Administration of Bacterial Lipopolysaccharide during Early Postnatal Ontogenesis Induces Transient Impairment of Long-Term Synaptic Plasticity Associated with Behavioral Abnormalities in Young Rats


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
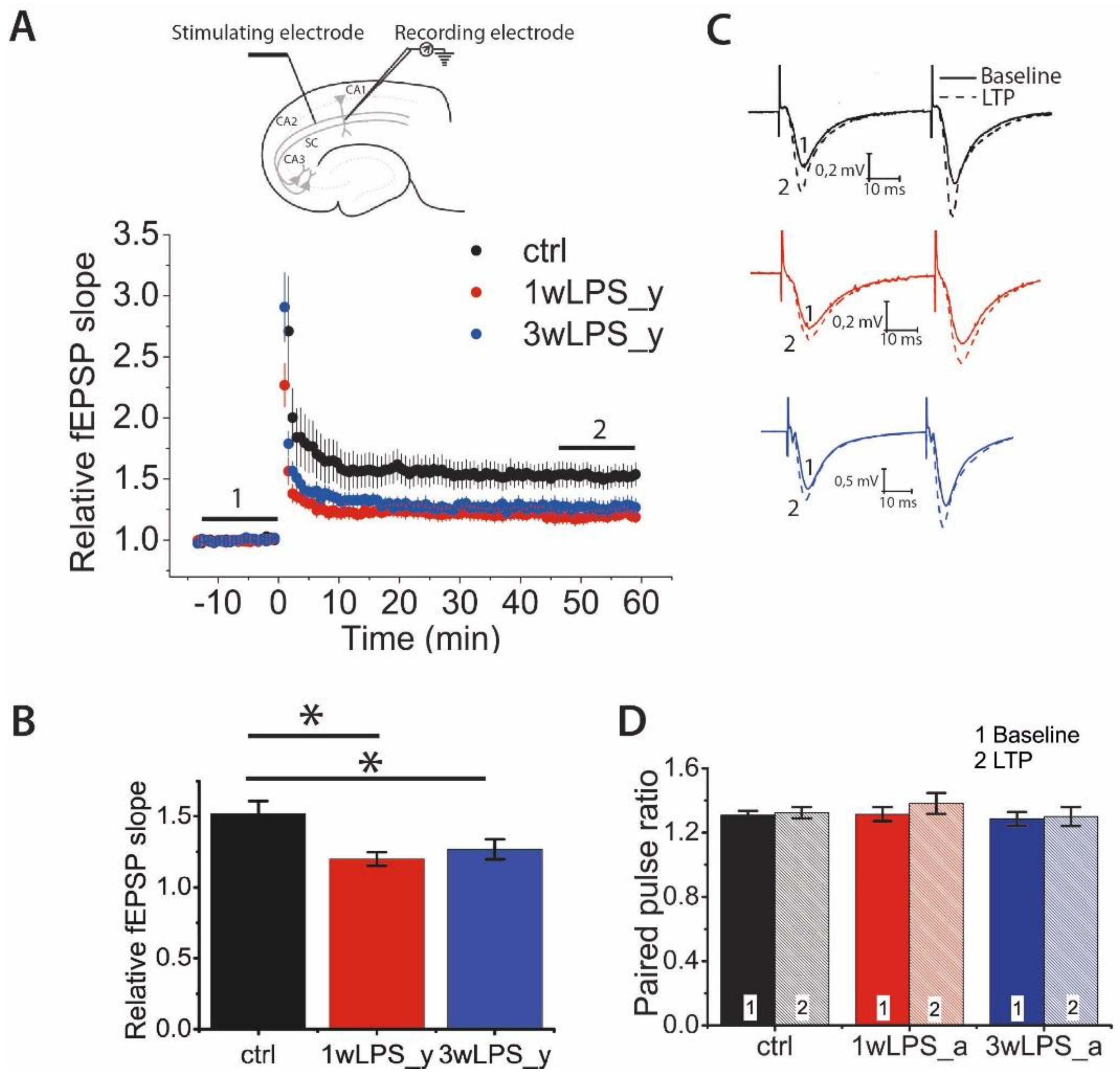
2.1. Hippocampal LTP in Young Rats is Weakened after the Administration of LPS in Early Postnatal Ontogenesis
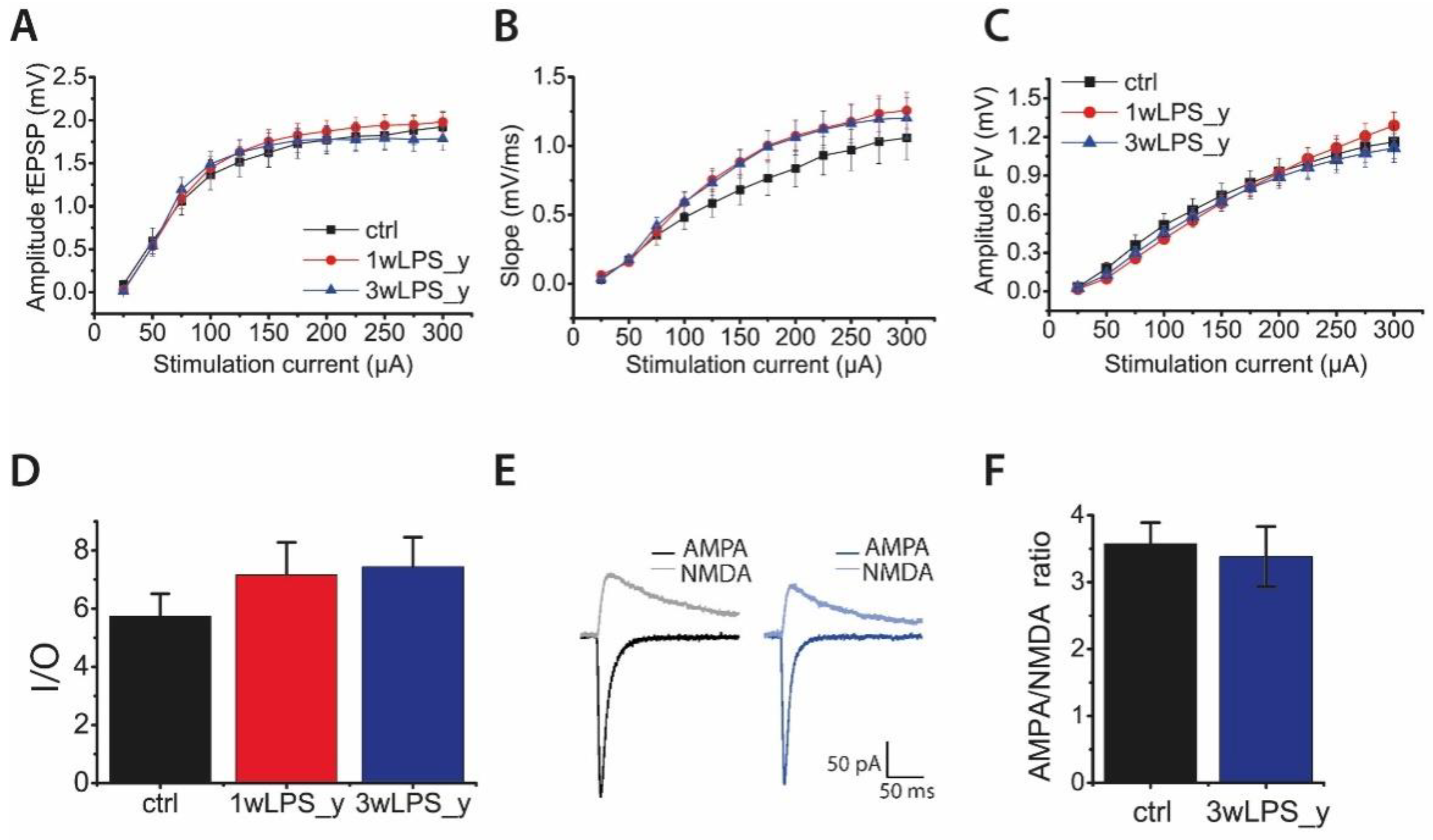
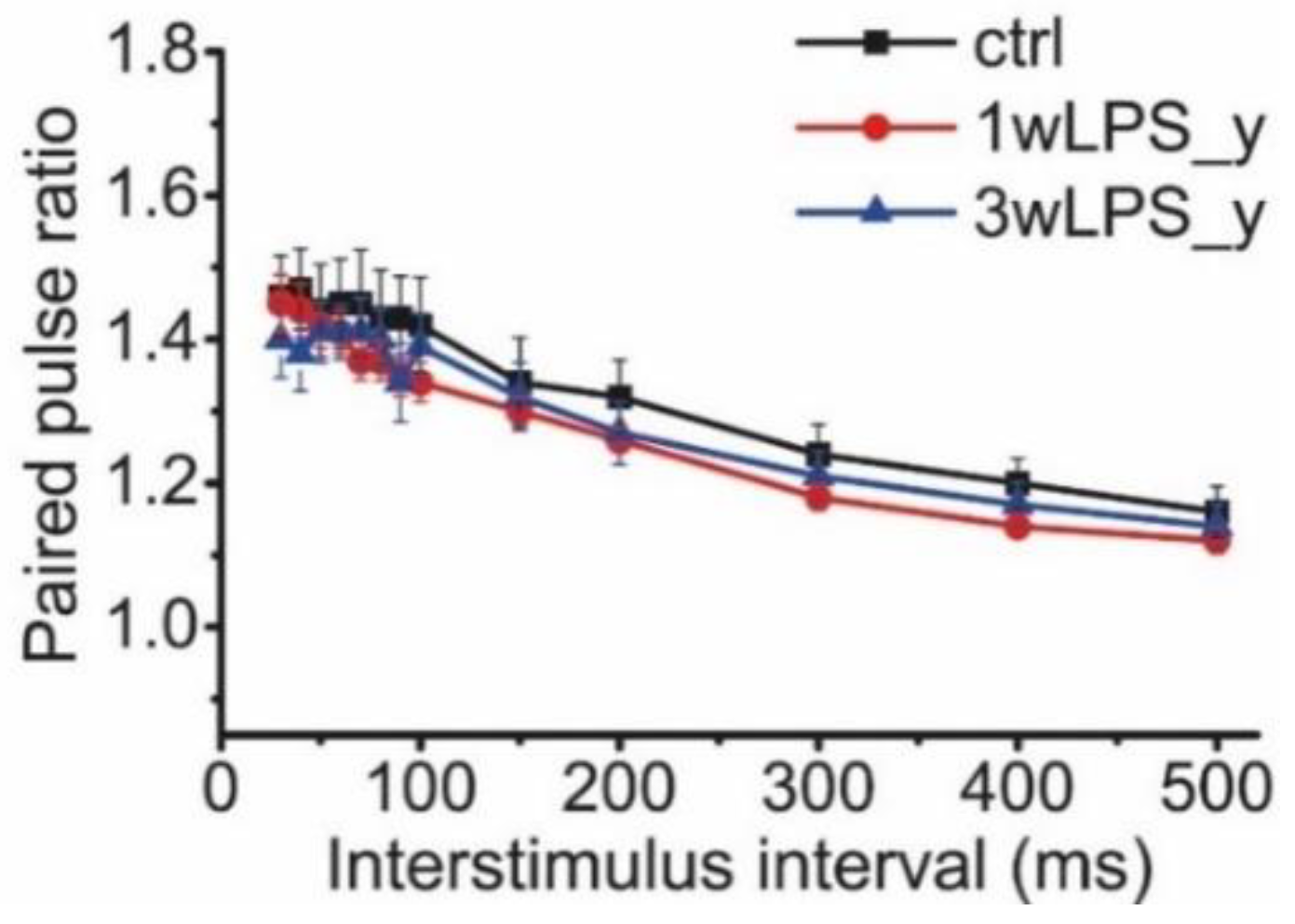
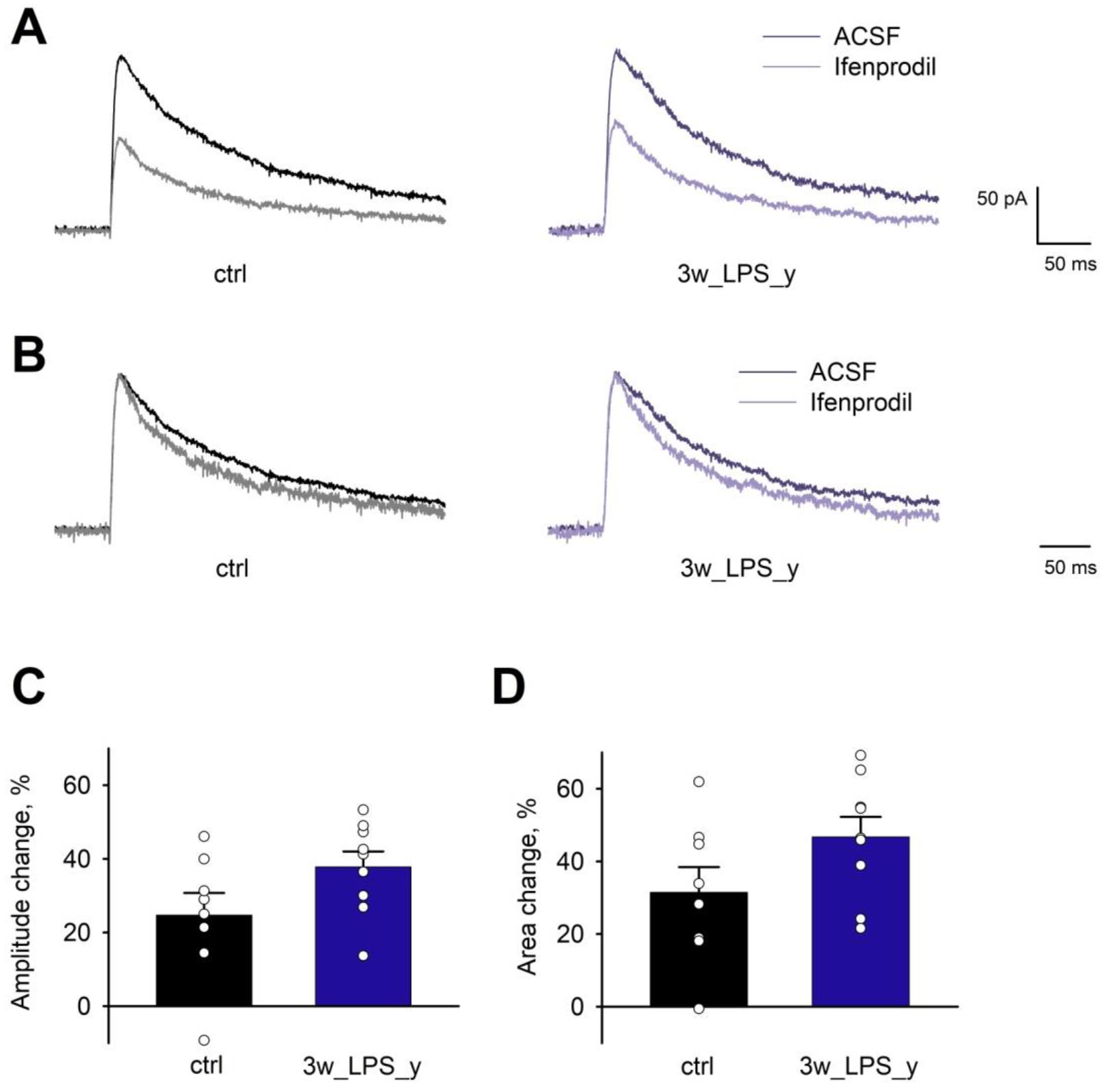
2.2. Main Properties of Excitatory Synaptic Transmission in Hippocampal Pyramidal Neurons are not Altered after LPS Treatment
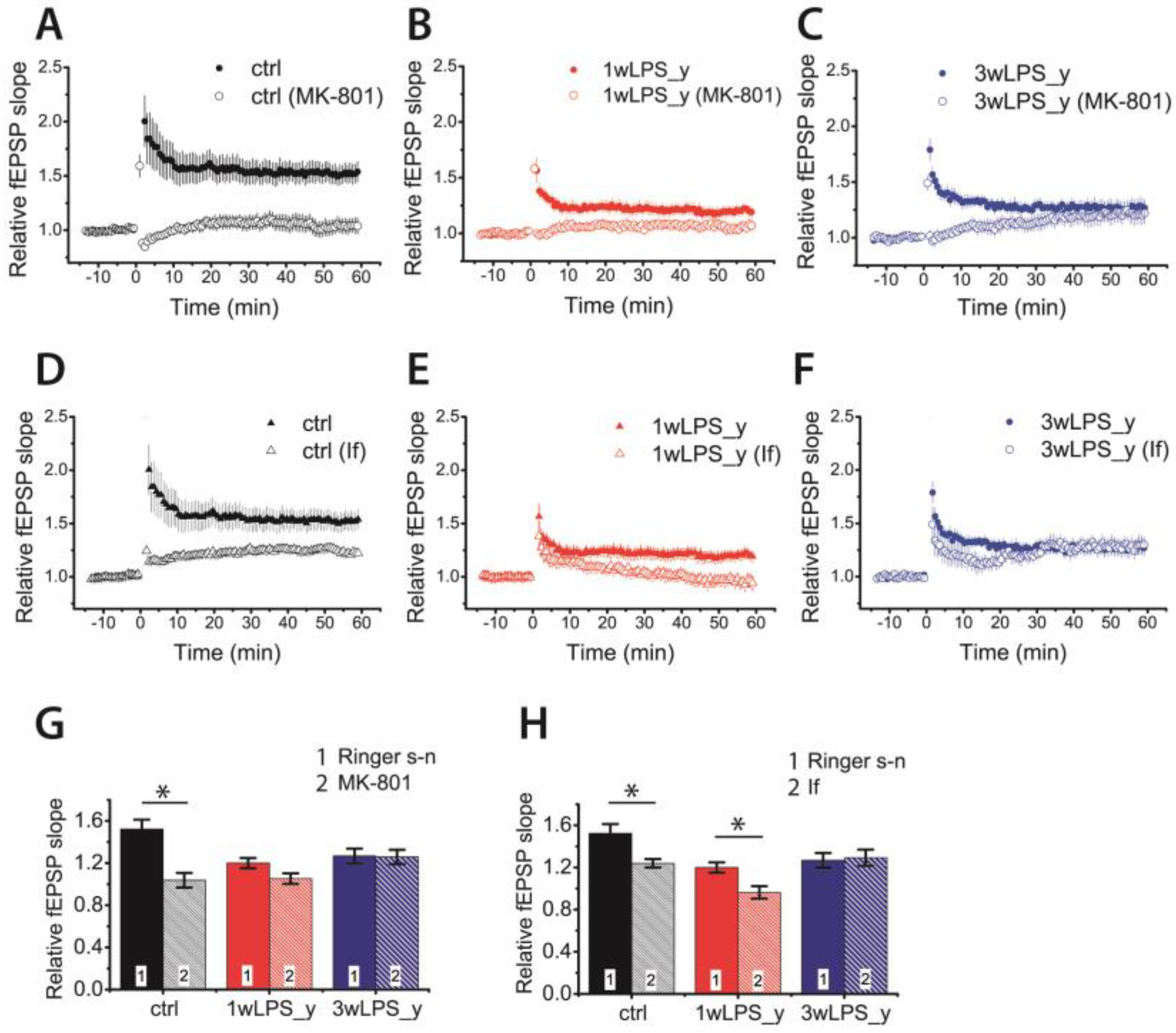
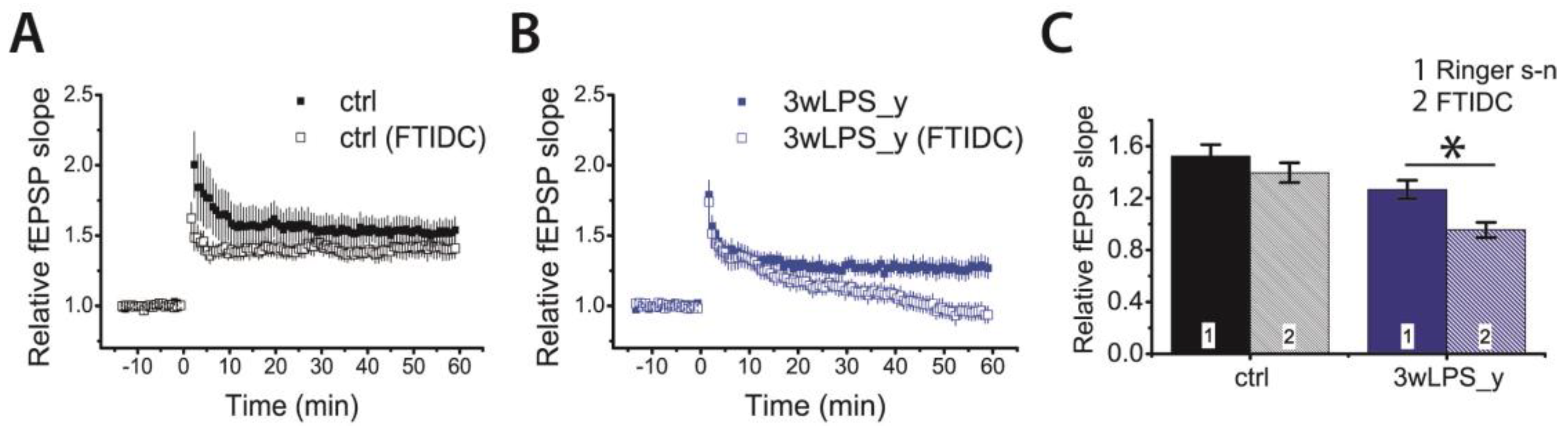
2.3. Pharmacological Properties of LTP Changed in Young Rats Following LPS Treatment
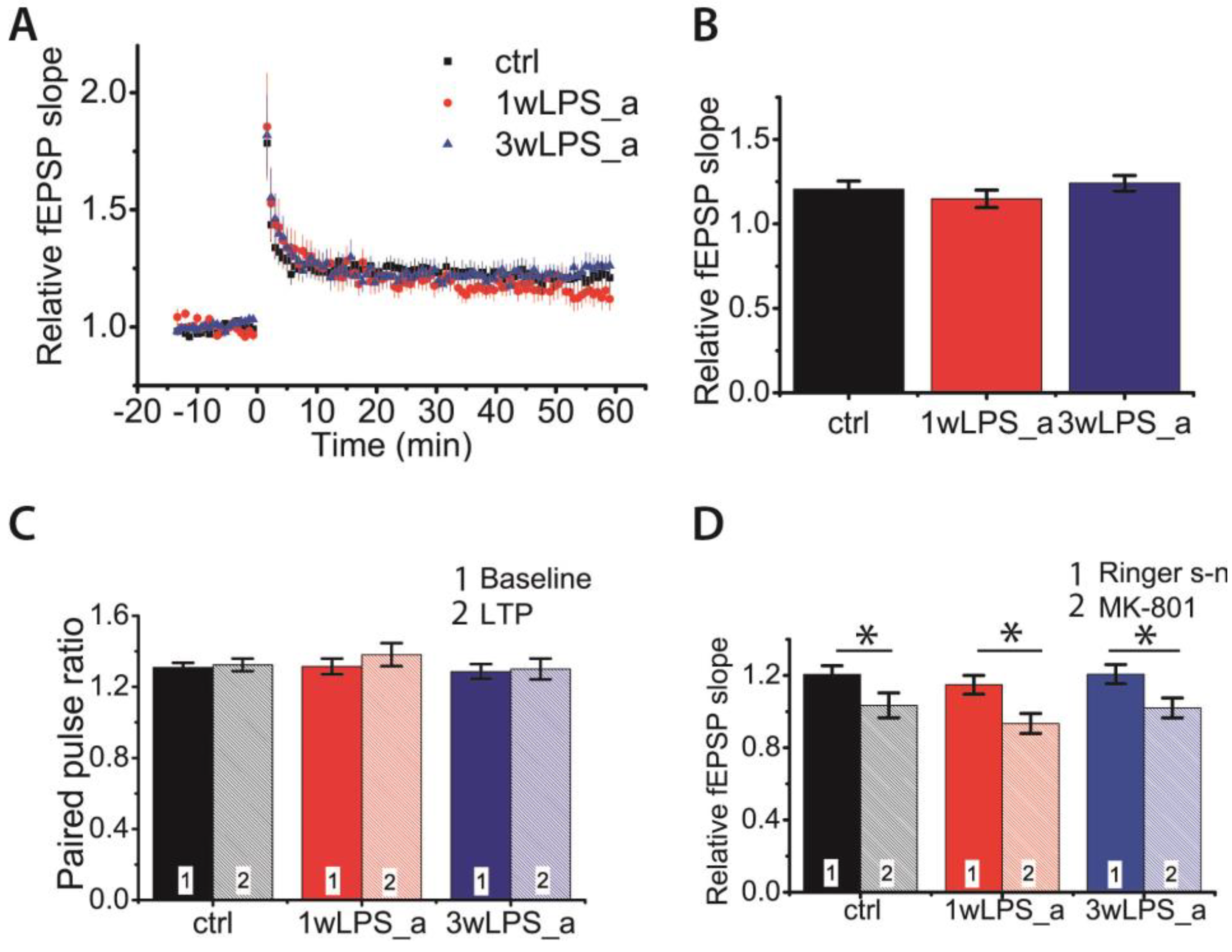
2.4. LPS Administration in Early Postnatal Ontogenesis Does Not Have a Delayed Impact on Synaptic Plasticity in the Hippocampus of Adolescent Rats
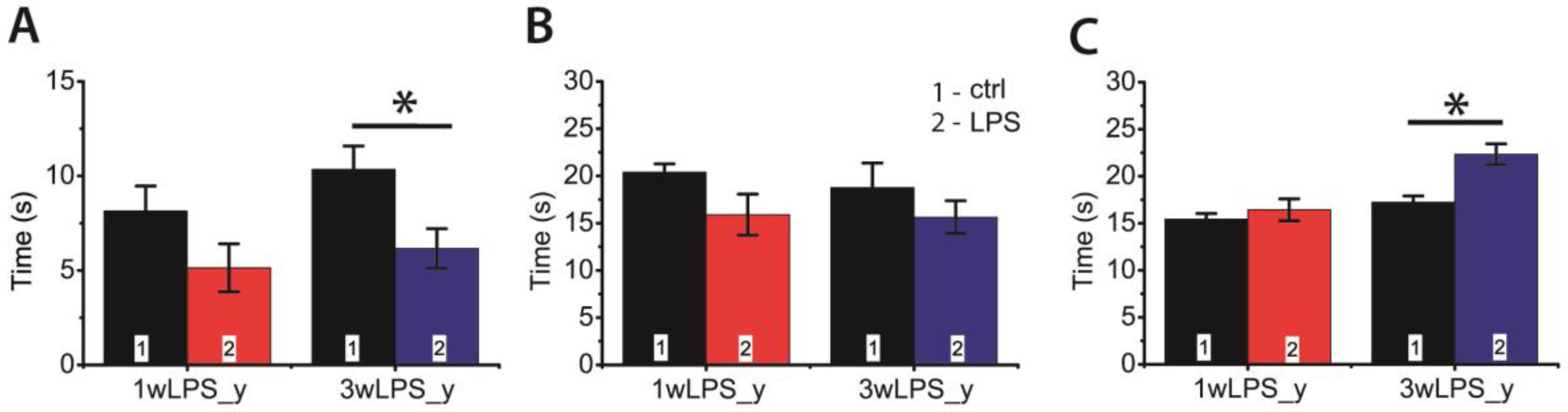
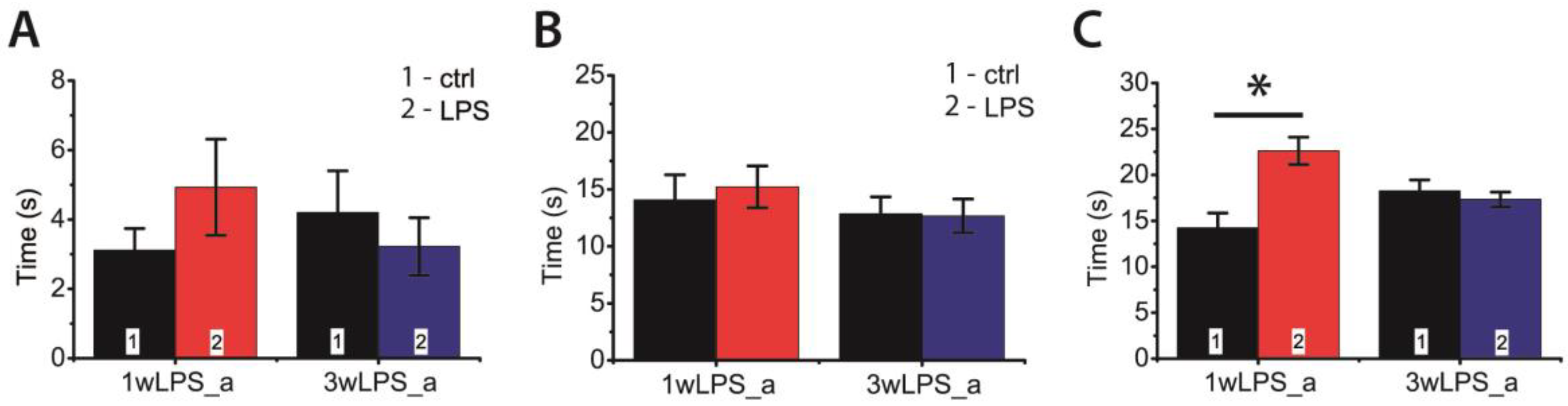
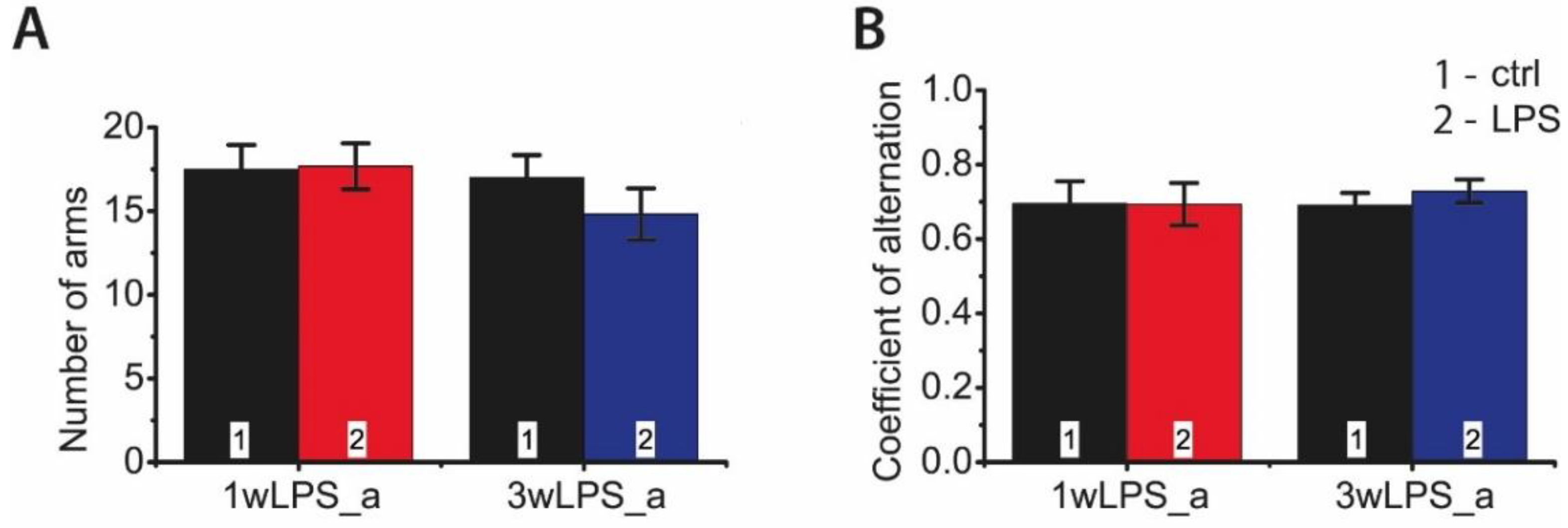
2.5. LPS Administration in Early Ontogenesis Induces Some Changes in the Behavior of Young and Adult Rats
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Hippocampal Brain Slice Preparation
4.3. Electrophysiology
4.4. Patch-Clamp Experiments
4.5. Drugs
4.6. Behavioral Testing
4.6.1. Open Field Test
4.6.2. Y-Maze Spontaneous Alternation Test
4.7. Data Analysis and Statistics
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DNQX | 6,7-dinitroquinoxaline-2,3-dione |
| eEPSC | evoked excitatory postsynaptic current |
| fEPSP | field excitatory postsynaptic potential |
| FV | fiber volley |
| FTIDC | 4-[1-(2-fluoropyridin-3-yl)-5-methyltriazol-4-yl]-N-methylN-propan-2-yl-3,6dihydro-2H-pyridine-1-carboxamide |
| IL-1β | interleukin-1β |
| IL-6 | interleukin 6 |
| LPS | lipopolysaccharide |
| LTP | long-term potentiation |
| NMDA | N-methyl-D-aspartate |
| NMDAR | NMDA receptor |
| PPR | paired-pulse ratio |
| TBS | theta-burst stimulation |
| TNF-α | tumor necrosis factor alpha |
References
- Carter, J.A.; Neville, B.G.; Newton, C.R. Neuro-cognitive impairment following acquired central nervous system infections in childhood: A systematic review. Brain Res. Brain Res. Rev. 2003, 43, 57–69. [Google Scholar] [CrossRef]
- Harre, E.M.; Galic, M.A.; Mouihate, A.; Noorbakhsh, F.; Pittman, Q.J. Neonatal inflammation produces selective behavioural deficits and alters N-methyl-D-aspartate receptor subunit mRNA in the adult rat brain. Eur. J. Neurosci. 2008, 27, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Rousset, C.I.; Hagberg, H.; Mallard, C. Lipopolysaccharide-induced inflammation and perinatal brain injury. Semin. Fetal Neonatal Med. 2006, 11, 343–353. [Google Scholar] [CrossRef]
- Golia, M.T.; Poggini, S.; Alboni, S.; Garofalo, S.; Albanese, N.C.; Viglione, A.; Ajmone-Cat, M.A.; St-Pierre, A.; Brunello, N.; Limatola, C.; et al. Interplay between inflammation and neural plasticity: Both immune activation and suppression impair LTP and BDNF expression. Brain Behav. Immun. 2019, 81, 484–494. [Google Scholar] [CrossRef]
- Kubera, M.; Curzytek, K.; Duda, W.; Leskiewicz, M.; Basta-Kaim, A.; Budziszewska, B.; Roman, A.; Zajicova, A.; Holan, V.; Szczesny, E.; et al. A new animal model of (chronic) depression induced by repeated and intermittent lipopolysaccharide administration for 4 months. Brain Behav. Immun. 2013, 31, 96–104. [Google Scholar] [CrossRef]
- Trotta, T.; Porro, C.; Calvello, R.; Panaro, M.A. Biological role of Toll-like receptor-4 in the brain. J. Neuroimmunol. 2014, 268, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, M.M.; Hutchinson, M.; Watkins, L.R.; Yin, H. Toll-like receptor 4 in CNS pathologies. J. Neurochem. 2010, 114, 13–27. [Google Scholar] [CrossRef]
- Onufriev, M.V.; Freiman, S.V.; Peregud, D.I.; Kudryashova, I.V.; Tishkina, A.O.; Stepanichev, M.Y.; Gulyaeva, N.V. Neonatal Proinflammatory Stress Induces Accumulation of Corticosterone and Interleukin-6 in the Hippocampus of Juvenile Rats: Potential Mechanism of Synaptic Plasticity Impairments. Biochem. Biokhimiia 2017, 82, 275–281. [Google Scholar] [CrossRef]
- Dias, P.; Freiberger, V.; Ventura, L.; Bragagnolo, D.; Dutra, M.L.; Horewicz, V.V.; Comim, C.M. Late Brain Involvement after Neonatal Immune Activation. Biomed. Res. Int. 2019, 2019, 9573248. [Google Scholar] [CrossRef]
- Trofimov, A.; Strekalova, T.; Mortimer, N.; Zubareva, O.; Schwarz, A.; Svirin, E.; Umriukhin, A.; Svistunov, A.; Lesch, K.P.; Klimenko, V. Postnatal LPS Challenge Impacts Escape Learning and Expression of Plasticity Factors Mmp9 and Timp1 in Rats: Effects of Repeated Training. Neurotox. Res. 2017, 32, 175–186. [Google Scholar] [CrossRef]
- Zubareva, O.E.; Klimenko, V.M. Long-term disorders of behavior in rats induced by administration of tumor necrosis factor during early postnatal ontogenesis. Neurosci. Behav. Physiol. 2009, 39, 21–24. [Google Scholar] [CrossRef]
- Zubareva, O.E.; Eliseeva, A.P.; Simbirtsev, A.S.; Klimenko, V.M. The effects of proinflammatory cytokines on the formation of behavior in early postnatal ontogenesis. Neurosci. Behav. Physiol. 2006, 36, 367–372. [Google Scholar] [CrossRef]
- Bliss, T.V.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Malenka, R.C.; Bear, M.F. LTP and LTD: An embarrassment of riches. Neuron 2004, 44, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Ko, G.Y.; Kelly, P.T. Cellular and molecular bases of memory: Synaptic and neuronal plasticity. J. Clin. Neurophysiol. 1997, 14, 264–293. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Malenka, R.C. Synaptic plasticity: Multiple forms, functions, and mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef] [PubMed]
- Larson, J.; Wong, D.; Lynch, G. Patterned stimulation at the theta frequency is optimal for the induction of hippocampal long-term potentiation. Brain Res. 1986, 368, 347–350. [Google Scholar] [CrossRef]
- Raymond, C.R. LTP forms 1, 2 and 3: Different mechanisms for the “long” in long-term potentiation. Trends Neurosci. 2007, 30, 167–175. [Google Scholar] [CrossRef]
- Cunningham, A.J.; Murray, C.A.; O’Neill, L.A.; Lynch, M.A.; O’Connor, J.J. Interleukin-1 beta (IL-1 beta) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci. Lett. 1996, 203, 17–20. [Google Scholar] [CrossRef]
- Rizzo, F.R.; Musella, A.; De Vito, F.; Fresegna, D.; Bullitta, S.; Vanni, V.; Guadalupi, L.; Stampanoni Bassi, M.; Buttari, F.; Mandolesi, G.; et al. Tumor Necrosis Factor and Interleukin-1beta Modulate Synaptic Plasticity during Neuroinflammation. Neural Plast. 2018, 2018, 8430123. [Google Scholar] [CrossRef]
- Bellinger, F.P.; Madamba, S.; Siggins, G.R. Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 1993, 628, 227–234. [Google Scholar] [CrossRef]
- Ross, F.M.; Allan, S.M.; Rothwell, N.J.; Verkhratsky, A. A dual role for interleukin-1 in LTP in mouse hippocampal slices. J. Neuroimmunol. 2003, 144, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Stampanoni Bassi, M.; Iezzi, E.; Mori, F.; Simonelli, I.; Gilio, L.; Buttari, F.; Sica, F.; De Paolis, N.; Mandolesi, G.; Musella, A.; et al. Interleukin-6 Disrupts Synaptic Plasticity and Impairs Tissue Damage Compensation in Multiple Sclerosis. Neurorehabil. Neural Repair 2019. [Google Scholar] [CrossRef] [PubMed]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-alpha. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Lewitus, G.M.; Pribiag, H.; Duseja, R.; St-Hilaire, M.; Stellwagen, D. An adaptive role of TNFalpha in the regulation of striatal synapses. J. Neurosci. 2014, 34, 6146–6155. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, L.J.; Wang, J.; Li, D.; Ren, W.J.; Peng, J.; Wei, X.; Xu, T.; Xin, W.J.; Pang, R.P.; et al. TNF-alpha Differentially Regulates Synaptic Plasticity in the Hippocampus and Spinal Cord by Microglia-Dependent Mechanisms after Peripheral Nerve Injury. J. Neurosci. 2017, 37, 871–881. [Google Scholar] [CrossRef]
- Ren, W.J.; Liu, Y.; Zhou, L.J.; Li, W.; Zhong, Y.; Pang, R.P.; Xin, W.J.; Wei, X.H.; Wang, J.; Zhu, H.Q.; et al. Peripheral nerve injury leads to working memory deficits and dysfunction of the hippocampus by upregulation of TNF-alpha in rodents. Neuropsychopharmacology 2011, 36, 979–992. [Google Scholar] [CrossRef]
- Tancredi, V.; D’Arcangelo, G.; Grassi, F.; Tarroni, P.; Palmieri, G.; Santoni, A.; Eusebi, F. Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci. Lett. 1992, 146, 176–178. [Google Scholar] [CrossRef]
- Caraci, F.; Gulisano, W.; Guida, C.A.; Impellizzeri, A.A.; Drago, F.; Puzzo, D.; Palmeri, A. A key role for TGF-beta1 in hippocampal synaptic plasticity and memory. Sci. Rep. 2015, 5, 11252. [Google Scholar] [CrossRef]
- Lynch, M.A. Long-term potentiation and memory. Physiol. Rev. 2004, 84, 87–136. [Google Scholar] [CrossRef]
- Cavazzini, M.; Bliss, T.; Emptage, N. Ca2+ and synaptic plasticity. Cell Calcium 2005, 38, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Raymond, C.R.; Redman, S.J. Different calcium sources are narrowly tuned to the induction of different forms of LTP. J. Neurophysiol. 2002, 88, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ardiles, A.O.; Yang, S.; Tran, T.; Posada-Duque, R.; Valdivia, G.; Baek, M.; Chuang, Y.A.; Palacios, A.G.; Gallagher, M.; et al. Metabotropic Glutamate Receptors Induce a Form of LTP Controlled by Translation and Arc Signaling in the Hippocampus. J. Neurosci. 2016, 36, 1723–1729. [Google Scholar] [CrossRef] [PubMed]
- Malenka, R.C.; Nicoll, R.A. NMDA-receptor-dependent synaptic plasticity: Multiple forms and mechanisms. Trends Neurosci. 1993, 16, 521–527. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Monyer, H.; Burnashev, N.; Laurie, D.J.; Sakmann, B.; Seeburg, P.H. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 1994, 12, 529–540. [Google Scholar] [CrossRef]
- Cull-Candy, S.; Brickley, S.; Farrant, M. NMDA receptor subunits: Diversity, development and disease. Curr. Opin. Neurobiol. 2001, 11, 327–335. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Liu, L.; Wong, T.P.; Pozza, M.F.; Lingenhoehl, K.; Wang, Y.; Sheng, M.; Auberson, Y.P.; Wang, Y.T. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 2004, 304, 1021–1024. [Google Scholar] [CrossRef]
- Sakimura, K.; Kutsuwada, T.; Ito, I.; Manabe, T.; Takayama, C.; Kushiya, E.; Yagi, T.; Aizawa, S.; Inoue, Y.; Sugiyama, H.; et al. Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature 1995, 373, 151–155. [Google Scholar] [CrossRef]
- Wenzel, A.; Fritschy, J.M.; Mohler, H.; Benke, D. NMDA receptor heterogeneity during postnatal development of the rat brain: Differential expression of the NR2A, NR2B, and NR2C subunit proteins. J. Neurochem. 1997, 68, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Postnikova, T.Y.; Trofimova, A.M.; Ergina, J.L.; Zubareva, O.E.; Kalemenev, S.V.; Zaitsev, A.V. Transient Switching of NMDA-Dependent Long-Term Synaptic Potentiation in CA3-CA1 Hippocampal Synapses to mGluR1-Dependent Potentiation After Pentylenetetrazole-Induced Acute Seizures in Young Rats. Cell. Mol. Neurobiol. 2019, 39, 287–300. [Google Scholar] [CrossRef]
- Selig, D.K.; Nicoll, R.A.; Malenka, R.C. Hippocampal long-term potentiation preserves the fidelity of postsynaptic responses to presynaptic bursts. J. Neurosci. 1999, 19, 1236–1246. [Google Scholar] [CrossRef]
- Buonomano, D.V. Distinct functional types of associative long-term potentiation in neocortical and hippocampal pyramidal neurons. J. Neurosci. 1999, 19, 6748–6754. [Google Scholar] [CrossRef]
- Zaitsev, A.V.; Anwyl, R. Inhibition of the slow afterhyperpolarization restores the classical spike timing-dependent plasticity rule obeyed in layer 2/3 pyramidal cells of the prefrontal cortex. J. Neurophysiol. 2012, 107, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Postnikova, T.Y.; Amakhin, D.V.; Trofimova, A.M.; Smolensky, I.V.; Zaitsev, A.V. Changes in Functional Properties of Rat Hippocampal Neurons Following Pentylenetetrazole-induced Status Epilepticus. Neuroscience 2019, 399, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Amakhin, D.V.; Malkin, S.L.; Ergina, J.L.; Kryukov, K.A.; Veniaminova, E.A.; Zubareva, O.E.; Zaitsev, A.V. Alterations in Properties of Glutamatergic Transmission in the Temporal Cortex and Hippocampus Following Pilocarpine-Induced Acute Seizures in Wistar Rats. Front. Cell. Neurosci. 2017, 11, 264. [Google Scholar] [CrossRef]
- Woolley, C.S.; Weiland, N.G.; McEwen, B.S.; Schwartzkroin, P.A. Estradiol increases the sensitivity of hippocampal CA1 pyramidal cells to NMDA receptor-mediated synaptic input: Correlation with dendritic spine density. J. Neurosci. 1997, 17, 1848–1859. [Google Scholar] [CrossRef]
- Dobrunz, L.E.; Stevens, C.F. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron 1997, 18, 995–1008. [Google Scholar] [CrossRef]
- Zucker, R.S.; Regehr, W.G. Short-term synaptic plasticity. Annu. Rev. Physiol. 2002, 64, 355–405. [Google Scholar] [CrossRef]
- Neves, G.; Cooke, S.F.; Bliss, T.V. Synaptic plasticity, memory and the hippocampus: A neural network approach to causality. Nat. Rev. Neurosci. 2008, 9, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Grover, L.M.; Kim, E.; Cooke, J.D.; Holmes, W.R. LTP in hippocampal area CA1 is induced by burst stimulation over a broad frequency range centered around delta. Learn. Mem. 2009, 16, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Mayford, M.; Siegelbaum, S.A.; Kandel, E.R. Synapses and memory storage. Cold Spring Harb. Perspect. Biol. 2012, 4, a005751. [Google Scholar] [CrossRef] [PubMed]
- Massey, P.V.; Johnson, B.E.; Moult, P.R.; Auberson, Y.P.; Brown, M.W.; Molnar, E.; Collingridge, G.L.; Bashir, Z.I. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J. Neurosci. 2004, 24, 7821–7828. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Hansen, K.B.; Jahr, C.E. Allosteric Interactions between NMDA Receptor Subunits Shape the Developmental Shift in Channel Properties. Neuron 2017, 94, 58–64. [Google Scholar] [CrossRef]
- Bortolotto, Z.A.; Collingridge, G.L. Characterisation of LTP induced by the activation of glutamate metabotropic receptors in area CA1 of the hippocampus. Neuropharmacology 1993, 32, 1–9. [Google Scholar] [CrossRef]
- Suzuki, G.; Kimura, T.; Satow, A.; Kaneko, N.; Fukuda, J.; Hikichi, H.; Sakai, N.; Maehara, S.; Kawagoe-Takaki, H.; Hata, M.; et al. Pharmacological characterization of a new, orally active and potent allosteric metabotropic glutamate receptor 1 antagonist, 4-[1-(2-fluoropyridin-3-yl)-5-methyl-1H-1,2,3-triazol-4-yl]-N-isopropyl-N-methyl- 3,6-dihydropyridine-1(2H)-carboxamide (FTIDC). J. Pharmacol. Exp. Ther. 2007, 321, 1144–1153. [Google Scholar] [CrossRef]
- Zhang, Z.W. Maturation of layer V pyramidal neurons in the rat prefrontal cortex: Intrinsic properties and synaptic function. J. Neurophysiol. 2004, 91, 1171–1182. [Google Scholar] [CrossRef]
- Harris, K.M.; Jensen, F.E.; Tsao, B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: Implications for the maturation of synaptic physiology and long-term potentiation. J. Neurosci. 1992, 12, 2685–2705. [Google Scholar] [CrossRef]
- Iwai, T.; Sawabe, T.; Tanimitsu, K.; Suzuki, M.; Sasaki-Hamada, S.; Oka, J. Glucagon-like peptide-1 protects synaptic and learning functions from neuroinflammation in rodents. J. Neurosci. Res. 2014, 92, 446–454. [Google Scholar] [CrossRef]
- O’Donnell, E.; Vereker, E.; Lynch, M.A. Age-related impairment in LTP is accompanied by enhanced activity of stress-activated protein kinases: Analysis of underlying mechanisms. Eur. J. Neurosci. 2000, 12, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Correa, S.A.; Eales, K.L. The Role of p38 MAPK and Its Substrates in Neuronal Plasticity and Neurodegenerative Disease. J. Signal Transduct. 2012, 2012, 649079. [Google Scholar] [CrossRef] [PubMed]
- Diering, G.H.; Huganir, R.L. The AMPA Receptor Code of Synaptic Plasticity. Neuron 2018, 100, 314–329. [Google Scholar] [CrossRef]
- Tanaka, S.; Ide, M.; Shibutani, T.; Ohtaki, H.; Numazawa, S.; Shioda, S.; Yoshida, T. Lipopolysaccharide-induced microglial activation induces learning and memory deficits without neuronal cell death in rats. J. Neurosci. Res. 2006, 83, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, X.; Ai, S.; Ouyang, W.; Le, Y.; Tong, J. Sepsis-induced selective loss of NMDA receptors modulates hippocampal neuropathology in surviving septic mice. PLoS ONE 2017, 12, e0188273. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Choi, B.R.; Chung, C.; Min, S.S.; Jeon, W.K.; Han, J.S. Chronic brain inflammation causes a reduction in GluN2A and GluN2B subunits of NMDA receptors and an increase in the phosphorylation of mitogen-activated protein kinases in the hippocampus. Mol. Brain 2014, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R.; McGlothan, J.L. Hippocampal NMDA receptor mRNA undergoes subunit specific changes during developmental lead exposure. Brain Res. 1998, 790, 98–107. [Google Scholar] [CrossRef]
- Gonzalez-Burgos, G.; Kroener, S.; Zaitsev, A.V.; Povysheva, N.V.; Krimer, L.S.; Barrionuevo, G.; Lewis, D.A. Functional maturation of excitatory synapses in layer 3 pyramidal neurons during postnatal development of the primate prefrontal cortex. Cereb. Cortex 2008, 18, 626–637. [Google Scholar] [CrossRef]
- Qiu, S.; Weeber, E.J. Reelin signaling facilitates maturation of CA1 glutamatergic synapses. J. Neurophysiol. 2007, 97, 2312–2321. [Google Scholar] [CrossRef]
- Degos, V.; Peineau, S.; Nijboer, C.; Kaindl, A.M.; Sigaut, S.; Favrais, G.; Plaisant, F.; Teissier, N.; Gouadon, E.; Lombet, A.; et al. G protein-coupled receptor kinase 2 and group I metabotropic glutamate receptors mediate inflammation-induced sensitization to excitotoxic neurodegeneration. Ann. Neurol. 2013, 73, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, F.M.; Ferreira, L.T.; Paquet, M.; Cregan, T.; Ding, Q.; Gros, R.; Ferguson, S.S. Phosphorylation-independent regulation of metabotropic glutamate receptor 5 desensitization and internalization by G protein-coupled receptor kinase 2 in neurons. J. Biol. Chem. 2009, 284, 23444–23453. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.N.; Cummins, R.A. The open-field test: A critical review. Psychol. Bull. 1976, 83, 482–504. [Google Scholar] [CrossRef] [PubMed]
- Sarter, M.; Bodewitz, G.; Stephens, D.N. Attenuation of scopolamine-induced impairment of spontaneous alteration behaviour by antagonist but not inverse agonist and agonist beta-carbolines. Psychopharmacology 1988, 94, 491–495. [Google Scholar] [CrossRef] [PubMed]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Postnikova, T.Y.; Griflyuk, A.V.; Ergina, J.L.; Zubareva, O.E.; Zaitsev, A.V. Administration of Bacterial Lipopolysaccharide during Early Postnatal Ontogenesis Induces Transient Impairment of Long-Term Synaptic Plasticity Associated with Behavioral Abnormalities in Young Rats. Pharmaceuticals 2020, 13, 48. https://doi.org/10.3390/ph13030048
Postnikova TY, Griflyuk AV, Ergina JL, Zubareva OE, Zaitsev AV. Administration of Bacterial Lipopolysaccharide during Early Postnatal Ontogenesis Induces Transient Impairment of Long-Term Synaptic Plasticity Associated with Behavioral Abnormalities in Young Rats. Pharmaceuticals. 2020; 13(3):48. https://doi.org/10.3390/ph13030048
Chicago/Turabian StylePostnikova, Tatyana Y., Alexandra V. Griflyuk, Julia L. Ergina, Olga E. Zubareva, and Aleksey V. Zaitsev. 2020. "Administration of Bacterial Lipopolysaccharide during Early Postnatal Ontogenesis Induces Transient Impairment of Long-Term Synaptic Plasticity Associated with Behavioral Abnormalities in Young Rats" Pharmaceuticals 13, no. 3: 48. https://doi.org/10.3390/ph13030048
APA StylePostnikova, T. Y., Griflyuk, A. V., Ergina, J. L., Zubareva, O. E., & Zaitsev, A. V. (2020). Administration of Bacterial Lipopolysaccharide during Early Postnatal Ontogenesis Induces Transient Impairment of Long-Term Synaptic Plasticity Associated with Behavioral Abnormalities in Young Rats. Pharmaceuticals, 13(3), 48. https://doi.org/10.3390/ph13030048