Radiolabelled Peptides for Positron Emission Tomography and Endoradiotherapy in Oncology †


Abstract
1. Introduction
2. Peptides as Targeting Probes—Pros and Cons
3. Improvement of Metabolic Stability
4. Labelling Strategies
4.1. Halogens
4.1.1. Fluorine
4.1.2. Bromine
4.1.3. Iodine
4.1.4. Astatine
4.2. Radiometals
4.2.1. Copper
4.2.2. Gallium
4.2.3. Zirconium
4.2.4. Yttrium, Lutetium
4.2.5. Actinium
5. Clinically Investigated or Routinely Used Radiolabelled Peptides
5.1. Somatostatin Receptor
5.2. Prostate-Specific Membrane Antigen
5.3. Integrins
5.4. Chemokine Receptor
5.5. Other Target Structures Expressed on Tumour Cells
6. Summary and Conclusions
Funding
Conflicts of Interest
References
- De Jong, M.; Bakker, W.H.; Krenning, E.P.; Breeman, W.A.; van der Pluijm, M.E.; Bernard, B.F.; Visser, T.J.; Jermann, E.; Behe, M.; Powell, P.; et al. Yttrium-90 and indium-111 labelling, receptor binding and biodistribution of [DOTA0,d-Phe1,Tyr3]octreotide, a promising somatostatin analogue for radionuclide therapy. Eur. J. Nucl. Med. 1997, 24, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Henze, M.; Schuhmacher, J.; Hipp, P.; Kowalski, J.; Becker, D.W.; Doll, J.; Macke, H.R.; Hofmann, M.; Debus, J.; Haberkorn, U. PET imaging of somatostatin receptors using [68Ga]DOTA-D-Phe1-Tyr3-octreotide: First results in patients with meningiomas. J. Nucl. Med. 2001, 42, 1053–1056. [Google Scholar] [PubMed]
- Haubner, R.; Decristoforo, C. Radiolabelled RGD peptides and peptidomimetics for tumour targeting. Front. Biosci. 2009, 14, 872–886. [Google Scholar] [CrossRef] [PubMed]
- Haubner, R.; Maschauer, S.; Prante, O. PET radiopharmaceuticals for imaging integrin expression: Tracers in clinical studies and recent developments. BioMed Res. Int. 2014, 2014, 871609. [Google Scholar] [CrossRef] [PubMed]
- Haubner, R.; Finkenstedt, A.; Stegmayr, A.; Rangger, C.; Decristoforo, C.; Zoller, H.; Virgolini, I.J. [68Ga]NODAGA-RGD—Metabolic stability, biodistribution, and dosimetry data from patients with hepatocellular carcinoma and liver cirrhosis. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 2005–2013. [Google Scholar] [CrossRef] [PubMed]
- Nock, B.A.; Kaloudi, A.; Lymperis, E.; Giarika, A.; Kulkarni, H.R.; Klette, I.; Singh, A.; Krenning, E.P.; de Jong, M.; Maina, T.; et al. Theranostic Perspectives in Prostate Cancer with the Gastrin-Releasing Peptide Receptor Antagonist NeoBOMB1: Preclinical and First Clinical Results. J. Nucl. Med. 2017, 58, 75–80. [Google Scholar] [CrossRef]
- Valverde, I.E.; Vomstein, S.; Mindt, T.L. Toward the Optimization of Bombesin-Based Radiotracers for Tumor Targeting. J. Med. Chem. 2016, 59, 3867–3877. [Google Scholar] [CrossRef]
- Froidevaux, S.; Eberle, A.N. Somatostatin analogs and radiopeptides in cancer therapy. Biopolymers 2002, 66, 161–183. [Google Scholar] [CrossRef]
- Murza, A.; Parent, A.; Besserer-Offroy, E.; Tremblay, H.; Karadereye, F.; Beaudet, N.; Leduc, R.; Sarret, P.; Marsault, E. Elucidation of the structure-activity relationships of apelin: Influence of unnatural amino acids on binding, signaling, and plasma stability. Chem. Med. Chem. 2012, 7, 318–325. [Google Scholar] [CrossRef]
- Klingler, M.; Decristoforo, C.; Rangger, C.; Summer, D.; Foster, J.; Sosabowski, J.K.; von Guggenberg, E. Site-specific stabilization of minigastrin analogs against enzymatic degradation for enhanced cholecystokinin-2 receptor targeting. Theranostics 2018, 8, 2896–2908. [Google Scholar] [CrossRef]
- Haubner, R.; Kuhnast, B.; Mang, C.; Weber, W.A.; Kessler, H.; Wester, H.J.; Schwaiger, M. [18F]Galacto-RGD: Synthesis, radiolabeling, metabolic stability, and radiation dose estimates. Bioconjug. Chem. 2004, 15, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Haubner, R.; Finsinger, D.; Kessler, H. Stereoisomeric Peptide Libraries and Peptidomimetics for Designing Selective Inhibitors of the αvβ3 Integrin for a New Cancer Therapy. Angew. Chem. Int. Ed. Engl. 1997, 36, 1374–1389. [Google Scholar] [CrossRef]
- Nicole, P.; Lins, L.; Rouyer-Fessard, C.; Drouot, C.; Fulcrand, P.; Thomas, A.; Couvineau, A.; Martinez, J.; Brasseur, R.; Laburthe, M. Identification of key residues for interaction of vasoactive intestinal peptide with human VPAC1 and VPAC2 receptors and development of a highly selective VPAC1 receptor agonist. Alanine scanning and molecular modeling of the peptide. J. Biol. Chem. 2000, 275, 24003–24012. [Google Scholar] [CrossRef] [PubMed]
- Vagner, J.; Qu, H.; Hruby, V.J. Peptidomimetics, a synthetic tool of drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 292–296. [Google Scholar] [CrossRef]
- Garcia-Garayoa, E.; Blauenstein, P.; Blanc, A.; Maes, V.; Tourwe, D.; Schubiger, P.A. A stable neurotensin-based radiopharmaceutical for targeted imaging and therapy of neurotensin receptor-positive tumours. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 37–47. [Google Scholar] [CrossRef]
- Becaud, J.; Mu, L.; Karramkam, M.; Schubiger, P.A.; Ametamey, S.M.; Graham, K.; Stellfeld, T.; Lehmann, L.; Borkowski, S.; Berndorff, D.; et al. Direct one-step 18F-labeling of peptides via nucleophilic aromatic substitution. Bioconjug. Chem. 2009, 20, 2254–2261. [Google Scholar] [CrossRef]
- Okarvi, S.M. Recent progress in fluorine-18 labelled peptide radiopharmaceuticals. Eur. J. Nucl. Med. 2001, 28, 929–938. [Google Scholar] [CrossRef]
- Wester, H.J.; Schottelius, M. Fluorine-18 Labeling of Peptides and Proteins. PET Chemistry. In Ernst Schering Research Foundation Workshop; Schubiger, P.A., Lehmann, L., Friebe, M., Eds.; Springer: Berlin, Heidelberg, 2007; Volume 64, pp. 79–111. [Google Scholar] [CrossRef]
- Vaidyanathan, G.; Zalutsky, M.R. Improved synthesis of N-succinimidyl 4-[18F]fluorobenzoate and its application to the labeling of a monoclonal antibody fragment. Bioconjug. Chem. 1994, 5, 352–356. [Google Scholar] [CrossRef]
- Schirrmacher, R.; Wangler, B.; Bailey, J.; Bernard-Gauthier, V.; Schirrmacher, E.; Wangler, C. Small Prosthetic Groups in 18F-Radiochemistry: Useful Auxiliaries for the Design of 18F-PET Tracers. Semin. Nucl. Med. 2017, 47, 474–492. [Google Scholar] [CrossRef]
- Wust, F.; Hultsch, C.; Bergmann, R.; Johannsen, B.; Henle, T. Radiolabelling of isopeptide N epsilon-(gamma-glutamyl)-L-lysine by conjugation with N-succinimidyl-4-[18F]fluorobenzoate. Appl. Radiat. Isot. 2003, 59, 43–48. [Google Scholar] [CrossRef]
- Lang, L.; Eckelman, W.C. One-step synthesis of 18F labeled [18F]-N-succinimidyl 4-(fluoromethyl)benzoate for protein labeling. Appl. Radiat. Isot. 1994, 45, 1155–1163. [Google Scholar] [CrossRef]
- Olberg, D.E.; Arukwe, J.M.; Grace, D.; Hjelstuen, O.K.; Solbakken, M.; Kindberg, G.M.; Cuthbertson, A. One step radiosynthesis of 6-[18F]fluoronicotinic acid 2,3,5,6-tetrafluorophenyl ester ([18F]F-Py-TFP): A new prosthetic group for efficient labeling of biomolecules with fluorine-18. J. Med. Chem. 2010, 53, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Koudih, R.; Kostikov, A.; Kovacevic, M.; Jolly, D.; Bernard-Gauthier, V.; Chin, J.; Jurkschat, K.; Wangler, C.; Wangler, B.; Schirrmacher, R. Automated radiosynthesis of N-succinimidyl 3-(di-tert-butyl[18F]fluorosilyl)benzoate ([18F]SiFB) for peptides and proteins radiolabeling for positron emission tomography. Appl. Radiat. Isot. 2014, 89, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Kluba, C.A.; Mindt, T.L. Click-to-Chelate: Development of technetium and rhenium-tricarbonyl labeled radiopharmaceuticals. Molecules 2013, 18, 3206–3226. [Google Scholar] [CrossRef]
- Marik, J.; Sutcliffe, J.L. Rapid preparation of [18F]fluoropeptides using Cu(I) catalyzed 1,3-dipolar cycloaddition. Tetrahedron Lett. 2006, 47, 6681–6684. [Google Scholar] [CrossRef]
- Glaser, M.; Arstad, E. “Click labeling” with 2-[18f]fluoroethylazide for positron emission tomography. Bioconjug Chem. 2007, 18, 989–993. [Google Scholar] [CrossRef]
- Arumugam, S.; Chin, J.; Schirrmacher, R.; Popik, V.V.; Kostikov, A.P. [18F]azadibenzocyclooctyne ([18F]ADIBO): A biocompatible radioactive labeling synthon for peptides using catalyst free [3+2] cycloaddition. Bioorg Med. Chem. Lett. 2011, 21, 6987–6991. [Google Scholar] [CrossRef]
- Campbell-Verduyn, L.S.; Mirfeizi, L.; Schoonen, A.K.; Dierckx, R.A.; Elsinga, P.H.; Feringa, B.L. Strain-promoted copper-free “click” chemistry for 18F radiolabeling of bombesin. Angew. Chem. Int. Ed. Engl. 2011, 50, 11117–11120. [Google Scholar] [CrossRef]
- Iwata, R.; Pascali, C.; Bogni, A.; Horvath, G.; Kovacs, Z.; Yanai, K.; Ido, T. A new, convenient method for the preparation of 4-[18F]fluorobenzyl halides. Appl. Radiat. Isot. 2000, 52, 87–92. [Google Scholar] [CrossRef]
- Poethko, T.; Schottelius, M.; Thumshirn, G.; Hersel, U.; Herz, M.; Henriksen, G.; Kessler, H.; Schwaiger, M.; Wester, H.J. Two-step methodology for high-yield routine radiohalogenation of peptides: 18F-labeled RGD and octreotide analogs. J. Nucl. Med. 2004, 45, 892–902. [Google Scholar]
- Bruus-Jensen, K.; Poethko, T.; Schottelius, M.; Hauser, A.; Schwaiger, M.; Wester, H.J. Chemoselective hydrazone formation between HYNIC-functionalized peptides and 18F-fluorinated aldehydes. Nucl. Med. Biol. 2006, 33, 173–183. [Google Scholar] [CrossRef]
- Hultsch, C.; Schottelius, M.; Auernheimer, J.; Alke, A.; Wester, H.J. 18F-Fluoroglucosylation of peptides, exemplified on cyclo(RGDfK). Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1469–1474. [Google Scholar] [CrossRef]
- Wuest, F.; Hultsch, C.; Berndt, M.; Bergmann, R. Direct labelling of peptides with 2-[18F]fluoro-2-deoxy-d-glucose ([18F]FDG). Bioorg. Med. Chem. Lett. 2009, 19, 5426–5428. [Google Scholar] [CrossRef]
- Prante, O.; Einsiedel, J.; Haubner, R.; Gmeiner, P.; Wester, H.J.; Kuwert, T.; Maschauer, S. 3,4,6-Tri-O-acetyl-2-deoxy-2-[18F]fluoroglucopyranosyl phenylthiosulfonate: A thiol-reactive agent for the chemoselective 18F-glycosylation of peptides. Bioconjug. Chem. 2007, 18, 254–262. [Google Scholar] [CrossRef]
- Maschauer, S.; Prante, O. A series of 2-O-trifluoromethylsulfonyl-d-mannopyranosides as precursors for concomitant 18F-labeling and glycosylation by click chemistry. Carbohydr. Res. 2009, 344, 753–761. [Google Scholar] [CrossRef]
- Shiue, C.Y.; Wolf, A.; Hainfeld, J.F. Synthesis of 18F-labelled N-(p-[18F]fluorophenyl)maleimide and its derivatives for labelling monoclonal antibody with 18F. J. Label. Compd. Radiopharm. 1989, 26, 287–289. [Google Scholar] [CrossRef]
- Toyokuni, T.; Walsh, J.C.; Dominguez, A.; Phelps, M.E.; Barrio, J.R.; Gambhir, S.S.; Satyamurthy, N. Synthesis of a new heterobifunctional linker, N-[4-(aminooxy)butyl]maleimide, for facile access to a thiol-reactive 18F-labeling agent. Bioconjug. Chem. 2003, 14, 1253–1259. [Google Scholar] [CrossRef]
- Wuest, F.; Berndt, M.; Bergmann, R.; van den Hoff, J.; Pietzsch, J. Synthesis and application of [18F]FDG-maleimidehexyloxime ([18F]FDG-MHO): A [18F]FDG-based prosthetic group for the chemoselective 18F-labeling of peptides and proteins. Bioconjug. Chem. 2008, 19, 1202–1210. [Google Scholar] [CrossRef]
- Iovkova, L.; Wangler, B.; Schirrmacher, E.; Schirrmacher, R.; Quandt, G.; Boening, G.; Schurmann, M.; Jurkschat, K. para-Functionalized aryl-di-tert-butylfluorosilanes as potential labeling synthons for 18F radiopharmaceuticals. Chemistry 2009, 15, 2140–2147. [Google Scholar] [CrossRef]
- Chansaenpak, K.; Wang, M.; Wu, Z.; Zaman, R.; Li, Z.; Gabbai, F.P. [18F]-NHC-BF3 adducts as water stable radio-prosthetic groups for PET imaging. Chem. Commun. 2015, 51, 12439–12442. [Google Scholar] [CrossRef]
- Glaser, M.; Karlsen, H.; Solbakken, M.; Arukwe, J.; Brady, F.; Luthra, S.K.; Cuthbertson, A. 18F-fluorothiols: A new approach to label peptides chemoselectively as potential tracers for positron emission tomography. Bioconjug. Chem. 2004, 15, 1447–1453. [Google Scholar] [CrossRef]
- Wangler, B.; Quandt, G.; Iovkova, L.; Schirrmacher, E.; Wangler, C.; Boening, G.; Hacker, M.; Schmoeckel, M.; Jurkschat, K.; Bartenstein, P.; et al. Kit-like 18F-labeling of proteins: Synthesis of 4-(di-tert-butyl[18F]fluorosilyl)benzenethiol (Si[18F]FA-SH) labeled rat serum albumin for blood pool imaging with PET. Bioconjug. Chem. 2009, 20, 317–321. [Google Scholar] [CrossRef]
- Kumar, K.; Ghosh, A. 18F-AlF Labeled Peptide and Protein Conjugates as Positron Emission Tomography Imaging Pharmaceuticals. Bioconjug. Chem. 2018, 29, 953–975. [Google Scholar] [CrossRef]
- Wan, W.; Guo, N.; Pan, D.; Yu, C.; Weng, Y.; Luo, S.; Ding, H.; Xu, Y.; Wang, L.; Lang, L.; et al. First experience of 18F-alfatide in lung cancer patients using a new lyophilized kit for rapid radiofluorination. J. Nucl. Med. 2013, 54, 691–698. [Google Scholar] [CrossRef]
- Wu, C.; Yue, X.; Lang, L.; Kiesewetter, D.O.; Li, F.; Zhu, Z.; Niu, G.; Chen, X. Longitudinal PET imaging of muscular inflammation using 18F-DPA-714 and 18F-Alfatide II and differentiation with tumors. Theranostics 2014, 4, 546–555. [Google Scholar] [CrossRef]
- Yu, C.; Pan, D.; Mi, B.; Xu, Y.; Lang, L.; Niu, G.; Yang, M.; Wan, W.; Chen, X. 18F-Alfatide II PET/CT in healthy human volunteers and patients with brain metastases. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 2021–2028. [Google Scholar] [CrossRef]
- Long, T.; Yang, N.; Zhou, M.; Chen, D.; Li, Y.; Li, J.; Tang, Y.; Liu, Z.; Li, Z.; Hu, S. Clinical Application of 18F-AlF-NOTA-Octreotide PET/CT in Combination With 18F-FDG PET/CT for Imaging Neuroendocrine Neoplasms. Clin. Nucl. Med. 2019, 44, 452–458. [Google Scholar] [CrossRef]
- Niedermoser, S.; Chin, J.; Wangler, C.; Kostikov, A.; Bernard-Gauthier, V.; Vogler, N.; Soucy, J.P.; McEwan, A.J.; Schirrmacher, R.; Wangler, B. In Vivo Evaluation of 18F-SiFAlin-Modified TATE: A Potential Challenge for 68Ga-DOTATATE, the Clinical Gold Standard for Somatostatin Receptor Imaging with PET. J. Nucl. Med. 2015, 56, 1100–1105. [Google Scholar] [CrossRef]
- Sundin, J.; Tolmachev, V.; Koziorowski, J.; Carlsson, J.; Lundqvist, H.; Welt, S.; Larson, S.; Sundin, A. High yield direct 76Br-bromination of monoclonal antibodies using chloramine-T. Nucl. Med. Biol. 1999, 26, 923–929. [Google Scholar] [CrossRef]
- McElvany, K.D.; Welch, M.J. Characterization of bromine-77-labeled proteins prepared using bromoperoxidase. J. Nucl. Med. 1980, 21, 953–960. [Google Scholar]
- Knight, L.C.; Harwig, S.L.; Welch, M.J. In vitro stability and in vivo clearance of fibrinogen or serum albumin labeled with 77Br, 131I, or 125I by direct or indirect synthetic methods. J. Nucl. Med. 1977, 18, 282–288. [Google Scholar]
- Wilbur, D.S.; Hylarides, M.D. Radiolabeling of a monoclonal antibody with N-succinimidyl para-[77Br]bromobenzoate. Int. J. Rad. Appl. Instrum. B 1991, 18, 363–365. [Google Scholar] [CrossRef]
- Lang, L.; Li, W.; Jia, H.M.; Fang, D.C.; Zhang, S.; Sun, X.; Zhu, L.; Ma, Y.; Shen, B.; Kiesewetter, D.O.; et al. New Methods for Labeling RGD Peptides with Bromine-76. Theranostics 2011, 1, 341–353. [Google Scholar] [CrossRef][Green Version]
- Tolmachev, V. Radiobromine-labelled tracers for positron emission tomography: Possibilities and pitfalls. Curr. Radiopharm. 2011, 4, 76–89. [Google Scholar] [CrossRef]
- Coenen, H.H.; Mertens, J.; Maziere, B. Radioiodination Reactions for Pharmaceuticals—Compendium for Effective Synthesis Strategies; Springer: Dordrecht, The Netherlands, 2006. [Google Scholar]
- Langone, J.J. Radioiodination by use of the Bolton-Hunter and related reagents. Methods Enzym. 1980, 70, 221–247. [Google Scholar]
- Garg, P.K.; Garg, S.; Zalutsky, M.R. N-succinimidyl 4-methyl-3-(tri-n-butylstannyl)benzoate: Synthesis and potential utility for the radioiodination of monoclonal antibodies. Nucl. Med. Biol. 1993, 20, 379–387. [Google Scholar] [CrossRef]
- Smit, J.A.; Myburgh, J.A.; Neirinckx, R.D. Specific inactivation of sensitized lymphocytes in vitro using antigens labelled with astatine-211. Clin. Exp. Immunol. 1973, 14, 107–116. [Google Scholar]
- Zalutsky, M.R.; Reardon, D.A.; Akabani, G.; Coleman, R.E.; Friedman, A.H.; Friedman, H.S.; McLendon, R.E.; Wong, T.Z.; Bigner, D.D. Clinical experience with α--particle emitting 211At: Treatment of recurrent brain tumor patients with 211At-labeled chimeric antitenascin monoclonal antibody 81C6. J. Nucl. Med. 2008, 49, 30–38. [Google Scholar] [CrossRef]
- Zalutsky, M.R.; Zhao, X.G.; Alston, K.L.; Bigner, D. High-level production of α-particle-emitting 211At and preparation of 211At-labeled antibodies for clinical use. J. Nucl. Med. 2001, 42, 1508–1515. [Google Scholar]
- Andersson, H.; Cederkrantz, E.; Back, T.; Divgi, C.; Elgqvist, J.; Himmelman, J.; Horvath, G.; Jacobsson, L.; Jensen, H.; Lindegren, S.; et al. Intraperitoneal α-particle radioimmunotherapy of ovarian cancer patients: Pharmacokinetics and dosimetry of 211At-MX35 F(ab’)2—A phase I study. J. Nucl Med. 2009, 50, 1153–1160. [Google Scholar] [CrossRef]
- Hallqvist, A.; Bergmark, K.; Back, T.; Andersson, H.; Dahm-Kahler, P.; Johansson, M.; Lindegren, S.; Jensen, H.; Jacobsson, L.; Hultborn, R.; et al. Intraperitoneal alpha-Emitting Radioimmunotherapy with 211At in Relapsed Ovarian Cancer: Long-Term Follow-up with Individual Absorbed Dose Estimations. J. Nucl. Med. 2019, 60, 1073–1079. [Google Scholar] [CrossRef]
- Lindegren, S.; Frost, S.; Back, T.; Haglund, E.; Elgqvist, J.; Jensen, H. Direct procedure for the production of 211At-labeled antibodies with an epsilon-lysyl-3-(trimethylstannyl)benzamide immunoconjugate. J. Nucl. Med. 2008, 49, 1537–1545. [Google Scholar] [CrossRef]
- Guerard, F.; Gestin, J.F.; Brechbiel, M.W. Production of [211At]-astatinated radiopharmaceuticals and applications in targeted alpha-particle therapy. Cancer Biother. Radiopharm. 2013, 28, 1–20. [Google Scholar] [CrossRef]
- Ludwig, R.; Fischer, S.; Dreyer, R.; Jacobi, R.; Berger, J. Complex formation equilibria between astatine(I) and sulphur-cotaining chelating ligands. Polyhedron 1991, 10, 11–17. [Google Scholar] [CrossRef]
- Yordanov, A.T.; Deal, K.; Germestani, K.; Kobayashi, H.; Herring, B.; Waldmann, T.A.; Brechbiel, M.W. Synthesis and biodistribution study of a new 211At-calix[4]arene complex. J. Lab. Compd. Radiopharm. 2000, 43, 1219–1225. [Google Scholar] [CrossRef]
- McLendon, R.E.; Archer, G.E.; Larsen, R.H.; Akabani, G.; Bigner, D.D.; Zalutsky, M.R. Radiotoxicity of systemically administered 211At-labeled human/mouse chimeric monoclonal antibody: A long-term survival study with histologic analysis. Int. J. Radiat Oncol. Biol. Phys. 1999, 45, 491–499. [Google Scholar] [CrossRef]
- Kiess, A.P.; Minn, I.; Vaidyanathan, G.; Hobbs, R.F.; Josefsson, A.; Shen, C.; Brummet, M.; Chen, Y.; Choi, J.; Koumarianou, E.; et al. (2S)-2-(3-(1-Carboxy-5-(4-211At-Astatobenzamido)Pentyl)Ureido)-Pentanedioic Acid for PSMA-Targeted alpha-Particle Radiopharmaceutical Therapy. J. Nucl. Med. 2016, 57, 1569–1575. [Google Scholar] [CrossRef]
- Wilbur, D.S.; Chyan, M.K.; Hamlin, D.K.; Vessella, R.L.; Wedge, T.J.; Hawthorne, M.F. Reagents for astatination of biomolecules. 2. Conjugation of anionic boron cage pendant groups to a protein provides a method for direct labeling that is stable to in vivo deastatination. Bioconjug Chem. 2007, 18, 1226–1240. [Google Scholar] [CrossRef]
- Anderson, C.J.; Ferdani, R. Copper-64 radiopharmaceuticals for PET imaging of cancer: Advances in preclinical and clinical research. Cancer Biother. Radiopharm. 2009, 24, 379–393. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, X.; Xiong, Z.; Cheng, Z.; Fisher, D.R.; Liu, S.; Gambhir, S.S.; Chen, X. microPET imaging of glioma integrin αvβ3 expression using 64Cu-labeled tetrameric RGD peptide. J. Nucl. Med. 2005, 46, 1707–1718. [Google Scholar]
- Chen, X.; Hou, Y.; Tohme, M.; Park, R.; Khankaldyyan, V.; Gonzales-Gomez, I.; Bading, J.R.; Laug, W.E.; Conti, P.S. Pegylated Arg-Gly-Asp peptide: 64Cu labeling and PET imaging of brain tumor αvβ3-integrin expression. J. Nucl. Med. 2004, 45, 1776–1783. [Google Scholar] [PubMed]
- Bass, L.A.; Wang, M.; Welch, M.J.; Anderson, C.J. In vivo transchelation of copper-64 from TETA-octreotide to superoxide dismutase in rat liver. Bioconjug. Chem. 2000, 11, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Anderson, C.J. Chelators for copper radionuclides in positron emission tomography radiopharmaceuticals. J. Label. Compd. Radiopharm. 2014, 57, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Dumont, R.A.; Deininger, F.; Haubner, R.; Maecke, H.R.; Weber, W.A.; Fani, M. Novel 64Cu- and 68Ga-labeled RGD conjugates show improved PET imaging of αvβ3 integrin expression and facile radiosynthesis. J. Nucl. Med. 2011, 52, 1276–1284. [Google Scholar] [CrossRef]
- Ghosh, S.C.; Pinkston, K.L.; Robinson, H.; Harvey, B.R.; Wilganowski, N.; Gore, K.; Sevick-Muraca, E.M.; Azhdarinia, A. Comparison of DOTA and NODAGA as chelators for 64Cu-labeled immunoconjugates. Nucl. Med. Biol. 2015, 42, 177–183. [Google Scholar] [CrossRef]
- Sevcenco, S.; Klingler, H.C.; Eredics, K.; Friedl, A.; Schneeweiss, J.; Knoll, P.; Kunit, T.; Lusuardi, L.; Mirzaei, S. Application of Cu-64 NODAGA-PSMA PET in Prostate Cancer. Adv. Ther. 2018, 35, 779–784. [Google Scholar] [CrossRef]
- Boal, A.K.; Rosenzweig, A.C. Structural biology of copper trafficking. Chem. Rev. 2009, 109, 4760–4779. [Google Scholar] [CrossRef]
- Zeglis, B.M.; Lewis, J.S. A practical guide to the construction of radiometallated bioconjugates for positron emission tomography. Dalton Trans. 2011, 40, 6168–6195. [Google Scholar] [CrossRef]
- Dos Santos, J.C.; Beijer, B.; Bauder-Wust, U.; Schafer, M.; Leotta, K.; Eder, M.; Benesova, M.; Kleist, C.; Giesel, F.; Kratochwil, C.; et al. Development of Novel PSMA Ligands for Imaging and Therapy with Copper Isotopes. J. Nucl. Med. 2019. [Google Scholar] [CrossRef]
- Price, E.W.; Orvig, C. Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef]
- Liu, S.; Vorobyova, I.; Park, R.; Conti, P.S. Biodistribution and Radiation Dosimetry of the Integrin Marker 64Cu-BaBaSar-RGD2 Determined from Whole-Body PET/CT in a Non-human Primate. Front. Phys. 2017. [Google Scholar] [CrossRef]
- International Atomic Energy Agency (IEAE). Gallium-68 Cyclotron Production, IAEA-TECDOC-1863; IAEA: Vienna, Austria, 2019. [Google Scholar]
- Eder, M.; Schafer, M.; Bauder-Wust, U.; Hull, W.E.; Wangler, C.; Mier, W.; Haberkorn, U.; Eisenhut, M. 68Ga-complex lipophilicity and the targeting property of a urea-based PSMA inhibitor for PET imaging. Bioconjug. Chem. 2012, 23, 688–697. [Google Scholar] [CrossRef]
- Knetsch, P.A.; Petrik, M.; Griessinger, C.M.; Rangger, C.; Fani, M.; Kesenheimer, C.; von Guggenberg, E.; Pichler, B.J.; Virgolini, I.; Decristoforo, C.; et al. [68Ga]NODAGA-RGD for imaging αvβ3 integrin expression. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1303–1312. [Google Scholar] [CrossRef]
- Notni, J.; Simecek, J.; Hermann, P.; Wester, H.J. TRAP, a powerful and versatile framework for gallium-68 radiopharmaceuticals. Chemistry 2011, 17, 14718–14722. [Google Scholar] [CrossRef]
- Notni, J.; Pohle, K.; Wester, H.J. Be spoilt for choice with radiolabelled RGD peptides: Preclinical evaluation of 68Ga-TRAP(RGD)3. Nucl. Med. Biol. 2013, 40, 33–41. [Google Scholar] [CrossRef]
- Simecek, J.; Zemek, O.; Hermann, P.; Notni, J.; Wester, H.J. Tailored Gallium(III) chelator NOPO: Synthesis, characterization, bioconjugation, and application in preclinical Ga-68-PET imaging. Mol. Pharm. 2014, 11, 3893–3903. [Google Scholar] [CrossRef]
- Waldron, B.P.; Parker, D.; Burchardt, C.; Yufit, D.S.; Zimny, M.; Roesch, F. Structure and stability of hexadentate complexes of ligands based on AAZTA for efficient PET labelling with gallium-68. Chem. Commun. 2013, 49, 579–581. [Google Scholar] [CrossRef]
- Knetsch, P.A.; Zhai, C.; Rangger, C.; Blatzer, M.; Haas, H.; Kaeopookum, P.; Haubner, R.; Decristoforo, C. [68Ga]FSC-(RGD)3 a trimeric RGD peptide for imaging αvβ3 integrin expression based on a novel siderophore derived chelating scaffold-synthesis and evaluation. Nucl. Med. Biol. 2015, 42, 115–122. [Google Scholar] [CrossRef]
- Kaeopookum, P.; Petrik, M.; Summer, D.; Klinger, M.; Zhai, C.; Rangger, C.; Haubner, R.; Haas, H.; Hajduch, M.; Decristoforo, C. Comparison of 68Ga-labeled RGD mono- and multimers based on a clickable siderophore-based scaffold. Nucl. Med. Biol. 2019, 78-79, 1–10. [Google Scholar] [CrossRef]
- Dijkers, E.C.; Kosterink, J.G.; Rademaker, A.P.; Perk, L.R.; van Dongen, G.A.; Bart, J.; de Jong, J.R.; de Vries, E.G.; Lub-de Hooge, M.N. Development and characterization of clinical-grade 89Zr-trastuzumab for HER2/neu immunoPET imaging. J. Nucl. Med. 2009, 50, 974–981. [Google Scholar] [CrossRef]
- Aerts, H.J.; Dubois, L.; Perk, L.; Vermaelen, P.; van Dongen, G.A.; Wouters, B.G.; Lambin, P. Disparity between in vivo EGFR expression and 89Zr-labeled cetuximab uptake assessed with PET. J. Nucl. Med. 2009, 50, 123–131. [Google Scholar] [CrossRef]
- Nagengast, W.B.; de Korte, M.A.; Oude Munnink, T.H.; Timmer-Bosscha, H.; den Dunnen, W.F.; Hollema, H.; de Jong, J.R.; Jensen, M.R.; Quadt, C.; Garcia-Echeverria, C.; et al. 89Zr-bevacizumab PET of early antiangiogenic tumor response to treatment with HSP90 inhibitor NVP-AUY922. J. Nucl. Med. 2010, 51, 761–767. [Google Scholar] [CrossRef]
- Zhang, Y.; Hong, H.; Cai, W. PET tracers based on Zirconium-89. Curr. Radiopharm. 2011, 4, 131–139. [Google Scholar] [CrossRef]
- Heskamp, S.; Raave, R.; Boerman, O.; Rijpkema, M.; Goncalves, V.; Denat, F. 89Zr-Immuno-Positron Emission Tomography in Oncology: State-of-the-Art 89Zr Radiochemistry. Bioconjug. Chem. 2017, 28, 2211–2223. [Google Scholar]
- Patra, M.; Bauman, A.; Mari, C.; Fischer, C.A.; Blacque, O.; Häussinger, D.; Gasser, G.; Mindt, T.L. An octadentate bifuntional chelating agent for the development of stable zirconium-89 based molecular imaging probes. Chem. Commun. 2014, 50, 11523–11525. [Google Scholar] [CrossRef]
- Vugts, D.J.; Klaver, C.; Sewing, C.; Poot, A.J.; Adamzek, K.; Huegli, S.; Mari, C.; Visser, G.W.M.; Valverde, I.E.; Gasser, G.; et al. Comparison of the octadentate bifunctional chelator DFO*-pPhe-NCS and the clinically used hexadentate bifunctional chelator DFO-pPhe-NCS for 89Zr-immuno-PET. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 286–295. [Google Scholar] [CrossRef]
- Petrik, M.; Zhai, C.; Novy, Z.; Urbanek, L.; Haas, H.; Decristoforo, C. In Vitro and In Vivo Comparison of Selected Ga-68 and Zr-89 Labelled Siderophores. Mol. Imaging Biol. 2016, 18, 344–352. [Google Scholar] [CrossRef]
- Deri, M.A.; Ponnala, S.; Zeglis, B.M.; Pohl, G.; Dannenberg, J.J.; Lewis, J.S.; Francesconi, L.C. Alternative chelator for 89Zr radiopharmaceuticals: Radiolabeling and evaluation of 3,4,3-(LI-1,2-HOPO). J. Med. Chem. 2014, 57, 4849–4860. [Google Scholar] [CrossRef]
- Pandya, D.N.; Bhatt, N.; Yuan, H.; Day, C.S.; Ehrmann, B.M.; Wright, M.; Bierbach, U.; Wadas, T.J. Zirconium tetraazamacrocycle complexes display extraordinary stability and provide a new strategy for zirconium-89-based radiopharmaceutical development. Chem. Sci. 2017, 8, 2309–2314. [Google Scholar] [CrossRef]
- Emmett, L.; Willowson, K.; Violet, J.; Shin, J.; Blanksby, A.; Lee, J. Lutetium (177) PSMA radionuclide therapy for men with prostate cancer: A review of the current literature and discussion of practical aspects of therapy. J. Med. Radiat. Sci. 2017, 64, 52–60. [Google Scholar] [CrossRef]
- Sinnes, J.P.; Nagel, J.; Rosch, F. AAZTA5/AAZTA5-TOC: Synthesis and radiochemical evaluation with 68Ga, 44Sc and 177Lu. EJNMMI Radiopharm. Chem. 2019, 4, 18. [Google Scholar] [CrossRef]
- Price, E.W.; Zeglis, B.M.; Cawthray, J.F.; Ramogida, C.F.; Ramos, N.; Lewis, J.S.; Adam, M.J.; Orvig, C. H4octapa-trastuzumab: Versatile acyclic chelate system for 111In and 177Lu imaging and therapy. J. Am. Chem. Soc. 2013, 135, 12707–12721. [Google Scholar] [CrossRef]
- Kang, C.S.; Sun, X.; Jia, F.; Song, H.A.; Chen, Y.; Lewis, M.; Chong, H.S. Synthesis and preclinical evaluation of bifunctional ligands for improved chelation chemistry of 90Y and 177Lu for targeted radioimmunotherapy. Bioconjug. Chem. 2012, 23, 1775–1782. [Google Scholar] [CrossRef]
- Ferrier, M.G.; Radchenko, V.; Wilbur, D.S. Radiochemical aspects of alpha emitting radionuclides for medical application. Radiochim. Acta 2019, 107, 1065–1085. [Google Scholar] [CrossRef]
- Chappell, L.L.; Deal, K.A.; Dadachova, E.; Brechbiel, M.W. Synthesis, conjugation, and radiolabeling of a novel bifunctional chelating agent for 225Ac radioimmunotherapy applications. Bioconjug. Chem. 2000, 11, 510–519. [Google Scholar] [CrossRef]
- Morgenstern, A.; Apostolidis, C.; Kratochwil, C.; Sathekge, M.; Krolicki, L.; Bruchertseifer, F. An Overview of Targeted Alpha Therapy with 225Actinium and 213Bismuth. Curr. Radiopharm. 2018, 11, 200–208. [Google Scholar] [CrossRef]
- Scheinberg, D.A.; McDevitt, M.R. Actinium-225 in targeted alpha-particle therapeutic applications. Curr. Radiopharm. 2011, 4, 306–320. [Google Scholar] [CrossRef]
- Kratochwil, C.; Bruchertseifer, F.; Giesel, F.L.; Weis, M.; Verburg, F.A.; Mottaghy, F.; Kopka, K.; Apostolidis, C.; Haberkorn, U.; Morgenstern, A. 225Ac-PSMA-617 for PSMA-Targeted α-Radiation Therapy of Metastatic Castration-Resistant Prostate Cancer. J. Nucl. Med. 2016, 57, 1941–1944. [Google Scholar] [CrossRef]
- Thiele, N.A.; Brown, V.; Kelly, J.M.; Amor-Coarasa, A.; Jermilova, U.; MacMillan, S.N.; Nikolopoulou, A.; Ponnala, S.; Ramogida, C.F.; Robertson, A.K.H.; et al. An Eighteen-Membered Macrocyclic Ligand for Actinium-225 Targeted Alpha Therapy. Angew. Chem. Int. Ed. Engl. 2017, 56, 14712–14717. [Google Scholar] [CrossRef]
- Wei, L.; Ye, Y.; Wadas, T.J.; Lewis, J.S.; Welch, M.J.; Achilefu, S.; Anderson, C.J. 64Cu-labeled CB-TE2A and diamsar-conjugated RGD peptide analogs for targeting angiogenesis: Comparison of their biological activity. Nucl. Med. Biol. 2009, 36, 277–285. [Google Scholar] [CrossRef][Green Version]
- Schjoeth-Eskesen, C.; Nielsen, C.H.; Heissel, S.; Hojrup, P.; Hansen, P.R.; Gillings, N.; Kjaer, A. [64Cu]-labelled trastuzumab: Optimisation of labelling by DOTA and NODAGA conjugation and initial evaluation in mice. J. Label. Compd. Radiopharm. 2015, 58, 227–233. [Google Scholar] [CrossRef]
- Craft, J.M.; De Silva, R.A.; Lears, K.A.; Andrews, R.; Liang, K.; Achilefu, S.; Rogers, B.E. In vitro and in vivo evaluation of a 64Cu-labeled NOTA-Bn-SCN-Aoc-bombesin analogue in gastrin-releasing peptide receptor expressing prostate cancer. Nucl. Med. Biol 2012, 39, 609–616. [Google Scholar] [CrossRef][Green Version]
- Spang, P.; Herrmann, C.; Roesch, F. Bifunctional Gallium-68 Chelators: Past, Present, and Future. Semin. Nucl. Med. 2016, 46, 373–394. [Google Scholar] [CrossRef]
- Haubner, R.; Vera, D.R.; Farshchi-Heydari, S.; Helbok, A.; Rangger, C.; Putzer, D.; Virgolini, I.J. Development of 68Ga-labelled DTPA galactosyl human serum albumin for liver function imaging. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 1245–1255. [Google Scholar] [CrossRef]
- Jeong, J.M.; Hong, M.K.; Chang, Y.S.; Lee, Y.S.; Kim, Y.J.; Cheon, G.J.; Lee, D.S.; Chung, J.K.; Lee, M.C. Preparation of a Promising Angiogenesis PET Imaging Agent: 68Ga-Labeled c(RGDyK)-Isothiocyanatobenzyl-1,4,7-Triazacyclononane-1,4,7-Triacetic Acid and Feasibility Studies in Mice. J. Nucl. Med. 2008, 49, 830–836. [Google Scholar] [CrossRef]
- Verel, I.; Visser, G.W.; Boellaard, R.; Stigter-van Walsum, M.; Snow, G.B.; van Dongen, G.A. 89Zr immuno-PET: Comprehensive procedures for the production of 89Zr-labeled monoclonal antibodies. J. Nucl. Med. 2003, 44, 1271–1281. [Google Scholar]
- Zhai, C.; He, S.; Ye, Y.; Rangger, C.; Kaeopookum, P.; Summer, D.; Haas, H.; Kremser, L.; Lindner, H.; Foster, J.; et al. Rational Design, Synthesis and Preliminary Evaluation of Novel Fusarinine C-Based Chelators for Radiolabeling with Zirconium-89. Biomolecules 2019, 9, 91. [Google Scholar] [CrossRef]
- Breeman, W.A.; De Jong, M.; Visser, T.J.; Erion, J.L.; Krenning, E.P. Optimising conditions for radiolabelling of DOTA-peptides with 90Y, 111In and 177Lu at high specific activities. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 917–920. [Google Scholar] [CrossRef]
- Robertson, A.K.H.; Ramogida, C.F.; Schaffer, P.; Radchenko, V. Development of 225Ac Radiopharmaceuticals: TRIUMF Perspectives and Experiences. Curr. Radiopharm. 2018, 11, 156–172. [Google Scholar]
- Sun, X.; Li, Y.; Liu, T.; Li, Z.; Zhang, X.; Chen, X. Peptide-based imaging agents for cancer detection. Adv. Drug Deliv. Rev. 2017, 110–111, 38–51. [Google Scholar] [CrossRef]
- Jackson, I.M.; Scott, P.J.H.; Thompson, S. Clinical Applications of Radiolabeled Peptides for PET. Semin. Nucl. Med. 2017, 47, 493–523. [Google Scholar] [CrossRef]
- Johnbeck, C.B.; Knigge, U.; Kjaer, A. PET tracers for somatostatin receptor imaging of neuroendocrine tumors: Current status and review of the literature. Future Oncol. 2014, 10, 2259–2277. [Google Scholar] [CrossRef]
- Hofmann, M.; Maecke, H.; Borner, R.; Weckesser, E.; Schoffski, P.; Oei, L.; Schumacher, J.; Henze, M.; Heppeler, A.; Meyer, J.; et al. Biokinetics and imaging with the somatostatin receptor PET radioligand 68Ga-DOTATOC: Preliminary data. Eur. J. Nucl. Med. 2001, 28, 1751–1757. [Google Scholar] [CrossRef]
- Poeppel, T.D.; Binse, I.; Petersenn, S.; Lahner, H.; Schott, M.; Antoch, G.; Brandau, W.; Bockisch, A.; Boy, C. 68Ga-DOTATOC versus 68Ga-DOTATATE PET/CT in functional imaging of neuroendocrine tumors. J. Nucl. Med. 2011, 52, 1864–1870. [Google Scholar] [CrossRef]
- Kabasakal, L.; Demirci, E.; Ocak, M.; Decristoforo, C.; Araman, A.; Ozsoy, Y.; Uslu, I.; Kanmaz, B. Comparison of 68Ga-DOTATATE and 68Ga-DOTANOC PET/CT imaging in the same patient group with neuroendocrine tumours. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 1271–1277. [Google Scholar] [CrossRef]
- Wild, D.; Schmitt, J.S.; Ginj, M.; Macke, H.R.; Bernard, B.F.; Krenning, E.; De Jong, M.; Wenger, S.; Reubi, J.C. DOTA-NOC, a high-affinity ligand of somatostatin receptor subtypes 2, 3 and 5 for labelling with various radiometals. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 1338–1347. [Google Scholar] [CrossRef]
- Pauwels, E.; Cleeren, F.; Bormans, G.; Deroose, C.M. Somatostatin receptor PET ligands—The next generation for clinical practice. Am. J. Nucl. Med. Mol. Imaging 2018, 8, 311–331. [Google Scholar]
- Anderson, C.J.; Dehdashti, F.; Cutler, P.D.; Schwarz, S.W.; Laforest, R.; Bass, L.A.; Lewis, J.S.; McCarthy, D.W. 64Cu-TETA-octreotide as a PET imaging agent for patients with neuroendocrine tumors. J. Nucl. Med. 2001, 42, 213–221. [Google Scholar]
- Pfeifer, A.; Knigge, U.; Mortensen, J.; Oturai, P.; Berthelsen, A.K.; Loft, A.; Binderup, T.; Rasmussen, P.; Elema, D.; Klausen, T.L.; et al. Clinical PET of neuroendocrine tumors using 64Cu-DOTATATE: First-in-humans study. J. Nucl. Med. 2012, 53, 1207–1215. [Google Scholar] [CrossRef]
- Johnbeck, C.B.; Knigge, U.; Loft, A.; Berthelsen, A.K.; Mortensen, J.; Oturai, P.; Langer, S.W.; Elema, D.R.; Kjaer, A. Head-to-Head Comparison of 64Cu-DOTATATE and 68Ga-DOTATOC PET/CT: A Prospective Study of 59 Patients with Neuroendocrine Tumors. J. Nucl. Med. 2017, 58, 451–457. [Google Scholar] [CrossRef]
- Schottelius, M.; Poethko, T.; Herz, M.; Reubi, J.C.; Kessler, H.; Schwaiger, M.; Wester, H.J. First 18F-labeled tracer suitable for routine clinical imaging of sst receptor-expressing tumors using positron emission tomography. Clin. Cancer Res. 2004, 10, 3593–3606. [Google Scholar] [CrossRef]
- Meisetschlager, G.; Poethko, T.; Stahl, A.; Wolf, I.; Scheidhauer, K.; Schottelius, M.; Herz, M.; Wester, H.J.; Schwaiger, M. Gluc-Lys([18F]FP)-TOCA PET in patients with SSTR-positive tumors: Biodistribution and diagnostic evaluation compared with [111In]DTPA-octreotide. J. Nucl. Med. 2006, 47, 566–573. [Google Scholar]
- Iddon, L.; Leyton, J.; Indrevoll, B.; Glaser, M.; Robins, E.G.; George, A.J.; Cuthbertson, A.; Luthra, S.K.; Aboagye, E.O. Synthesis and in vitro evaluation of [18F]fluoroethyl triazole labelled [Tyr3]octreotate analogues using click chemistry. Bioorg. Med. Chem. Lett. 2011, 21, 3122–3127. [Google Scholar] [CrossRef]
- Dubash, S.R.; Keat, N.; Mapelli, P.; Twyman, F.; Carroll, L.; Kozlowski, K.; Al-Nahhas, A.; Saleem, A.; Huiban, M.; Janisch, R.; et al. Clinical Translation of a Click-Labeled 18F-Octreotate Radioligand for Imaging Neuroendocrine Tumors. J. Nucl. Med. 2016, 57, 1207–1213. [Google Scholar] [CrossRef]
- Waldmann, C.M.; Stuparu, A.D.; van Dam, R.M.; Slavik, R. The Search for an Alternative to [68Ga]Ga-DOTA-TATE in Neuroendocrine Tumor Theranostics: Current State of 18F-labeled Somatostatin Analog Development. Theranostics 2019, 9, 1336–1347. [Google Scholar] [CrossRef]
- Ilhan, H.; Lindner, S.; Todica, A.; Cyran, C.C.; Tiling, R.; Auernhammer, C.J.; Spitzweg, C.; Boeck, S.; Unterrainer, M.; Gildehaus, F.J.; et al. Biodistribution and first clinical results of 18F-SiFAlin-TATE PET: A novel 18F-labeled somatostatin analog for imaging of neuroendocrine tumors. Eur. J. Nucl. Med. Mol. Imaging 2019. [Google Scholar] [CrossRef]
- Ginj, M.; Zhang, H.; Waser, B.; Cescato, R.; Wild, D.; Wang, X.; Erchegyi, J.; Rivier, J.; Macke, H.R.; Reubi, J.C. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc. Natl. Acad. Sci. USA 2006, 103, 16436–16441. [Google Scholar] [CrossRef]
- Fani, M.; Braun, F.; Waser, B.; Beetschen, K.; Cescato, R.; Erchegyi, J.; Rivier, J.E.; Weber, W.A.; Maecke, H.R.; Reubi, J.C. Unexpected sensitivity of sst2 antagonists to N-terminal radiometal modifications. J. Nucl. Med. 2012, 53, 1481–1489. [Google Scholar] [CrossRef]
- Nicolas, G.P.; Schreiter, N.; Kaul, F.; Uiters, J.; Bouterfa, H.; Kaufmann, J.; Erlanger, T.E.; Cathomas, R.; Christ, E.; Fani, M.; et al. Sensitivity Comparison of 68Ga-OPS202 and 68Ga-DOTATOC PET/CT in Patients with Gastroenteropancreatic Neuroendocrine Tumors: A Prospective Phase II Imaging Study. J. Nucl. Med. 2018, 59, 915–921. [Google Scholar] [CrossRef]
- Basu, S.; Parghane, R.V.; Banerjee, S. Availability of both [177Lu]Lu-DOTA-TATE and [90Y]Y-DOTATATE as PRRT agents for neuroendocrine tumors: Can we evolve a rational sequential duo-PRRT protocol for large volume resistant tumors? Eur. J. Nucl. Med. Mol. Imaging 2019. [Google Scholar] [CrossRef]
- Ballal, S.; Yadav, M.P.; Bal, C.; Sahoo, R.K.; Tripathi, M. Broadening horizons with 225Ac-DOTATATE targeted alpha therapy for gastroenteropancreatic neuroendocrine tumour patients stable or refractory to 177Lu-DOTATATE PRRT: First clinical experience on the efficacy and safety. Eur. J. Nucl. Med. Mol. Imaging 2019. [Google Scholar] [CrossRef]
- Dalm, S.U.; Nonnekens, J.; Doeswijk, G.N.; de Blois, E.; van Gent, D.C.; Konijnenberg, M.W.; de Jong, M. Comparison of the Therapeutic Response to Treatment with a 177Lu-Labeled Somatostatin Receptor Agonist and Antagonist in Preclinical Models. J. Nucl. Med. 2016, 57, 260–265. [Google Scholar] [CrossRef]
- Reidy-Lagunes, D.; Pandit-Taskar, N.; O’Donoghue, J.A.; Krebs, S.; Staton, K.D.; Lyashchenko, S.K.; Lewis, J.S.; Raj, N.; Gonen, M.; Lohrmann, C.; et al. Phase I Trial of Well-Differentiated Neuroendocrine Tumors (NETs) with Radiolabeled Somatostatin Antagonist 177Lu-Satoreotide Tetraxetan. Clin. Cancer Res. 2019, 25, 6939–6947. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Zhang, J.; Nan, F.; Petukhov, P.A.; Grajkowska, E.; Wroblewski, J.T.; Yamamoto, T.; Bzdega, T.; Wroblewska, B.; Neale, J.H. Synthesis of urea-based inhibitors as active site probes of glutamate carboxypeptidase II: Efficacy as analgesic agents. J. Med. Chem. 2004, 47, 1729–1738. [Google Scholar] [CrossRef]
- Foss, C.A.; Mease, R.C.; Fan, H.; Wang, Y.; Ravert, H.T.; Dannals, R.F.; Olszewski, R.T.; Heston, W.D.; Kozikowski, A.P.; Pomper, M.G. Radiolabeled small-molecule ligands for prostate-specific membrane antigen: In vivo imaging in experimental models of prostate cancer. Clin. Cancer Res. 2005, 11, 4022–4028. [Google Scholar] [CrossRef]
- Banerjee, S.R.; Foss, C.A.; Castanares, M.; Mease, R.C.; Byun, Y.; Fox, J.J.; Hilton, J.; Lupold, S.E.; Kozikowski, A.P.; Pomper, M.G. Synthesis and evaluation of technetium-99m- and rhenium-labeled inhibitors of the prostate-specific membrane antigen (PSMA). J. Med. Chem. 2008, 51, 4504–4517. [Google Scholar] [CrossRef]
- Chen, Y.; Foss, C.A.; Byun, Y.; Nimmagadda, S.; Pullambhatla, M.; Fox, J.J.; Castanares, M.; Lupold, S.E.; Babich, J.W.; Mease, R.C.; et al. Radiohalogenated prostate-specific membrane antigen (PSMA)-based ureas as imaging agents for prostate cancer. J. Med. Chem. 2008, 51, 7933–7943. [Google Scholar] [CrossRef]
- Benesova, M.; Schafer, M.; Bauder-Wust, U.; Afshar-Oromieh, A.; Kratochwil, C.; Mier, W.; Haberkorn, U.; Kopka, K.; Eder, M. Preclinical Evaluation of a Tailor-Made DOTA-Conjugated PSMA Inhibitor with Optimized Linker Moiety for Imaging and Endoradiotherapy of Prostate Cancer. J. Nucl. Med. 2015, 56, 914–920. [Google Scholar] [CrossRef]
- Cardinale, J.; Schafer, M.; Benesova, M.; Bauder-Wust, U.; Leotta, K.; Eder, M.; Neels, O.C.; Haberkorn, U.; Giesel, F.L.; Kopka, K. Preclinical Evaluation of 18F-PSMA-1007, a New Prostate-Specific Membrane Antigen Ligand for Prostate Cancer Imaging. J. Nucl. Med. 2017, 58, 425–431. [Google Scholar] [CrossRef]
- Wester, H.J.; Schottelius, M. PSMA-Targeted Radiopharmaceuticals for Imaging and Therapy. Semin. Nucl Med. 2019, 49, 302–312. [Google Scholar] [CrossRef]
- Afshar-Oromieh, A.; Holland-Letz, T.; Giesel, F.L.; Kratochwil, C.; Mier, W.; Haufe, S.; Debus, N.; Eder, M.; Eisenhut, M.; Schafer, M.; et al. Diagnostic performance of 68Ga-PSMA-11 (HBED-CC) PET/CT in patients with recurrent prostate cancer: Evaluation in 1007 patients. Eur J. Nucl. Med. Mol. Imaging 2017, 44, 1258–1268. [Google Scholar] [CrossRef]
- Afshar-Oromieh, A.; Malcher, A.; Eder, M.; Eisenhut, M.; Linhart, H.G.; Hadaschik, B.A.; Holland-Letz, T.; Giesel, F.L.; Kratochwil, C.; Haufe, S.; et al. PET imaging with a [68Ga]gallium-labelled PSMA ligand for the diagnosis of prostate cancer: Biodistribution in humans and first evaluation of tumour lesions. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 486–495. [Google Scholar] [CrossRef]
- Virgolini, I.; Decristoforo, C.; Haug, A.; Fanti, S.; Uprimny, C. Current status of theranostics in prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 471–495. [Google Scholar] [CrossRef]
- De Visschere, P.J.L.; Standaert, C.; Futterer, J.J.; Villeirs, G.M.; Panebianco, V.; Walz, J.; Maurer, T.; Hadaschik, B.A.; Lecouvet, F.E.; Giannarini, G.; et al. A Systematic Review on the Role of Imaging in Early Recurrent Prostate Cancer. Eur. Urol. Oncol. 2019, 2, 47–76. [Google Scholar] [CrossRef]
- Weineisen, M.; Schottelius, M.; Simecek, J.; Baum, R.P.; Yildiz, A.; Beykan, S.; Kulkarni, H.R.; Lassmann, M.; Klette, I.; Eiber, M.; et al. 68Ga- and 177Lu-Labeled PSMA I&T: Optimization of a PSMA-Targeted Theranostic Concept and First Proof-of-Concept Human Studies. J. Nucl. Med. 2015, 56, 1169–1176. [Google Scholar]
- Zhang, A.X.; Murelli, R.P.; Barinka, C.; Michel, J.; Cocleaza, A.; Jorgensen, W.L.; Lubkowski, J.; Spiegel, D.A. A remote arene-binding site on prostate specific membrane antigen revealed by antibody-recruiting small molecules. J. Am. Chem. Soc. 2010, 132, 12711–12716. [Google Scholar] [CrossRef]
- McCarthy, M.; Langton, T.; Kumar, D.; Campbell, A. Comparison of PSMA-HBED and PSMA-I&T as diagnostic agents in prostate carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1455–1462. [Google Scholar]
- Derlin, T.; Schmuck, S.; Juhl, C.; Teichert, S.; Zorgiebel, J.; Wester, H.J.; Schneefeld, S.M.; Walte, A.C.A.; Thackeray, J.T.; Ross, T.L.; et al. Imaging Characteristics and First Experience of [68Ga]THP-PSMA, a Novel Probe for Rapid Kit-Based Ga-68 Labeling and PET Imaging: Comparative Analysis with [68Ga]PSMA I&T. Mol. Imaging Biol. 2018, 20, 650–658. [Google Scholar]
- Green, M.A.; Hutchins, G.D.; Bahler, C.D.; Tann, M.; Mathias, C.J.; Territo, W.; Sims, J.; Polson, H.; Alexoff, D.; Eckelman, W.C.; et al. [68Ga]Ga-P16-093 as a PSMA-Targeted PET Radiopharmaceutical for Detection of Cancer: Initial Evaluation and Comparison with [68Ga]Ga-PSMA-11 in Prostate Cancer Patients Presenting with Biochemical Recurrence. Mol. Imaging Biol. 2019. [Google Scholar] [CrossRef]
- Mease, R.C.; Dusich, C.L.; Foss, C.A.; Ravert, H.T.; Dannals, R.F.; Seidel, J.; Prideaux, A.; Fox, J.J.; Sgouros, G.; Kozikowski, A.P.; et al. N-[N-[(S)-1,3-Dicarboxypropyl]carbamoyl]-4-[18F]fluorobenzyl-L-cysteine, [18F]DCFBC: A new imaging probe for prostate cancer. Clin. Cancer Res. 2008, 14, 3036–3043. [Google Scholar] [CrossRef]
- Chen, Y.; Pullambhatla, M.; Foss, C.A.; Byun, Y.; Nimmagadda, S.; Senthamizhchelvan, S.; Sgouros, G.; Mease, R.C.; Pomper, M.G. 2-(3-{1-Carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl)-amino]-pentyl}-ureido)-pen tanedioic acid, [18F]DCFPyL, a PSMA-based PET imaging agent for prostate cancer. Clin. Cancer Res. 2011, 17, 7645–7653. [Google Scholar] [CrossRef]
- Malik, N.; Baur, B.; Winter, G.; Reske, S.N.; Beer, A.J.; Solbach, C. Radiofluorination of PSMA-HBED via Al18F(2+) Chelation and Biological Evaluations In Vitro. Mol. Imaging Biol. 2015, 17, 777–785. [Google Scholar] [CrossRef]
- Behr, S.C.; Aggarwal, R.; VanBrocklin, H.F.; Flavell, R.R.; Gao, K.; Small, E.J.; Blecha, J.; Jivan, S.; Hope, T.A.; Simko, J.P.; et al. Phase I Study of CTT1057, an 18F-Labeled Imaging Agent with Phosphoramidate Core Targeting Prostate-Specific Membrane Antigen in Prostate Cancer. J. Nucl. Med. 2019, 60, 910–916. [Google Scholar] [CrossRef]
- Zlatopolskiy, B.D.; Endepols, H.; Krapf, P.; Guliyev, M.; Urusova, E.A.; Richarz, R.; Hohberg, M.; Dietlein, M.; Drzezga, A.; Neumaier, B. Discovery of 18F-JK-PSMA-7, a PET Probe for the Detection of Small PSMA-Positive Lesions. J. Nucl. Med. 2019, 60, 817–823. [Google Scholar] [CrossRef]
- Dietlein, F.; Kobe, C.; Neubauer, S.; Schmidt, M.; Stockter, S.; Fischer, T.; Schomacker, K.; Heidenreich, A.; Zlatopolskiy, B.D.; Neumaier, B.; et al. PSA-Stratified Performance of 18F- and 68Ga-PSMA PET in Patients with Biochemical Recurrence of Prostate Cancer. J. Nucl. Med. 2017, 58, 947–952. [Google Scholar] [CrossRef]
- Giesel, F.L.; Knorr, K.; Spohn, F.; Will, L.; Maurer, T.; Flechsig, P.; Neels, O.; Schiller, K.; Amaral, H.; Weber, W.A.; et al. Detection Efficacy of 18F-PSMA-1007 PET/CT in 251 Patients with Biochemical Recurrence of Prostate Cancer After Radical Prostatectomy. J. Nucl. Med. 2019, 60, 362–368. [Google Scholar] [CrossRef]
- Giesel, F.L.; Will, L.; Lawal, I.; Lengana, T.; Kratochwil, C.; Vorster, M.; Neels, O.; Reyneke, F.; Haberkon, U.; Kopka, K.; et al. Intraindividual Comparison of 18F-PSMA-1007 and 18F-DCFPyL PET/CT in the Prospective Evaluation of Patients with Newly Diagnosed Prostate Carcinoma: A Pilot Study. J. Nucl. Med. 2018, 59, 1076–1080. [Google Scholar] [CrossRef]
- Dannoon, S.; Ganguly, T.; Cahaya, H.; Geruntho, J.J.; Galliher, M.S.; Beyer, S.K.; Choy, C.J.; Hopkins, M.R.; Regan, M.; Blecha, J.E.; et al. Structure-Activity Relationship of 18F-Labeled Phosphoramidate Peptidomimetic Prostate-Specific Membrane Antigen (PSMA)-Targeted Inhibitor Analogues for PET Imaging of Prostate Cancer. J. Med. Chem. 2016, 59, 5684–5694. [Google Scholar] [CrossRef]
- Dietlein, F.; Hohberg, M.; Kobe, C.; Zlatopolskiy, B.D.; Krapf, P.; Endepols, H.; Tager, P.; Hammes, J.; Heidenreich, A.; Neumaier, B.; et al. A novel 18F-labeled PSMA ligand for PET/CT imaging of prostate cancer patients: First-in-man observational study and clinical experience with 18F-JK-PSMA-7 during the first year of application. J. Nucl. Med. 2019. [Google Scholar] [CrossRef]
- Kersemans, K.; De Man, K.; Courtyn, J.; Van Royen, T.; Piron, S.; Moerman, L.; Brans, B.; De Vos, F. Automated radiosynthesis of Al[18F]PSMA-11 for large scale routine use. Appl. Radiat. Isot. 2018, 135, 19–27. [Google Scholar] [CrossRef]
- Piron, S.; De Man, K.; Van Laeken, N.; D’Asseler, Y.; Bacher, K.; Kersemans, K.; Ost, P.; Decaestecker, K.; Deseyne, P.; Fonteyne, V.; et al. Radiation Dosimetry and Biodistribution of 18F-PSMA-11 for PET Imaging of Prostate Cancer. J. Nucl. Med. 2019, 60, 1736–1742. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, K.; Ahmadzadehfar, H.; Boegemann, M. 177Lu-PSMA-617 radioligand therapy in mCRPC: Ready for phase III trial? Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 513–514. [Google Scholar] [CrossRef] [PubMed]
- Heck, M.M.; Tauber, R.; Schwaiger, S.; Retz, M.; D’Alessandria, C.; Maurer, T.; Gafita, A.; Wester, H.J.; Gschwend, J.E.; Weber, W.A.; et al. Treatment Outcome, Toxicity, and Predictive Factors for Radioligand Therapy with 177Lu-PSMA-I&T in Metastatic Castration-resistant Prostate Cancer. Eur. Urol. 2019, 75, 920–926. [Google Scholar] [PubMed]
- Lau, J.; Jacobson, O.; Niu, G.; Lin, K.S.; Benard, F.; Chen, X. Bench to Bedside: Albumin Binders for Improved Cancer Radioligand Therapies. Bioconjug Chem. 2019, 30, 487–502. [Google Scholar] [CrossRef]
- Wang, Z.; Tian, R.; Niu, G.; Ma, Y.; Lang, L.; Szajek, L.P.; Kiesewetter, D.O.; Jacobson, O.; Chen, X. Single Low-Dose Injection of Evans Blue Modified PSMA-617 Radioligand Therapy Eliminates Prostate-Specific Membrane Antigen Positive Tumors. Bioconjug Chem. 2018, 29, 3213–3221. [Google Scholar] [CrossRef]
- Zang, J.; Fan, X.; Wang, H.; Liu, Q.; Wang, J.; Li, H.; Li, F.; Jacobson, O.; Niu, G.; Zhu, Z.; et al. First-in-human study of 177Lu-EB-PSMA-617 in patients with metastatic castration-resistant prostate cancer. Eur J. Nucl. Med. Mol. Imaging 2019, 46, 148–158. [Google Scholar] [CrossRef]
- Chakravarty, R.; Siamof, C.M.; Dash, A.; Cai, W. Targeted alpha-therapy of prostate cancer using radiolabeled PSMA inhibitors: A game changer in nuclear medicine. Am. J. Nucl. Med. Mol. Imaging 2018, 8, 247–267. [Google Scholar]
- Sathekge, M.; Knoesen, O.; Meckel, M.; Modiselle, M.; Vorster, M.; Marx, S. 213Bi-PSMA-617 targeted alpha-radionuclide therapy in metastatic castration-resistant prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1099–1100. [Google Scholar] [CrossRef]
- Khreish, F.; Ebert, N.; Ries, M.; Maus, S.; Rosar, F.; Bohnenberger, H.; Stemler, T.; Saar, M.; Bartholoma, M.; Ezziddin, S. 225Ac-PSMA-617/177Lu-PSMA-617 tandem therapy of metastatic castration-resistant prostate cancer: Pilot experience. Eur. J. Nucl. Med. Mol. Imaging 2019. [Google Scholar] [CrossRef]
- Fendler, W.P.; Rahbar, K.; Herrmann, K.; Kratochwil, C.; Eiber, M. 177Lu-PSMA Radioligand Therapy for Prostate Cancer. J. Nucl. Med. 2017, 58, 1196–1200. [Google Scholar] [CrossRef]
- Chen, H.; Niu, G.; Wu, H.; Chen, X. Clinical Application of Radiolabeled RGD Peptides for PET Imaging of Integrin alphavbeta3. Theranostics 2016, 6, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Debordeaux, F.; Chansel-Debordeaux, L.; Pinaquy, J.B.; Fernandez, P.; Schulz, J. What about αvβ3integrins in molecular imaging in oncology? Nucl. Med. Biol 2018, 62-63, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Aumailley, M.; Gurrath, M.; Muller, G.; Calvete, J.; Timpl, R.; Kessler, H. Arg-Gly-Asp constrained within cyclic pentapeptides. Strong and selective inhibitors of cell adhesion to vitronectin and laminin fragment P1. FEBS Lett. 1991, 291, 50–54. [Google Scholar] [CrossRef]
- Haubner, R.; Wester, H.J.; Reuning, U.; Senekowitsch-Schmidtke, R.; Diefenbach, B.; Kessler, H.; Stocklin, G.; Schwaiger, M. Radiolabeled αvβ3 integrin antagonists: A new class of tracers for tumor targeting. J. Nucl. Med. 1999, 40, 1061–1071. [Google Scholar]
- Haubner, R.; Wester, H.J.; Weber, W.A.; Mang, C.; Ziegler, S.I.; Goodman, S.L.; Senekowitsch-Schmidtke, R.; Kessler, H.; Schwaiger, M. Noninvasive imaging of αvβ3 integrin expression using 18F-labeled RGD-containing glycopeptide and positron emission tomography. Cancer Res. 2001, 61, 1781–1785. [Google Scholar]
- Haubner, R.; Weber, W.A.; Beer, A.J.; Vabuliene, E.; Reim, D.; Sarbia, M.; Becker, K.F.; Goebel, M.; Hein, R.; Wester, H.J.; et al. Noninvasive visualization of the activated αvβ3 integrin in cancer patients by positron emission tomography and [18F]Galacto-RGD. PLoS Med. 2005, 2, e70. [Google Scholar] [CrossRef]
- Beer, A.J.; Haubner, R.; Goebel, M.; Luderschmidt, S.; Spilker, M.E.; Wester, H.J.; Weber, W.A.; Schwaiger, M. Biodistribution and pharmacokinetics of the αvβ3-selective tracer 18F-galacto-RGD in cancer patients. J. Nucl. Med. 2005, 46, 1333–1341. [Google Scholar]
- Beer, A.J.; Grosu, A.L.; Carlsen, J.; Kolk, A.; Sarbia, M.; Stangier, I.; Watzlowik, P.; Wester, H.J.; Haubner, R.; Schwaiger, M. [18F]galacto-RGD positron emission tomography for imaging of αvβ3 expression on the neovasculature in patients with squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2007, 13, 6610–6616. [Google Scholar] [CrossRef]
- Beer, A.J.; Haubner, R.; Sarbia, M.; Goebel, M.; Luderschmidt, S.; Grosu, A.L.; Schnell, O.; Niemeyer, M.; Kessler, H.; Wester, H.J.; et al. Positron emission tomography using [18F]Galacto-RGD identifies the level of integrin αvβ3 expression in man. Clin. Cancer Res. 2006, 12, 3942–3949. [Google Scholar] [CrossRef]
- Mirfeizi, L.; Walsh, J.; Kolb, H.; Campbell-Verduyn, L.; Dierckx, R.A.; Feringa, B.L.; Elsinga, P.H.; de Groot, T.; Sannen, I.; Bormans, G.; et al. Synthesis of [18F]RGD-K5 by catalyzed [3 + 2] cycloaddition for imaging integrin αvβ3 expression in vivo. Nucl. Med. Biol. 2013, 40, 710–716. [Google Scholar] [CrossRef]
- Glaser, M.; Morrison, M.; Solbakken, M.; Arukwe, J.; Karlsen, H.; Wiggen, U.; Champion, S.; Kindberg, G.M.; Cuthbertson, A. Radiosynthesis and biodistribution of cyclic RGD peptides conjugated with novel [18F]fluorinated aldehyde-containing prosthetic groups. Bioconjug. Chem. 2008, 19, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Haubner, R.; Wester, H.J. Radiolabeled tracers for imaging of tumor angiogenesis and evaluation of anti-angiogenic therapies. Curr. Pharm. Des. 2004, 10, 1439–1455. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, Z.; Chen, K.; Yan, Y.; Watzlowik, P.; Wester, H.J.; Chin, F.T.; Chen, X. 18F-labeled galacto and PEGylated RGD dimers for PET imaging of αvβ3 integrin expression. Mol. Imaging Biol. 2010, 12, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Lang, L.; Li, W.; Guo, N.; Ma, Y.; Zhu, L.; Kiesewetter, D.O.; Shen, B.; Niu, G.; Chen, X. Comparison study of [18F]FAl-NOTA-PRGD2, [18F]FPPRGD2, and [68Ga]Ga-NOTA-PRGD2 for PET imaging of U87MG tumors in mice. Bioconjug. Chem. 2011, 22, 2415–2422. [Google Scholar] [CrossRef] [PubMed]
- Schnell, O.; Krebs, B.; Carlsen, J.; Miederer, I.; Goetz, C.; Goldbrunner, R.H.; Wester, H.J.; Haubner, R.; Popperl, G.; Holtmannspotter, M.; et al. Imaging of integrin αvβ3 expression in patients with malignant glioma by [18F] Galacto-RGD positron emission tomography. Neuro Oncol. 2009, 11, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Dumont, R.A.; Hildebrandt, I.; Su, H.; Haubner, R.; Reischl, G.; Czernin, J.G.; Mischel, P.S.; Weber, W.A. Noninvasive imaging of αvβ3 function as a predictor of the antimigratory and antiproliferative effects of dasatinib. Cancer Res. 2009, 69, 3173–3179. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yang, M.; Gao, H.; Yan, Y.; Sun, X.; Chen, K.; Quan, Q.; Lang, L.; Kiesewetter, D.; Niu, G.; Chen, X. PET imaging of early response to the tyrosine kinase inhibitor ZD4190. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1237–1247. [Google Scholar] [CrossRef][Green Version]
- Ji, S.; Zheng, Y.; Shao, G.; Zhou, Y.; Liu, S. Integrin αvβ3-targeted radiotracer 99mTc-3P-RGD2 useful for noninvasive monitoring of breast tumor response to antiangiogenic linifanib therapy but not anti-integrin αvβ3 RGD2 therapy. Theranostics 2013, 3, 816–830. [Google Scholar] [CrossRef]
- Shi, J.; Jin, Z.; Liu, X.; Fan, D.; Sun, Y.; Zhao, H.; Zhu, Z.; Liu, Z.; Jia, B.; Wang, F. PET imaging of neovascularization with 68Ga-3PRGD2 for assessing tumor early response to Endostar antiangiogenic therapy. Mol. Pharm. 2014, 11, 3915–3922. [Google Scholar] [CrossRef]
- Bao, X.; Wang, M.W.; Luo, J.M.; Wang, S.Y.; Zhang, Y.P.; Zhang, Y.J. Optimization of Early Response Monitoring and Prediction of Cancer Antiangiogenesis Therapy via Noninvasive PET Molecular Imaging Strategies of Multifactorial Bioparameters. Theranostics 2016, 6, 2084–2098. [Google Scholar] [CrossRef]
- Minamimoto, R.; Karam, A.; Jamali, M.; Barkhodari, A.; Gambhir, S.S.; Dorigo, O.; Iagaru, A. Pilot prospective evaluation of 18F-FPPRGD2 PET/CT in patients with cervical and ovarian cancer. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ma, L.; Shang, D.; Liu, Z.; Yu, Q.; Wang, S.; Teng, X.; Zhang, Q.; Hu, X.; Zhao, W.; et al. Pretreatment PET/CT imaging of angiogenesis based on 18F-RGD tracer uptake may predict antiangiogenic response. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Nieberler, M.; Reuning, U.; Reichart, F.; Notni, J.; Wester, H.J.; Schwaiger, M.; Weinmuller, M.; Rader, A.; Steiger, K.; Kessler, H. Exploring the Role of RGD-Recognizing Integrins in Cancer. Cancers 2017, 9, 116. [Google Scholar] [CrossRef] [PubMed]
- McParland, B.J.; Miller, M.P.; Spinks, T.J.; Kenny, L.M.; Osman, S.; Khela, M.K.; Aboagye, E.; Coombes, R.C.; Hui, A.M.; Cohen, P.S. The biodistribution and radiation dosimetry of the Arg-Gly-Asp peptide 18F-AH111585 in healthy volunteers. J. Nucl. Med. 2008, 49, 1664–1667. [Google Scholar] [CrossRef] [PubMed]
- Doss, M.; Kolb, H.C.; Zhang, J.J.; Belanger, M.J.; Stubbs, J.B.; Stabin, M.G.; Hostetler, E.D.; Alpaugh, R.K.; von Mehren, M.; Walsh, J.C.; et al. Biodistribution and radiation dosimetry of the integrin marker 18F-RGD-K5 determined from whole-body PET/CT in monkeys and humans. J. Nucl. Med. 2012, 53, 787–795. [Google Scholar] [CrossRef]
- Mittra, E.S.; Goris, M.L.; Iagaru, A.H.; Kardan, A.; Burton, L.; Berganos, R.; Chang, E.; Liu, S.; Shen, B.; Chin, F.T.; et al. Pilot pharmacokinetic and dosimetric studies of 18F-FPPRGD2: A PET radiopharmaceutical agent for imaging αvβ3 integrin levels. Radiology 2011, 260, 182–191. [Google Scholar] [CrossRef]
- Zheng, K.; Liang, N.; Zhang, J.; Lang, L.; Zhang, W.; Li, S.; Zhao, J.; Niu, G.; Li, F.; Zhu, Z.; et al. 68Ga-NOTA-PRGD2 PET/CT for Integrin Imaging in Patients with Lung Cancer. J. Nucl. Med. 2015, 56, 1823–1827. [Google Scholar] [CrossRef]
- Kircher, M.; Herhaus, P.; Schottelius, M.; Buck, A.K.; Werner, R.A.; Wester, H.J.; Keller, U.; Lapa, C. CXCR4-directed theranostics in oncology and inflammation. Ann. Nucl. Med. 2018, 32, 503–511. [Google Scholar] [CrossRef]
- Zlotnik, A.; Burkhardt, A.M.; Homey, B. Homeostatic chemokine receptors and organ-specific metastasis. Nat. Rev. Immunol. 2011, 11, 597–606. [Google Scholar] [CrossRef]
- Domanska, U.M.; Kruizinga, R.C.; Nagengast, W.B.; Timmer-Bosscha, H.; Huls, G.; de Vries, E.G.; Walenkamp, A.M. A review on CXCR4/CXCL12 axis in oncology: No place to hide. Eur. J. Cancer 2013, 49, 219–230. [Google Scholar] [CrossRef]
- Scala, S. Molecular Pathways: Targeting the CXCR4-CXCL12 Axis - Untapped Potential in the Tumor Microenvironment. Clin. Cancer Res. 2015, 21, 4278–4285. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, M.; Wang, L.; Wang, S.; Kang, F.; Li, G.; Jacobson, O.; Niu, G.; Yang, W.; Wang, J.; et al. Prospective Study of 68Ga-NOTA-NFB: Radiation Dosimetry in Healthy Volunteers and First Application in Glioma Patients. Theranostics 2015, 5, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Vag, T.; Gerngross, C.; Herhaus, P.; Eiber, M.; Philipp-Abbrederis, K.; Graner, F.P.; Ettl, J.; Keller, U.; Wester, H.J.; Schwaiger, M. First Experience with Chemokine Receptor CXCR4-Targeted PET Imaging of Patients with Solid Cancers. J. Nucl. Med. 2016, 57, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Werner, R.A.; Kircher, S.; Higuchi, T.; Kircher, M.; Schirbel, A.; Wester, H.J.; Buck, A.K.; Pomper, M.G.; Rowe, S.P.; Lapa, C. CXCR4-Directed Imaging in Solid Tumors. Front. Oncol. 2019, 9, 770. [Google Scholar] [CrossRef]
- Philipp-Abbrederis, K.; Herrmann, K.; Knop, S.; Schottelius, M.; Eiber, M.; Luckerath, K.; Pietschmann, E.; Habringer, S.; Gerngross, C.; Franke, K.; et al. In vivo molecular imaging of chemokine receptor CXCR4 expression in patients with advanced multiple myeloma. EMBO Mol. Med. 2015, 7, 477–487. [Google Scholar] [CrossRef]
- Lapa, C.; Schreder, M.; Schirbel, A.; Samnick, S.; Kortum, K.M.; Herrmann, K.; Kropf, S.; Einsele, H.; Buck, A.K.; Wester, H.J.; et al. [68Ga]Pentixafor-PET/CT for imaging of chemokine receptor CXCR4 expression in multiple myeloma - Comparison to [18F]FDG and laboratory values. Theranostics 2017, 7, 205–212. [Google Scholar] [CrossRef]
- Herhaus, P.; Habringer, S.; Philipp-Abbrederis, K.; Vag, T.; Gerngross, C.; Schottelius, M.; Slotta-Huspenina, J.; Steiger, K.; Altmann, T.; Weisser, T.; et al. Targeted positron emission tomography imaging of CXCR4 expression in patients with acute myeloid leukemia. Haematologica 2016, 101, 932–940. [Google Scholar] [CrossRef]
- Mayerhoefer, M.E.; Jaeger, U.; Staber, P.; Raderer, M.; Wadsak, W.; Pfaff, S.; Kornauth, C.; Senn, D.; Weber, M.; Wester, H.J.; et al. [68Ga]Ga-Pentixafor PET/MRI for CXCR4 Imaging of Chronic Lymphocytic Leukemia: Preliminary Results. Invest. Radiol. 2018, 53, 403–408. [Google Scholar] [CrossRef]
- Schottelius, M.; Osl, T.; Poschenrieder, A.; Hoffmann, F.; Beykan, S.; Hanscheid, H.; Schirbel, A.; Buck, A.K.; Kropf, S.; Schwaiger, M.; et al. [177Lu]pentixather: Comprehensive Preclinical Characterization of a First CXCR4-directed Endoradiotherapeutic Agent. Theranostics 2017, 7, 2350–2362. [Google Scholar] [CrossRef]
- Lapa, C.; Herrmann, K.; Schirbel, A.; Hanscheid, H.; Luckerath, K.; Schottelius, M.; Kircher, M.; Werner, R.A.; Schreder, M.; Samnick, S.; et al. CXCR4-directed endoradiotherapy induces high response rates in extramedullary relapsed Multiple Myeloma. Theranostics 2017, 7, 1589–1597. [Google Scholar] [CrossRef]
- Lapa, C.; Hanscheid, H.; Kircher, M.; Schirbel, A.; Wunderlich, G.; Werner, R.A.; Samnick, S.; Kotzerke, J.; Einsele, H.; Buck, A.K.; et al. Feasibility of CXCR4-Directed Radioligand Therapy in Advanced Diffuse Large B-Cell Lymphoma. J. Nucl Med. 2019, 60, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Habringer, S.; Lapa, C.; Herhaus, P.; Schottelius, M.; Istvanffy, R.; Steiger, K.; Slotta-Huspenina, J.; Schirbel, A.; Hanscheid, H.; Kircher, S.; et al. Dual Targeting of Acute Leukemia and Supporting Niche by CXCR4-Directed Theranostics. Theranostics 2018, 8, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Wester, H.J.; Keller, U.; Schottelius, M.; Beer, A.; Philipp-Abbrederis, K.; Hoffmann, F.; Simecek, J.; Gerngross, C.; Lassmann, M.; Herrmann, K.; et al. Disclosing the CXCR4 expression in lymphoproliferative diseases by targeted molecular imaging. Theranostics 2015, 5, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Majkowska-Pilip, A.; Halik, P.K.; Gniazdowska, E. The Significance of NK1 Receptor Ligands and Their Application in Targeted Radionuclide Tumour Therapy. Pharmaceutics 2019, 11, 443. [Google Scholar] [CrossRef]
- Krolicki, L.; Bruchertseifer, F.; Kunikowska, J.; Koziara, H.; Krolicki, B.; Jakucinski, M.; Pawlak, D.; Apostolidis, C.; Mirzadeh, S.; Rola, R.; et al. Prolonged survival in secondary glioblastoma following local injection of targeted alpha therapy with 213Bi-substance P analogue. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1636–1644. [Google Scholar] [CrossRef]
- Raderer, M.; Kurtaran, A.; Leimer, M.; Angelberger, P.; Niederle, B.; Vierhapper, H.; Vorbeck, F.; Hejna, M.H.; Scheithauer, W.; Pidlich, J.; et al. Value of peptide receptor scintigraphy using 123I-vasoactive intestinal peptide and 111In-DTPA-D-Phe1-octreotide in 194 carcinoid patients: Vienna University Experience, 1993 to 1998. J. Clin. Oncol. 2000, 18, 1331–1336. [Google Scholar] [CrossRef]
- Tripathi, S.; Trabulsi, E.J.; Gomella, L.; Kim, S.; McCue, P.; Intenzo, C.; Birbe, R.; Gandhe, A.; Kumar, P.; Thakur, M. VPAC1 Targeted 64Cu-TP3805 Positron Emission Tomography Imaging of Prostate Cancer: Preliminary Evaluation in Man. Urology 2016, 88, 111–118. [Google Scholar] [CrossRef]
- Thakur, M.L.; Zhang, K.; Berger, A.; Cavanaugh, B.; Kim, S.; Channappa, C.; Frangos, A.J.; Wickstrom, E.; Intenzo, C.M. VPAC1 receptors for imaging breast cancer: A feasibility study. J. Nucl. Med. 2013, 54, 1019–1025. [Google Scholar] [CrossRef]
- Klingler, M.; Hörmann, A.A.; von Guggenberg, E. Cholecystokinin-2 receptor targeting with radiolabeled peptides: Current status and future directions. Curr. Med. Chem. 2020; Submitted. [Google Scholar]
- Kunikowska, J.; Ziemnicka, K.; Pawlak, D.; Ruchala, M.; Kolasa, A.; Janicka-Jedynska, M.; Wozniak, A.; Mikolajczak, R.; Krolicki, L. Medullary thyroid carcinoma - PET/CT imaging with 68Ga-labelled gastrin and somatostatin analogues. Endokrynol. Pol. 2016, 67, 68–71. [Google Scholar] [CrossRef]
- Behe, M.; Behr, T.M. Cholecystokinin-B (CCK-B)/gastrin receptor targeting peptides for staging and therapy of medullary thyroid cancer and other CCK-B receptor expressing malignancies. Biopolymers 2002, 66, 399–418. [Google Scholar] [CrossRef]
- Sauter, A.W.; Mansi, R.; Hassiepen, U.; Muller, L.; Panigada, T.; Wiehr, S.; Wild, A.M.; Geistlich, S.; Behe, M.; Rottenburger, C.; et al. Targeting of the Cholecystokinin-2 Receptor with the Minigastrin Analog 177Lu-DOTA-PP-F11N: Does the Use of Protease Inhibitors Further Improve In Vivo Distribution? J. Nucl. Med. 2019, 60, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Walters, J.; Pop, C.; Scott, F.L.; Drag, M.; Swartz, P.; Mattos, C.; Salvesen, G.S.; Clark, A.C. A constitutively active and uninhibitable caspase-3 zymogen efficiently induces apoptosis. Biochem. J. 2009, 424, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Chen, G.; Gangadharmath, U.; Gomez, L.F.; Liang, Q.; Mu, F.; Mocharla, V.P.; Szardenings, A.K.; Walsh, J.C.; Xia, C.F.; et al. Evaluation of [18F]-CP18 as a PET imaging tracer for apoptosis. Mol. Imaging Biol. 2013, 15, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Doss, M.; Kolb, H.C.; Walsh, J.C.; Mocharla, V.; Fan, H.; Chaudhary, A.; Zhu, Z.; Alpaugh, R.K.; Lango, M.N.; Yu, J.Q. Biodistribution and radiation dosimetry of 18F-CP-18, a potential apoptosis imaging agent, as determined from PET/CT scans in healthy volunteers. J. Nucl. Med. 2013, 54, 2087–2092. [Google Scholar] [CrossRef]
- Ferreira, C.A.; Fuscaldi, L.L.; Townsend, D.M.; Rubello, D.; Barros, A.L.B. Radiolabeled bombesin derivatives for preclinical oncological imaging. Biomed. Pharm. 2017, 87, 58–72. [Google Scholar] [CrossRef]
- Baidoo, K.E.; Lin, K.S.; Zhan, Y.; Finley, P.; Scheffel, U.; Wagner, H.N., Jr. Design, synthesis, and initial evaluation of high-affinity technetium bombesin analogues. Bioconjug. Chem. 1998, 9, 218–225. [Google Scholar] [CrossRef]
- Dimitrakopoulou-Strauss, A.; Seiz, M.; Tuettenberg, J.; Schmieder, K.; Eisenhut, M.; Haberkorn, U.; Strauss, L.G. Pharmacokinetic studies of 68Ga-labeled Bombesin (68Ga-BZH3) and F-18 FDG PET in patients with recurrent gliomas and comparison to grading: Preliminary results. Clin. Nucl. Med. 2011, 36, 101–108. [Google Scholar] [CrossRef]
- Wieser, G.; Mansi, R.; Grosu, A.L.; Schultze-Seemann, W.; Dumont-Walter, R.A.; Meyer, P.T.; Maecke, H.R.; Reubi, J.C.; Weber, W.A. Positron emission tomography (PET) imaging of prostate cancer with a gastrin releasing peptide receptor antagonist - from mice to men. Theranostics 2014, 4, 412–419. [Google Scholar] [CrossRef]
- Maina, T.; Bergsma, H.; Kulkarni, H.R.; Mueller, D.; Charalambidis, D.; Krenning, E.P.; Nock, B.A.; de Jong, M.; Baum, R.P. Preclinical and first clinical experience with the gastrin-releasing peptide receptor-antagonist [68Ga]SB3 and PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 964–973. [Google Scholar] [CrossRef]
- Kahkonen, E.; Jambor, I.; Kemppainen, J.; Lehtio, K.; Gronroos, T.J.; Kuisma, A.; Luoto, P.; Sipila, H.J.; Tolvanen, T.; Alanen, K.; et al. In vivo imaging of prostate cancer using [68Ga]-labeled bombesin analog BAY86-7548. Clin. Cancer Res. 2013, 19, 5434–5443. [Google Scholar] [CrossRef]
- Stoykow, C.; Erbes, T.; Maecke, H.R.; Bulla, S.; Bartholoma, M.; Mayer, S.; Drendel, V.; Bronsert, P.; Werner, M.; Gitsch, G.; et al. Gastrin-releasing Peptide Receptor Imaging in Breast Cancer Using the Receptor Antagonist 68Ga-RM2 And PET. Theranostics 2016, 6, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Minamimoto, R.; Hancock, S.; Schneider, B.; Chin, F.T.; Jamali, M.; Loening, A.; Vasanawala, S.; Gambhir, S.S.; Iagaru, A. Pilot Comparison of 68Ga-RM2 PET and 68Ga-PSMA-11 PET in Patients with Biochemically Recurrent Prostate Cancer. J. Nucl. Med. 2016, 57, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Ploug, M.; Behrendt, N.; Lober, D.; Dano, K. Protein structure and membrane anchorage of the cellular receptor for urokinase-type plasminogen activator. Semin. Thromb. Hemost. 1991, 17, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Persson, M.; Skovgaard, D.; Brandt-Larsen, M.; Christensen, C.; Madsen, J.; Nielsen, C.H.; Thurison, T.; Klausen, T.L.; Holm, S.; Loft, A.; et al. First-in-human uPAR PET: Imaging of Cancer Aggressiveness. Theranostics 2015, 5, 1303–1316. [Google Scholar] [CrossRef]
- Skovgaard, D.; Persson, M.; Kjaer, A. Urokinase Plasminogen Activator Receptor-PET with 68Ga-NOTA-AE105: First Clinical Experience with a Novel PET Ligand. PET Clin. 2017, 12, 311–319. [Google Scholar] [CrossRef]
- Korner, M.; Stockli, M.; Waser, B.; Reubi, J.C. GLP-1 receptor expression in human tumors and human normal tissues: Potential for in vivo targeting. J. Nucl. Med. 2007, 48, 736–743. [Google Scholar] [CrossRef]
- Gotthardt, M.; Fischer, M.; Naeher, I.; Holz, J.B.; Jungclas, H.; Fritsch, H.W.; Behe, M.; Goke, B.; Joseph, K.; Behr, T.M. Use of the incretin hormone glucagon-like peptide-1 (GLP-1) for the detection of insulinomas: Initial experimental results. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 597–606. [Google Scholar] [CrossRef]
- Hubalewska-Dydejczyk, A.; Sowa-Staszczak, A.; Tomaszuk, M.; Stefanska, A. GLP-1 and exendin-4 for imaging endocrine pancreas. A review. Labelled glucagon-like peptide-1 analogues: Past, present and future. Q. J. Nucl. Med. Mol. Imaging 2015, 59, 152–160. [Google Scholar]
- Antwi, K.; Fani, M.; Nicolas, G.; Rottenburger, C.; Heye, T.; Reubi, J.C.; Gloor, B.; Christ, E.; Wild, D. Localization of Hidden Insulinomas with 68Ga-DOTA-Exendin-4 PET/CT: A Pilot Study. J. Nucl. Med. 2015, 56, 1075–1078. [Google Scholar] [CrossRef] [PubMed]
- Antwi, K.; Fani, M.; Heye, T.; Nicolas, G.; Rottenburger, C.; Kaul, F.; Merkle, E.; Zech, C.J.; Boll, D.; Vogt, D.R.; et al. Comparison of glucagon-like peptide-1 receptor (GLP-1R) PET/CT, SPECT/CT and 3T MRI for the localisation of occult insulinomas: Evaluation of diagnostic accuracy in a prospective crossover imaging study. Eur J. Nucl Med. Mol. Imaging 2018, 45, 2318–2327. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Pan, Q.; Yao, S.; Yu, M.; Wu, W.; Xue, H.; Kiesewetter, D.O.; Zhu, Z.; Li, F.; Zhao, Y.; et al. Glucagon-Like Peptide-1 Receptor PET/CT with 68Ga-NOTA-Exendin-4 for Detecting Localized Insulinoma: A Prospective Cohort Study. J. Nucl. Med. 2016, 57, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Boss, M.; Buitinga, M.; Jansen, T.J.; Brom, M.; Visser, E.P.; Gotthardt, M. PET-based dosimetry of [68Ga]Ga-NODAGA-exendin-4 in humans, a tracer for beta cell imaging. J. Nucl. Med. 2019. [Google Scholar] [CrossRef]
- Kratochwil, C.; Flechsig, P.; Lindner, T.; Abderrahim, L.; Altmann, A.; Mier, W.; Adeberg, S.; Rathke, H.; Rohrich, M.; Winter, H.; et al. 68Ga-FAPI PET/CT: Tracer Uptake in 28 Different Kinds of Cancer. J. Nucl. Med. 2019, 60, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Giesel, F.L.; Kratochwil, C.; Lindner, T.; Marschalek, M.M.; Loktev, A.; Lehnert, W.; Debus, J.; Jager, D.; Flechsig, P.; Altmann, A.; et al. 68Ga-FAPI PET/CT: Biodistribution and Preliminary Dosimetry Estimate of 2 DOTA-Containing FAP-Targeting Agents in Patients with Various Cancers. J. Nucl. Med. 2019, 60, 386–392. [Google Scholar] [CrossRef]
- Lindner, T.; Loktev, A.; Altmann, A.; Giesel, F.; Kratochwil, C.; Debus, J.; Jager, D.; Mier, W.; Haberkorn, U. Development of Quinoline-Based Theranostic Ligands for the Targeting of Fibroblast Activation Protein. J. Nucl. Med. 2018, 59, 1415–1422. [Google Scholar] [CrossRef]





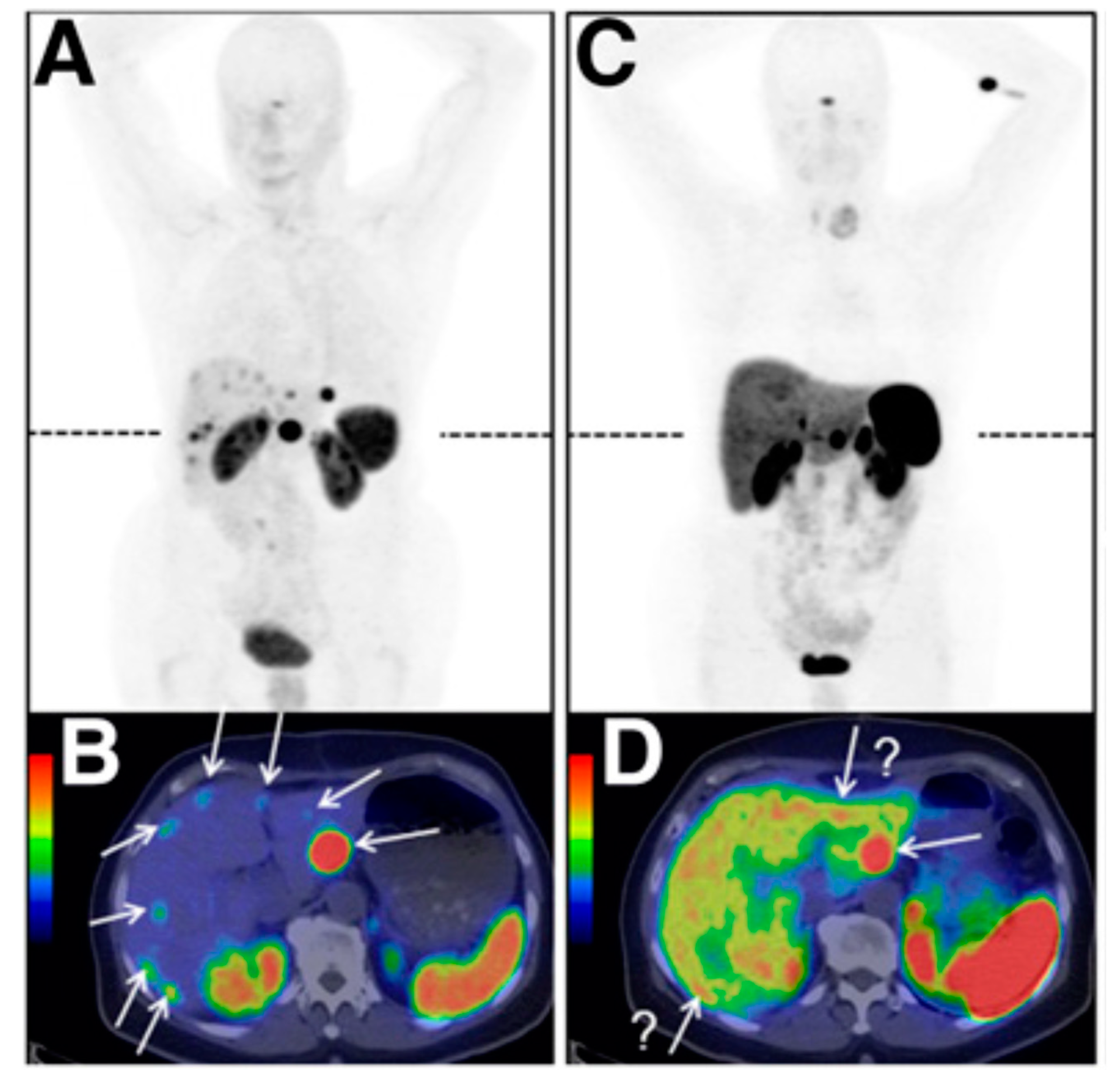{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isotope | Half-Life [h] | Decay Mode | Energy (keV) |
|---|---|---|---|
| Halogens | |||
| [18F] | 1.82 | β+ (96.9%) EC a (3.1%) | β+: 633.9 |
| [76Br] | 16.2 | β+ (54%) EC a (46%) | β+: 4963 |
| [124I] | 4.176 d | β+ (23%) EC a (77%) | β++EC a: 3160 |
| [131I] | 8.0233 d | β− (100%) | β−: 970.8 |
| [211At] | 7.22 | α (41.8%) EC a (58.2%) | α: 4000–8500 |
| Radiometals | |||
| [64Cu] | 12.7 | β+ (17.8%) β- (38.4%) EC a (43.5%) | β+: 653 β−: 579 |
| [68Ga] | 1.1 | β+ (88.9%) EC a (11.1%) | β+: 1899.1 |
| [89Zr] | 78.42 | β+ (23%) EC a (77%) | β+: 902 |
| [90Y] | 64.1 | β− (100%) | β−: 2279 |
| [177Lu] | 159.4 | β− (100%) | β−: 498 |
| [225Ac] | 240 | α (100%) | α: 5800–8400 |
| Isotope | Chelator | Labelling Conditions | Reference |
|---|---|---|---|
| [64Cu] | CB-TE2A a | 95 °C, 1 h | [113] |
| CB-TE2P | RT | [80] | |
| DOTA b | RT, 15 min | [114] | |
| NOTA c | RT, 45 min | [115] | |
| PCB-TE2A | RT | [75] | |
| TETA d | RT, 45 min | [74] | |
| Sarcophagine-based chelators | RT, within min | [75,82] | |
| [68Ga] | AAZTA e | RT, 10 min | [116] |
| DFO f | RT, 5 min | [116] | |
| DOTA b | 95 °C, 5 min | [116] | |
| DTPA g | RT, 10 min | [117] | |
| HBED-CC h | 80–100 °C, 5–20 min | [85] | |
| NODAGA i | RT, 10 min | [86] | |
| NOTA c | RT, 10 min | [118] | |
| TRAPj/NOPO k | 95–100 °C, 5 min; RT or 95 °C, 5 min | [87,89] | |
| [89Zr] | DFO f | RT, 30 min | [119] |
| DOTA b | 95 °C, 60 min | [97] | |
| FSC l | RT, 90 min | [120] | |
| [90Y] | 3p-C-NETA-NCS m | RT, 60 min | [106] |
| CHX-A’’-DTPA n | 37–75 °C, 30–60 min | [82] | |
| DOTA b | 80 °C, 20 min | [121] | |
| [177Lu] | 3p-C-NETA-NCS m | RT, 60 min | [106] |
| AAZTA-5 | RT, 10 min | [104] | |
| DOTA b | 80 °C, 20 min | [121] | |
| H4octapa o | RT, 15 min | [105] | |
| [224Ac] | DOTA b | 95 °C, 5 min | [109] |
| H2macropa p | RT, within several min | [112] | |
| HEHA q | 37 °C, 30 min | [122] |
| Peptide/Chelator | Isotope | Target Receptor | Tumour Types | Reference |
|---|---|---|---|---|
| AlF-NOTA-octreotide | [18F] | SSTR a | NET b | [48] |
| DOTA-JR11 (OPS201) | [177Lu] | SSTR a (antagonist) | NET b | [145] |
| DOTANOC | [68Ga] | SSTR-2/3/5 a | NET b | [127] |
| DOTATATE | [68Ga] | SSTR-2 a | NET b | [125] |
| [177Lu]/[90Y] | [143] | |||
| [64Cu] | [132] | |||
| [225Ac] | [144] | |||
| DOTATOC | [68Ga] | SSTR-2/5 a | NET b | [1] |
| [90Y] | [143] | |||
| FET-βAG-TOCA | [18F] | SSTR a | NET b | [136] |
| Gluc-Lys-[18F]FP-TOCA | [18F] | SSTR a | NET b | [134] |
| NODAGA-JR11 (OPS202) | [68Ga] | SSTR-2 a (antagonist) | NET b | [142] |
| SiFAlin-TATE | [18F] | SSTR a | NET b | [139] |
| TETA-octreotide | [64Cu] | SSTR-2 a | NET b | [131] |
| CTT1057 | [18F] | PSMA c | prostate cancer | [166] |
| DCFPyL | [18F] | PSMA c | prostate cancer | [164] |
| EB-PSMA-617 | [177Lu] | PSMA c | prostate cancer | [178] |
| HBED-CC-PSMA (PSMA 11) | [68Ga] | PSMA c | prostate cancer | [85] |
| [18F] | [165,174] | |||
| JK-PSMA-7 | [18F] | PSMA c | prostate cancer | [167] |
| PSMA-I&T | [68Ga] | PSMA c | prostate cancer | [158] |
| [177Lu] | [176] | |||
| PSMA 617 | [177Lu] | PSMA c | prostate cancer | [151] |
| [213Bi] | [181] | |||
| [225Ac] | [182] | |||
| P16-093 | [68Ga] | PSMA c | prostate cancer | [162] |
| PSMA-1007 | [18F] | PSMA c | prostate cancer | [152] |
| THP-PSMA | [68Ga] | PSMA c | prostate cancer | [161] |
| Alfatide | [18F] | integrin αvβ3 | tumour angiogenesis | [197] |
| Alfatide II | [18F] | integrin αvβ3 | tumour angiogenesis | [46] |
| Fluciclatide | [18F] | integrin αvβ3 | tumour angiogenesis | [194] |
| FPPRGD2 | [18F] | integrin αvβ3 | tumour angiogenesis | [196] |
| Galacto-RGD | [18F] | integrin αvβ3 | tumour angiogenesis | [11,188] |
| NODAGA-RGD | [68Ga] | integrin αvβ3 | tumour angiogenesis | [5] |
| NOTA-RGD | [68Ga] | integrin αvβ3 | tumour angiogenesis | [118] |
| NOTA-PRGD2 | [68Ga] | integrin αvβ3 | tumour angiogenesis | [197] |
| RGD-5K | [18F] | integrin αvβ3 | tumour angiogenesis | [193] |
| NOTA-NFB | [68Ga] | chemokine receptor-4 | glioblastoma | [215] |
| Pentixafor | [68Ga] | chemokine receptor-4 | glioblastoma | [211] |
| Pentixather | [177Lu] [90Y] | chemokine receptor-4 | multiple myeloma, diffuse large B cell lymphoma, acute myeloid leukemia | [222] |
| TP3805 | [64Cu] | VPAC1 d | prostate cancer | [230] |
| DOTA-MG0 | [68Ga] | CCK2-R e | MTC f | [233] |
| DTPA-MG0 | [90Y] | CCK2-R e | MTC f | [234] |
| DOTA-PP-F11N | [177Lu] | CCK2-R e | MTC f | [235] |
| CP-18 | [18F] | caspases | apoptosis | [237,238] |
| CB-TE2A-AR06 | [64Cu] | bombesin | prostate cancer | [242] |
| BAY86-7548/RM2 | [68Ga] | bombesin | prostate/breast cancer | [244,245] |
| BZH3 | [68Ga] | bombesin | glioma | [241] |
| SB3 | [68Ga] | bombesin | prostate/breast cancer | [243] |
| DOTA-AE105 | [64Cu] | uPAR g | breast, prostate, bladder cancer | [248] |
| NOTA-AE105 | [68Ga] | uPAR g | breast, prostate, bladder cancer | [249] |
| DOTA-exendin-4 | [68Ga] | GLP-1 h | insulinoma | [253] |
| NOTA-exendin-4 | [68Ga] | GLP-1 h | insulinoma | [255] |
| NODAGA-exendin-4 | [68Ga] | GLP-1 h | Hyperinsulinemic, hypoglycemia | [256] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rangger, C.; Haubner, R. Radiolabelled Peptides for Positron Emission Tomography and Endoradiotherapy in Oncology. Pharmaceuticals 2020, 13, 22. https://doi.org/10.3390/ph13020022
Rangger C, Haubner R. Radiolabelled Peptides for Positron Emission Tomography and Endoradiotherapy in Oncology. Pharmaceuticals. 2020; 13(2):22. https://doi.org/10.3390/ph13020022
Chicago/Turabian StyleRangger, Christine, and Roland Haubner. 2020. "Radiolabelled Peptides for Positron Emission Tomography and Endoradiotherapy in Oncology" Pharmaceuticals 13, no. 2: 22. https://doi.org/10.3390/ph13020022
APA StyleRangger, C., & Haubner, R. (2020). Radiolabelled Peptides for Positron Emission Tomography and Endoradiotherapy in Oncology. Pharmaceuticals, 13(2), 22. https://doi.org/10.3390/ph13020022