Effects of an Adaptogenic Extract on Electrical Activity of the Brain in Elderly Subjects with Mild Cognitive Impairment: A Randomized, Double-Blind, Placebo-Controlled, Two-Armed Cross-Over Study



Abstract
1. Introduction
2. Results
2.1. Baseline Data of Study Participants
2.2. Efficacy of Treatment
- Objective recording of quantitative EEG (efficacy primary outcomes) during performance of several psychometric tasks,
- Objective assessment of cognitive functions and mental performance in several psychometric tests (efficacy secondary outcomes), and
- Subjective assessment of mental health and quality of sleep based on validated questionnaires (efficacy secondary outcomes).
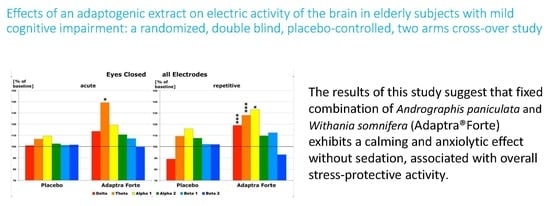
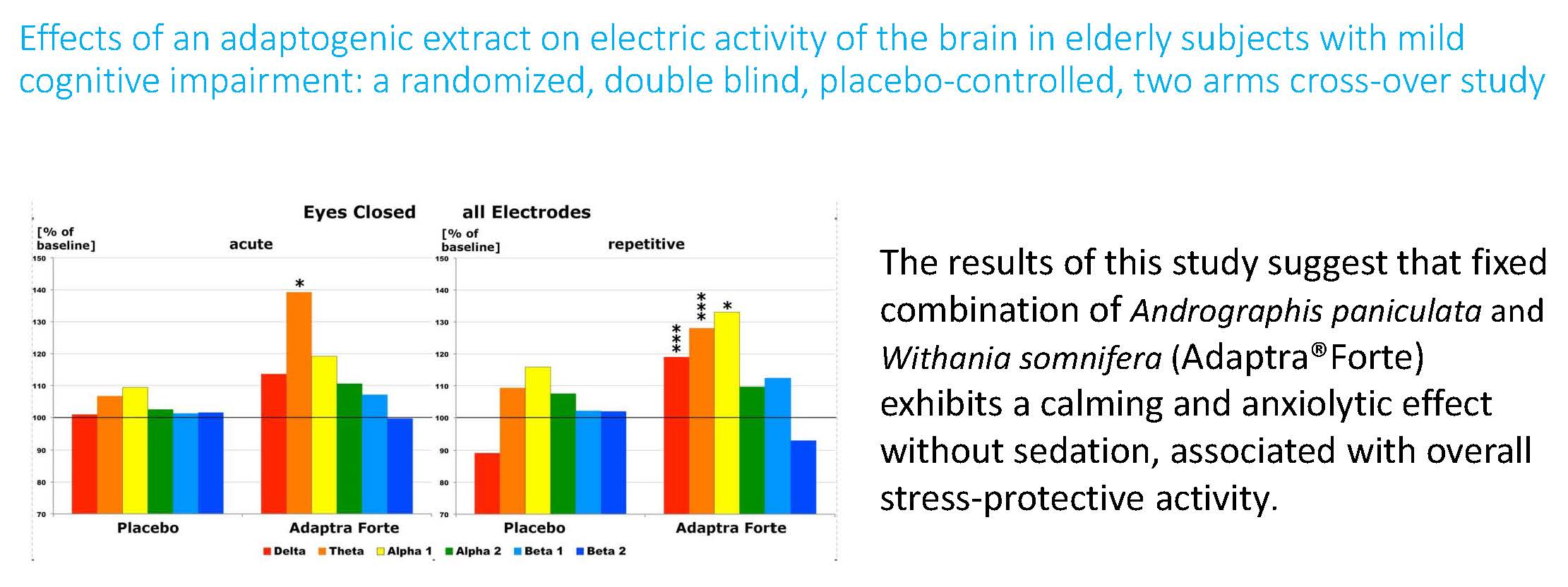
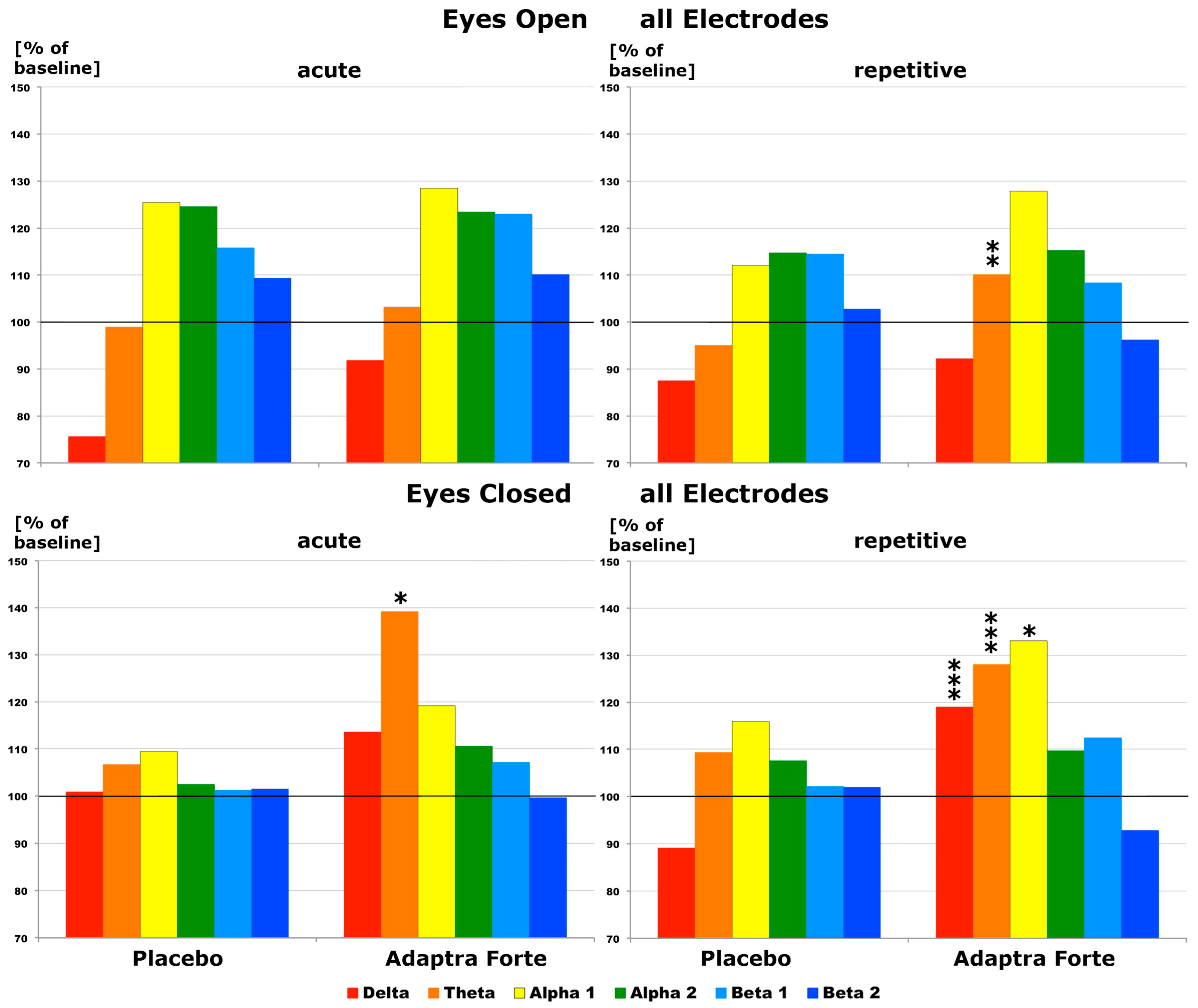
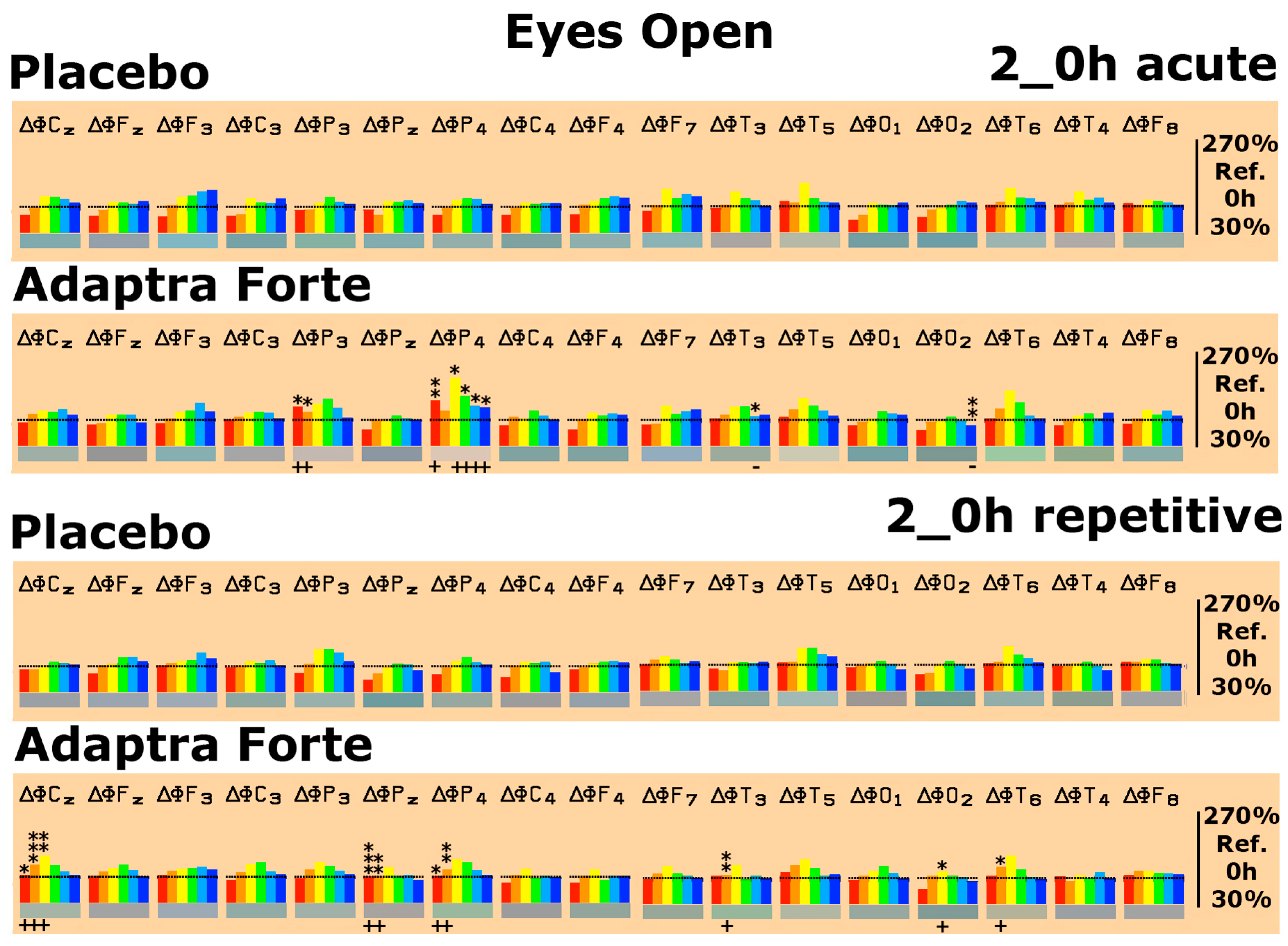
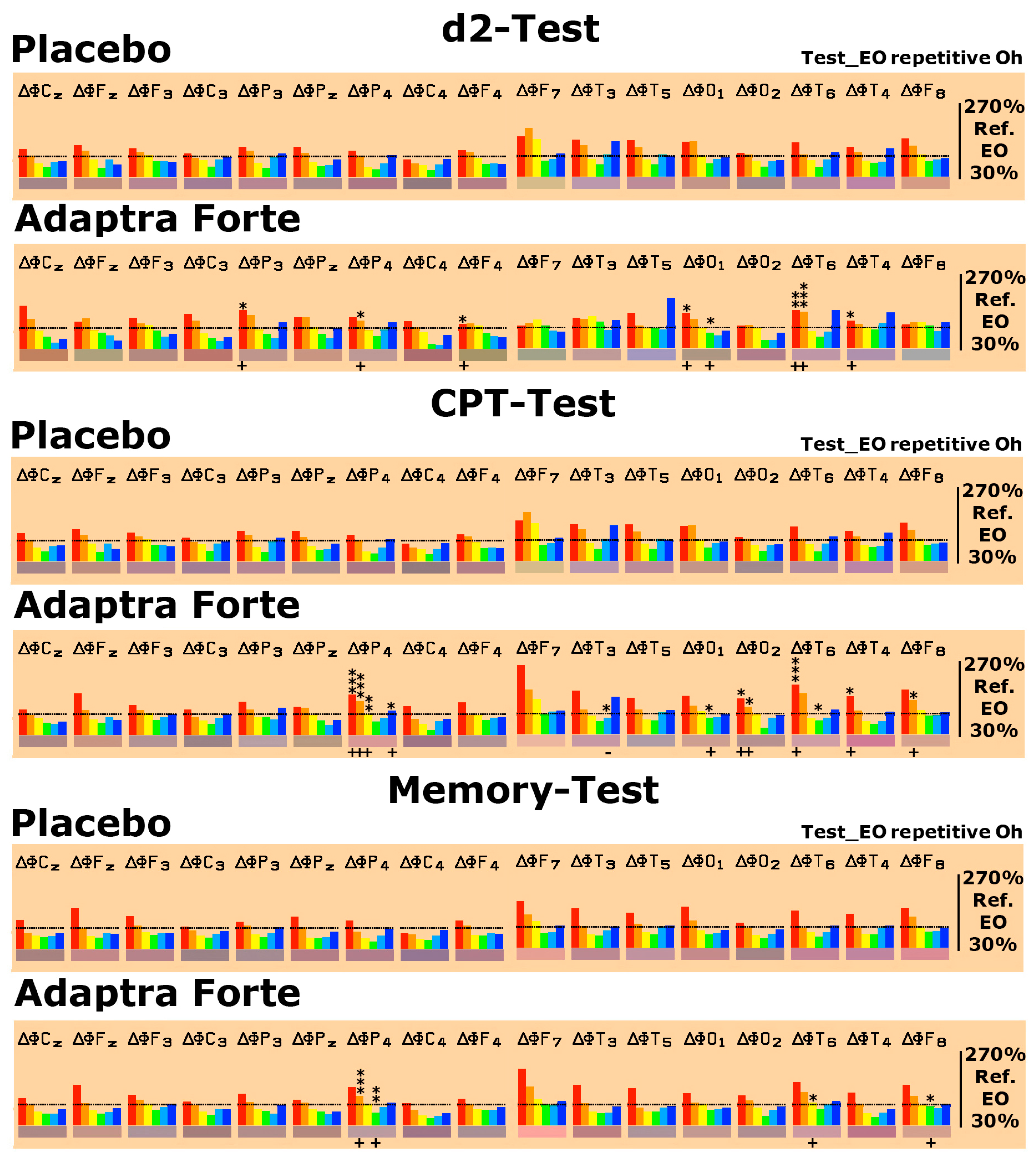
2.2.1. Efficacy Primary Endpoint
Quantitative Electroencephalogram (EEG) Results after Acute and Repetitive Dosing
Follow-Up Two Weeks after the Last Experimental Day
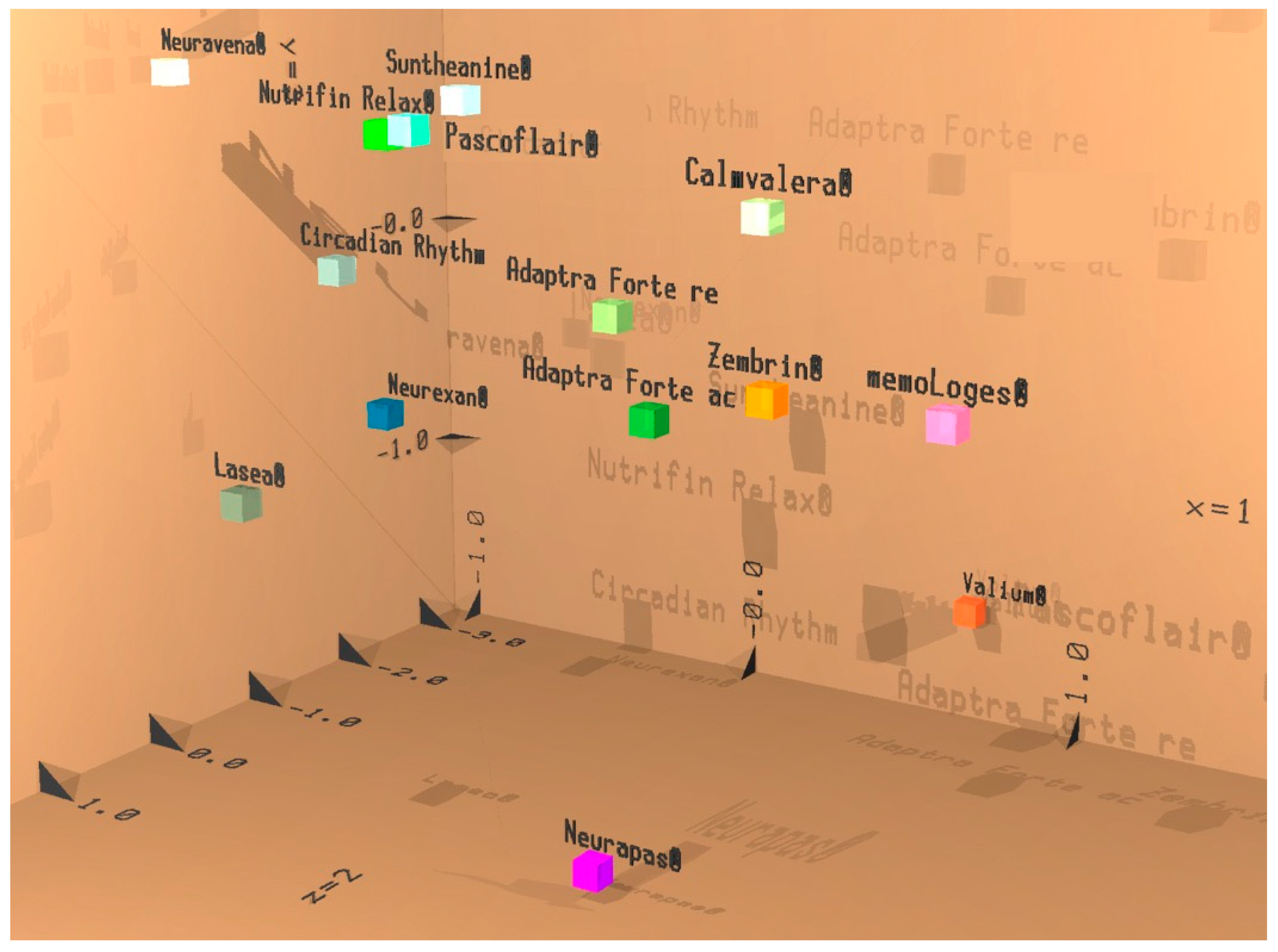
Efficacy of Adaptra® Forte Documented by Discriminant Analysis
2.2.2. Efficacy Primary Endpoints
Results from Performance of Psychometric Tests
Subjective Assessment of Effect of Adaptra® Forte Mood State and Quality of Sleep Based on Questionnaires
2.3. Safety Evaluation
3. Discussion
4. Materials and Methods
4.1. Participant Eligibility
4.2. Selection of the Study Population
4.2.1. Screening and Recruitment
4.2.2. Inclusion Criteria
- Male and female volunteers suffering from cognitive deficits.
- Questionnaire-DemTect. “DemTect” (for pre-selection of subjects) score values of 8–12 were regarded as conclusive.
- Age between 60 and 75 years inclusive.
- Subjects should be right-handed.
- Subject must be capable of providing informed consent.
- Acceptance of written consent to participate in the study after instruction in written and oral form (informed consent).
4.2.3. Exclusion Criteria
- Clinically relevant acute or chronic diseases as determined by case history or clinical examination.
- Clinically relevant allergic symptoms.
- Intake of clinically relevant medication in the last two weeks prior to screening (SC) as well as during the active study period based on notification by the subject or their case history.
- Intake of medication with primarily central nervous action (e.g., psychopharmaceuticals or centrally acting antihypertensives, antiepileptics, antidepressants).
- Known intolerance/hypersensitivity (allergy) to plant-derived extracts or any other of the ingredients of the investigational product (anamnestic survey).
- Presence of a rare genetic disease, such as fructose intolerance, glucose–galactose malabsorption, or sucrose–isomaltase deficiency (anamnestic).
- Intake of unusual quantities or abuse of coffee (more than 4 cups a day), tea (more than 4 cups a day), or tobacco (more than 20 cigarettes per day).
- Detection of alcohol at the time of the initial examination (day SC) or on study days A, B, C, D, and FU (positive alcohol test) or by case history.
- Smoking in the study center on study days A, B, C, D, and FU.
- Results of the DemTect Questionnaire score being <8 or >12.
- Participation in another clinical trial within the last 60 days.
- Poor compliance.
- Cancellation of informed consent.
4.2.4. Withdrawal of Subjects
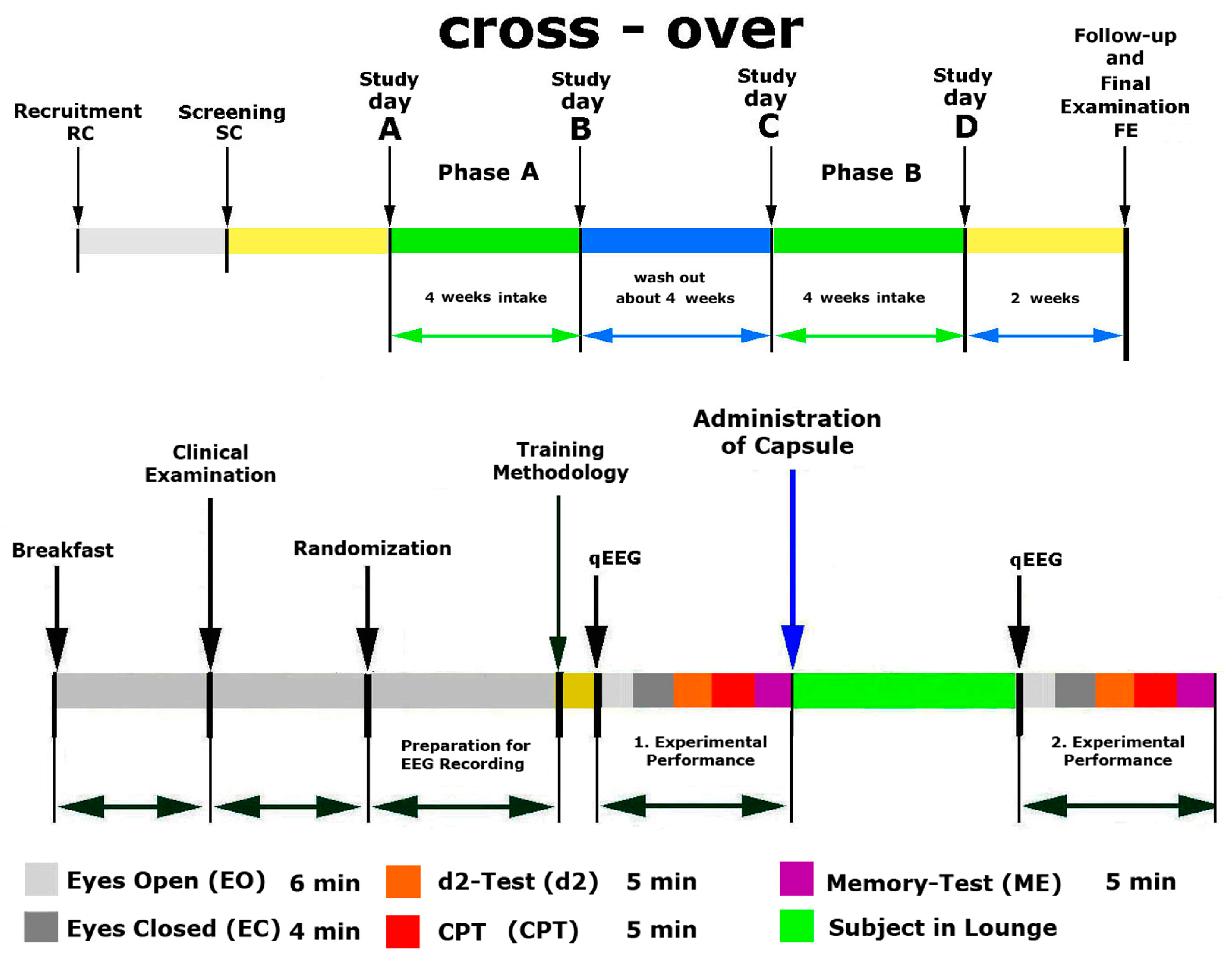
4.3. Study Design
4.4. Intervention and Comparator
Doses and Treatment Regimens
4.5. Allocation, Study Procedures, and Follow-Up
4.5.1. Randomization, Blinding, and Allocation Concealment
4.5.2. Evaluation of Compliance
4.5.3. Efficacy Measurement
Phase A, Screening and Training
Phase A, Treatment and Assessment
Phase B, Treatment and Assessment
4.6. Efficacy and Safety Evaluation
4.6.1. Efficacy Primary Outcome
4.6.2. Efficacy Secondary Outcomes
- The accuracy of processing the test for attention, concentration, and visual scanning speed, which is expressed as an error rate (ER) score (E%), is defined as the ratio of errors made to the total number of correct responses during 4 min and 40 s
- “Accuracy score” obtained in ME-Test
- Concentration performance score in CPT
- Profile of Mood States (POMS) using a validated POMS questionnaire [54,55,56]. The questionnaire contains 65 words/statements that describe people’s feelings. The test requires the patient to indicate for each word or statement how they have been feeling in the past week including today. Scores are: S1 = dejection (Niedergeschlagenheit); S2 = sullenness (Missmut); S3 = fatigue (Müdigkeit), S4 = thirst for action (Tatendrang).
- Quality of sleep score obtained by SF-B/R (Schlaffragebogen) questionnaire (a subjective assessment of sleep quality), used for quantitative and qualitative description and evaluation of sleep behavior and sleep experience. The SF-B/R comprises 31 questions and refers to the last two weeks [57,58,59]. The following subscores were used for the evaluation of the sleep questionnaire SF-B/R: SQ = sleep quality (Schlafqualität); GES = feeling refreshed after sleep/feeling of being recovered after sleep (Gefühl des Erholtseins nach dem Schlaf); PSYA = psychic well-balanced feeling in the evening (psychische Ausgeglichenheit vor dem Schlafenlegen); PSYE = psychic exhausted feeling in the evening (psychisches Erschöpftsein vor dem Schlafenlegen); PSS = psychosomatic problems during sleep (psychosomatische Symptome in der Schlafphase); TRME = dream recall (Traumerinnerung); SWR = sleep–wake regulation (Schlaf-Wach-Regulation).
4.7. Safety Outcomes
4.8. Sample Size Considerations
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethics Approval
References
- Panossian, A.G. Understanding adaptogenic activity: Specificity of the pharmacological action of adaptogens and other phytochemicals. Ann. N. Y. Acad. Sci. 2017, 1401, 49–64. [Google Scholar] [CrossRef]
- Panossian, A.G. Adaptogens: Tonic herbs for fatigue and stress. Altern. Compliment. Ther. 2003, 9, 327–332. [Google Scholar] [CrossRef]
- Samuelsson, G.; Bohlin, L. Drugs of Natural Origin: A Treatise of Pharmacognosy, 6th ed.; Swedish Academy of Pharmaceutical Sciences: Stockholm, Sweden, 2009. [Google Scholar]
- Panossian, A.; Seo, E.J.; Efferth, T. Novel molecular mechanisms for the adaptogenic effects of herbal extracts on isolated brain cells using systems biology. Phytomedicine 2018, 50, 257–284. [Google Scholar] [CrossRef]
- Panossian, A.; Hamm, R.; Wikman, G.; Kadioglu, O.; Efferth, T. Synergy and antagonism of active constituents of ADAPT-232 on transcriptional level of metabolic regulation in isolated neuroglia cells. Front. Neurosci. 2013, 20, 7–16. [Google Scholar]
- Panossian, A.; Hamm, R.; Wikman, G.; Efferth, T. Mechanism of action of Rhodiola, salidroside, tyrosol and triandrin in isolated neuroglial cells: An interactive pathway analysis of the downstream effects using RNA microarray data. Phytomedicine 2014, 21, 1325–1348. [Google Scholar] [CrossRef] [PubMed]
- Panossian, A.; Wikman, G.; Kaur, P.; Asea, A. Adaptogens exert a stress-protective effect by modulation of expression of molecular chaperones. Phytomedicine 2009, 16, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Panossian, A.G.; Wikman, G.; Kaur, P.; Asea, A. Adaptogens Stimulate Neuropeptide Y and Hsp72 Expression and Release in Neuroglia Cells. Front. Neurosci. 2012, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Panossian, A.; Hambartsumyan, M.; Hovanissian, A.; Gabrielyan, E.; Wilkman, G. The Adaptogens Rhodiola and Schizandra modify the response to immobilization stress in rabbits by suppressing the increase of phosphorylated stress-activated protein kinase, nitric oxide and cortisol. Drug. Targets Insights 2007, 2, 39–54. [Google Scholar] [CrossRef]
- Mohanan, P.; Subramaniyam, S.; Mathiyalagan, R.; Yang, D.C. Molecular signaling of ginsenosides Rb1, Rg1, and Rg3 and their mode of actions. J. Ginseng. Res. 2018, 42, 123–132. [Google Scholar] [CrossRef]
- Olsson, E.M.; von Schéele, B.; Panossian, A.G. A randomized double-blind placebo controlled parallel group study of SHR-5 extract of Rhodiola Rosea roots as treatment for patients with stress related fatigue. Planta Med. 2009, 75, 105–112. [Google Scholar] [CrossRef]
- Darbinyan, V.; Aslanyan, G.; Amroyan, E.; Gabrielyan, E.; Malmström, C.; Panossian, A. Clinical trial of Rhodiola rosea L. extract SHR-5 in the treatment of mild to moderate depression. Nord. J. Psychiatry 2007, 61, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Darbinyan, V.; Kteyan, A.; Panossian, A.; Gabrielian, E.; Wikman, G.; Wagner, H. Rhodiola rosea in stress induced fatigue—A double blind cross-over study of a standardized extract SHR-5 with a repeated low-dose regimen on the mental performance of healthy physicians during night duty. Phytomedicine 2000, 7, 365–371. [Google Scholar] [CrossRef]
- Panossian, A.; Gerbarg, P. Potential Use of Plant Adaptogens in Age-related Disorders. In Complementary, Alternative, and Integrative Interventions in Mental Health and Aging; Lavretsky, H., Sajatovic, M., Reynolds, C.F., III, Eds.; Oxford University Press: New York, NY, USA, 2016; pp. 197–211. [Google Scholar]
- Panossian, A.; Wikman, G. Evidence-based efficacy of adaptogens in fatigue, and molecular mechanisms related to their stress-protective activity. Curr. Clin. Pharmacol. 2009, 4, 198–219. [Google Scholar] [CrossRef] [PubMed]
- Panossian, A.; Wikman, G. Effects of Adaptogens on the Central Nervous System and the Molecular Mechanisms Associated with Their Stress—Protective Activity. Pharmaceuticals 2010, 3, 188–224. [Google Scholar] [CrossRef] [PubMed]
- Amsterdam, J.D.; Panossian, A.G. Rhodiola rosea L. as a putative botanical antidepressant. Phytomedicine 2016, 23, 770–783. [Google Scholar] [CrossRef]
- Panossian, A.; Amsterdam, J. Adaptogens in Psychiatric Practice. In Complementary and Integrative Treatments in Psychiatric Practice; Gerbarg, P.L., Muskin, P.R., Brown, R.P., Eds.; American Psychiatric Publishing: Arlington, VA, USA, 2017; pp. 155–181. [Google Scholar]
- Panossian, A.; Wikman, G. Evidence Based Efficacy and Effectiveness of Rhodiola SHR-5 Extract in Treating Stress- and Age-Associated Disorders. In Rhodiola Rosea, Series: Traditional Herbal Medicines for Modern Times; Cuerrier, A., Ampong-Nyarko, K., Eds.; CRC Press: Boca Raton, FL, USA; London, UK; New York, NY, USA, 2014; pp. 203–221. [Google Scholar]
- Kumar, R.; Gupta, K.; Saharia, K.; Pradhan, D.; Subramaniam, J.R. Withania somnifera root extract extends lifespan of Caenorhabditis elegans. Ann. Neurosci. 2013, 20, 13–16. [Google Scholar] [CrossRef]
- Jafari, M.; Felgner, J.S.; Bussel, I.I.; Hutchili, T.; Khodayari, B.; Rose, M.R.; Vince-Cruz, C.; Mueller, L.D. Rhodiola: A promising anti-aging Chinese herb. Rejuvenation Res. 2007, 10, 587–602. [Google Scholar] [CrossRef]
- Lee, J.H.; Choi, S.H.; Kwon, O.S.; Shin, T.J.; Lee, J.H.; Lee, B.H.; Yoon, I.S.; Pyo, M.K.; Rhim, H.; Lim, Y.H.; et al. Effects of ginsenosides, active ingredients of Panax ginseng, on development, growth, and life span of Caenorhabditis elegans. Biol. Pharm. Bull. 2007, 30, 2126–2134. [Google Scholar] [CrossRef]
- Narimanian, M.; Badalyan, M.; Panosyan, V.; Gabrielyan, E.; Panossian, A.; Wikman, G.; Wagner, H. Impact of Chisan (ADAPT 232) on the Quality- of -Life and its efficacy as an adjuvant in the treatment of acute non-specific pneumonia. Phytomedicine 2005, 12, 723–729. [Google Scholar] [CrossRef]
- Facchinetti, F.; Neri, I.; Tarbusi, M. Eleutherococcus senticocus reduces cardiovascular stress response in healthy subjects: Randomized, placebo-controlled trial. Stress Health 2002, 18, 11–17. [Google Scholar] [CrossRef]
- Kulkarni, S.K.; Dhir, A. Withania somnifera: An Indian ginseng. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1093–1105. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.K.; Bhattacharya, A.; Sairam, K.; Ghosal, S. Anxiolytic-antidepressant activity of Withania somnifera glycowithanolides: An experimental study. Phytomedicine 2000, 7, 463–469. [Google Scholar] [CrossRef]
- Bhattacharya, S.K.; Muruganandam, A.V. Adaptogenic activity of Withania somnifera: An experimental study using a rat model of chronic stress. Pharmacol. Biochem. Behav. 2003, 75, 547–555. [Google Scholar] [CrossRef]
- Singh, N.; Nath, R.; Lata, A.; Singh, S.P.; Kohli, R.P.; Bhargava, K.P. Withania somnifera (Ashwagandha), a rejuvenating herbal drug which enhances survival during stress (an adaptogen). Int. J. Crude Drug. Res. 1982, 20, 29–35. [Google Scholar] [CrossRef]
- Singh, R.H.; Malviya, P.C.; Sarkar, F.H.; Udupa, K.N. Studies on the psychotropic effect of an indigenous rasayana drug, Asvagandha (Withania somnifera dunol) Part II: (Experimental studies). J. Res. Ind. Med. Yoga Homeo. 1979, 14, 49–54. [Google Scholar]
- Yidya Prabhu, M.; Rao, A. Neuropharmacological activity of Withania somnifera. Fitoter 1990, 66, 237–240. [Google Scholar]
- Candelaria, M.; Cuellar, E.; Reyes-Ruiz, J.M.; Darabedian, N.; Feimeng, Z.; Miledi, R.; Russo-Neustadt, A.; Limon, A. Direct evidence for GABAergic activity of Withania somnifera on mammalian ionotropic GABAA and GABAp receptors. J. Ethnopharmacol. 2015, 171, 264–272. [Google Scholar] [CrossRef]
- Chandrasekhar, K.; Kapoor, J.; Anishetty, S. A prospective, randomized double-blind, placebo-controlled study of safety and efficacy of a high-concentration full-spectrum extract of ashwagandha root in reducing stress and anxiety in adults. Indian J. Psychol. Med. 2012, 34, 255–262. [Google Scholar] [CrossRef]
- Thakur, A.K.; Chatterjee, S.S.; Kumar, V. Adaptogenic potential of andrographolide: An active principle of the king of bitters (Andrographis paniculata). J. Tradit. Complementary Med. 2015, 5, 42–50. [Google Scholar] [CrossRef]
- Panossian, A.; Wikman, G. Efficacy of Andrographis paniculata in upper respiratory tract (URT) infectious diseases and the mechanism of action. In Evidence and Rational Based Research on Chinese Drugs; Wagner, H., Ulrich Merzenich, G., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 137–180. [Google Scholar]
- EMA/HMPC/320433/2012. Based on Article 10a of Directive 2001/83/EC as amended (well-established use). Based on Article 16d (1), Article 16f and Article 16h of Directive 2001/83/EC as amended (traditional use). In Assessment Report on Andrographis Paniculata Nees, Folium; European Medicines Agency: London, UK, 2014; pp. 1–28. [Google Scholar]
- Iruretagoyena, M.I.; Tobar, J.A.; Gonzalez, P.A.; Sepulveda, S.E.; Figueroa, C.A.; Burgos, R.A.; Hancke, J.L.; Kalergis, A.M. Andrographolide interferes with T cell activation and reduces experimental autoimmune encephalomyelitis in the mouse. J. Pharmacol. Exp. Ther. 2005, 312, 366–372. [Google Scholar] [CrossRef]
- Panossian, A.; Seo, E.J.; Wikman, G.; Efferth, T. Synergy assessment of fixed combinations of Herba Andrographidis and Radix Eleutherococci extracts by transcriptome-wide microarray profiling. Phytomedicine 2015, 22, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Mishra, K.P.; Ganju, L.; Singh, S.B. Andrographolide—A promising therapeutic agent, negatively regulates glial cell derived neurodegeneration of prefrontal cortex, hippocampus and working memory impairment. J. Neuroimmunol. 2017, 313, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Dimpfel, W.; Schombert, L.; Vega-Morales, T.; Wiebe, J.C. Neuropharmacological Characterization of Extracts from Rhodiola rosea, Oenothera paradoxa and Paullinia cupana in Comparison to Caffeine. Pharmacol. Pharm. 2016, 7, 290–303. [Google Scholar] [CrossRef]
- LaFrance, C.; Dumont, M. Diurnal variations in the waking EEG: Comparisons with Sleep latencies and subjective alertness. J. Sleep Res. 2000, 9, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Dimpfel, W.; Schober, F. Norepinephrine, EEG theta waves and sedation. Brain Pharmacol. 2001, 1, 89–97. [Google Scholar]
- Kim, W.H.; Cho, D.; Lee, B.; Song, J.J.; Shin, T.J. Changes in brain activation during sedation induced by dexmedetomidine. J. Int. Med. Res. 2017, 45, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Dimpfel, W. Classification of Herbal Drug Effects by Discriminant analysis of Qunatitative Human EEG Data. Neurosci. Med. 2019, 10, 101–117. [Google Scholar] [CrossRef]
- Anderer, P.; Saletu, B.; Pascual-Marqui, R.D. Effect of the 5-HT (1A) partial agonist buspirone on regional brain electrical activity in man: A functional neuroimaging study using low-resolution electromagnetic tomography (LORETA). Psychiatry Res. 2000, 100, 81–96. [Google Scholar] [CrossRef]
- Sauseng, P.; Hoppe, J.; Klimesch, W.; Gerloff, C.; Humel, F.C. Dissociation of sustained attention from central executiv functions: Local activity and interregional connectivity in the theta range. Eur. J. Neurosci. 2007, 25, 587–593. [Google Scholar] [CrossRef]
- Pratte, M.A.; Nanavati, K.B.; Young, V.; Morley, C.P. An alternative treatment for anxiety: A systematic review of human trial results reported for the Ayurvedic herb ashwagandha (Withania somnifera). J. Altern. Complementary Med. 2014, 20, 901–908. [Google Scholar] [CrossRef]
- Kaur, T.; Singh, H.; Mishra, R.; Manchanda, S.; Gupta, M.; Saini, V.; Sharma, A.; Kaur, G. Withania somnifera as a potential anxiolytic and immunomodulatory agent in acute sleep deprived female Wistar rats. Mol. Cell Biochem. 2017, 427, 91–101. [Google Scholar] [CrossRef]
- Thakur, A.K.; Soni, U.K.; Rai, G.; Chatterjee, S.S.; Kumar, V. Protective effects of Andrographyis paniculata extract and pure andrographolide against chronic stress-triggered pathologies in rats. Cell Mol. Neurobiol. 2014, 34, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Schober, F.; Schellenberg, R.; Dimpfel, W. Reflection of Mental Exercise in the Dynamic Quantitative Topographical EEG. Neuropsychobiolgy 1995, 31, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Harmony, T.; Fernandez-Bouzas, A.; Marosi, E.; Fernandez, T.; Bernal, J.; Rodriguez, M.; Reyes, A.; Silva, J.; Alonso, M.; Casian, G. Correlation between Computed Tomography and Voltage and Current Source Density Spectral. Parameters in Patients with Brain Lesions. Electroencephalogr. Clin. Neurophysiol. 1993, 87, 196–205. [Google Scholar] [CrossRef]
- Dimpfel, W.; Wedekind, W.; Keplinger, I. Efficacy of Dimethylaminoethanol (DMAE) Containing Vitamin-mineral Drug Combination on EEG Patterns in the Presence of Different Emotional States. Eur. J. Med. Res. 2003, 8, 183–191. [Google Scholar]
- Dimpfel, W.; Wedekind, W.; Keplinger, I. Gender Difference in Electrical Brain Activity during Presentation of Various Film Excerpts with Different Emotional Content. Eur. J. Med. Res. 2003, 8, 192–198. [Google Scholar]
- Dimpfel, W.; Hofmann, H.C.; Prohaska, A.; Schober, F.; Schellenberg, R. Source density analysis of functional topographical EEG: Monitoring of cognitive drug action. Eur. J. Med. Res. 1996, 1, 283–290. [Google Scholar]
- Grulke, N.; Bailer, H.; Schmutzer, G.; Brähler, E.; Blaser, G.; Geyer, M.; Albani, C. Normierung der deutschen Kurzform des Fragebogens “Profile of Mood States” (POMS) anhand einer repräsentativen Bevölkerungsstichprobe—Kurzbericht (Standardization of the German Short Version of “Profile of Mood States” (POMS) in a Representative Sample-Short). Psychother. Psych. Med. 2006, 56, 1–5. [Google Scholar]
- Terry, P.C.; Lane, A.M.; Fogarty, G.J. Construct validity of the Profile of Mood States-Adolescents for use with adults. Psychol. Sport Exerc. 2003, 4, 125–139. [Google Scholar] [CrossRef]
- Albani, C.; Blaser, G.; Geyer, M.; Schmutzer, G.; Brähler, E.; Bailer, H.; Grulke, N. Überprüfung der Gütekriterien der deutschen Kurzform des Fragebogens “Profile of Mood States” (POMS) in einer repräsentativen Bevölkerungsstichprobe. Psychother. Psych. Med. 2005, 55, 324–330. [Google Scholar] [CrossRef]
- Görtelmeyer, R. Schlaffragebogen SF-A und SF-B. Internationale Skalen für Psychiatrie. In CIPS Collegium Internationale Psychiatriae Scalarum (Hrsg.); Beltz: Weinheim, Germany, 1986. [Google Scholar]
- Görtelmeyer, R. Typologie des Schlafverhaltens. Eine Untersuchung an berufstätigen Personen. In Beiträge zur Klinischen Psychologie und Psychotherapie (Band 4); Wittchen, H.U., Ed.; Roderer: Regensburg, Germany, 1988. [Google Scholar]
- Görtelmeyer, R. SF-A/R und SF-B/R—Schlaffragebogen A und B—Revidierte Fassung. In PSYNDEX Tests Info; Hogrefe: Göttingen, Germany, 2011. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Demographic Characteristics | n | Mean | SD |
|---|---|---|---|
| Age | |||
| Total | 16 | 63.81 | 2.04 |
| Male | 8 | 63.88 | 1.81 |
| Female | 8 | 63.75 | 2.38 |
| Gender | |||
| Total | 16 | 11.00 | 1.11 |
| Male | 8 | 10.86 | 1.21 |
| Female | 8 | 11.14 | 1.07 |
| Body height (cm) | |||
| Total | 16 | 173 | 0.10 |
| Male | 8 | 181 | 0.06 |
| Female | 8 | 164 | 0.06 |
| Weight (kg) | |||
| Total | 16 | 78.03 | 14.92 |
| Male | 8 | 89.60 | 7.96 |
| Female | 8 | 66.50 | 10.46 |
| Body Mass Index (BMI) (kg/cm2) | |||
| Total | 16 | 25.99 | 3.49 |
| Male | 8 | 27.33 | 2.60 |
| Female | 8 | 24.64 | 3.91 |
| Absolute Values for “Eyes Open” at 0 h—First Day (Acute) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δ | θ | α1 | α2 | β1 | β2 | |||||||
| E | Placebo n = 15 | Adaptra® n = 15 | Placebo n = 15 | Adaptra® n = 15 | Placebo n = 15 | Adaptra® n = 15 | Placebo n = 15 | Adaptra® n = 15 | Placebo n = 15 | Adaptra® n = 15 | Placebo n = 15 | Adaptra® n = 15 |
| Cz | 1.73 | 2.00 | 0.59 | 0.59 | 0.71 | 0.57 | 0.73 | 0.70 | 1.30 | 1.19 | 1.42 | 1.30 |
| Fz | 1.90 | 2.40 | 0.61 | 0.64 | 0.66 | 0.84 | 0.72 | 0.73 | 1.06 | 0.91 | 1.19 | 1.43 |
| F3 | 2.05 | 3.09 | 0.79 | 0.78 | 1.00 | 1.18 | 1.04 | 0.98 | 2.38 | 2.15 | 4.29 | 4.39 |
| C3 | 1.23 | 1.38 | 0.41 | 0.43 | 0.74 | 0.81 | 0.89 | 1.00 | 1.99 | 1.57 | 1.45 | 1.44 |
| P3 | 1.01 | 1.78 | 0.35 | 0.49 | 0.76 | 0.96 | 1.02 | 1.15 | 1.08 | 1.13 | 0.84 | 0.85 |
| Pz | 0.99 | 1.17 | 0.35 | 0.33 | 0.51 | 0.53 | 0.64 | 0.62 | 0.80 | 0.69 | 0.55 | 0.55 |
| P4 | 1.19 | 1.47 | 0.30 | 0.35 | 0.91 | 0.69 | 0.85 | 0.74 | 0.86 | 0.81 | 0.54 | 0.65 |
| C4 | 1.18 | 1.46 | 0.38 | 0.38 | 0.72 | 0.82 | 1.08 | 1.31 | 2.07 | 2.37 | 1.78 | 2.22 |
| F4 | 2.80 | 3.23 | 0.97 | 0.77 | 1.28 | 1.08 | 1.17 | 0.85 | 2.44 | 1.72 | 5.46 | 3.57 |
| F7 | 7.23 | 10.74 | 1.33 | 1.76 | 3.38 | 2.59 | 2.82 | 2.02 | 3.26 | 3.68 | 4.23 | 6.45 |
| T3 | 3.38 | 5.24 | 0.91 | 1.07 | 2.26 | 3.43 | 2.31 | 2.11 | 3.02 | 2.57 | 3.76 | 3.12 |
| T5 | 2.75 | 3.46 | 1.08 | 1.23 | 3.17 | 3.00 | 2.90 | 2.30 | 2.53 | 2.95 | 2.07 | 1.99 |
| O1 | 2.16 | 2.70 | 0.59 | 0.67 | 0.70 | 1.02 | 0.86 | 1.17 | 1.92 | 1.95 | 2.42 | 2.43 |
| O2 | 2.07 | 2.57 | 0.61 | 0.58 | 0.83 | 0.87 | 1.14 | 1.28 | 2.39 | 2.56 | 2.48 | 1.97 |
| T6 | 2.61 | 2.80 | 0.97 | 0.88 | 1.60 | 1.83 | 1.78 | 1.97 | 2.59 | 1.81 | 1.36 | 1.91 |
| T4 | 2.66 | 3.38 | 0.85 | 0.87 | 1.97 | 2.45 | 2.11 | 2.77 | 3.98 | 3.68 | 3.77 | 4.90 |
| F8 | 7.34 | 6.75 | 1.55 | 1.32 | 3.45 | 2.35 | 2.42 | 1.66 | 3.97 | 2.81 | 7.02 | 4.70 |
| Med | 2.16 | 2.40 | 0.79 | 0.67 | 0.91 | 1.19 | 1.02 | 1.28 | 1.68 | 1.72 | 2.16 | 2.34 |
| Absolute Values for “Eyes Closed” at 0 h—First Day (Acute) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δ | θ | α1 | α2 | β1 | β2 | |||||||
| E | Placebo n = 15 | Adaptra® n = 15 | Placebo n = 15 | Adaptra® n = 15 | Placebo n = 15 | Adaptra® n = 15 | Placebo n = 15 | Adaptra® n = 15 | Placebo n = 15 | Adaptra® n = 15 | Placebo n = 15 | Adaptra® n = 15 |
| Cz | 2.00 | 1.96 | 0.71 | 0.73 | 0.81 | 0.98 | 0.99 | 0.94 | 1.81 | 1.50 | 1.39 | 1.55 |
| Fz | 2.25 | 2.45 | 0.72 | 0.87 | 0.96 | 1.25 | 0.84 | 1.09 | 0.95 | 1.09 | 0.90 | 0.97 |
| F3 | 2.57 | 3.30 | 0.78 | 0.92 | 1.57 | 1.64 | 1.13 | 1.26 | 1.24 | 1.56 | 1.56 | 1.52 |
| C3 | 1.23 | 1.58 | 0.50 | 0.52 | 0.79 | 1.21 | 1.12 | 1.35 | 1.70 | 1.92 | 1.37 | 1.45 |
| P3 | 1.28 | 1.80 | 0.40 | 0.51 | 1.12 | 1.83 | 1.38 | 1.95 | 1.27 | 1.15 | 0.78 | 0.80 |
| Pz | 1.04 | 1.40 | 0.35 | 0.50 | 1.04 | 1.47 | 1.12 | 1.00 | 0.77 | 0.97 | 0.59 | 0.54 |
| P4 | 1.13 | 1.64 | 0.38 | 0.47 | 1.05 | 1.39 | 0.97 | 1.51 | 0.85 | 0.77 | 0.48 | 0.58 |
| C4 | 1.29 | 1.49 | 0.42 | 0.49 | 0.89 | 1.07 | 1.43 | 1.88 | 2.46 | 2.49 | 1.41 | 1.46 |
| F4 | 2.76 | 2.87 | 0.97 | 0.87 | 1.44 | 1.72 | 1.08 | 1.30 | 1.48 | 1.47 | 1.56 | 1.30 |
| F7 | 9.44 | 7.78 | 1.52 | 1.63 | 3.54 | 3.78 | 2.45 | 2.50 | 1.86 | 2.39 | 2.08 | 2.22 |
| T3 | 3.19 | 3.56 | 1.00 | 1.14 | 2.38 | 2.99 | 2.27 | 2.25 | 1.98 | 2.54 | 1.67 | 2.00 |
| T5 | 2.86 | 3.32 | 1.41 | 1.67 | 5.40 | 7.11 | 2.85 | 4.59 | 2.82 | 2.74 | 1.77 | 1.40 |
| O1 | 2.49 | 2.19 | 0.77 | 0.74 | 1.06 | 1.92 | 1.29 | 1.81 | 2.08 | 1.89 | 1.55 | 1.54 |
| O2 | 2.01 | 2.99 | 0.65 | 0.68 | 1.55 | 1.79 | 1.73 | 2.28 | 2.06 | 2.66 | 1.52 | 1.50 |
| T6 | 2.77 | 3.40 | 1.31 | 1.57 | 5.87 | 10.60 | 3.81 | 3.51 | 2.28 | 2.62 | 1.67 | 1.43 |
| T4 | 3.10 | 3.90 | 0.95 | 1.06 | 2.70 | 3.01 | 2.42 | 2.08 | 1.98 | 2.36 | 1.98 | 2.37 |
| F8 | 8.07 | 6.00 | 1.83 | 1.46 | 3.72 | 3.41 | 2.98 | 2.67 | 2.07 | 2.22 | 2.38 | 2.23 |
| Med | 2.35 | 2.86 | 0.77 | 0.87 | 1.83 | 1.71 | 1.72 | 1.60 | 1.67 | 1.77 | 1.28 | 1.45 |
| Performance of Psychometric Tests | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| d2 Test | CPT | Memory Test | |||||||
| 0 h | 2 h | 2 h vs. Baseline Two-tailed t-Test p < | 0 h | 2 h | 2 h vs. Baseline Two-tailedt-Test p < | 0h | 2h | 2 h vs. Baseline Two-tailed t-Test p < | |
| n = 15 | n = 15 | n = 15 | n = 15 | n = 15 | n = 15 | ||||
| Placebo repetitive–acute | 0.48 (0.65) | 0.39 (1.10) | 0.799 | −0.82 (2.45) | 0.88 (2.45) | 0.076 | −1.14 (3.39) | −0.62 (4.68) | 0.729 |
| Adaptra® Forte repetitive–acute | 1.31 (1.16) | 1.48 (1.44) | 0.719 | 0.74 (3.73) | 1.28 (3.68) | 0.693 | 2.20 (3.24) | 1.19 (4.28) | 0.481 |
| Placebo vs. Adaptra® Forte two-tailed t-test p < | 0.029 | 0.027 | 0.194 | 0.726 | 0.012 | 0.278 | |||
| Follow-up | 12.01 (2.59) | 4.49 (3.21) | 9.34 (3.49) | ||||||
| Values are means (SD) | |||||||||
| Evaluation of the SF-B/R Questionnaire (Schlaffragebogen) | |||||||
|---|---|---|---|---|---|---|---|
| SQ | GES | PSYA | PSYE | PSS | TRME | SWR | |
| Placebo repetitive–acute (n = 15) | 0.24 (0.35) | 0.11 (0.24) | 0.03 (0.41) | −0.18 (0.56) | −0.10 (0.44) | −0.13 (0.35) | 0.40 (0.82) |
| Adaptra® Forte repetitive–acute (n = 15) | −0.22 (0.72) | −0.25 (0.53) | −0.12 (0.77) | 0.22 (0.69) | 0.23 (0.31) | 0.10 (0.57) | −0.05 (0.81) |
| Placebo vs. Adaptra® Forte two-tailed t-test p < | 0.039 | 0.028 | 0.510 | 0.094 | 0.024 | 0.192 | 0.145 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dimpfel, W.; Schombert, L.; Keplinger-Dimpfel, I.K.; Panossian, A. Effects of an Adaptogenic Extract on Electrical Activity of the Brain in Elderly Subjects with Mild Cognitive Impairment: A Randomized, Double-Blind, Placebo-Controlled, Two-Armed Cross-Over Study. Pharmaceuticals 2020, 13, 45. https://doi.org/10.3390/ph13030045
Dimpfel W, Schombert L, Keplinger-Dimpfel IK, Panossian A. Effects of an Adaptogenic Extract on Electrical Activity of the Brain in Elderly Subjects with Mild Cognitive Impairment: A Randomized, Double-Blind, Placebo-Controlled, Two-Armed Cross-Over Study. Pharmaceuticals. 2020; 13(3):45. https://doi.org/10.3390/ph13030045
Chicago/Turabian StyleDimpfel, Wilfried, Leonie Schombert, Ingrid K. Keplinger-Dimpfel, and Alexander Panossian. 2020. "Effects of an Adaptogenic Extract on Electrical Activity of the Brain in Elderly Subjects with Mild Cognitive Impairment: A Randomized, Double-Blind, Placebo-Controlled, Two-Armed Cross-Over Study" Pharmaceuticals 13, no. 3: 45. https://doi.org/10.3390/ph13030045
APA StyleDimpfel, W., Schombert, L., Keplinger-Dimpfel, I. K., & Panossian, A. (2020). Effects of an Adaptogenic Extract on Electrical Activity of the Brain in Elderly Subjects with Mild Cognitive Impairment: A Randomized, Double-Blind, Placebo-Controlled, Two-Armed Cross-Over Study. Pharmaceuticals, 13(3), 45. https://doi.org/10.3390/ph13030045