CDK4/6 and MAPK—Crosstalk as Opportunity for Cancer Treatment


Abstract
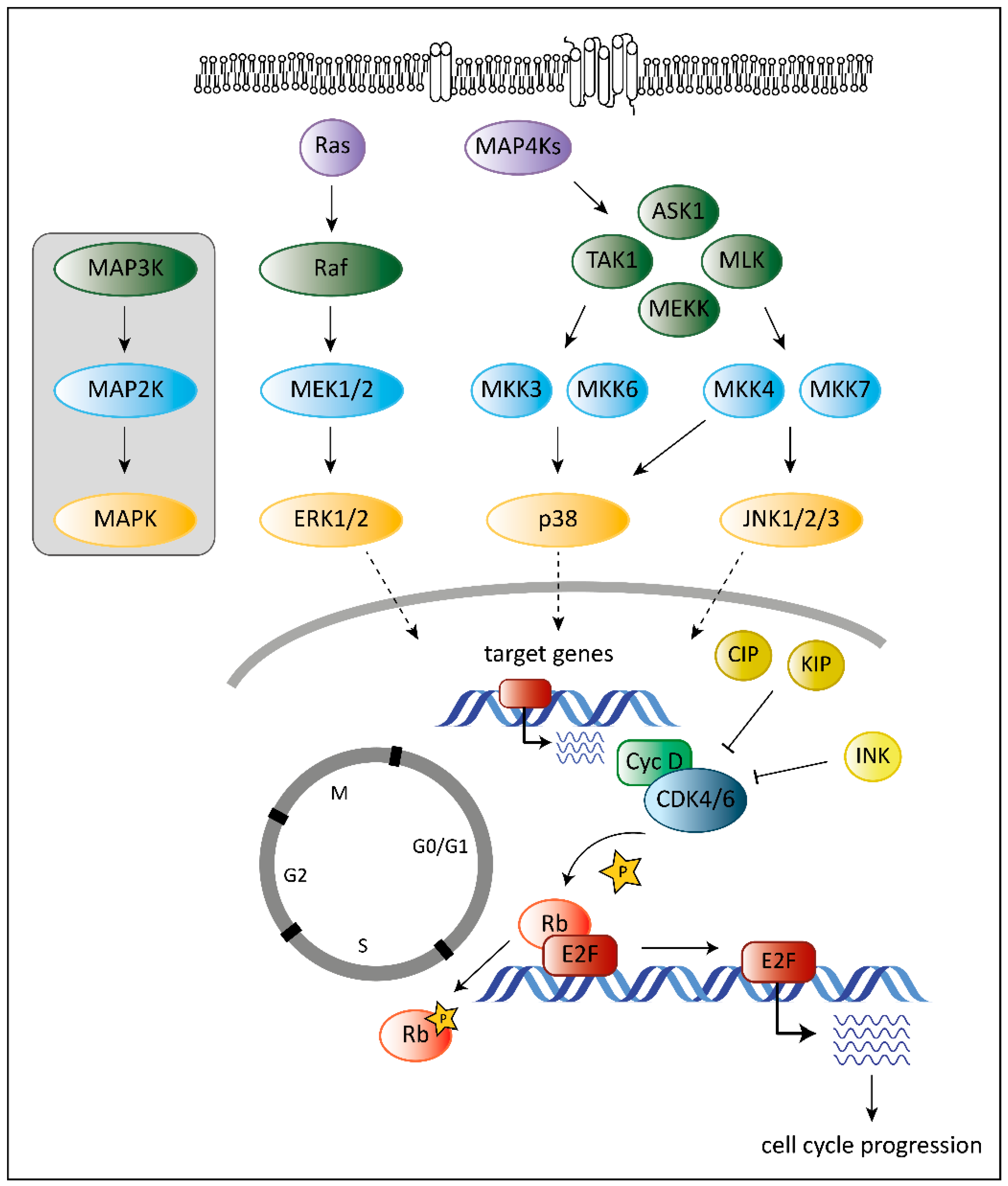
1. Introduction
2. The Role of CDK6 in Cancer
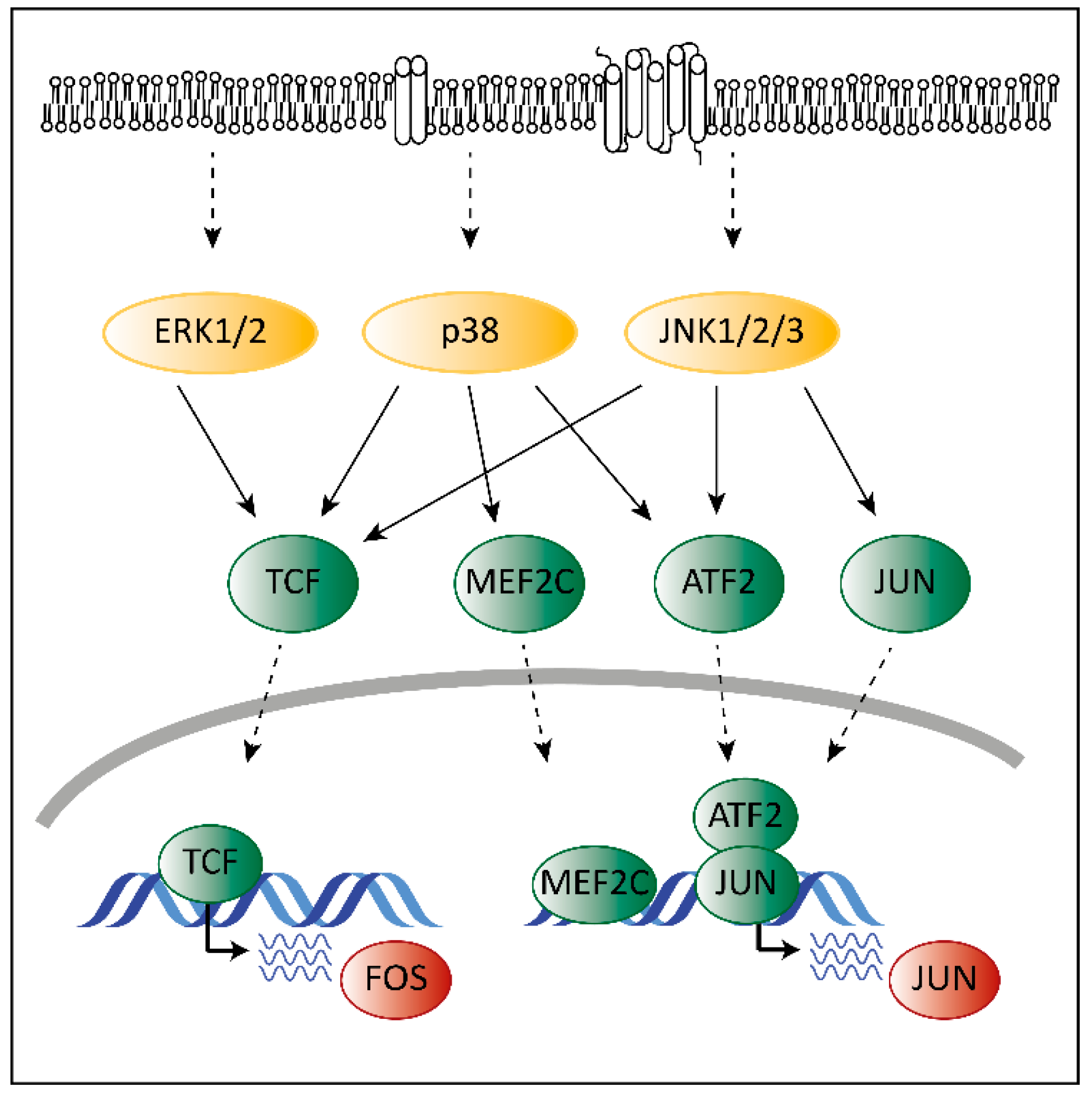
3. The Role of MAPKs in Cancer
4. The Unique Dual Role of p38
5. CDK6 as a Target in Cancer Therapy
6. Resistance to CDK4/6 Inhibitors
7. MAPK as Target in Cancer Therapy
8. Combinatorial Treatment with CDK4/6 and MAPK Inhibitors
9. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef]
- Lavoie, J.N.; L’Allemain, G.; Brunei, A.; Müller, R.; Pouysségur, J. Cyclin D1 expression is regulated positively by the p42/p44(MAPK) and negatively by the p38/HOG(MAPK) pathway. J. Biol. Chem. 1996, 271, 20608–20616. [Google Scholar] [CrossRef] [PubMed]
- Meyerson, M.; Harlow, E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol. Cell. Biol. 1994, 14, 2077–2086. [Google Scholar] [CrossRef] [PubMed]
- Tigan, A.S.; Bellutti, F.; Kollmann, K.; Tebb, G.; Sexl, V. CDK6-a review of the past and a glimpse into the future: From cell-cycle control to transcriptional regulation. Oncogene 2016, 35, 3083–3091. [Google Scholar] [CrossRef] [PubMed]
- Classon, M.; Harlow, E. The retinoblastoma tumour suppressor in development and cancer. Nat. Rev. Cancer 2002, 2, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Roovers, K.; Assoian, R.K. Integrating the MAP kinase signal into the G1 phase cell cycle machinery. BioEssays 2000, 22, 818–826. [Google Scholar] [CrossRef]
- Sewing, A.; Wiseman, B.; Lloyd, A.C.; Land, H. High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1. Mol. Cell. Biol. 1997, 17, 5588–5597. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.Y.; Mercer, S.E.; Ewton, D.Z.; Yan, Z.; Jin, K.; Friedman, E. The stress-activated protein kinases p38 alpha and JNK1 stabilize p21(Cip1) by phosphorylation. J. Biol. Chem. 2002, 277, 29792–29802. [Google Scholar] [CrossRef]
- Yan, Z.; Kim, G.Y.; Deng, X.; Friedman, E. Transforming growth factor β1 induces proliferation in colon carcinoma cells by ras-dependent, smad-independent down-regulation of p21cip1. J. Biol. Chem. 2002, 277, 9870–9879. [Google Scholar] [CrossRef]
- Bulavin, D.V.; Higashimoto, Y.; Popoff, I.J.; Gaarde, W.A.; Basrur, V.; Potapova, O.; Appella, E.; Fornace, A.J. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 2001, 411, 102–107. [Google Scholar] [CrossRef]
- Munshi, A.; Ramesh, R. Mitogen-Activated Protein Kinases and Their Role in Radiation Response. Genes Cancer 2013, 4, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Manzano, J.L.; Layos, L.; Bugés, C.; De los Llanos Gil, M.; Vila, L.; Martínez-Balibrea, E.; Martínez-Cardús, A. Resistant mechanisms to BRAF inhibitors in melanoma. Ann. Transl. Med. 2016, 4, 237. [Google Scholar] [CrossRef]
- Khatib, Z.A.; Matsushime, H.; Valentine, M.; Shapiro, D.N.; Sherr, C.J.; Look, A.T. Coamplification of the CDK4 gene with MDM2 and GLI in human sarcomas. Cancer Res. 1993, 53, 5535–5541. [Google Scholar] [PubMed]
- Schmidt, E.E.; Ichimura, K.; Reifenberger, G.; Collins, V.P. CDKN2 (p16/MTS1) gene deletion or CDK4 amplification occurs in the majority of glioblastomas. Cancer Res. 1994, 54, 6321–6324. [Google Scholar]
- Wölfel, T.; Hauer, M.; Schneider, J.; Serrano, M.; Wölfel, C.; Klehmann-Hieb, E.; De Plaen, E.; Hankeln, T.; Meyer zum Büschenfelde, K.H.; Beach, D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 1995, 269, 1281–1284. [Google Scholar] [CrossRef]
- Chilosi, M.; Doglioni, C.; Yan, Z.; Lestani, M.; Menestrina, F.; Sorio, C.; Benedetti, A.; Vinante, F.; Pizzolo, G.; Inghirami, G. Differential expression of cyclin-dependent kinase 6 in cortical thymocytes and T-cell lymphoblastic lymphoma/leukemia. Am. J. Pathol. 1998, 152, 209–217. [Google Scholar] [PubMed]
- Nagel, S.; Leich, E.; Quentmeier, H.; Meyer, C.; Kaufmann, M.; Drexler, H.G.; Zettl, A.; Rosenwald, A.; MacLeod, R.A.F. Amplification at 7q22 targets cyclin-dependent kinase 6 in T-cell lymphoma. Leukemia 2008, 22, 387–392. [Google Scholar] [CrossRef]
- Kollmann, K.; Heller, G.; Schneckenleithner, C.; Warsch, W.; Scheicher, R.; Ott, R.G.; Schäfer, M.; Fajmann, S.; Schlederer, M.; Schiefer, A.-I.; et al. A Kinase-Independent Function of CDK6 Links the Cell Cycle to Tumor Angiogenesis. Cancer Cell 2013, 24, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef]
- Kollmann, K.; Sexl, V. CDK6 and p16INK4A in lymphoid malignancies. Oncotarget 2013, 4, 1858–1859. [Google Scholar] [CrossRef]
- Kollmann, K.; Briand, C.; Bellutti, F.; Schicher, N.; Blunder, S.; Zojer, M.; Hoeller, C.; Kollmann, K.; Briand, C.; Bellutti, F.; et al. The interplay of CDK4 and CDK6 in melanoma. Oncotarget 2019, 10, 1346–1359. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Sicinska, E.; Geng, Y.; Ahnström, M.; Zagozdzon, A.; Kong, Y.; Gardner, H.; Kiyokawa, H.; Harris, L.N.; Stål, O.; et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell 2006, 9, 23–32. [Google Scholar] [CrossRef]
- Bartkova, J.; Lukas, J.; Müller, H.; Lützhøt, D.; Strauss, M.; Bartek, J. Cyclin D1 protein expression and function in human breast cancer. Int. J. Cancer 1994, 57, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Dickson, C.; Fantl, V.; Gillett, C.; Brookes, S.; Bartek, J.; Smith, R.; Fisher, C.; Barnes, D.; Peters, G. Amplification of chromosome band 11q13 and a role for cyclin D1 in human breast cancer. Cancer Lett. 1995, 90, 43–50. [Google Scholar] [CrossRef]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Foster, J.S.; Wimalasena, J. Estrogen regulates activity of cyclin-dependent kinases and retinoblastoma protein phosphorylation in breast cancer cells. Mol. Endocrinol. 1996, 10, 488–498. [Google Scholar] [PubMed]
- Lim, J.T.E.; Mansukhani, M.; Weinstein, I.B. Cyclin-dependent kinase 6 associates with the androgen receptor and enhances its transcriptional activity in prostate cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5156–5161. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Anderson, K.; Jacobsen, S.E.W.; Nishikawa, S.; Nerlov, C. Cdk6 blocks myeloid differentiation by interfering with Runx1 DNA binding and Runx1-C/EBPα interaction. Embo J. 2007, 26, 2361–2370. [Google Scholar] [CrossRef]
- Placke, T.; Faber, K.; Nonami, A.; Putwain, S.L.; Salih, H.R.; Heidel, F.H.; Krämer, A.; Root, D.E.; Barbie, D.A.; Krivtsov, A.V.; et al. Requirement for CDK6 in MLL-rearranged acute myeloid leukemia. Blood 2014, 124, 13–23. [Google Scholar] [CrossRef]
- Sebolt-Leopold, J.S.; Herrera, R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat. Rev. Cancer 2004, 4, 937–947. [Google Scholar] [CrossRef]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Map kinase signaling pathways and hematologic malignancies. Blood 2003, 101, 4667–4679. [Google Scholar] [CrossRef] [PubMed]
- Towatari, M.; Iida, H.; Tanimoto, M.; Iwata, H.; Hamaguchi, M.; Saito, H. Constitutive activation of mitogen-activated protein kinase pathway in acute leukemia cells. Leukemia 1997, 11, 479–484. [Google Scholar] [CrossRef]
- Yu, C.; Krystal, G.; Varticovksi, L.; McKinstry, R.; Rahmani, M.; Dent, P.; Grant, S. Pharmacologic mitogen-activated protein/extracellular signal-regulated kinase kinase/mitogen-activated protein kinase inhibitors interact synergistically with STI571 to induce apoptosis in Bcr/Abl-expressing human leukemia cells. Cancer Res. 2002, 62, 188–199. [Google Scholar]
- Whitmarsh, A.J.; Davis, R.J. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med. 1996, 74, 589–607. [Google Scholar] [CrossRef]
- Raingeaud, J.; Gupta, S.; Rogers, J.S.; Dickens, M.; Han, J.; Ulevitch, R.J.; Davis, R.J. Pro-inflammatory Cytokines and Environmental Stress Cause p38 Mitogen-Activated Protein Kinase Activation by Dual Phosphorylation on Tyrosine and Threonine. J. Biol. Chem. 1995, 270, 7420–7426. [Google Scholar] [CrossRef]
- Van Dam, H.; Wilhelm, D.; Herr, I.; Steffen, A.; Herrlich, P.; Angel, P. ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents. Embo J. 1995, 14, 1798–1811. [Google Scholar] [CrossRef]
- Kollmann, K.; Heller, G.; Sexl, V. C-JUN prevents methylation of p16INK4a (and Cdk6): The villain turned bodyguard. Oncotarget 2011, 2, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Igea, A.; Nebreda, A.R. The stress kinase p38α as a target for cancer therapy. Cancer Res. 2015, 75, 3997–4002. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Sun, P. The pathways to tumor suppression via route p38. Trends Biochem. Sci. 2007, 32, 364–371. [Google Scholar] [CrossRef]
- Lee, A.C.; Fenster, B.E.; Ito, H.; Takeda, K.; Bae, N.S.; Hirai, T.; Yu, Z.-X.; Ferrans, V.J.; Howard, B.H.; Finkel, T. Ras Proteins Induce Senescence by Altering the Intracellular Levels of Reactive Oxygen Species. J. Biol. Chem. 1999, 274, 7936–7940. [Google Scholar] [CrossRef]
- Nicke, B.; Bastien, J.; Khanna, S.J.; Warne, P.H.; Cowling, V.; Cook, S.J.; Peters, G.; Delpuech, O.; Schulze, A.; Berns, K.; et al. Involvement of MINK, a ste20 family kinase, in ras oncogene-induced growth arrest in human ovarian surface epithelial cells. Mol. Cell 2005, 20, 673–685. [Google Scholar] [CrossRef]
- Dolado, I.; Swat, A.; Ajenjo, N.; De Vita, G.; Cuadrado, A.; Nebreda, A.R. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell 2007, 11, 191–205. [Google Scholar] [CrossRef]
- Iwasa, H.; Han, J.; Ishikawa, F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells 2003, 8, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, J.X.; Liao, R.; Deng, Q.; Zhou, J.J.; Huang, S.; Sun, P. Sequential Activation of the MEK–Extracellular Signal-Regulated Kinase and MKK3/6–p38 Mitogen-Activated Protein Kinase Pathways Mediates Oncogenic ras-Induced Premature Senescence. Mol. Cell. Biol. 2002, 22, 3389–3403. [Google Scholar] [CrossRef]
- Bulavin, D.V.; Phillips, C.; Nannenga, B.; Timofeev, O.; Donehower, L.A.; Anderson, C.W.; Appella, E.; Fornace, A.J. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16Ink4a-p19 Arf pathway. Nat. Genet. 2004, 36, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Esteva, F.J.; Sahin, A.A.; Smith, T.L.; Yang, Y.; Pusztai, L.; Nahta, R.; Buchholz, T.A.; Buzdar, A.U.; Hortobagyi, G.N.; Bacus, S.S. Prognostic Significance of Phosphorylated P38 Mitogen-Activated Protein Kinase and HER-2 Expression in Lymph Node-Positive Breast Carcinoma. Cancer 2004, 100, 499–506. [Google Scholar] [CrossRef]
- Benhar, M.; Dalyot, I.; Engelberg, D.; Levitzki, A. Enhanced ROS Production in Oncogenically Transformed Cells Potentiates c-Jun N-Terminal Kinase and p38 Mitogen-Activated Protein Kinase Activation and Sensitization to Genotoxic Stress. Mol. Cell. Biol. 2001, 21, 6913–6926. [Google Scholar] [CrossRef] [PubMed]
- Engelberg, D. Stress-activated protein kinases—Tumor suppressors or tumor initiators? Semin. Cancer Biol. 2004, 14, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Bragado, P.; Armesilla, A.; Silva, A.; Porras, A. Apoptosis by cisplatin requires p53 mediated p38α MAPK activation through ROS generation. Apoptosis 2007, 12, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Parmar, S.; Katsoulidis, E.; Verma, A.; Li, Y.; Sassano, A.; Lal, L.; Majchrzak, B.; Ravandi, F.; Tallman, M.S.; Fish, E.N.; et al. Role of the p38 mitogen-activated protein kinase pathway in the generation of the effects of imatinib mesylate (STI571) in BCR-ABL-expressing cells. J. Biol. Chem. 2004, 279, 25345–25352. [Google Scholar] [CrossRef] [PubMed]
- Giafis, N.; Katsoulidis, E.; Sassano, A.; Tallman, M.S.; Higgins, L.S.; Nebreda, A.R.; Davis, R.J.; Platanias, L.C. Role of the p38 mitogen-activated protein kinase pathway in the generation of arsenic trioxide-dependent cellular responses. Cancer Res. 2006, 66, 6763–6771. [Google Scholar] [CrossRef]
- Cánovas, B.; Igea, A.; Sartori, A.A.; Gomis, R.R.; Paull, T.T.; Isoda, M.; Pérez-Montoyo, H.; Serra, V.; González-Suárez, E.; Stracker, T.H.; et al. Targeting p38α Increases DNA Damage, Chromosome Instability, and the Anti-tumoral Response to Taxanes in Breast Cancer Cells. Cancer Cell 2018, 33, 1094–1110.e8. [Google Scholar]
- Paillas, S.; Boissière, F.; Bibeau, F.; Denouel, A.; Mollevi, C.; Causse, A.; Denis, V.; Vezzio-Vié, N.; Marzi, L.; Cortijo, C.; et al. Targeting the p38 MAPK pathway inhibits irinotecan resistance in colon adenocarcinoma. Cancer Res. 2011, 71, 1041–1049. [Google Scholar] [CrossRef]
- Sánchez-Martínez, C.; Gelbert, L.M.; Lallena, M.J.; De Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs. Bioorganic Med. Chem. Lett. 2015, 25, 3420–3435. [Google Scholar] [CrossRef]
- Malínková, V.; Vylíčil, J.; Kryštof, V. Cyclin-dependent kinase inhibitors for cancer therapy: A patent review (2009–2014). Expert Opin. Pat. 2015, 25, 953–970. [Google Scholar] [CrossRef]
- Toogood, P.L.; Harvey, P.J.; Repine, J.T.; Sheehan, D.J.; VanderWel, S.N.; Zhou, H.; Keller, P.R.; McNamara, D.J.; Sherry, D.; Zhu, T.; et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J. Med. Chem. 2005, 48, 2388–2406. [Google Scholar] [CrossRef]
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; del Prado, M.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Wishart, G.N.; Flack, R.S.; et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: In-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investig. New Drugs 2014, 32, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Loo, A.; Chopra, R.; Caponigro, G.; Huang, A.; Vora, S.; Parasuraman, S.; Howard, S.; Keen, N.; Sellers, W.; et al. LEE011: An orally bioavailable, selective small molecule inhibitor of CDK4/6– Reactivating Rb in cancer. Mol. Cancer Ther. 2013, 12, 2528. [Google Scholar]
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol. 2018, 28, 911–925. [Google Scholar] [CrossRef]
- Gong, X.; Litchfield, L.M.; Webster, Y.; Chio, L.C.; Wong, S.S.; Stewart, T.R.; Dowless, M.; Dempsey, J.; Zeng, Y.; Torres, R.; et al. Genomic Aberrations that Activate D-type Cyclins Are Associated with Enhanced Sensitivity to the CDK4 and CDK6 Inhibitor Abemaciclib. Cancer Cell 2017, 32, 761–776.e6. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2014, 16, 25–35. [Google Scholar] [CrossRef]
- Turner, N.C.; Ro, J.; André, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Huang Bartlett, C.; Zhang, K.; et al. Palbociclib in Hormone-Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.-A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Yardley, D.A. MONALEESA clinical program: A review of ribociclib use in different clinical settings. Futur. Oncol. 2019, 15, 2673–2686. [Google Scholar] [CrossRef]
- Hortobagyi, G.N. Ribociclib for the first-line treatment of advanced hormone receptor-positive breast cancer: A review of subgroup analyses from the MONALEESA-2 trial 11 Medical and Health Sciences 1112 Oncology and Carcinogenesis 11 Medical and Health Sciences 1103 Clinical Sciences. Breast Cancer Res. 2018, 20, 123. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.-A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martín, M.; et al. Phase III Randomized Study of Ribociclib and Fulvestrant in Hormone Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Advanced Breast Cancer: MONALEESA-3. J. Clin. Oncol. 2018, 36, 2465–2472. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and safety of Abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non–small cell lung cancer, and other solid tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.W.; Mazieres, J.; Barlesi, F.; Dragnev, K.H.; Koczywas, M.; Göskel, T.; Cortot, A.B.; Girard, N.; Wesseler, C.; Bischoff, H.; et al. A Randomized Phase III Study of Abemaciclib Versus Erlotinib in Patients with Stage IV Non-small Cell Lung Cancer With a Detectable KRAS Mutation Who Failed Prior Platinum-Based Therapy: JUNIPER. Front. Oncol. 2020, 10, 2338. [Google Scholar] [CrossRef]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.-C.; Manso, L.; et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef]
- Dickler, M.N.; Tolaney, S.M.; Rugo, H.S.; Cortes, J.; Dieras, V.; Patt, D.; Wildiers, H.; Hudis, C.A.; O’Shaughnessy, J.; Zamora, E.; et al. MONARCH 1, a phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as a single agent, n patients with refractory HR+/HER2- metastatic breast cancer. Clin. Cancer Res. 2017, 23, 5218–5224. [Google Scholar] [CrossRef]
- Sledge, G.W.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in combination with fulvestrant in women with HR+/HER2-advanced breast cancer who had progressed while receiving endocrine therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef]
- Hafner, M.; Mills, C.E.; Subramanian, K.; Chen, C.; Chung, M.; Boswell, S.A.; Everley, R.A.; Liu, C.; Walmsley, C.S.; Juric, D.; et al. Multiomics Profiling Establishes the Polypharmacology of FDA-Approved CDK4/6 Inhibitors and the Potential for Differential Clinical Activity. Cell Chem. Biol. 2019, 26, 1067–1080.e8. [Google Scholar] [CrossRef]
- Schoninger, S.F.; Blain, S.W. The ongoing search for biomarkers of CDK4/6 inhibitor responsiveness in breast cancer. Mol. Cancer Ther. 2020, 19, 3–12. [Google Scholar] [CrossRef]
- Flaherty, K.T.; LoRusso, P.M.; DeMichele, A.; Abramson, V.G.; Courtney, R.; Randolph, S.S.; Shaik, M.N.; Wilner, K.D.; O’Dwyer, P.J.; Schwartz, G.K. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 568–576. [Google Scholar] [CrossRef]
- Bozhenko, V.K.; Kulinich, T.M.; Kudinova, E.A.; Boldyrev, A.; Solodkij, V.A. New targeted anti CDK4/6 peptide MM-D37K. J. Clin. Oncol. 2013, 31, e13545. [Google Scholar] [CrossRef]
- Bisi, J.E.; Sorrentino, J.A.; Roberts, P.J.; Tavares, F.X.; Strum, J.C. Preclinical Characterization of G1T28: A Novel CDK4/6 Inhibitor for Reduction of Chemotherapy-Induced Myelosuppression. Mol. Cancer Ther. 2016, 15, 783–793. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Roberts, P.J.; Sorrentino, J.A.; Bisi, J.E.; Storrie-White, H.; Tiessen, R.G.; Makhuli, K.M.; Wargin, W.A.; Tadema, H.; Van Hoogdalem, E.J.; et al. Transient CDK4/6 inhibition protects hematopoietic stem cells from chemotherapy-induced exhaustion. Sci. Transl. Med. 2017, 9, aal3986. [Google Scholar] [CrossRef] [PubMed]
- Hart, L.L.; Ferrarotto, R.; Andric, Z.G.; Beck, J.T.; Subramanian, J.; Radosavljevic, D.Z.; Zaric, B.; Hanna, W.T.; Aljumaily, R.; Owonikoko, T.K.; et al. Myelopreservation with Trilaciclib in Patients Receiving Topotecan for Small Cell Lung Cancer: Results from a Randomized, Double-Blind, Placebo-Controlled Phase II Study. Adv. Ther. Available online: https://link.springer.com/article/10.1007/s12325-020-01538-0 (accessed on 15 October 2020). [CrossRef]
- Weiss, J.M.; Csoszi, T.; Maglakelidze, M.; Hoyer, R.J.; Beck, J.T.; Domine Gomez, M.; Lowczak, A.; Aljumaily, R.; Rocha Lima, C.M.; Boccia, R.V.; et al. Myelopreservation with the CDK4/6 inhibitor trilaciclib in patients with small-cell lung cancer receiving first-line chemotherapy: A phase Ib/randomized phase II trial. Ann. Oncol. 2019, 30, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; He, Y.; Fu, H.; Li, Y.; Bao, X.; Wang, Q.; Wang, Y.; Xie, C.; Lou, L. Preclinical characterization of SHR6390, a novel CDK 4/6 inhibitor, in vitro and in human tumor xenograft models. Cancer Sci. 2019, 110, 1420–1430. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Q.; Yuan, J.; Wang, J.; Chen, Z.; Liu, Z.; Li, Z.; Lai, Y.; Gao, J.; Shen, L. CDK4/6 inhibitor-SHR6390 exerts potent antitumor activity in esophageal squamous cell carcinoma by inhibiting phosphorylated Rb and inducing G1 cell cycle arrest. J. Transl. Med. 2017, 15, 127. [Google Scholar] [CrossRef]
- Watts, C.K.W.; Sweeney, K.J.E.; Warlters, A.; Musgrove, E.A.; Sutherland, R.L. Antiestrogen regulation of cell cycle progression and cyclin D1 gene expression in MCF-7 human breast cancer cells. Breast Cancer Res. Treat. 1994, 31, 95–105. [Google Scholar] [CrossRef]
- Prall, O.W.J.; Sarcevic, B.; Musgrove, E.A.; Watts, C.K.W.; Sutherland, R.L. Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J. Biol. Chem. 1997, 272, 10882–10894. [Google Scholar] [CrossRef]
- Miller, T.W.; Balko, J.M.; Fox, E.M.; Ghazoui, Z.; Dunbier, A.; Anderson, H.; Dowsett, M.; Jiang, A.; Adam Smith, R.; Maira, S.M.; et al. ERα-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discov. 2011, 1, 338–351. [Google Scholar] [CrossRef]
- Geoerger, B.; Bourdeaut, F.; DuBois, S.G.; Fischer, M.; Geller, J.I.; Gottardo, N.G.; Marabelle, A.; Pearson, A.D.J.; Modak, S.; Cash, T.; et al. A phase I study of the CDK4/6 inhibitor ribociclib (LEE011) in pediatric patients with malignant rhabdoid tumors, neuroblastoma, and other solid tumors. Clin. Cancer Res. 2017, 23, 2433–2441. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.A.; Schwartz, G.K.; Louise Keohan, M.; D’Angelo, S.P.; Gounder, M.M.; Chi, P.; Antonescu, C.R.; Landa, J.; Qin, L.X.; Crago, A.M.; et al. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with cdk4 inhibitor palbociclib a phase 2 clinical trial. JAMA Oncol. 2016, 2, 937–940. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.P.; LaCasce, A.S.; Smith, M.R.; Noy, A.; Chirieac, L.R.; Rodig, S.J.; Yu, J.Q.; Vallabhajosula, S.; Schoder, H.; English, P.; et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood 2012, 119, 4597–4607. [Google Scholar] [CrossRef] [PubMed]
- Uras, I.Z.; Sexl, V.; Kollmann, K. CDK6 inhibition: A novel approach in AML management. Int. J. Mol. Sci. 2020, 21, 2528. [Google Scholar] [CrossRef]
- Wang, L.; Wang, J.; Blaser, B.W.; Duchemin, A.M.; Kusewitt, D.F.; Liu, T.; Caligiuri, M.A.; Briesewitz, R. Pharmacologic inhibition of CDK4/6: Mechanistic evidence for selective activity or acquired resistance in acute myeloid leukemia. Blood 2007, 110, 2075–2083. [Google Scholar] [CrossRef]
- Uras, I.Z.; Walter, G.J.; Scheicher, R.; Bellutti, F.; Prchal-Murphy, M.; Tigan, A.S.; Valent, P.; Heidel, F.H.; Kubicek, S.; Scholl, C.; et al. Palbociclib treatment of FLT3-ITD+ AML cells uncovers a kinase-dependent transcriptional regulation of FLT3 and PIM1 by CDK6. Blood 2016, 127, 2890–2902. [Google Scholar] [CrossRef]
- Keegan, K.; Li, C.; Li, Z.; Ma, J.; Ragains, M.; Coberly, S.; Hollenback, D.; Eksterowicz, J.; Liang, L.; Weidner, M.; et al. Preclinical evaluation of AMG 925, a FLT3/CDK4 dual kinase inhibitor for treating acute myeloid leukemia. Mol. Cancer Ther. 2014, 13, 880–889. [Google Scholar] [CrossRef]
- Li, Z.; Wang, X.; Eksterowicz, J.; Gribble, M.W.; Alba, G.Q.; Ayres, M.; Carlson, T.J.; Chen, A.; Chen, X.; Cho, R.; et al. Discovery of AMG 925, a FLT3 and CDK4 dual kinase inhibitor with preferential affinity for the activated state of FLT3. J. Med. Chem. 2014, 57, 3430–3449. [Google Scholar] [CrossRef]
- Uras, I.Z.; Maurer, B.; Nivarthi, H.; Jodl, P.; Kollmann, K.; Prchal-Murphy, M.; Milosevic Feenstra, J.D.; Zojer, M.; Lagger, S.; Grausenburger, R.; et al. CDK6 coordinates JAK2V617F mutant MPN via NF-kB and apoptotic networks. Blood 2019, 133, 1677–1690. [Google Scholar] [CrossRef]
- De Dominici, M.; Porazzi, P.; Xiao, Y.; Chao, A.; Tang, H.-Y.; Kumar, G.; Fortina, P.; Spinelli, O.; Rambaldi, A.; Peterson, L.F.; et al. Selective inhibition of Ph-positive ALL cell growth through kinase-dependent and independent effects by CDK6-specific PROTACs. Blood 2020, 135, 1560–1573. [Google Scholar] [CrossRef]
- Bellutti, F.; Tigan, A.-S.; Nebenfuehr, S.; Dolezal, M.; Zojer, M.; Grausenburger, R.; Hartenberger, S.; Kollmann, S.; Doma, E.; Prchal-Murphy, M.; et al. CDK6 Antagonizes p53-Induced Responses during Tumorigenesis. Cancer Discov. 2018, 8, 884–897. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.L.; McClendon, A.K.; Knudsen, E.S. Modification of the DNA damage response by therapeutic CDK4/6 inhibition. J. Biol. Chem. 2012, 287, 29075–29087. [Google Scholar] [CrossRef] [PubMed]
- Salvador-Barbero, B.; Álvarez-Fernández, M.; Zapatero-Solana, E.; El Bakkali, A.; del Menéndez, M.C.; López-Casas, P.P.; Di Domenico, T.; Xie, T.; VanArsdale, T.; Shields, D.J.; et al. CDK4/6 Inhibitors Impair Recovery from Cytotoxic Chemotherapy in Pancreatic Adenocarcinoma. Cancer Cell 2020, 37, 340–353.e6. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.; Dustin, D.; Fuqua, S.A. Targeted therapy for breast cancer and molecular mechanisms of resistance to treatment. Curr. Opin. Pharm. 2016, 31, 97–103. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Goel, S. CDK4/6 Inhibition in Breast Cancer: Mechanisms of Response and Treatment Failure. Curr. Breast Cancer Rep. 2017, 9, 26–33. [Google Scholar] [CrossRef]
- Comstock, C.E.S.; Augello, M.A.; Goodwin, J.F.; De Leeuw, R.; Schiewer, M.J.; Ostrander, W.F.; Burkhart, R.A.; McClendon, A.K.; McCue, P.A.; Trabulsi, E.J.; et al. Targeting cell cycle and hormone receptor pathways in cancer. Oncogene 2013, 32, 5481–5491. [Google Scholar] [CrossRef]
- Sharifi, M.N.; Anandan, A.; Grogan, P.; O’Regan, R.M. Therapy after cyclin-dependent kinase inhibition in metastatic hormone receptor-positive breast cancer: Resistance mechanisms and novel treatment strategies. Cancer 2020, 126, 3400–3416. [Google Scholar] [CrossRef]
- DeMichele, A.; Clark, A.S.; Tan, K.S.; Heitjan, D.F.; Gramlich, K.; Gallagher, M.; Lal, P.; Feldman, M.; Zhang, P.; Colameco, C.; et al. CDK 4/6 Inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: Phase II activity, safety, and predictive biomarker assessment. Clin. Cancer Res. 2015, 21, 995–1001. [Google Scholar] [CrossRef]
- Raspé, E.; Coulonval, K.; Pita, J.M.; Paternot, S.; Rothé, F.; Twyffels, L.; Brohée, S.; Craciun, L.; Larsimont, D.; Kruys, V.; et al. CDK 4 phosphorylation status and a linked gene expression profile predict sensitivity to palbociclib. Embo Mol. Med. 2017, 9, 1052–1066. [Google Scholar] [CrossRef]
- Finn, R.S.; Liu, Y.; Zhu, Z.; Martin, M.; Rugo, H.S.; Dieras, V.; Im, S.A.; Gelmon, K.A.; Harbeck, N.; Lu, D.R.; et al. Biomarker analyses of response to cyclin-dependent kinase 4/6 inhibition and endocrine therapy in women with treatment-naïve metastatic breast cancer. Clin. Cancer Res. 2020, 26, 110–121. [Google Scholar] [CrossRef]
- Yang, C.; Li, Z.; Bhatt, T.; Dickler, M.; Giri, D.; Scaltriti, M.; Baselga, J.; Rosen, N.; Chandarlapaty, S. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 2017, 36, 2255–2264. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.L.; Thangavel, C.; McClendon, A.K.; Reed, C.A.; Knudsen, E.S. Therapeutic CDK4/6 inhibition in breast cancer: Key mechanisms of response and failure. Oncogene 2010, 29, 4018–4032. [Google Scholar] [CrossRef]
- Malorni, L.; Piazza, S.; Ciani, Y.; Guarducci, C.; Bonechi, M.; Biagioni, C.; Hart, C.D.; Verardo, R.; Di Leo, A.; Migliaccio, I. A gene expression signature of retinoblastoma loss-of-function is a predictive biomarker of resistance to palbociclib in breast cancer cell lines and is prognostic in patients with ER positive early breast cancer. Oncotarget 2016, 7, 68012–68022. [Google Scholar] [CrossRef]
- Li, Z.; Razavi, P.; Li, Q.; Toy, W.; Liu, B.; Ping, C.; Hsieh, W.; Sanchez-Vega, F.; Brown, D.N.; Da Cruz Paula, A.F.; et al. Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell 2018, 34, 893–905.e8. [Google Scholar] [CrossRef] [PubMed]
- Alevizopoulos, K.; Vlach, J.; Hennecke, S.; Amati, B. Cyclin E and c-Myc promote cell proliferation in the presence of p16(INK4a) of hypophosphorylated retinoblastoma family proteins. Embo J. 1997, 16, 5322–5333. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Tsiperson, V.; Gottesman, S.R.S.; Somma, J.; Blain, S.W. Dual inhibition of CDK4 and CDK2 via targeting p27 tyrosine phosphorylation induces a potent and durable response in breast cancer cells. Mol. Cancer Res. 2018, 16, 361–377. [Google Scholar] [CrossRef]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef]
- Guarducci, C.; Bonechi, M.; Benelli, M.; Biagioni, C.; Boccalini, G.; Romagnoli, D.; Verardo, R.; Schiff, R.; Osborne, C.K.; De Angelis, C.; et al. Cyclin E1 and Rb modulation as common events at time of resistance to palbociclib in hormone receptor-positive breast cancer. Npj Breast Cancer 2018, 4, 38. [Google Scholar] [CrossRef]
- Turner, N.C.; Liu, Y.; Zhu, Z.; Loi, S.; Colleoni, M.; Loibl, S.; DeMichele, A.; Harbeck, N.; André, F.; Bayar, M.A.M.; et al. Cyclin E1 expression and palbociclib efficacy in previously treated hormone receptor-positive metastatic breast cancer. J. Clin. Oncol. 2019, 37, 1169–1178. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Gibbs, J.B.; Oliff, A.; Kohl, N.E. Farnesyltransferase inhibitors: Ras research yields a potential cancer therapeutic. Cell 1994, 77, 175–178. [Google Scholar] [CrossRef]
- Whyte, D.B.; Kirschmeier, P.; Hockenberry, T.N.; Nunez-Oliva, I.; James, L.; Catino, J.J.; Bishop, W.R.; Pai, J.K. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J. Biol. Chem. 1997, 272, 14459–14464. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Fakih, M.; O’Neil, B.; Price, T.J.; Falchook, G.S.; Desai, J.; Kuo, J.; Govindan, R.; Rasmussen, E.; Morrow, P.K.H.; Ngang, J.; et al. Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRAS G12C inhibitor, in advanced solid tumors. J. Clin. Oncol. 2019, 37, 3003. [Google Scholar] [CrossRef]
- Lyons, J.F.; Wilhelm, S.; Hibner, B.; Bollag, G. Discovery of a novel Raf kinase inhibitor. In Endocrine-Related Cancer; Bioscientifica: Bristol, UK, 2001; Volume 8, pp. 219–225. [Google Scholar]
- Lee, J.T.; McCubrey, J.A. BAY-43-9006 Bayer/Onyx. Curr. Opin. Investig. Drugs 2003, 4, 757–763. [Google Scholar] [PubMed]
- Wilhelm, S.M.; Carter, C.; Tang, L.Y.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Escudier, B.; Szczylik, C.; Eisen, T.; Stadler, W.M.; Schwartz, B.; Shan, M.; Bukowski, R.M. Randomized phase III trial of the Raf kinase and VEGFR inhibitor sorafenib (BAY 43–9006) in patients with advanced renal cell carcinoma (RCC). J. Clin. Oncol. 2005, 23, LBA4510. [Google Scholar] [CrossRef]
- Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; Testori, A.; Lorigan, P.C.; et al. Vemurafenib in patients with BRAFV600 mutation-positive metastatic melanoma: Final overall survival results of the randomized BRIM-3 study. Ann. Oncol. 2017, 28, 2581–2587. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, K.; Zhu, X.; Lin, G.; Song, F.; Zhao, Y.; Piao, Y.; Liu, J.; Cheng, W.; Bi, X.; et al. Encorafenib (LGX818), a potent BRAF inhibitor, induces senescence accompanied by autophagy in BRAFV600E melanoma cells. Cancer Lett. 2016, 370, 332–344. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Cuyle, P.-J.A.R.; Huijberts, S.; Schellens, J.H.M.; Elez, E.; Fakih, M.; Montagut Viladot, C.; Peeters, M.; et al. Updated results of the BEACON CRC safety lead-in: Encorafenib (ENCO) + binimetinib (BINI) + cetuximab (CETUX) for BRAFV600E-mutant metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2019, 37, 688. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandal, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Cabanillas, M.E.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Keam, B.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.; et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600–mutant anaplastic thyroid cancer. J. Clin. Oncol. 2018, 36, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Kim, T.M.; Mazieres, J.; Quoix, E.; Riely, G.; Barlesi, F.; Souquet, P.J.; Smit, E.F.; Groen, H.J.M.; Kelly, R.J.; et al. Dabrafenib in patients with BRAFV600E-positive advanced non-small-cell lung cancer: A single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 642–650. [Google Scholar] [CrossRef]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1315–1327. [Google Scholar] [CrossRef]
- Robert, C.; Flaherty, K.T.; Hersey, P.; Nathan, P.D.; Garbe, C.; Milhem, M.M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. METRIC phase III study: Efficacy of trametinib (T), a potent and selective MEK inhibitor (MEKi), in progression-free survival (PFS) and overall survival (OS), compared with chemotherapy (C) in patients (pts) with BRAF V600E/K mutant advanced or metastatic melanoma (MM). J. Clin. Oncol. 2012, 30, LBA8509. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef]
- Girotti, M.R.; Pedersen, M.; Sanchez-Laorden, B.; Viros, A.; Turajlic, S.; Niculescu-Duvaz, D.; Zambon, A.; Sinclair, J.; Hayes, A.; Gore, M.; et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013, 3, 158–167. [Google Scholar] [CrossRef]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef]
- Shi, H.; Moriceau, G.; Kong, X.; Lee, M.K.; Lee, H.; Koya, R.C.; Ng, C.; Chodon, T.; Scolyer, R.A.; Dahlman, K.B.; et al. Melanoma whole-exome sequencing identifies V600E B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun. 2012, 3, 724. [Google Scholar] [CrossRef] [PubMed]
- Trunzer, K.; Pavlick, A.C.; Schuchter, L.; Gonzalez, R.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; Kim, K.B.; Weber, J.S.; et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J. Clin. Oncol. 2013, 31, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Paraiso, K.H.T.; Fedorenko, I.V.; Cantini, L.P.; Munko, A.C.; Hall, M.; Sondak, V.K.; Messina, J.L.; Flaherty, K.T.; Smalley, K.S.M. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br. J. Cancer 2010, 102, 1724–1730. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.; Moffat, J.; Schaefer, G.; Chan, J.; Wang, X.; Orr, C.; Cheng, J.; Hunsaker, T.; Shao, L.; Wang, S.J.; et al. Combined MEK and ERK inhibition overcomes therapy-mediated pathway reactivation in RAS mutant tumors. PLoS ONE 2017, 12, e0185862. [Google Scholar] [CrossRef] [PubMed]
- Germann, U.A.; Furey, B.F.; Markland, W.; Hoover, R.R.; Aronov, A.M.; Roix, J.J.; Hale, M.; Boucher, D.M.; Sorrell, D.A.; Martinez-Botella, G.; et al. Targeting the MAPK signaling pathway in cancer: Promising preclinical activity with the novel selective ERK1/2 inhibitor BVD-523 (ulixertinib). Mol. Cancer Ther. 2017, 16, 2351–2363. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Jean Lee Wong, D.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov. 2018, 8, 184–195. [Google Scholar] [CrossRef]
- Chin, H.; Lai, D.; Falchook, G. Extracellular signal-regulated kinase (ERK) inhibitors in oncology clinical trials. J. Immunother. Precis. Oncol. 2019, 2, 10. [Google Scholar] [CrossRef]
- Sunaga, N.; Miura, Y.; Tsukagoshi, Y.; Kasahara, N.; Masuda, T.; Sakurai, R.; Kaira, K.; Hisada, T. Dual inhibition of MEK and p38 impairs tumor growth in KRAS-mutated non-small cell lung cancer. Oncol. Lett. 2019, 17, 3569. [Google Scholar] [CrossRef]
- Vitos-Faleato, J.; Real, S.M.; Gutierrez-Prat, N.; Villanueva, A.; Llonch, E.; Drosten, M.; Barbacid, M.; Nebreda, A.R. Requirement for epithelial p38α in KRAS-driven lung tumor progression. Proc. Natl. Acad. Sci. USA 2020, 117, 2588–2596. [Google Scholar] [CrossRef]
- Pranteda, A.; Piastra, V.; Stramucci, L.; Fratantonio, D.; Bossi, G. The p38 mapk signaling activation in colorectal cancer upon therapeutic treatments. Int. J. Mol. Sci. 2020, 21, 2773. [Google Scholar] [CrossRef]
- Pedersen, I.M.; Buhl, A.M.; Klausen, P.; Geisler, C.H.; Jurlander, J. The chimeric anti-CD20 antibody rituximab induces apoptosis in B-cell chronic lymphocytic leukemia cells through a p38 mitogen activated protein-kinase-dependent mechanism. Blood 2002, 99, 1314–1319. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Igea, A.; Canovas, B.; Dolado, I.; Nebreda, A.R. Inhibition of p38 MAPK sensitizes tumour cells to cisplatin-induced apoptosis mediated by reactive oxygen species and JNK. Embo Mol. Med. 2013, 5, 1759–1774. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Ebisuya, M.; Ashida, F.; Okamoto, K.; Yonehara, S.; Nishida, E. Continuous ERK Activation Downregulates Antiproliferative Genes throughout G1 Phase to Allow Cell-Cycle Progression. Curr. Biol. 2006, 16, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Gong, X.; Zhang, Y.; Van Horn, R.D.; Yin, T.; Huber, L.; Burke, T.F.; Manro, J.; Iversen, P.W.; Wu, W.; et al. RAF inhibitor LY3009120 sensitizes RAS or BRAF mutant cancer to CDK4/6 inhibition by abemaciclib via superior inhibition of phospho-RB and suppression of cyclin D1. Nat. Publ. Gr. 2018, 37, 821–832. [Google Scholar] [CrossRef]
- Yadav, V.; Burke, T.F.; Huber, L.; Van Horn, R.D.; Zhang, Y.; Buchanan, S.G.; Chan, E.M.; Starling, J.J.; Beckmann, R.P.; Peng, S. Bin The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregulation. Mol. Cancer 2014, 13, 2253–2263. [Google Scholar] [CrossRef]
- Li, Z.; Wang, B.; Gu, S.; Jiang, P.; Sahu, A.; Chen, C.H.; Han, T.; Shi, S.; Wang, X.; Traugh, N.; et al. CRISPR Screens Identify Essential Cell Growth Mediators in BRAF Inhibitor-resistant Melanoma. Genom. Proteom. Bioinform. 2020, 18, 26–40. [Google Scholar] [CrossRef]
- Ramsdale, R.; Jorissen, R.N.; Li, F.Z.; Al-Obaidi, S.; Ward, T.; Sheppard, K.E.; Bukczynska, P.E.; Young, R.J.; Boyle, S.E.; Shackleton, M.; et al. The transcription cofactor c-JUN mediates phenotype switching and BRAF inhibitor resistance in melanoma. Sci. Signal. 2015, 8, ra82. [Google Scholar] [CrossRef]
- Kim, H.S.; Jung, M.; Kang, H.N.; Kim, H.; Park, C.W.; Kim, S.M.; Shin, S.J.; Kim, S.H.; Kim, S.G.; Kim, E.K.; et al. Oncogenic BRAF fusions in mucosal melanomas activate the MAPK pathway and are sensitive to MEK/PI3K inhibition or MEK/CDK4/6 inhibition. Oncogene 2017, 36, 3334–3345. [Google Scholar] [CrossRef]
- Kwong, L.N.; Costello, J.C.; Liu, H.; Jiang, S.; Helms, T.L.; Langsdorf, A.E.; Jakubosky, D.; Genovese, G.; Muller, F.L.; Jeong, J.H.; et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat. Med. 2012, 18, 1503–1510. [Google Scholar] [CrossRef]
- Sosman, J.A.; Kittaneh, M.; Lolkema, M.P.J.K.; Postow, M.A.; Schwartz, G.; Franklin, C.; Matano, A.; Bhansali, S.; Parasuraman, S.; Kim, K. A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS -mutant melanoma: Early encouraging clinical activity. J. Clin. Oncol. 2014, 32, 9009. [Google Scholar] [CrossRef]
- Lee, M.S.; Helms, T.L.; Feng, N.; Gay, J.; Chang, Q.E.; Tian, F.; Wu, J.Y.; Toniatti, C.; Heffernan, T.P.; Powis, G.; et al. Efficacy of the combination of MEK and CDK4/6 inhibitors in vitro and in vivo in KRAS mutant colorectal cancer models. Oncotarget 2016, 7, 39595–39608. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, S.; Chen, X.; Zheng, X.; Yao, Y.; Lu, G.; Zhou, J. Palbociclib, a selective CDK4/6 inhibitor, enhances the effect of selumetinib in RAS-driven non-small cell lung cancer. Cancer Lett. 2017, 408, 130–137. [Google Scholar] [CrossRef]
- Guiley, K.Z.; Stevenson, J.W.; Lou, K.; Barkovich, K.J.; Kumarasamy, V.; Wijeratne, T.U.; Bunch, K.L.; Tripathi, S.; Knudsen, E.S.; Witkiewicz, A.K.; et al. P27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science 2019, 366, eaaw2106. [Google Scholar] [CrossRef]
- Nebenfuehr, S.; Kollmann, K.; Sexl, V. The role of CDK6 in cancer. Int. J. Cancer 2020, ijc.33054. [Google Scholar] [CrossRef]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Daage, L.C.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Hart, L.S.; Rader, J.A.; Raman, P.; Batra, V.; Russell, M.R.; Tsang, M.; Gagliardi, M.; Chen, L.; Martinez, D.; Li, Y.; et al. Preclinical therapeutic synergy of MEK1/2 and CDK4/6 inhibition in neuroblastoma. Clin. Cancer Res. 2017, 23, 1785–1796. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, C.; Wan, H.; Zhang, G.; Feng, J.; Zhang, L.; Chen, X.; Zhong, D.; Lou, L.; Tao, W.; et al. Discovery and development of pyrotinib: A novel irreversible EGFR/HER2 dual tyrosine kinase inhibitor with favorable safety profiles for the treatment of breast cancer. Eur. J. Pharm. Sci. 2017, 110, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, Q.; Chen, S.; Zhu, W.; Fan, Y.; Wang, J.; Luo, Y.; Xing, P.; Lan, B.; Li, M.; et al. Phase I Study and Biomarker Analysis of Pyrotinib, a Novel Irreversible Pan-ErbB Receptor Tyrosine Kinase Inhibitor, in Patients With Human Epidermal Growth Factor Receptor 2–Positive Metastatic Breast Cancer. J. Clin. Oncol. 2017, 35, 3105–3112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Hong, R.; Kaping, L.; Xu, F.; Xia, W.; Qin, G.; Zheng, Q.; Lu, Q.; Zhai, Q.; Shi, Y.; et al. CDK4/6 inhibitor palbociclib enhances the effect of pyrotinib in HER2-positive breast cancer. Cancer Lett. 2019, 447, 130–140. [Google Scholar] [CrossRef]
- Santarius, T.; Shipley, J.; Brewer, D.; Stratton, M.R.; Cooper, C.S. A census of amplified and overexpressed human cancer genes. Nat. Rev. Cancer 2010, 10, 59–64. [Google Scholar] [CrossRef]
- Yu, Q.; Geng, Y.; Sicinski, P. Specific protection against breast cancers by cyclin D1 ablation. Nature 2001, 411, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, R.; McNair, C.; Schiewer, M.J.; Neupane, N.P.; Brand, L.J.; Augello, M.A.; Li, Z.; Cheng, L.C.; Yoshida, A.; Courtney, S.M.; et al. MAPK Reliance via acquired CDK4/6 inhibitor resistance in cancer. Clin. Cancer Res. 2018, 24, 4201–4214. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinicaltrials.gov Identifier. | Phase | Treatment | Drug Target | Target Disease | Primary Results |
|---|---|---|---|---|---|
| NCT03041311 | II | trilaciclib plus etoposide/carboplatin/atezolizumab | CDK4/6 plus chemotherapy/immune therapy | small cell lung cancer | N/A |
| NCT02514447 | Ib/IIa | trilaciclib plus topotecan | CDK4/6 plus chemotherapy | small cell lung cancer | better tolerability of chemotherapy |
| NCT02499770 | Ib/IIa | trilaciclib plus etoposide/carboplatin | CDK4/6 plus chemotherapy | small cell lung cancer | better tolerability of chemotherapy |
| NCT02978716 | II | trilaciclib plus gemcitabine plus carboplatin | CDK4/6 plus chemotherapy | hormone receptor negative breast cancer | N/A |
| NCT03480256 | I | SHR6390 plus pyrotinib | CDK4/6 plus ErbB | ErbB2 positive gastric cancer | partial response |
| NCT04095390 | II | SHR6390 plus pyrotinib plus letrozole/capecitabine | CDK4/6 plus ErbB plus endocrine therapy/chemotherapy | ErbB2 positive breast cancer | N/A |
| NCT03772353 | Ib/II | SHR6390 plus pyrotinib plus letrozole | CDK4/6 plus ErbB plus endocrine therapy | hormone receptor positive, ErbB2 positive breast cancer | N/A |
| NCT03481998 | Ib/II | SHR6390 plus letrozole/anastrozole/fulvestrant | CDK4/6 plus endocrine therapy | hormone receptor positive, ErbB2 negative breast cancer | N/A |
| NCT03966898 | III | SHR6390 plus letrozole/anastrozole | CDK4/6 plus endocrine therapy | hormone receptor positive, ErbB2 negative breast cancer | N/A |
| Raf Inhibitor | MEK Inhibitor | Target Disease | References |
|---|---|---|---|
| dabrafenib | trametinib | melanoma, thyroid cancer, non-small cell lung cancer | [133,134,135] |
| vemurafenib | cobimetinib | melanoma | [136] |
| encorafenib | binimetinib | melanoma, metastatic colorectal cancer with BRAFV600E mutations | [132,137] |
| trametinib | advanced melanoma with BRAFV600E and BRAFV600K mutations | [138] |
| Clinicaltrials.gov Identifier | Phase | Treatment | Drug Target | Target Disease | Primary Results |
|---|---|---|---|---|---|
| NCT03600883 | I/II | AMG 510 | KRAS G12C | advanced solid tumors with KRAS p.G12C mutation | N/A |
| NCT01875705 | I | GDC-0994 | ERK | advanced or metastatic solid tumors | partial response |
| NCT02457793 | I | GDC-0994 plus cobimetinib | ERK plus MEK | advanced or metastatic solid tumors | overlapping adverse events |
| NCT01781429 | I | BVD-523 (ulixertinib) | ERK | advanced solid tumors | partial response |
| NCT02296242 | I/II | BVD-523 (ulixertinib) | ERK | AML, MDS | N/A |
| NCT04198818 | I/II | HH2710 | ERK | advanced solid tumors | N/A |
| NCT04081259 | I | LY3214996 | ERK | AML | N/A |
| NCT01393990 | I | LY2228820 (ralimetinib) monotherapy or LY2228820 plus midazolam/tamoxifen | p38/sedative/endocrine therapy | advanced tumors | N/A |
| NCT01663857 | Ib/II | LY2228820 (ralimetinib) plus gemcitabine plus carboplatin | p38 plus chemotherapy | recurrent platinum-sensitive ovarian cancer | improvement of progression free survival |
| NCT02860780 | I | LY2228820 (ralimetinib) plus prexasertib | p38 plus CHK1 | advanced or metastatic tumors | N/A |
| NCT00095680 | II | SCIO-469 monotherapy or SCIO-469 plus bortezomib | p38/proteasome | multiple myeloma | N/A |
| NCT00113893 | II | SCIO-469 | p38 | MDS, hematologic diseases | N/A |
| Clinicaltrials.gov Identifier | Phase | Treatment | Drug Target | Target Disease | Primary Results |
|---|---|---|---|---|---|
| NCT02159066 | II | LGX818 (encorafenib) plus binimetinib plus ribociclib | BRAF plus MEK1/2 plus CDK4/6 | BRAF-mutated melanoma | N/A |
| NCT01543698 | Ib/II | LGX818 (encorafenib) plus binimetinib plus ribociclib | BRAF plus MEK1/2 plus CDK4/6 | tumors with BRAFV600 mutations | N/A |
| NCT03981614 | II | binimetinib plus palbociclib | MEK1/2 plus CDK4/6 | advanced stage colorectal carcinoma with NRAS or KRAS mutations | N/A |
| NCT03170206 | I/II | binimetinib plus palbociclib | MEK1/2 plus CDK4/6 | advanced stage NSCLC with KRAS mutations | N/A |
| NCT02780128 | I | ceritinib plus trametinib plus ribociclib | ALK plus MEK1/2 plus CDK4/6 | relapsed neuroblastoma with ALK or MAPK mutations | N/A |
| NCT03454035 | I | BVD-523 (ulixertinib) plus palbociclib | ERK plus CDK4/6 | pancreatic cancer and other solid tumors | N/A |
| NCT02065063 | I/II | palbociclib plus trametinib | CDK4/6 plus MEK1/2 | solid tumors | N/A |
| NCT03132454 | I | palbociclib plus sorafenib/decitabine/dexamethasone | CDK4/6 plus Raf-1/chemotherapy | recurrent or refractory leukemia | N/A |
| NCT01781572 | Ib/II | ribociclib plus binimetinib | CDK4/6 plus MEK1/2 | advanced NRAS-mutant melanoma | partial response |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scheiblecker, L.; Kollmann, K.; Sexl, V. CDK4/6 and MAPK—Crosstalk as Opportunity for Cancer Treatment. Pharmaceuticals 2020, 13, 418. https://doi.org/10.3390/ph13120418
Scheiblecker L, Kollmann K, Sexl V. CDK4/6 and MAPK—Crosstalk as Opportunity for Cancer Treatment. Pharmaceuticals. 2020; 13(12):418. https://doi.org/10.3390/ph13120418
Chicago/Turabian StyleScheiblecker, Lisa, Karoline Kollmann, and Veronika Sexl. 2020. "CDK4/6 and MAPK—Crosstalk as Opportunity for Cancer Treatment" Pharmaceuticals 13, no. 12: 418. https://doi.org/10.3390/ph13120418
APA StyleScheiblecker, L., Kollmann, K., & Sexl, V. (2020). CDK4/6 and MAPK—Crosstalk as Opportunity for Cancer Treatment. Pharmaceuticals, 13(12), 418. https://doi.org/10.3390/ph13120418