Novel mTORC1 Inhibitors Kill Glioblastoma Stem Cells

 and
and 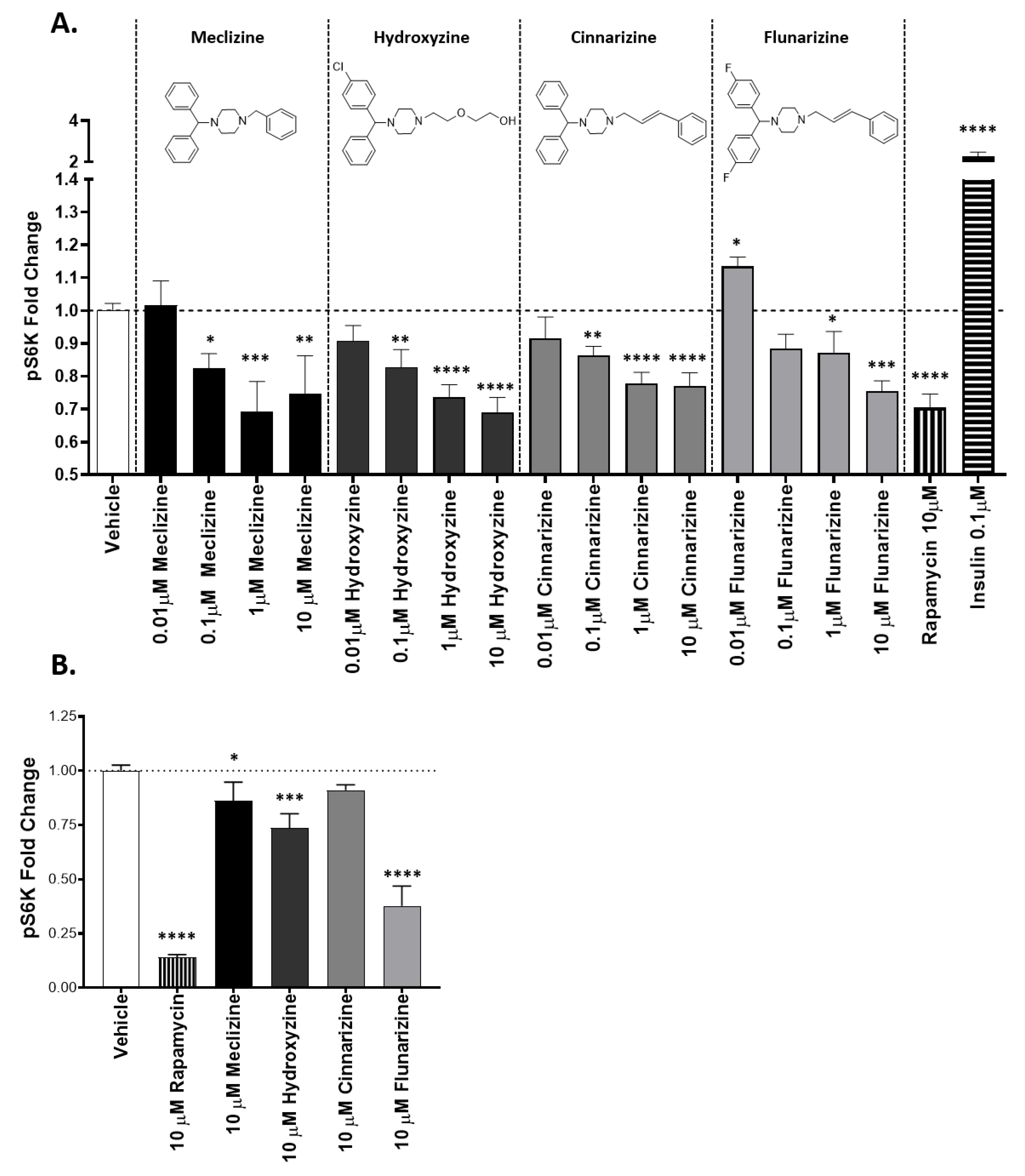{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Piperazine Compounds Dose-Dependently Inhibit mTORC1 Downstream Target
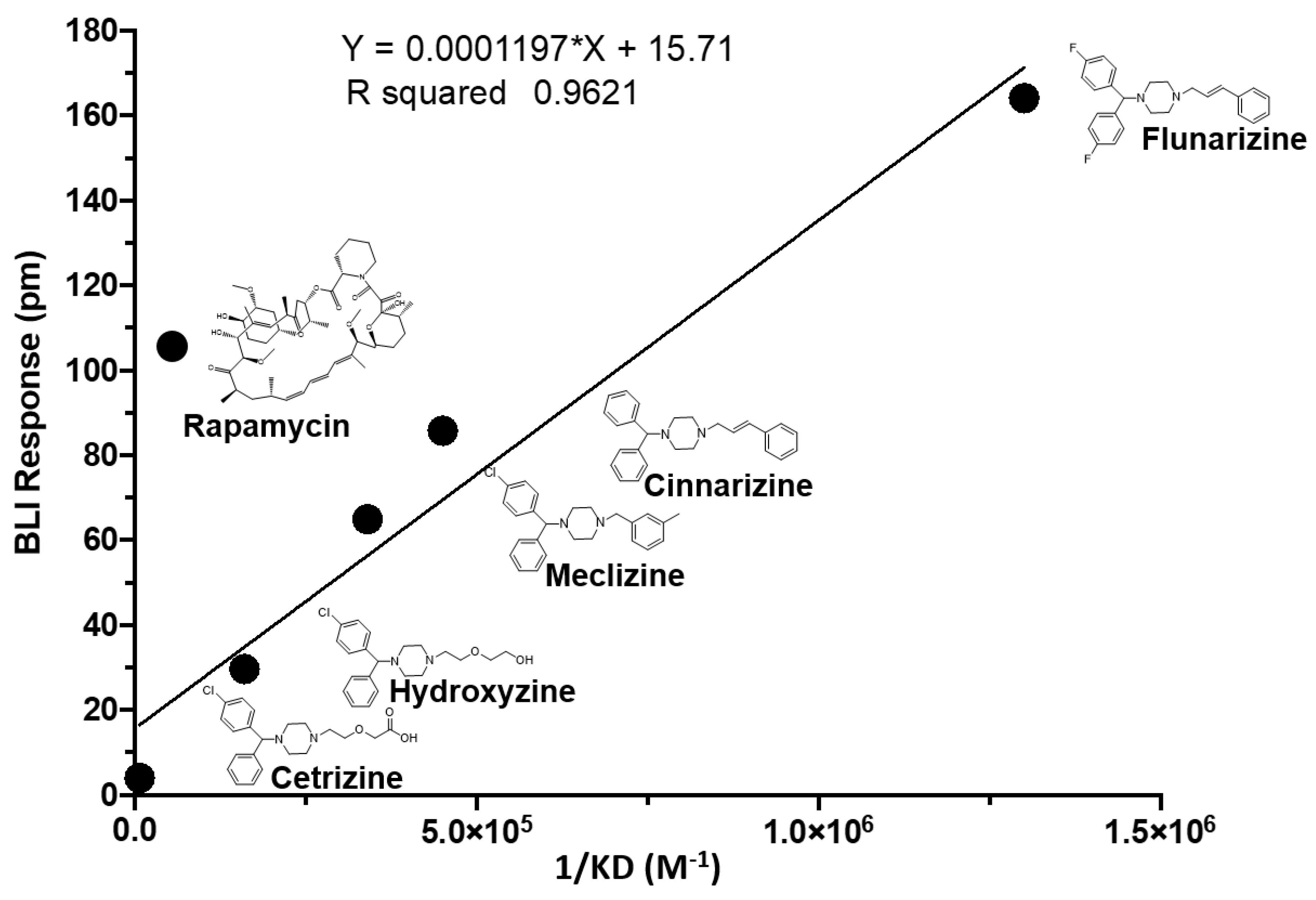
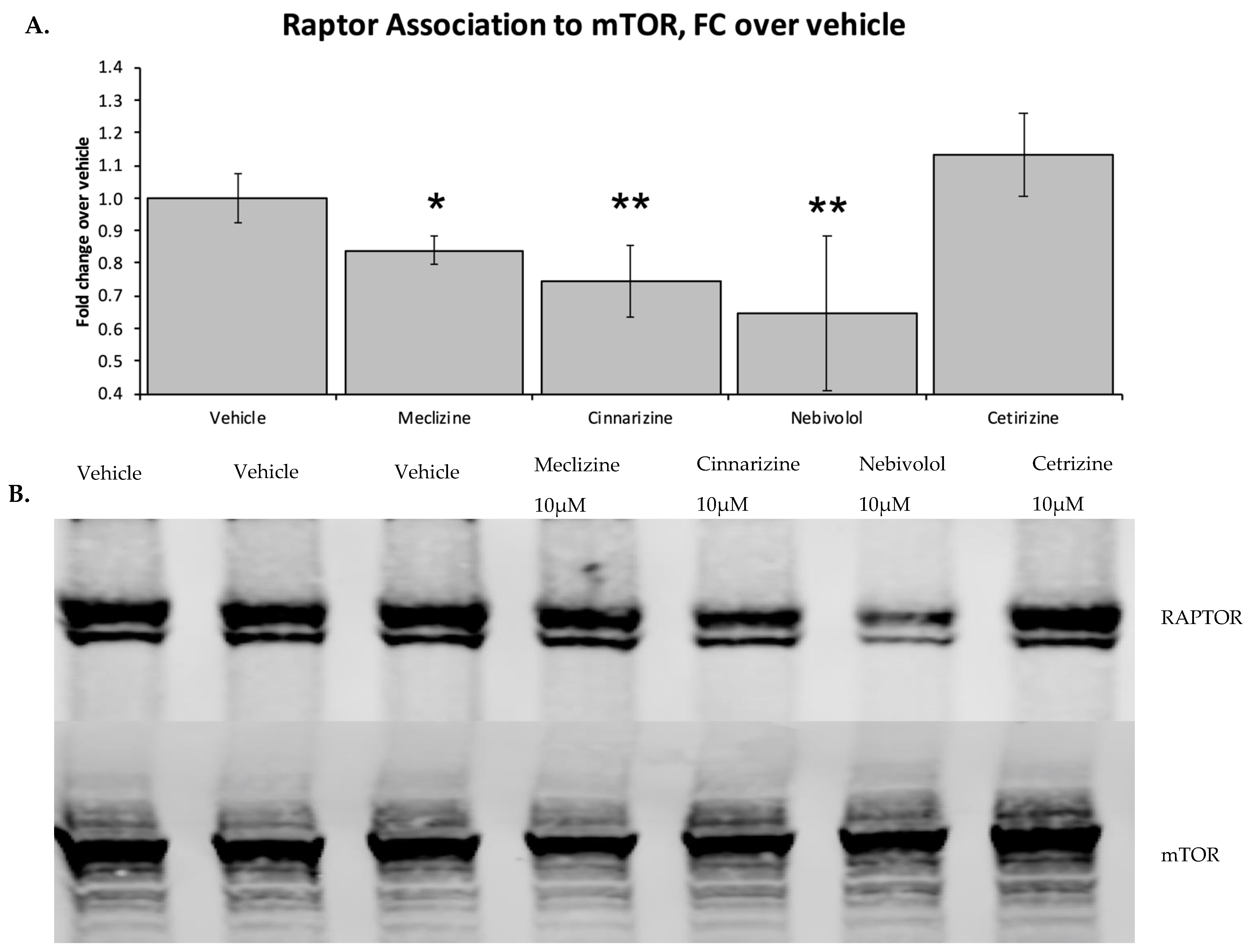
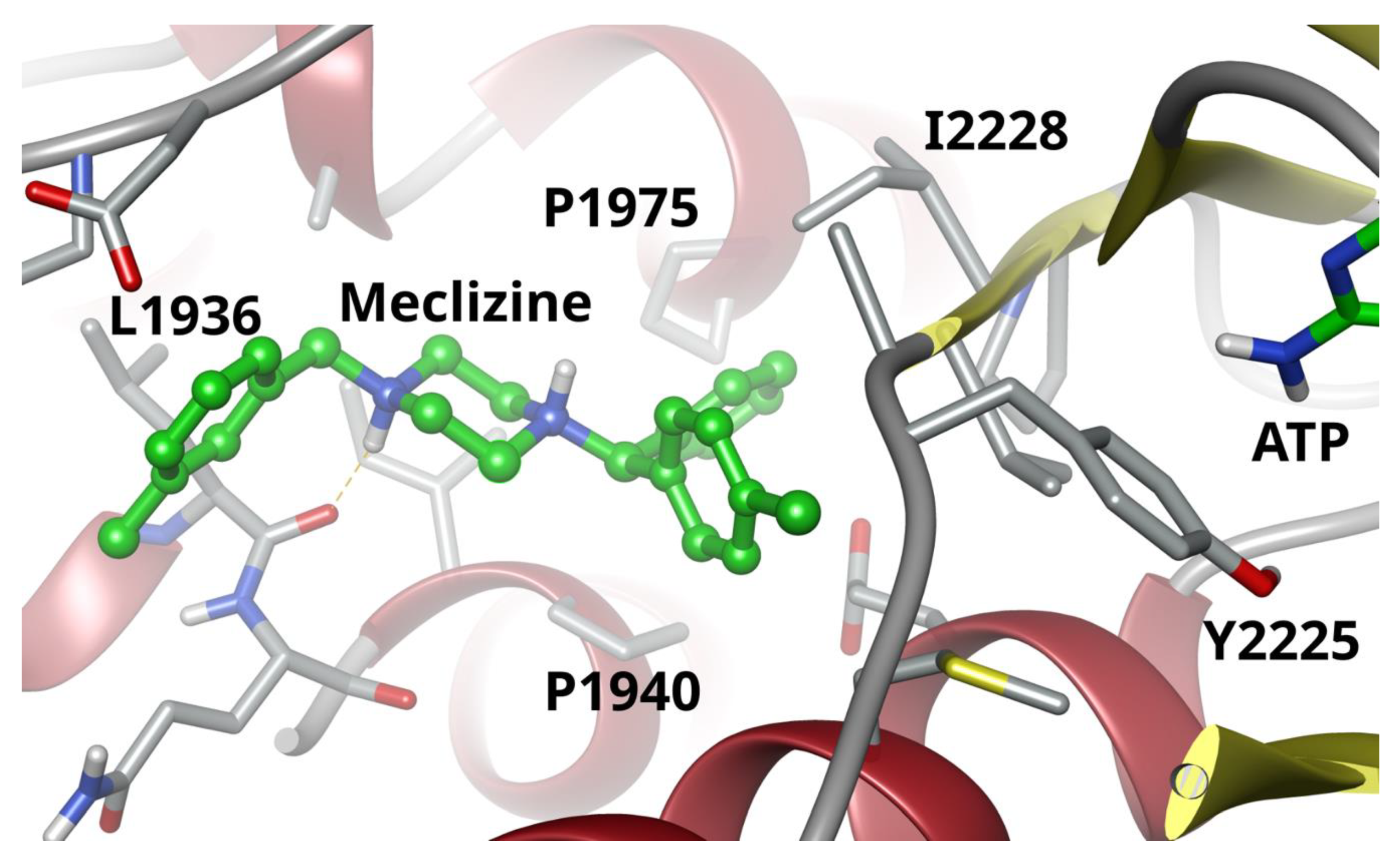
2.2. Two Biophysical Assays Confirm Piperazine Interaction with Human mTOR
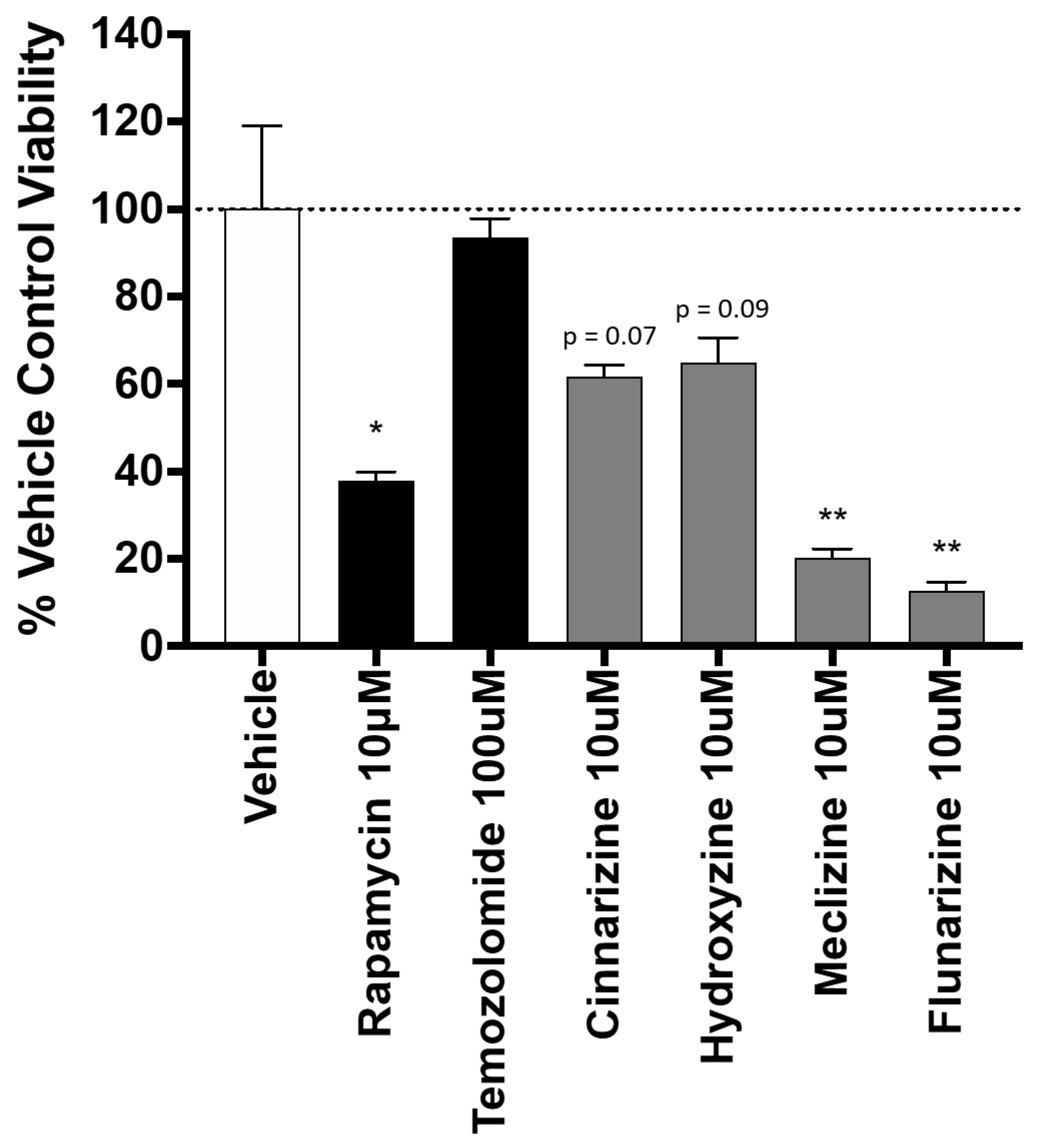
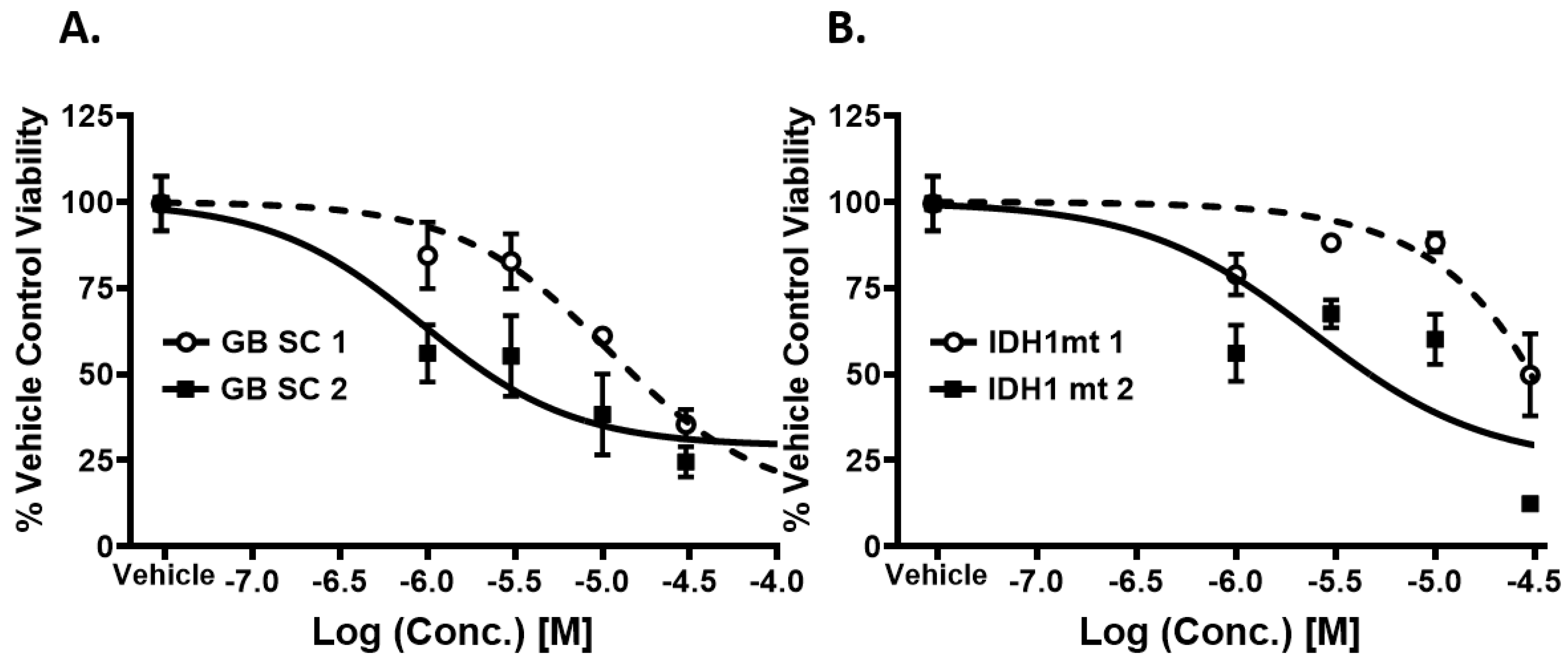
2.3. Piperazine Drugs Kill GBMSCs
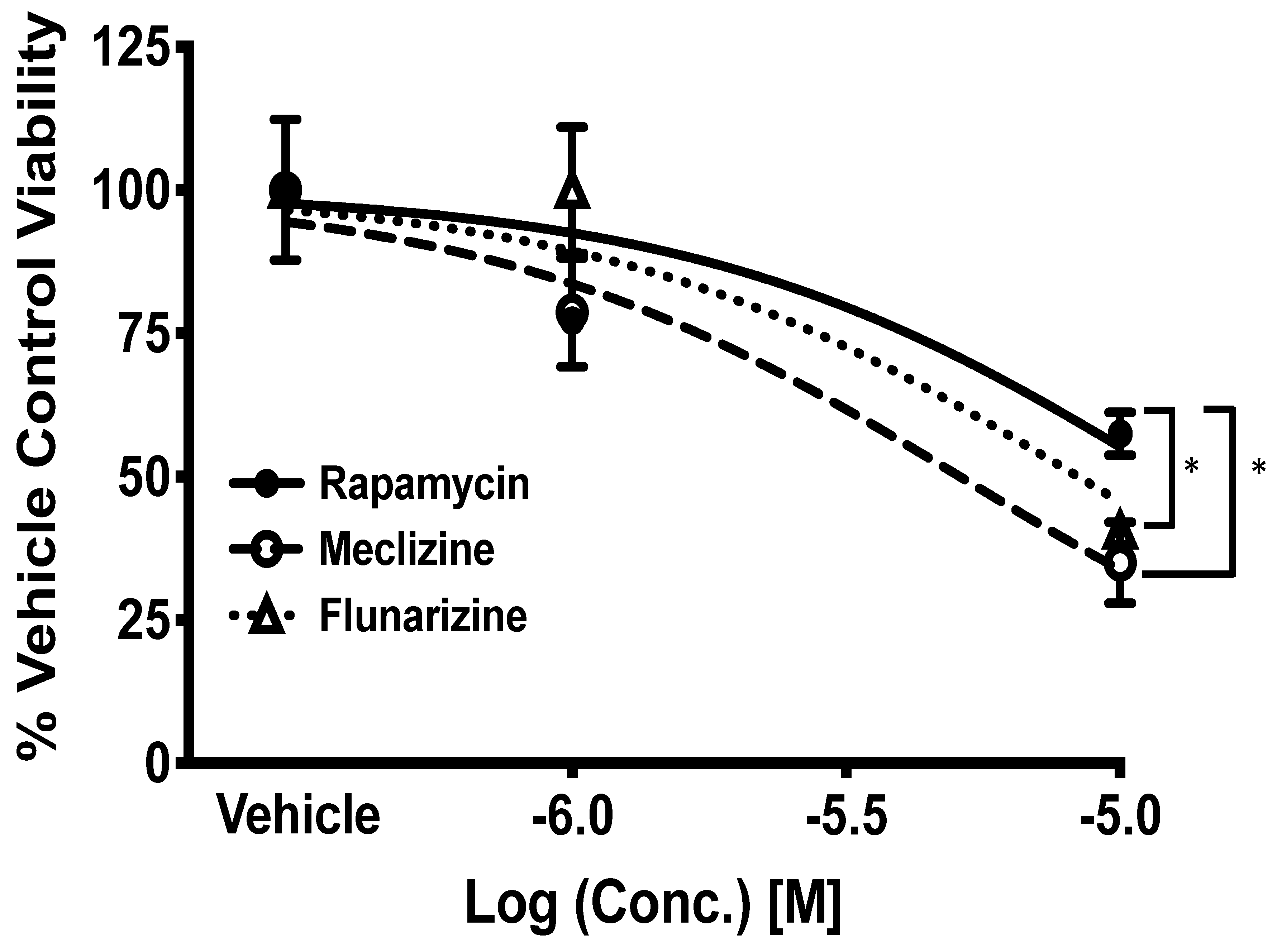
2.4. Comparison of GBM Cell Viability after Treatment with Meclizine, Flunarizine, or Rapamycin
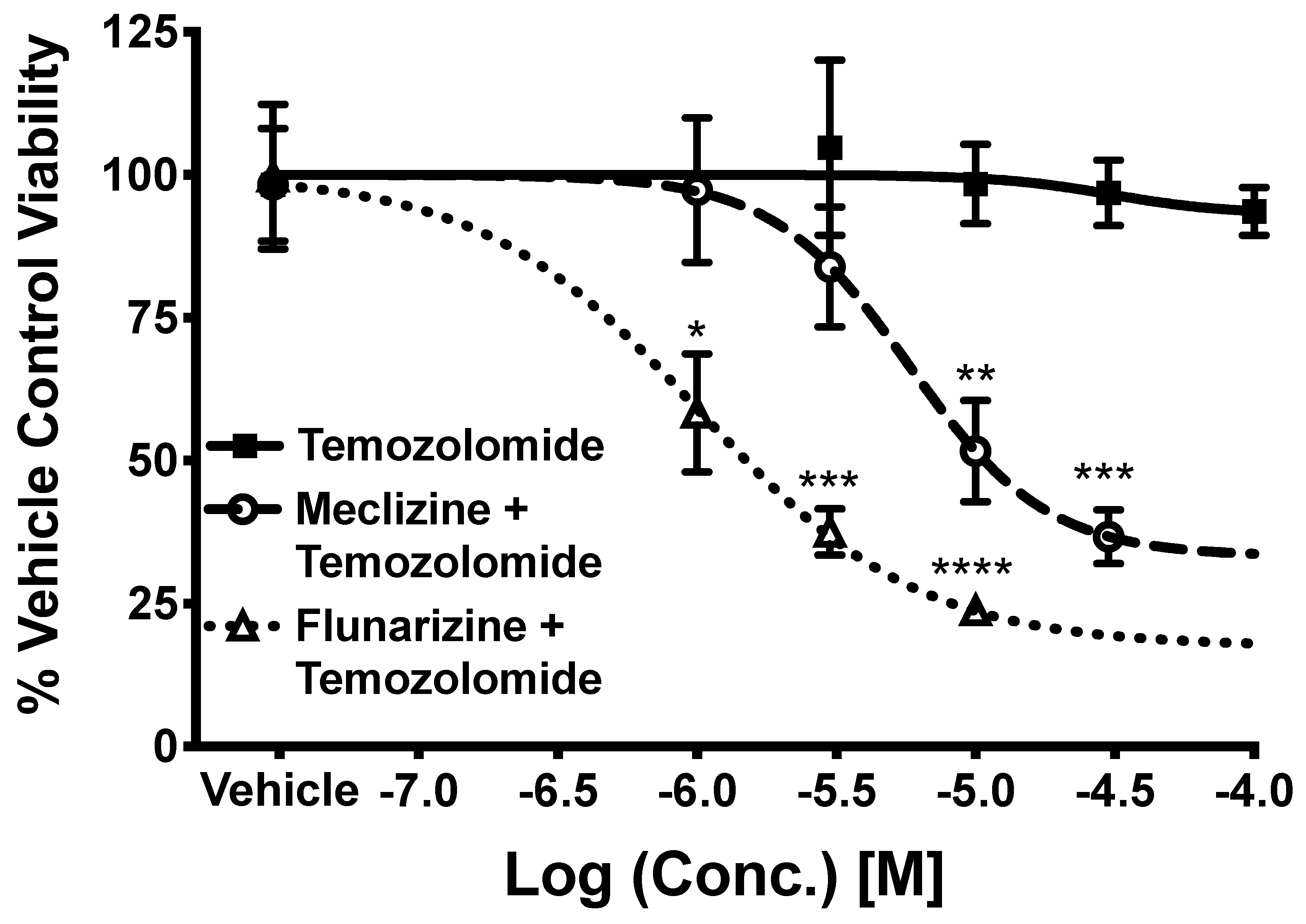
2.5. Co-Administration of Temozolomide with Meclizine or Flunarizine Potentiates GBMSC Killing
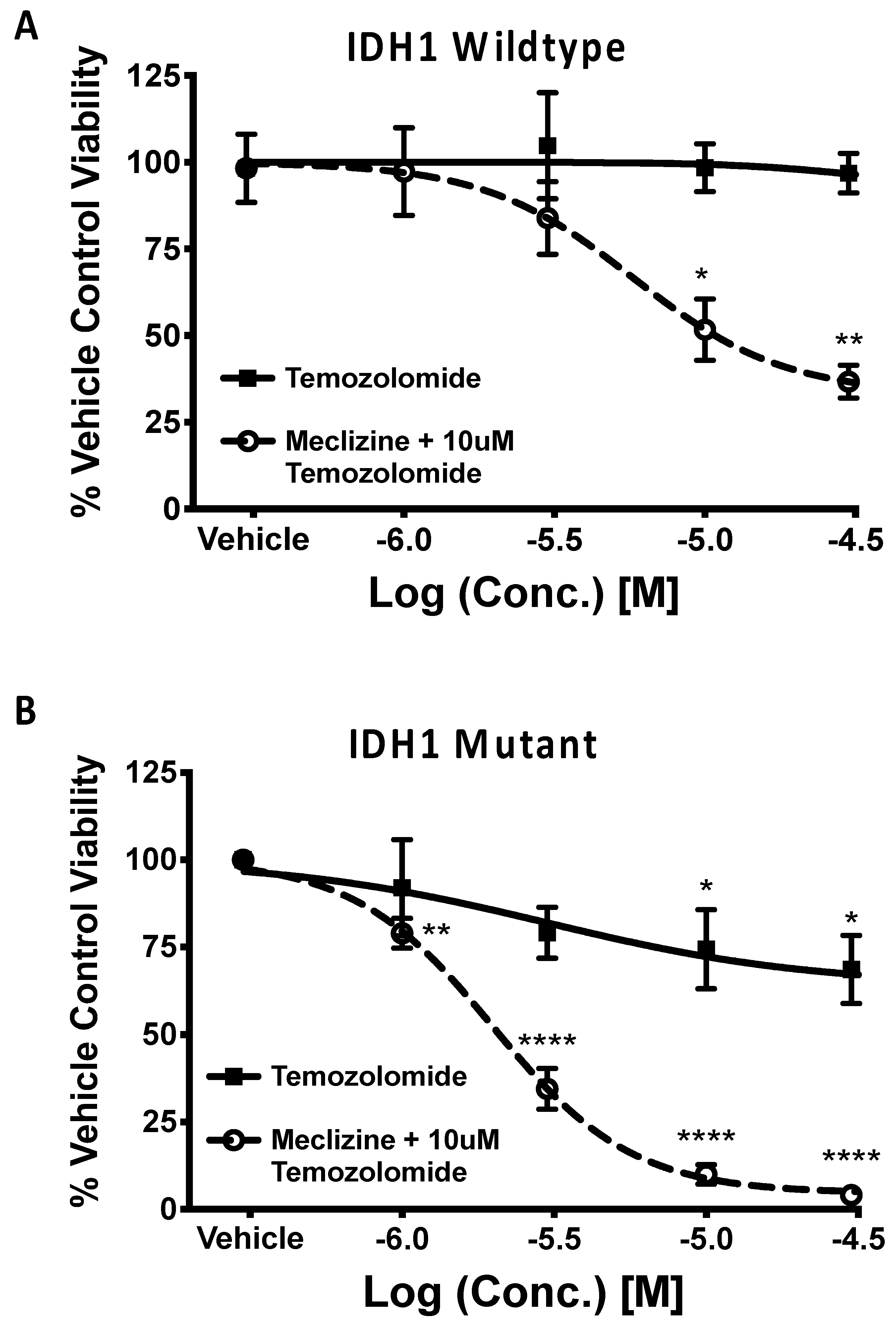
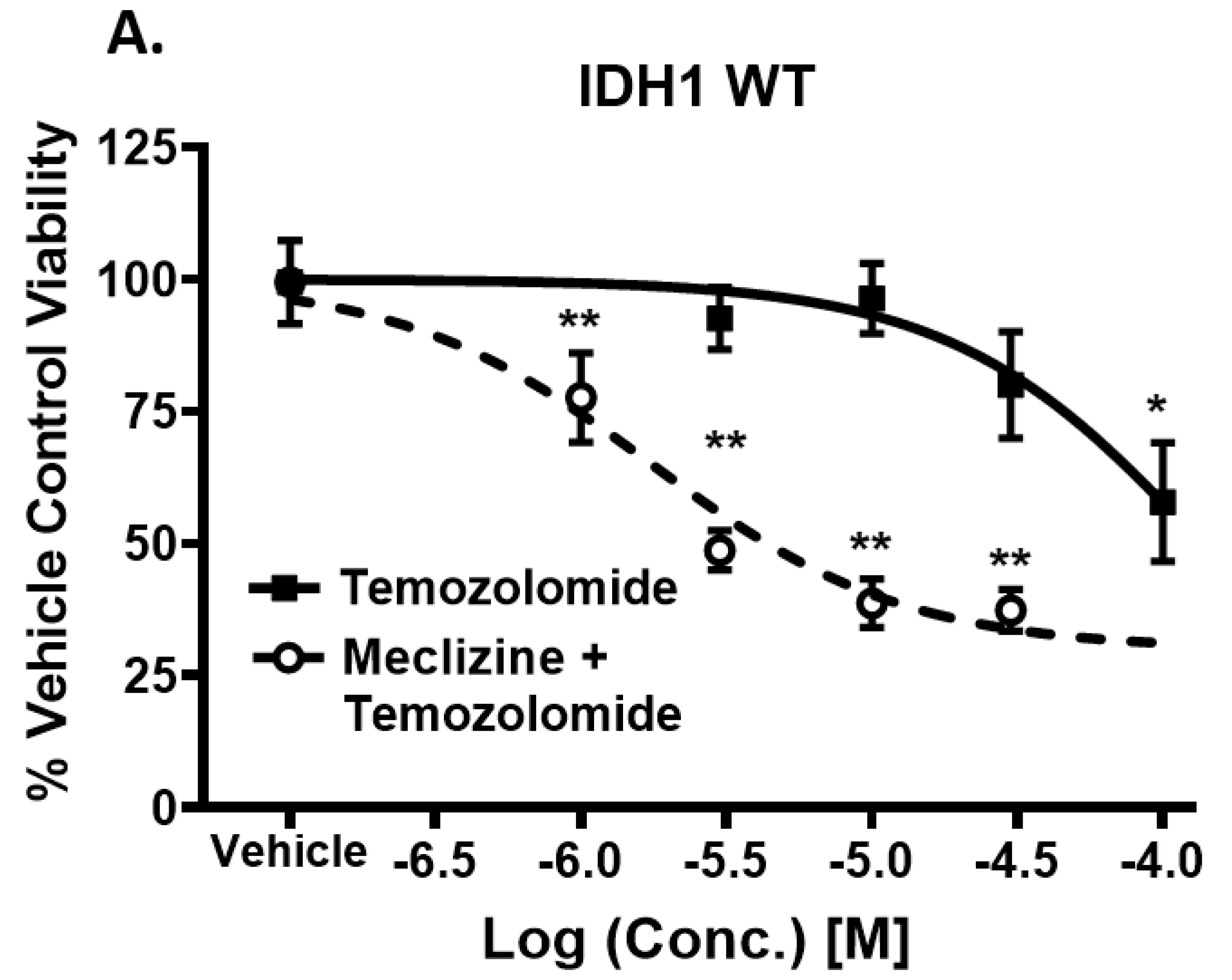
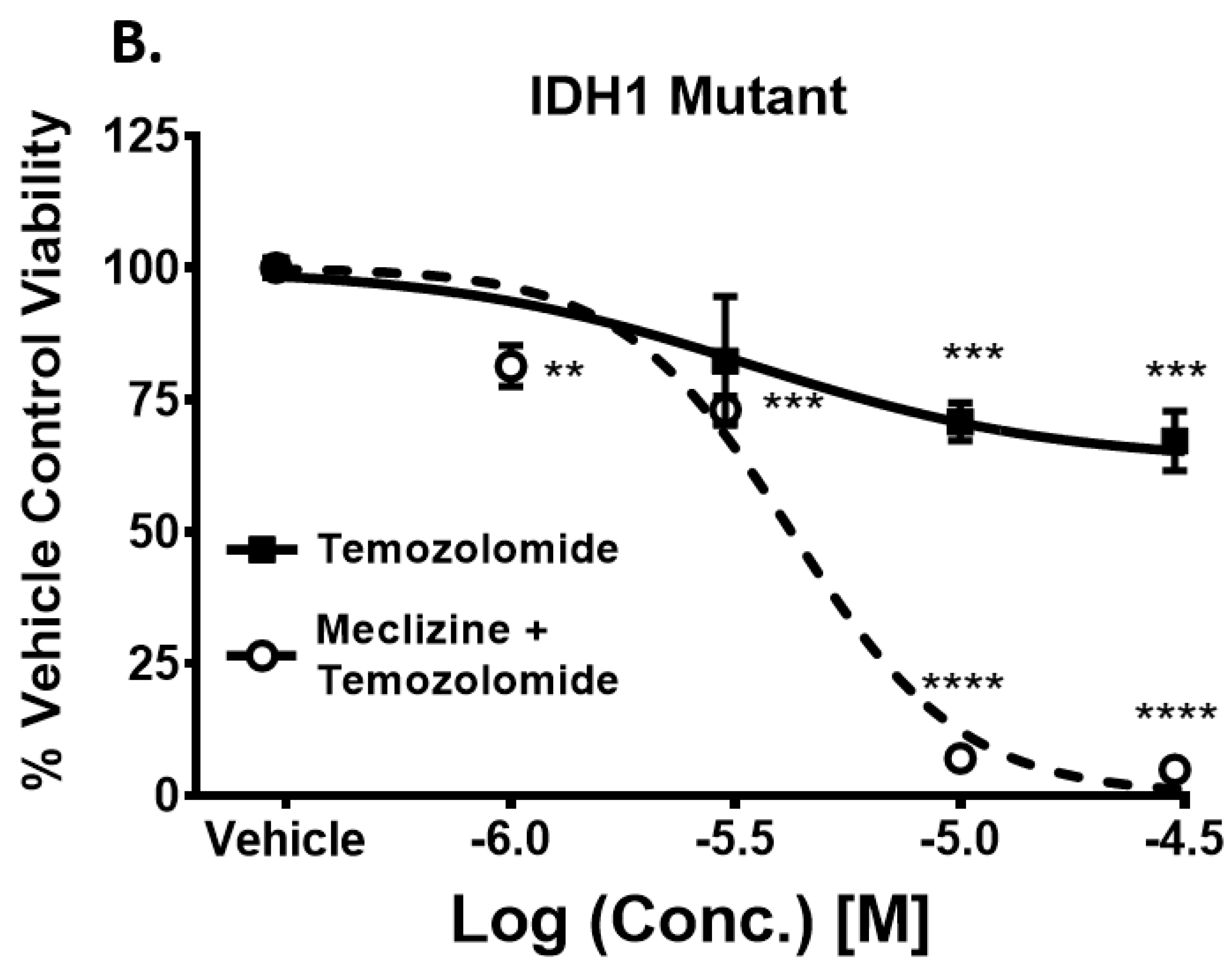
2.6. Meclizine Confers Synthetic Lethality on GBM Stem Cell Harboring R132H IDH1 Mutation
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Compounds
4.3. Bi-Layer Interferometry
4.4. Field-Effect Biosensing
4.5. Cell Viability Assay
4.6. pS6K ELISA
4.7. Co-Immunoprecipitation
4.8. Western Blot
4.9. Data Analysis
Author Contributions
Funding
Conflicts of Interest
Appendix A






References and Notes
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Begicevic, R.-R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Francipane, M.G.; Lagasse, E. Therapeutic potential of mTOR inhibitors for targeting cancer stem cells. Br. J. Clin. Pharmacol. 2016, 82, 1180–1188. [Google Scholar] [CrossRef]
- Karthik, G.-M.; Ma, R.; Lövrot, J.; Kis, L.L.; Lindh, C.; Blomquist, L.; Fredriksson, I.; Bergh, J.; Hartman, J. mTOR inhibitors counteract tamoxifen-induced activation of breast cancer stem cells. Cancer Lett. 2015, 367, 76–87. [Google Scholar] [CrossRef]
- Katsuno, Y.; Meyer, D.S.; Zhang, Z.; Shokat, K.M.; Akhurst, R.J.; Miyazono, K.; Derynck, R. Chronic TGF-β exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci. Signal. 2019, 12, eaau8544. [Google Scholar] [CrossRef]
- Kim, J.; Guan, K.-L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Hay, N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell 2005, 8, 179–183. [Google Scholar] [CrossRef]
- Fernandes, C.; Costa, A.; Osório, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017. [Google Scholar]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in Glioblastoma: Rationale and Preclinical/Clinical Evidence. Dis. Markers 2018, 2018, 9230479. [Google Scholar] [CrossRef]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.-L.; Schulze, A. SREBP Activity Is Regulated by mTORC1 and Contributes to Akt-Dependent Cell Growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef]
- Kim, J.E.; Chen, J. Regulation of Peroxisome Proliferator-Activated Receptor–Activity by Mammalian Target of Rapamycin and Amino Acids in Adipogenesis. Diabetes 2004, 53, 2748–2756. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Gravel, S.-P.; Chénard, V.; Sikström, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 Controls Mitochondrial Activity and Biogenesis through 4E-BP-Dependent Translational Regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.-P.; Enomoto, A.; Shiraki, Y.; Wang, S.-Q.; Wang, X.; Toyokuni, S.; Asai, N.; Ushida, K.; Ara, H.; Ohka, F.; et al. Significance of low mTORC1 activity in defining the characteristics of brain tumor stem cells. Neuro-Oncology 2016, 19, 636–647. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, J.; Yu, J.; Tu, L.; Huang, N.; Li, H.; Luo, Y. Isocitrate Dehydrogenase Mutations in Glioma: From Basic Discovery to Therapeutics Development. Front. Oncol. 2019, 9, 506. [Google Scholar] [CrossRef] [PubMed]
- Machado, L.E.; Alvarenga, A.W.; Da Silva, F.F.; Roffé, M.; Begnami, M.D.; Torres, L.F.B.; Da Cunha, I.W.; Martins, V.R.; Hajj, G.N.M. Overexpression of mTOR and p(240–244)S6 in IDH1 Wild-Type Human Glioblastomas Is Predictive of Low Survival. J. Histochem. Cytochem. 2018, 66, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Carbonneau, M.; Gagné, L.M.; LaLonde, M.-E.; Germain, M.-A.; Motorina, A.; Guiot, M.-C.; Secco, B.; Vincent, E.E.; Tumber, A.; Hulea, L.; et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat. Commun. 2016, 7, 12700. [Google Scholar] [CrossRef] [PubMed]
- Hujber, Z.; Petővári, G.; Szoboszlai, N.; Dankó, T.; Nagy, N.; Kriston, C.; Krencz, I.; Paku, S.; Ozohanics, O.; Drahos, L.; et al. Rapamycin (mTORC1 inhibitor) reduces the production of lactate and 2-hydroxyglutarate oncometabolites in IDH1 mutant fibrosarcoma cells. J. Exp. Clin. Cancer Res. 2017, 36, 74. [Google Scholar] [CrossRef]
- Allen, S.A.; Tomilov, A.; Cortopassi, G. Small molecules bind human mTOR protein and inhibit mTORC1 specifically. Biochem. Pharmacol. 2018, 155, 298–304. [Google Scholar] [CrossRef]
- Wood, P.J.; Hirst, D.G. Cinnarizine and flunarizine as radiation sensitisers in two murine tumours. Br. J. Cancer 1988, 58, 742–745. [Google Scholar] [CrossRef]
- Wood, P.J.; Hirst, D.G. Cinnarizine and flunarizine improve the tumour radiosensitisation induced by erythrocyte transfusion in anaemic mice. Br. J. Cancer 1989, 60, 36–40. [Google Scholar] [CrossRef]
- Chheda, M.G.; Wen, P.Y.; Hochberg, F.H.; Chi, A.S.; Drappatz, J.; Eichler, A.F.; Yang, D.; Beroukhim, R.; Norden, A.D.; Gerstner, E.R.; et al. Vandetanib plus sirolimus in adults with recurrent glioblastoma: Results of a phase I and dose expansion cohort study. J. Neuro-Oncol. 2015, 121, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Chinnaiyan, P.; Won, M.; Wen, P.Y.; Rojiani, A.M.; Werner-Wasik, M.; Shih, H.A.; Ashby, L.S.; Yu, H.-H.M.; Stieber, V.W.; Malone, S.C.; et al. A randomized phase II study of everolimus in combination with chemoradiation in newly diagnosed glioblastoma: Results of NRG Oncology RTOG 0913. Neuro-Oncology 2017, 20, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.J.; Galanis, E.; Anderson, S.K.; Schiff, D.; Kaufmann, T.J.; Peller, P.J.; Giannini, C.; Brown, P.D.; Uhm, J.H.; McGraw, S.; et al. A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neuro-Oncology 2015, 17, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Olmez, I.; Brenneman, B.; Xiao, A.; Serbulea, V.; Benamar, M.; Zhang, Y.; Manigat, L.; Abbas, T.; Lee, J.; Nakano, I.; et al. Combined CDK4/6 and mTOR Inhibition Is Synergistic against Glioblastoma via Multiple Mechanisms. Clin. Cancer Res. 2017, 23, 6958–6968. [Google Scholar] [CrossRef]
- Patel, P.N.; Ambizas, E.M. Meclizine: Safety and Efficacy in the Treatment and Prevention of Motion Sickness. Clin. Med. Insights Ther. 2011, 3, CMT.S6237. [Google Scholar] [CrossRef]
- Mohamed, B.P.; Goadsby, P.J.; Prabhakar, P. Safety and efficacy of flunarizine in childhood migraine: 11 years’ experience, with emphasis on its effect in hemiplegic migraine. Dev. Med. Child Neurol. 2012, 54, 274–277. [Google Scholar] [CrossRef]
- Scholtz, A.W.; Hahn, A.; Stefflova, B.; Medzhidieva, D.; Ryazantsev, S.V.; Paschinin, A.; Kunelskaya, N.; Schumacher, K.; Weisshaar, G. Efficacy and Safety of a Fixed Combination of Cinnarizine 20 mg and Dimenhydrinate 40 mg vs Betahistine Dihydrochloride 16 mg in Patients with Peripheral Vestibular Vertigo: A Prospective, Multinational, Multicenter, Double-Blind, Randomized, Non-inferiority Clinical Trial. Clin. Drug Investig. 2019, 39, 1045–1056. [Google Scholar] [CrossRef]
- Abbott, D.; Comby, P.; Charuel, C.; Graepel, P.; Hanton, G.; Leblanc, B.; Lodola, A.; Longeart, L.; Paulus, G.; Peters, C.; et al. Preclinical safety profile of sildenafil. Int. J. Impot. Res. 2004, 16, 498–504. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. National Library of Medicine (U.S.). ABI-009 (Nab-Rapamycin) in Recurrent High Grade Glioma and Newly Diagnosed Glioblastoma. August 2018.
- ClinicalTrials.gov. National Library of Medicine (U.S.). A Study of Temsirolimus and Bevacizumab in Recurrent Glioblastoma Multiforme.
- ClinicalTrials.gov. National Library of Medicine (U.S.). Everolimus, Temozolomide, and Radiation Therapy in Treating Patients with Newly Diagnosed Glioblastoma Multiforme (RTOG 0913).
- Rapamune Prescribing Information. United States Food and Drug Administration: Wyeth Pharmaceuticals, Inc.
- Saffari, A.; Brösse, I.; Wiemer-Kruel, A.; Wilken, B.; Kreuzaler, P.; Hahn, A.; Bernhard, M.K.; Van Tilburg, C.M.; Hoffmann, G.F.; Gorenflo, M.; et al. Safety and efficacy of mTOR inhibitor treatment in patients with tuberous sclerosis complex under 2 years of age—A multicenter retrospective study. Orphanet J. Rare Dis. 2019, 14, 96. [Google Scholar] [CrossRef]
- Franz, D.N.; Belousova, E.; Sparagana, S.; Bebin, E.M.; Frost, M.; Kuperman, R.; Witt, O.; Kohrman, M.H.; Flamini, J.R.; Wu, J.Y.; et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): A multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2013, 381, 125–132. [Google Scholar] [CrossRef]
- Schmeel, L.C.; Schmeel, F.C.; Kim, Y.; Blaum-Feder, S.; Endo, T.; Schmidt-Wolf, I.G.H. In vitro efficacy of cinnarizine against lymphoma and multiple myeloma. Anticancer. Res. 2015, 35, 835–841. [Google Scholar]
- Schmeel, L.C.; Schmeel, F.C.; Kim, Y.; Blaum-Feder, S.; Endo, T.; Schmidt-Wolf, I.G.H. Flunarizine exhibits in vitro efficacy against lymphoma and multiple myeloma cells. Anticancer. Res. 2015, 35, 1369–1376. [Google Scholar] [PubMed]
- Teive, H.A.G.; Troiano, A.R.; Germiniani, F.M.; Werneck, L.C. Flunarizine and cinnarizine-induced parkinsonism: A historical and clinical analysis. Park. Relat. Disord. 2004, 10, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Galanis, E.; Wu, W.; Peller, P.J.; Giannini, C.; Brown, P.D.; Uhm, J.H.; McGraw, S.; Jaeckle, K.A.; Buckner, J.C. North Central Cancer Treatment Group Phase I Trial N057K of Everolimus (RAD001) and Temozolomide in Combination With Radiation Therapy in Patients With Newly Diagnosed Glioblastoma Multiforme. Int. J. Radiat. Oncol. 2011, 81, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Bopp, E.J.; Estrada, T.J.L.; Kilday, J.M.; Spradling, J.C.; Daniel, C.; Pellegrini, J.E. Biphasic dosing regimen of meclizine for prevention of postoperative nausea and vomiting in a high-risk population. AANA J. 2010, 78, 55–62. [Google Scholar]
- Salmito, M.C.; Duarte, J.A.; Morganti, L.O.G.; Brandão, P.V.C.; Nakao, B.H.; Villa, T.R.; Ganança, F.F. Prophylactic treatment of vestibular migraine. Braz. J. Otorhinolaryngol. 2017, 83, 404–410. [Google Scholar] [CrossRef]
- Gohil, V.M.; Offner, N.; Walker, J.A.; Sheth, S.A.; Fossale, E.; Gusella, J.F.; MacDonald, M.E.; Neri, C.; Mootha, V.K. Meclizine is neuroprotective in models of Huntington’s disease. Hum. Mol. Genet. 2011, 20, 294–300. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Maciejewski, J.P.; Wilmink, J.W.; van Noorden, C.J.F. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene 2018, 37, 1949–1960. [Google Scholar] [CrossRef]
- Wang, P.; Wu, J.; Ma, S.; Zhang, L.; Yao, J.; Hoadley, K.A.; Wilkerson, M.D.; Perou, C.M.; Guan, K.-L.; Shen, H.-R.; et al. Oncometabolite D-2-Hydroxyglutarate Inhibits ALKBH DNA Repair Enzymes and Sensitizes IDH Mutant Cells to Alkylating Agents. Cell Rep. 2015, 13, 2353–2361. [Google Scholar] [CrossRef]
- Mallette, F.A.; Mattiroli, F.; Cui, G.; Young, L.C.; Hendzel, M.J.; Mer, G.; Sixma, T.K.; Richard, S. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012, 31, 1865–1878. [Google Scholar] [CrossRef]
- Jihong, Z.; Malcolm, F.G.S.; Tracey, D.B. Temozolomide: Mechanisms of Action, Repair and Resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef]
- Qi, S.; Yu, L.; Gui, S.; Ding, Y.; Han, H.; Zhang, X.; Wu, L.; Yao, F. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci. 2012, 103, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Nevo, I.; Woolard, K.; Cam, M.; Li, A.; Webster, J.D.; Kotliarov, Y.; Kim, H.S.; Ahn, S.; Walling, J.; Kotliarova, S.; et al. Identification of Molecular Pathways Facilitating Glioma Cell Invasion In Situ. PLoS ONE 2014, 9, e111783. [Google Scholar] [CrossRef] [PubMed]
- Son, M.J.; Woolard, K.; Nam, D.-H.; Lee, J.; Fine, H.A. SSEA-1 Is an Enrichment Marker for Tumor-Initiating Cells in Human Glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandoval, J.A.; Tomilov, A.; Datta, S.; Allen, S.; O’Donnell, R.; Sears, T.; Woolard, K.; Kovalskyy, D.; Angelastro, J.M.; Cortopassi, G. Novel mTORC1 Inhibitors Kill Glioblastoma Stem Cells. Pharmaceuticals 2020, 13, 419. https://doi.org/10.3390/ph13120419
Sandoval JA, Tomilov A, Datta S, Allen S, O’Donnell R, Sears T, Woolard K, Kovalskyy D, Angelastro JM, Cortopassi G. Novel mTORC1 Inhibitors Kill Glioblastoma Stem Cells. Pharmaceuticals. 2020; 13(12):419. https://doi.org/10.3390/ph13120419
Chicago/Turabian StyleSandoval, Jose A., Alexey Tomilov, Sandipan Datta, Sonia Allen, Robert O’Donnell, Thomas Sears, Kevin Woolard, Dmytro Kovalskyy, James M. Angelastro, and Gino Cortopassi. 2020. "Novel mTORC1 Inhibitors Kill Glioblastoma Stem Cells" Pharmaceuticals 13, no. 12: 419. https://doi.org/10.3390/ph13120419
APA StyleSandoval, J. A., Tomilov, A., Datta, S., Allen, S., O’Donnell, R., Sears, T., Woolard, K., Kovalskyy, D., Angelastro, J. M., & Cortopassi, G. (2020). Novel mTORC1 Inhibitors Kill Glioblastoma Stem Cells. Pharmaceuticals, 13(12), 419. https://doi.org/10.3390/ph13120419