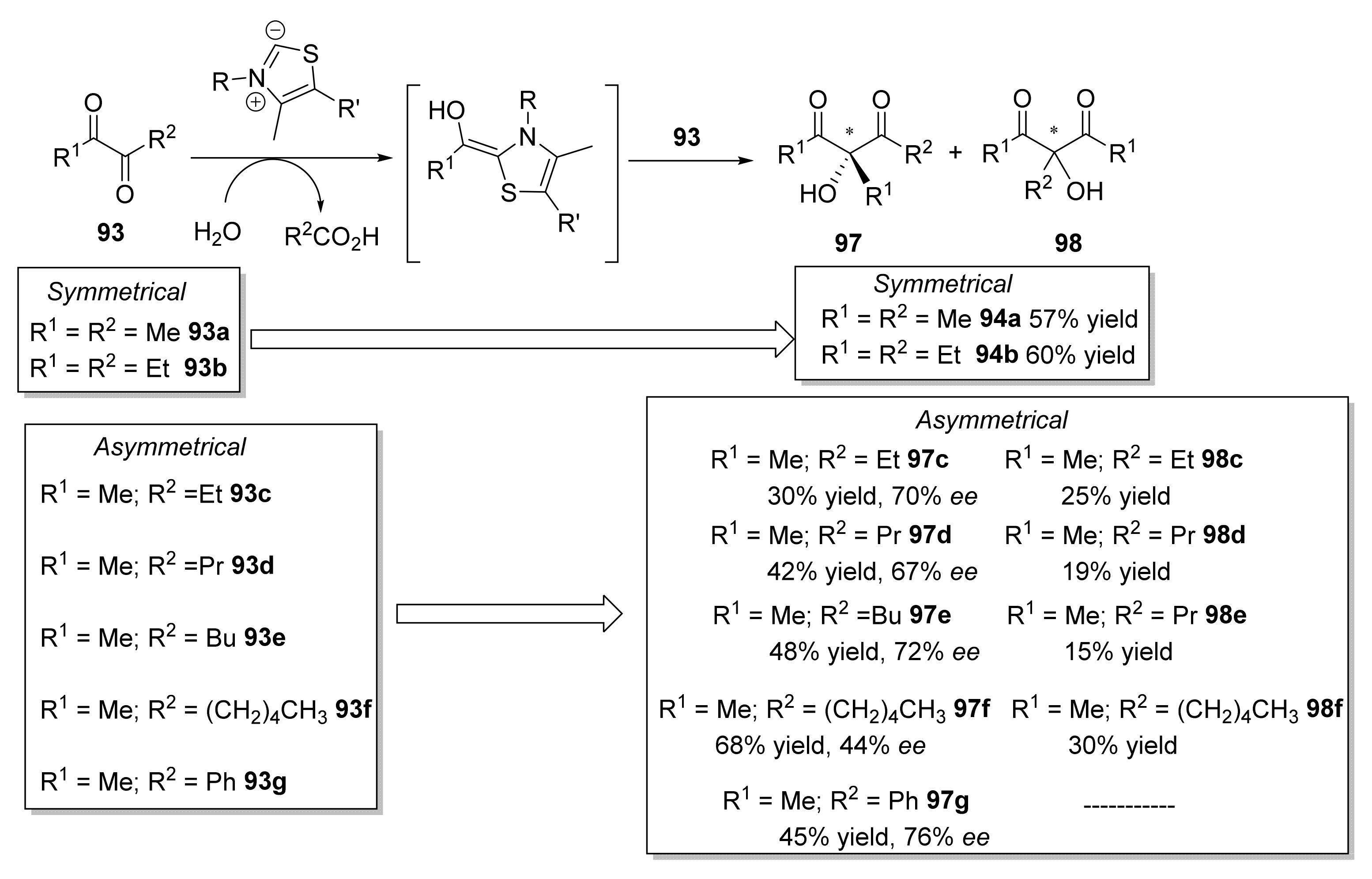
- Lehwald, P.; Richter, M.; Röhr, C.; Liu, H.W.; Müller, M. Enantioselective intermolecular aldehyde-ketone cross-coupling through an enzymatic carboligation reaction. Angew. Chem. Int. Ed. 2010, 49, 2389–2392. [Google Scholar] [CrossRef]
- Loschonsky, S.; Waltzer, S.; Fraas, S.; Wacker, T.; Andrade, S.L.A.; Kroneck, P.M.H.; Müller, M. Catalytic scope of the thiamine-dependent multifunctional enzyme cyclohexane-1,2-dione hydrolase. ChemBioChem 2014, 15, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Loschonsky, S.; Wacker, T.; Waltzer, S.; Giovannini, P.P.; McLeish, M.J.; Andrade, S.L.A.; Müller, M. Extended reaction scope of thiamine diphosphate dependent cyclohexane-1,2-dione hydrolase: From C-C bond cleavage to C-C bond ligation. Angew. Chem. Int. Ed. 2014, 53, 14402–14406. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, P.P.; Pedrini, P.; Venturi, V.; Fantin, G.; Medici, A. Bacillus stearothermophilus acetylacetoin synthase: A new catalyst for C-C bond formation. J. Mol. Catal. B Enzym. 2010, 64, 113–117. [Google Scholar] [CrossRef]
- Bortolini, O.; Giovannini, P.P.; Maietti, S.; Massi, A.; Pedrini, P.; Sacchetti, G.; Venturi, V. An enzymatic approach to the synthesis of optically pure (3R)- and (3S)-enantiomers of green tea flavor compound 3-hydroxy-3-methylnonane-2,4-dione. J. Mol. Catal. B Enzym. 2013, 85–86, 93–98. [Google Scholar] [CrossRef]
- Giovannini, P.P.; Fantin, G.; Massi, A.; Venturi, V.; Pedrini, P. Enzymatic diastereo- and enantioselective synthesis of α-alkyl- α,β-dihydroxyketones. Org. Biomol. Chem. 2011, 9, 8038–8045. [Google Scholar] [CrossRef]
- Giovannini, P.P.; Bortolini, O.; Cavazzini, A.; Greco, R.; Fantin, G.; Massi, A. Expanding the scope of enzymatic carboligation reactions in flow-mode: Production of optically active tertiary alcohols with packed-bed micro-bioreactors. Green Chem. 2014, 16, 3904–3915. [Google Scholar] [CrossRef]
- Bernacchia, G.; Bortolini, O.; De Bastiani, M.; Lerin, L.A.; Loschonsky, S.; Massi, A.; Müller, M.; Giovannini, P.P. Enzymatic Chemoselective Aldehyde-Ketone Cross-Couplings through the Polarity Reversal of Methylacetoin. Angew. Chem. Int. Ed. 2015, 54, 7171–7175. [Google Scholar] [CrossRef]
- Giovannini, P.P.; Lerin, L.A.; Müller, M.; Bernacchia, G.; Bastiani, M.D.; Catani, M.; Di Carmine, G.; Massi, A. (S)-Selectivity in Phenylacetyl Carbinol Synthesis Using the Wild-Type Enzyme Acetoin:Dichlorophenolindophenol Oxidoreductase from Bacillus licheniformis. Adv. Synth. Catal. 2016, 358, 2767–2776. [Google Scholar] [CrossRef]
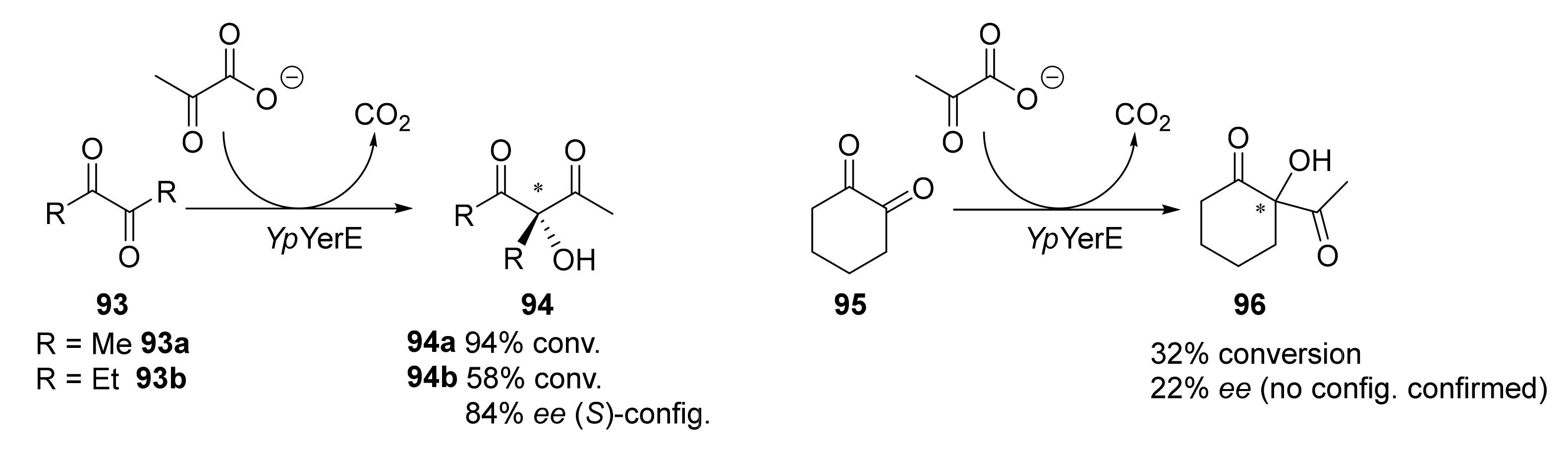
- Hampel, S.; Steitz, J.P.; Baierl, A.; Lehwald, P.; Wiesli, L.; Richter, M.; Fries, A.; Pohl, M.; Schneider, G.; Dobritzsch, D.; et al. Structural and Mutagenesis Studies of the Thiamine-Dependent, Ketone-Accepting YerE from Pseudomonas protegens. ChemBioChem 2018, 19, 2283–2292. [Google Scholar] [CrossRef]
- Oliveira, V.D.G.; Cardoso, M.F.D.C.; Forezi, L.D.S.M. Organocatalysis: A brief overview on its evolution and applications. Catalysts 2018, 8, 605. [Google Scholar] [CrossRef] [Green Version]
- Xiang, S.H.; Tan, B. Advances in asymmetric organocatalysis over the last 10 years. Nat. Commun. 2020, 11, 1–5. [Google Scholar] [CrossRef]
- Flanigan, D.M.; Romanov-Michailidis, F.; White, N.A.; Rovis, T. Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes. Chem. Rev. 2015, 115, 9307–9387. [Google Scholar] [CrossRef] [Green Version]
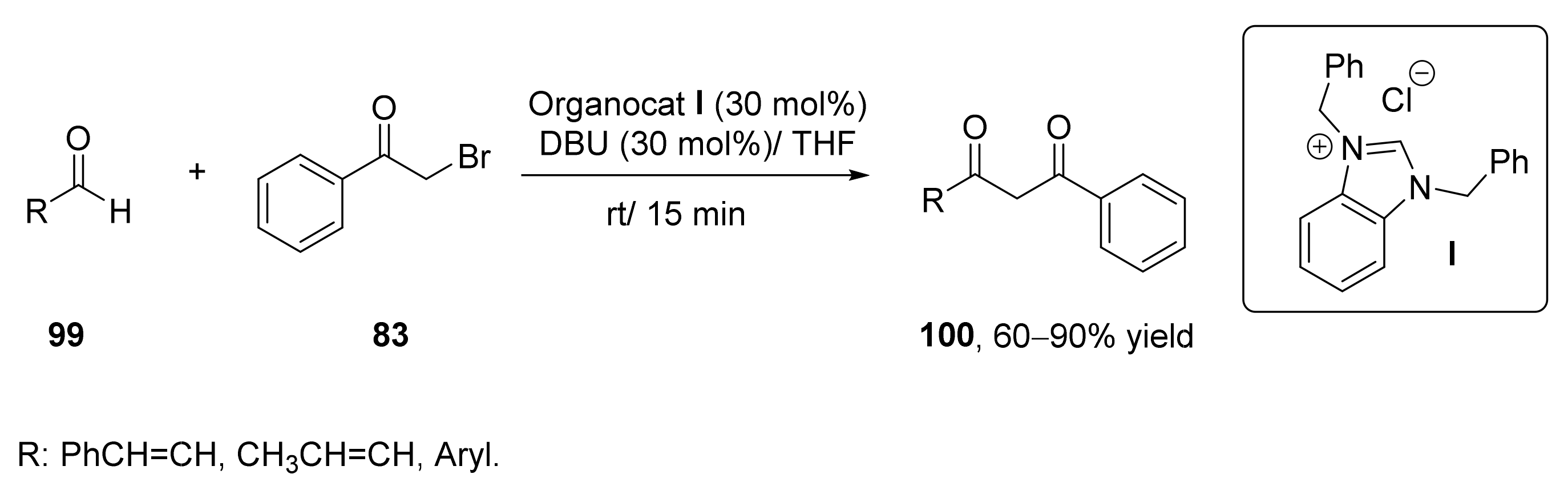
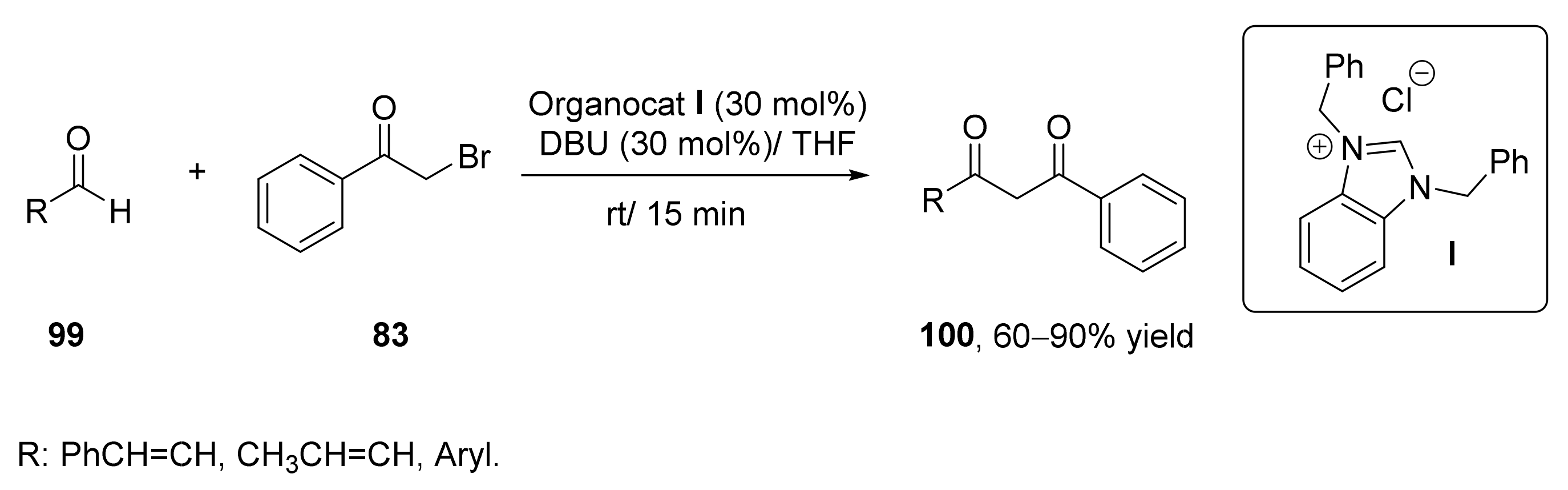
- Singh, S.; Singh, P.; Rai, V.K.; Kapoor, R.; Yadav, L.D.S. Nucleophilic acylation of α-haloketones with aldehydes: An umpolung strategy for the synthesis of 1,3-diketones. Tetrahedron Lett. 2011, 52, 125–128. [Google Scholar] [CrossRef]
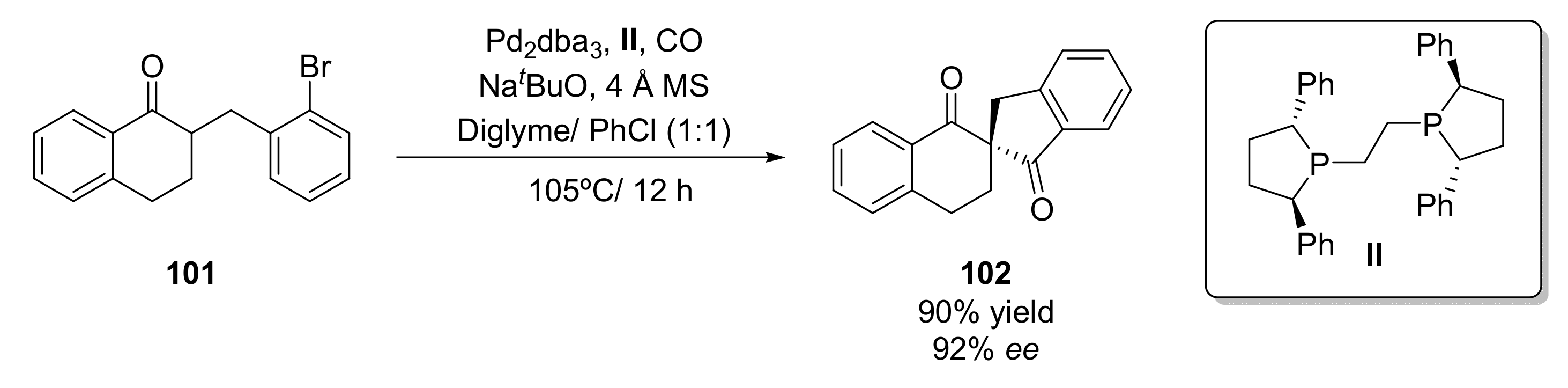
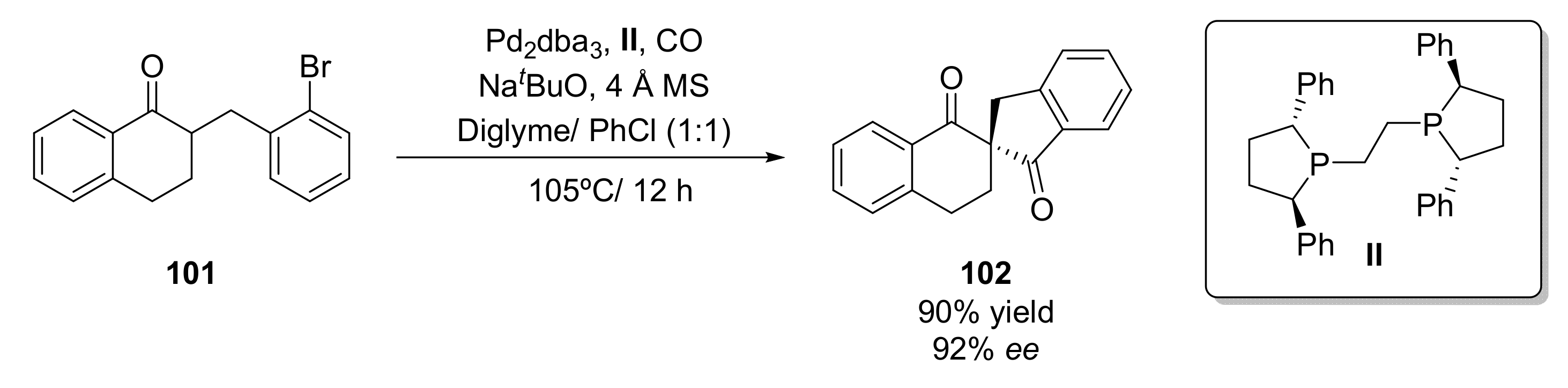
- Wu, T.; Zhou, Q.; Tang, W. Enantioselective α-Carbonylative Arylation for Facile Construction of Chiral Spirocyclic β,β′-Diketones. Angew. Chem. Int. Ed. 2021, 60, 9978–9983. [Google Scholar] [CrossRef] [PubMed]
- Ahumada, G.; Roisnel, T.; Sinbandhit, S.; Manzur, C.; Carrillo, D.; Hamon, J.R. Synthesis, characterization and X-ray crystal structures of chiral ferrocene-containing β-diketones. J. Organomet. Chem. 2013, 737, 1–6. [Google Scholar] [CrossRef] [Green Version]
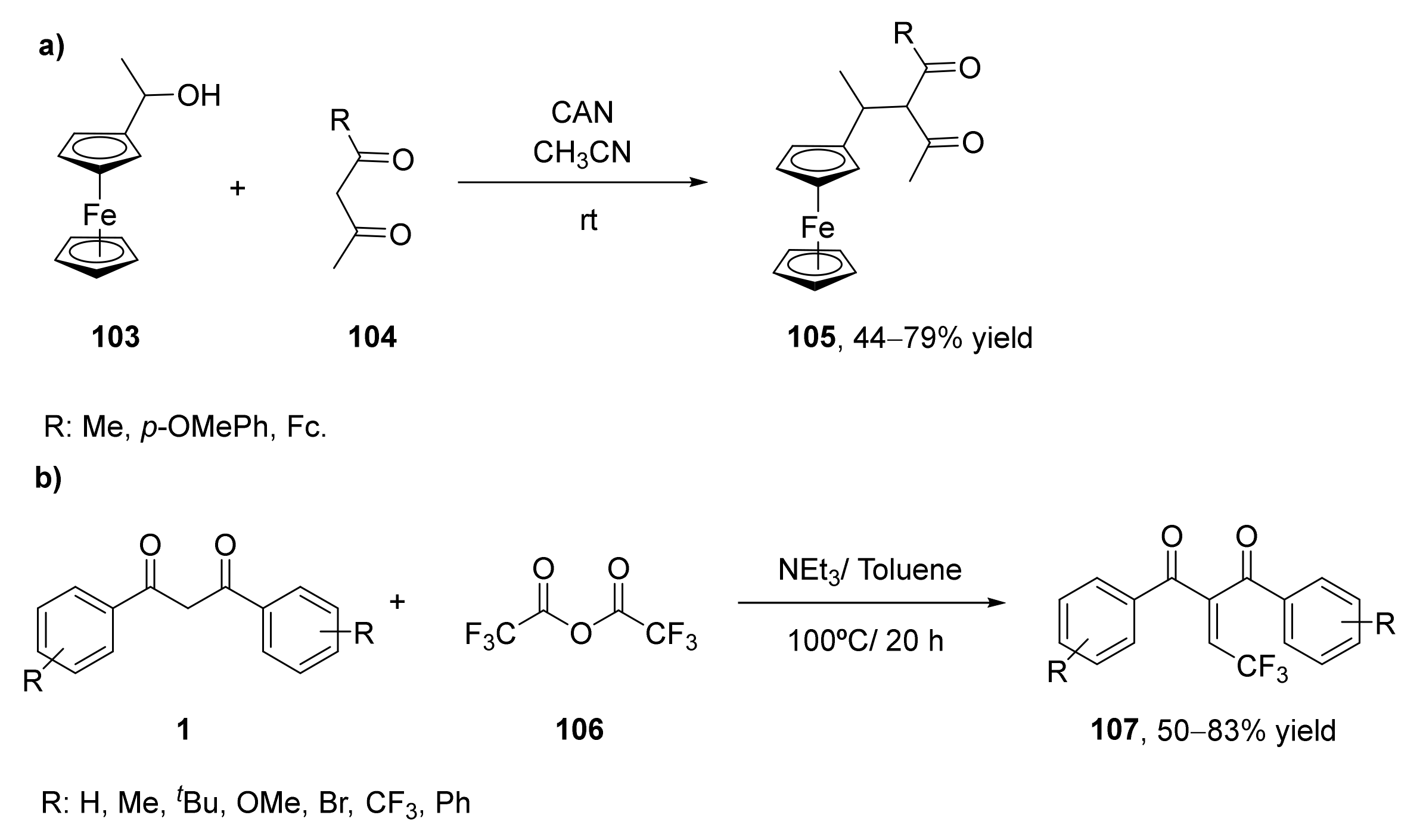
- Xu, X.; Jiang, R.; Zhou, X.; Liu, Y.; Ji, S.; Zhang, Y. Cerium ammonium nitrate: An efficient catalyst for carbon-carbon bond formation from ferrocenyl alcohol substrate. Tetrahedron 2009, 65, 877–882. [Google Scholar] [CrossRef]
- Podyachev, S.N.; Sudakova, S.N.; Gimazetdinova, G.S.; Shamsutdinova, N.A.; Syakaev, V.V.; Barsukova, T.A.; Iki, N.; Lapaev, D.V.; Mustafina, A.R. Synthesis, metal binding and spectral properties of novel bis-1,3-diketone calix[4]arenes. New J. Chem. 2017, 41, 1526–1537. [Google Scholar] [CrossRef]
- Chen, Y.; You, Y.; Weng, Z. Syntheses of 2-(2,2,2-trifluoroethylidene)/(2,2-difluoroethyl)-1,3-dicarbonyl compounds and their fungicidal activities. Org. Chem. Front. 2019, 6, 213–217. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Gonzalo, G.; Alcántara, A.R. Recent Developments in the Synthesis of β-Diketones. Pharmaceuticals 2021, 14, 1043. https://doi.org/10.3390/ph14101043
de Gonzalo G, Alcántara AR. Recent Developments in the Synthesis of β-Diketones. Pharmaceuticals. 2021; 14(10):1043. https://doi.org/10.3390/ph14101043
Chicago/Turabian Stylede Gonzalo, Gonzalo, and Andrés R. Alcántara. 2021. "Recent Developments in the Synthesis of β-Diketones" Pharmaceuticals 14, no. 10: 1043. https://doi.org/10.3390/ph14101043
APA Stylede Gonzalo, G., & Alcántara, A. R. (2021). Recent Developments in the Synthesis of β-Diketones. Pharmaceuticals, 14(10), 1043. https://doi.org/10.3390/ph14101043