Biological Characterization of 8-Cyclopropyl-2-(pyridin-3-yl)thiazolo[5,4-f]quinazolin-9(8H)-one, a Promising Inhibitor of DYRK1A

 ,
,  ,
, 
Abstract
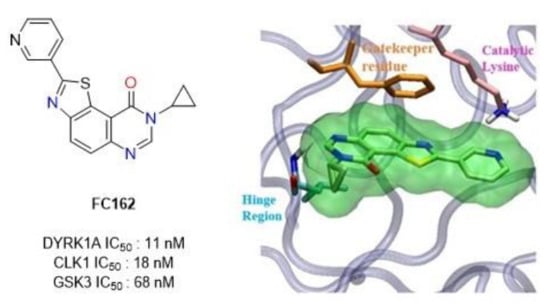
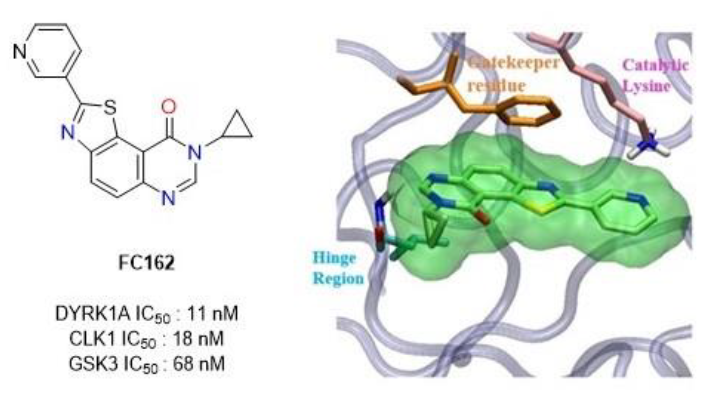
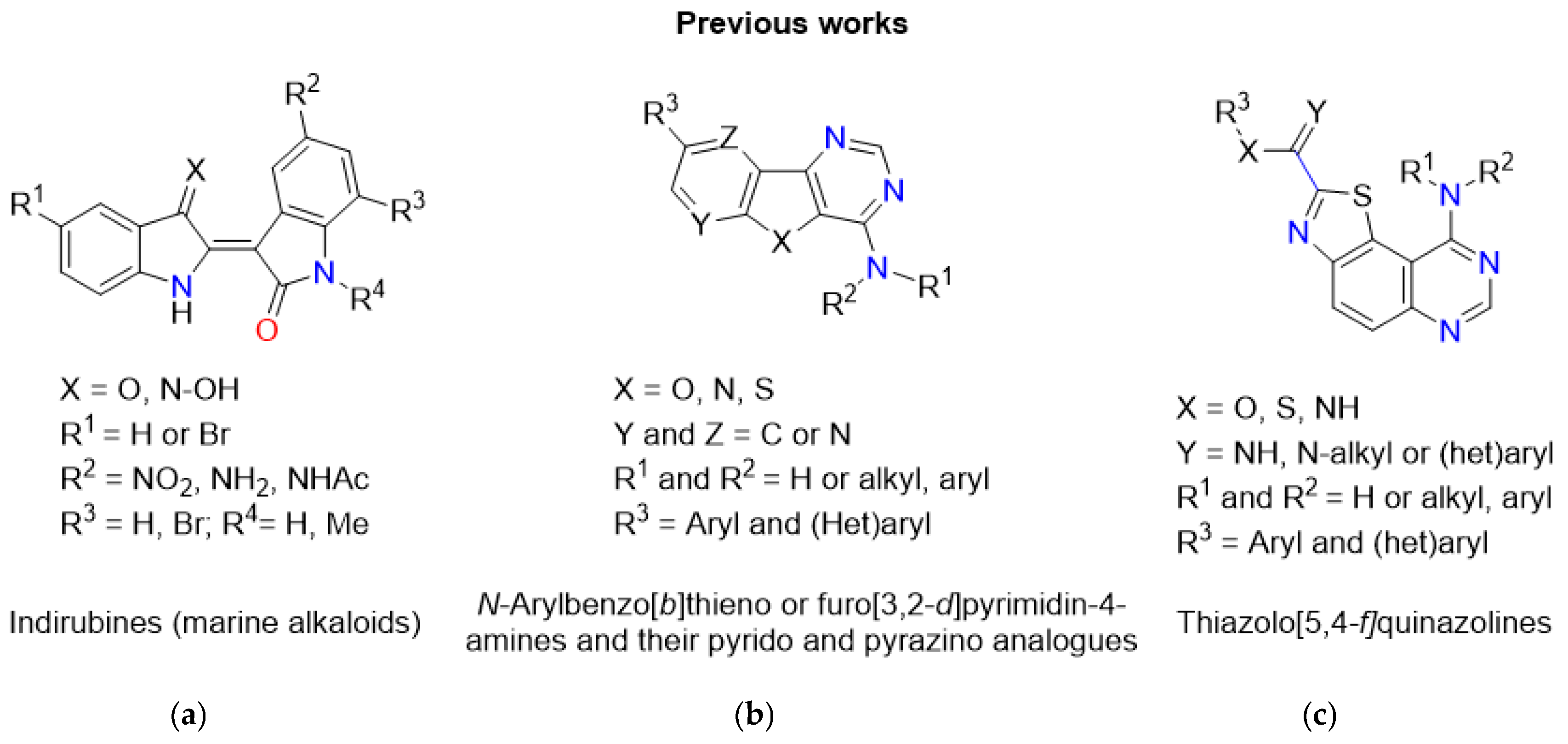
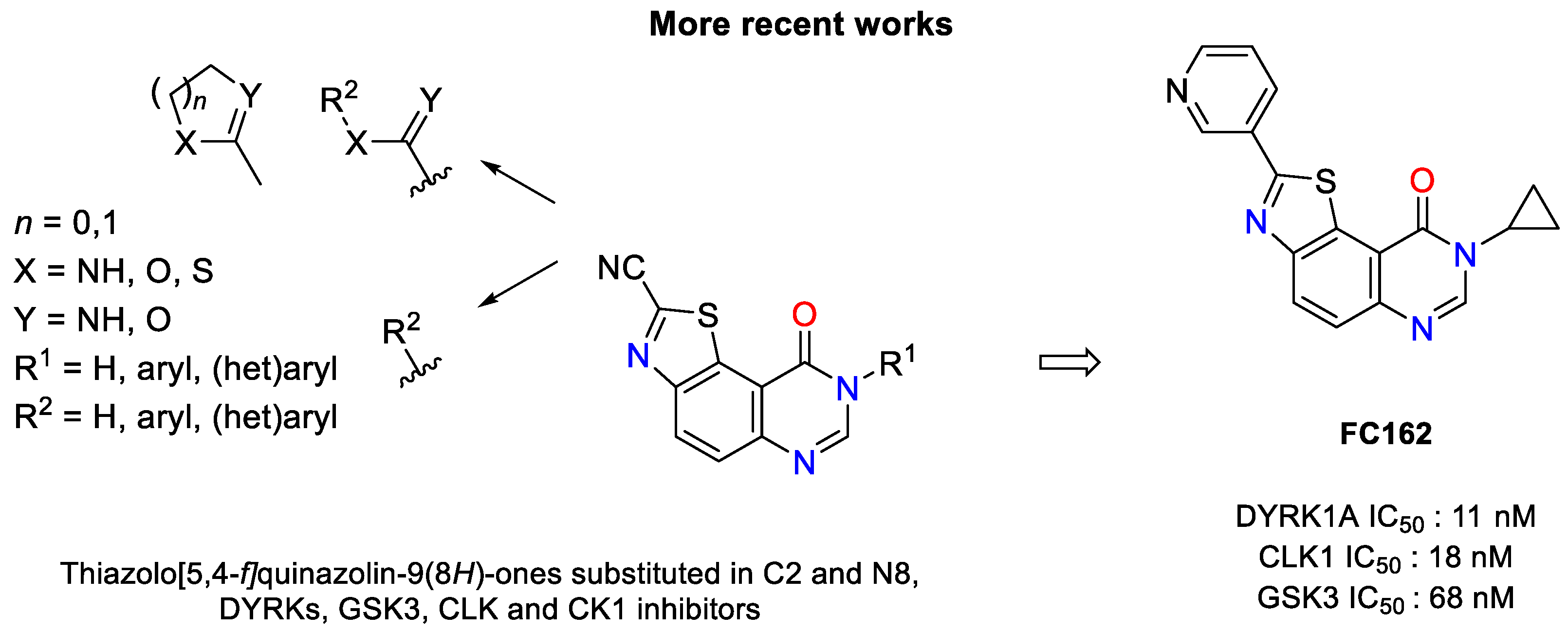
1. Introduction
2. Results
2.1. Chemistry
2.2. Biological Studies
2.2.1. BBB Permeability Assay
2.2.2. Effect of FC162 on Thr212-Tau Phosphorylation in SH-SY5Y Cells
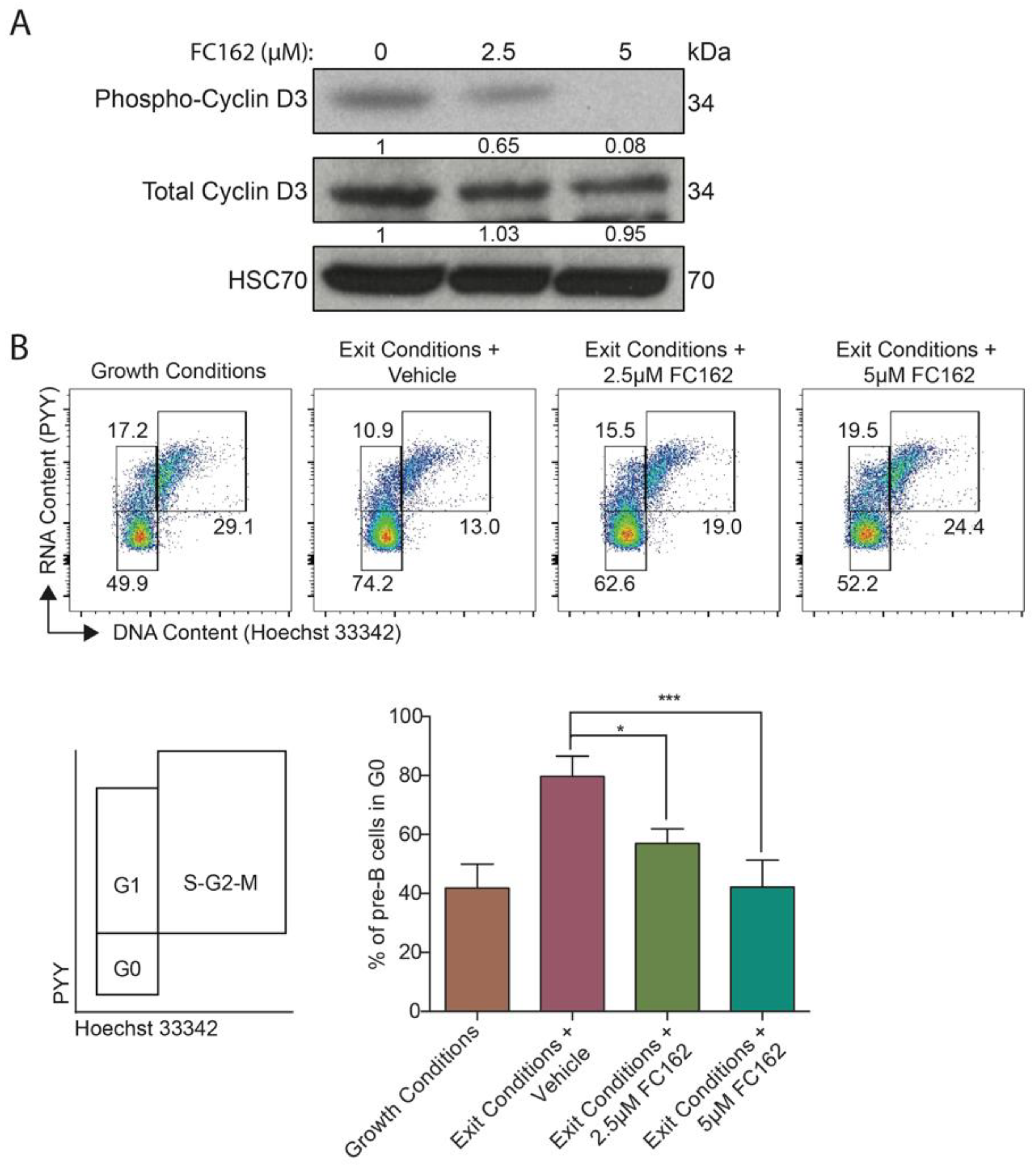
2.2.3. DYRK1A-Specific Inhibitory Activity in Pre-B Cells
3. Discussion
4. Material and Methods
4.1. PAMPA-BBB Permeability Assay
4.2. Effect of FC162 Thr212-Tau Phosphorylation in SH-SY5Y Cells
4.2.1. Culture and Treatment of Cell Lines
4.2.2. Cell Lysis, Electrophoresis, and Western Blotting
4.3. DYRK1A-Specific Inhibitory Activity in Pre-B Cells
4.3.1. Immunoblotting
4.3.2. Cell Cycle Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Dirice, E.; Walpita, D.; Vetere, A.; Meier, B.C.; Kahraman, S.; Hu, J.; Dancík, V.; Burns, S.M.; Gilbert, T.J.; Olson, D.E.; et al. Inhibition of DYRK1A stimulated human β-cell proliferation. Diabetes 2016, 65, 1660–1671. [Google Scholar] [CrossRef]
- Belgardt, B.F.; Lammert, E. DYRK1A: A promising drug target for islet transplant-based diabetes therapeutics. Diabetes 2016, 65, 1496–1498. [Google Scholar] [CrossRef] [PubMed]
- Ionescu, A.; Dufrasne, F.; Gelbcke, M.; Jabin, I.; Kiss, R.; Lamoral-Theys, D. DYRK1A kinase inhibitors with emphasis on cancer. Mini-Rev. Med. Chem. 2012, 12, 1315–1329. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Martinez, P.; Zahonero, C.; Sanchez-Gomez, P. DYRK: The double-edge kinase as a protagonist in cell growth and tumorigenesis. Mol. Cell. Oncol. 2015, 2, e970048. [Google Scholar] [CrossRef] [PubMed]
- Duchon, A.; Herault, Y. DYRK1A, a dosage-sensitive gene involved in neurodevelopment disorders, is a target for drug development in Down syndrome. Front. Behav. Neurosci. 2016, 10, 104–120. [Google Scholar] [CrossRef] [PubMed]
- Branca, C.; Shaw, D.M.; Belfiore, R.; Gokhale, V.; Shaw, A.Y.; Foley, C.; Smith, B.; Hulme, C.; Dunckley, T.; Meechoovet, B.; et al. Dyrk1 inhibition improves Alzheimer’s disease-like pathology. Aging Cell 2017, 16, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Stotani, S.; Giordanetto, F.; Medda, F. DYRK1A inhibition as potential treatment for Alzheimer’s disease. Future Med. Chem. 2016, 8, 681–696. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Fruit, C.; Herault, Y.; Meijer, L.; Besson, T. Dual-specificity tyrosine phosphorylation regulated kinase 1A (DYRK1A) inhibitors: A survey of recent patent literature. Expert Opin. Ther. Pat. 2017, 27, 1183–1199. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. Small-molecule kinase inhibitors: An analysis of FDA-approved drugs. Drug Discov. Today 2016, 21, 5–10. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [PubMed]
- Beauchard, A.; Ferandin, Y.; Frère, S.; Lozach, O.; Blairvacq, M.; Meijer, M.; Thiéry, V.; Besson, T. Synthesis of novel 5-substituted indirubins as protein kinases inhibitors. Bioorg. Med. Chem. 2006, 14, 6434–6443. [Google Scholar] [CrossRef] [PubMed]
- Beauchard, A.; Laborie, H.; Rouillard, H.; Ferandin, Y.; Lozach, O.; Le Guével, R.; Guillouzo, C.; Meijer, L.; Besson, T.; Thiéry, V. Synthesis and kinase inhibitory activity of novel substituted indigoids. Bioorg. Med. Chem. 2009, 17, 6257–6263. [Google Scholar] [CrossRef] [PubMed]
- Loidreau, Y.; Marchand, P.; Dubouilh-Benard, C.; Nourrisson, M.-R.; Duflos, M.; Lozach, O.; Loaëc, N.; Meijer, L.; Besson, T. Synthesis and biological evaluation of N-arylbenzo[b]thieno[3,2-d]pyrimidin-4-amines and their pyrido and pyrazino analogues as Ser/Thr kinase inhibitors. Eur. J. Med. Chem. 2012, 58, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Loidreau, Y.; Marchand, P.; Dubouilh-Benard, C.; Nourrisson, M.-R.; Duflos, M.; Lozach, O.; Meijer, L.; Besson, T. Synthesis and biological evaluation of N-aryl-7-methoxybenzo[b]furo[3,2-d]pyrimidin-4-amines and their N-arylbenzo[b]thieno[3,2-d]pyrimidin-4-amine analogues as dual inhibitors of CLK1 and DYRK1A kinases. Eur. J. Med. Chem. 2013, 59, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Loidreau, Y.; Deau, E.; Marchand, P.; Nourrisson, M.-R.; Logé, C.; Coadou, G.; Loaëc, N.; Meijer, L.; Besson, T. Synthesis and molecular modelling studies of 8-arylpyrido[3’,2’:4,5]thieno[3,2-d]pyrimidin-4-amines as multitarget Ser/Thr kinases inhibitors. Eur. J. Med. Chem. 2015, 92, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Testard, A.; Logé, C.; Léger, B.; Robert, J.-M.; Lozach, O.; Blairvacq, M.; Meijer, L.; Thiéry, V.; Besson, T. Thiazolo[5,4-f]quinazolin-9-ones, inhibitors of glycogen synthase kinase-3. Bioorg. Med. Chem. Lett. 2006, 16, 3419–3423. [Google Scholar] [CrossRef]
- Logé, C.; Testard, A.; Thiéry, V.; Lozach, O.; Blairvacq, M.; Robert, J.-M.; Meijer, L.; Besson, T. Novel 9-oxo-thiazolo[5,4-f]quinazoline-2-carbonitrile derivatives as dual cyclin-dependent kinase 1 (CDK1)/glycogen synthase kinase-3 (GSK-3) inhibitors: Synthesis, biological evaluation and molecular modeling studies. Eur. J. Med. Chem. 2008, 43, 1469–1477. [Google Scholar] [CrossRef]
- Deau, E.; Loidreau, Y.; Marchand, P.; Nourrisson, M.-R.; Loaëc, N.; Meijer, L.; Levacher, V.; Besson, T. Synthesis of novel 7-substituted pyrido[2’,3’:4,5]furo[3,2-d]pyrimidin-4-amines and their N-aryl analogues and evaluation of their inhibitory activity against Ser/Thr Kinases. Bioorg. Med. Chem. Lett. 2013, 23, 6784–6788. [Google Scholar] [CrossRef]
- Foucourt, A.; Hédou, D.; Dubouilh-Benard, C.; Désiré, L.; Casagrande, A.-S.; Leblond, B.; Loaëc, N.; Meijer, L.; Besson, T. Design and synthesis of thiazolo[5,4-f]quinazolines as DYRK1A inhibitors, Part I. Molecules 2014, 19, 15546–15571. [Google Scholar] [CrossRef]
- Foucourt, A.; Hédou, D.; Dubouilh-Benard, C.; Désiré, L.; Casagrande, A.-S.; Leblond, B.; Loaëc, N.; Meijer, L.; Besson, T. Design and synthesis of thiazolo[5,4-f]quinazolines as DYRK1A inhibitors, Part II. Molecules 2014, 19, 15411–15439. [Google Scholar] [CrossRef] [PubMed]
- Leblond, B.; Casagrande, A.-S.; Désiré, L.; Foucourt, A.; Besson, T. DYRK1 inhibitors and uses thereof WO2013026806. Chem. Abstr. 2013, 158, 390018. [Google Scholar]
- Chaikuad, A.; Diharce, J.; Schröder, M.; Foucourt, A.; Leblond, B.; Casagrande, A.-S.; Désiré, L.; Bonnet, P.; Knapp, S.; Besson, T. An unusual binding mode of the methyl 9-anilinothiazolo[5,4-f]quinazoline-2 carbimidates (EHT 1610 and EHT 5372) confers high selectivity for DYRK kinases. J. Med. Chem. 2016, 59, 10315–10321. [Google Scholar] [CrossRef] [PubMed]
- Coutadeur, S.; Benyamine, H.; Delalonde, L.; de Oliveira, C.; Leblond, B.; Foucourt, A.; Besson, T.; Casagrande, A.-S.; Taverne, T.; Girard, A.; et al. A Novel DYRK1A (dual specificity tyrosine phosphorylation-regulated kinase 1A) inhibitor for the treatment of Alzheimer’s disease: Effect on tau and amyloid pathologies in vitro. J. Neurochem. 2015, 133, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Hédou, D.; Godeau, J.; Loaëc, N.; Meijer, L.; Fruit, C.; Besson, T. Synthesis of thiazolo[5,4-f]quinazolin-9(8H)-ones as multi-target directed ligands of Ser/Thr kinases. Molecules 2016, 21, 578. [Google Scholar] [CrossRef] [PubMed]
- Hédou, D.; Dubouilh-Benard, C.; Loaëc, N.; Meijer, L.; Fruit, C.; Besson, T. Synthesis of bioactive 2-(arylamino)thiazolo[5,4-f]-quinazolin-9-ones via the Hügershoff reaction or Cu-catalyzed intramolecular C-S bond formation. Molecules 2016, 21, 794. [Google Scholar] [CrossRef]
- Couly, F.; Harari, M.; Dubouilh-Benard, C.; Bailly, L.; Petit, E.; Diharce, J.; Bonnet, P.; Meijer, L.; Fruit, C.; Besson, T. Development of kinase inhibitors via metal-catalyzed C–H arylation of 8-alkyl-thiazolo[5,4-f]-quinazolin-9-ones Designed by Fragment-Growing Studies. Molecules 2018, 23, 2181. [Google Scholar] [CrossRef]
- Becker, W.; Joost, H.G. Structural and functional characteristics of Dyrk, a novel subfamily of protein kinases with dual specificity. Prog. Nucleic Acid Res. Mol. Biol. 1998, 62, 1–17. [Google Scholar] [CrossRef]
- Aranda, S.; Laguna, A.; de La Luna, S. DYRK family of protein kinases: Evolutionary relationships, biochemical properties and functional roles. FASEB J. 2011, 25, 449–462. [Google Scholar] [CrossRef]
- Hanks, S.K.; Hunter, T. Protein kinases 6. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. 1995, 9, 576–596. [Google Scholar] [CrossRef]
- Jarhad, D.B.; Mashelkar, K.K.; Kim, H.-R.; Noh, M.; Jeong, L.S. Dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) inhibitors as potential therapeutics. J. Med. Chem. 2018, 61, 9791–9810. [Google Scholar] [CrossRef] [PubMed]
- Czarna, A.; Wang, J.; Zelencova, D.; Liu, Y.; Deng, X.; Choi, H.G.; Zhang, T.; Zhou, W.; Chang, J.W.; Kildalsen, H.; et al. Novel Scaffolds for Dual Specificity Tyrosine-Phosphorylation-Regulated Kinase (DYRK1A) Inhibitors. J. Med. Chem. 2018, 61, 7560–7572. [Google Scholar] [CrossRef] [PubMed]
- Harari, M.; Couly, F.; Fruit, C.; Besson, T. Pd-catalyzed and copper assisted regioselective sequential C2 and C7 arylation of thiazolo[5,4-f]quinazolin-9(8H)-one with aryl halides. Org. Lett. 2016, 18, 3282–3285. [Google Scholar] [CrossRef] [PubMed]
- Couly, F.; Dubouilh-Benard, C.; Besson, T.; Fruit, C. Arylation of thiazolo[5,4-f]quinazolin-9(8H)-one backbone: Synthesis of an array of potential kinase inhibitors. Synthesis 2017, 49, 4615–4622. [Google Scholar] [CrossRef]
- Primot, A.; Baratte, B.; Gompel, M.; Borgne, A.; Liabeuf, S.; Romette, J.L.; Jho, E.H.; Costantini, F.; Meijer, L. Purification of GSK-3 by affinity chromatography on immobilized axin. Protein Expr. Purif. 2000, 20, 394–404. [Google Scholar] [CrossRef]
- Reinhardt, J.; Ferandin, Y.; Meijer, L. Purification of CK1 by affinity chromatography on immobilised axin. Protein Expr. Purif. 2007, 54, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Debdab, M.; Carreaux, F.; Renault, S.; Soundararajan, M.; Fedorov, O.; Filippakopoulos, P.; Lozach, O.; Babault, L.; Tahtouh, T.; Baratte, B.; et al. Leucettines, a class of potent inhibitors of cdc2-like kinases and dual specificity, tyrosine phosphorylation regulated kinases derived from the marine sponge leucettamine B. Modulation of alternative pre-RNA splicing. J. Med. Chem. 2011, 54, 4172–4186. [Google Scholar] [CrossRef] [PubMed]
- Tahtouh, T.; Elkins, J.M.; Filippakopoulos, P.; Soundararajan, M.; Burgy, G.; Durieu, E.; Cochet, C.; Schmid, R.S.; Lo, D.C.; Delhommel, F.; et al. Selectivity, cocrystal structures and neuroprotective properties of leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J. Med. Chem. 2012, 55, 9312–9330. [Google Scholar] [CrossRef]
- Thompson, B.; Bhansali, R.; Diebold, L.; Cook, D.E.; Stolzenburg, L.; Casagrande, A.-S.; Besson, T.; Leblond, B.; Desire, L.; Malinge, S.; et al. DYRK1A controls the transition from proliferation to quiescence during lymphoid development by destabilizing Cyclin D3. J. Exp. Med. 2015, 21, 723–740; [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Duchon, A.; Manousopoulou, A.; Loaëc, N.; Villiers, B.; Pani, G.; Karatas, M.; Mechling, A.E.; Harsan, L.A.; Limanton, E.; et al. Correction of cognitive deficits in mouse models of Down syndrome by pharmacological inhibitor of DYRK1A. Dis. Model Mech. 2018, 11, dmm035634. [Google Scholar] [CrossRef]
- Ryoo, S.R.; Jeong, H.K.; Radnaabazar, C.; Yoo, J.J.; Cho, H.J.; Lee, H.W.; Kim, I.S.; Cheon, Y.H.; Ahn, Y.S.; Chung, S.H.; et al. DYRK1A-mediated hyperphosphorylation of Tau. A functional link between Down syndrome and Alzheimer disease. J. Biol. Chem. 2007, 282, 34850–34857. [Google Scholar] [CrossRef] [PubMed]
- Trushina, N.I.; Bakota, L.; Mulkidjanian, A.Y.; Brandt, R. The Evolution of Tau Phosphorylation and Interactions. Front Aging Neurosci. 2019, 11, 256. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Byers, H.L.; Wray, S.; Leung, K.Y.; Saxton, M.J.; Seereeram, A.; Reynolds, C.H.; Ward, M.A.; Anderton, B.H. Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J. Biol. Chem. 2007, 282, 23645–23654. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product Name | Concentration (μM) | logPe | Pe (10−6 cm−1) | BBB Cross |
|---|---|---|---|---|
| FC162 | 100 | −4.92 ± 0.04 | 12.18 ± 1.10 | YES |
| Theophylline | 250 | −6.26 ± 0.03 | 0.55 ± 0.03 | NO |
| Corticosterone | 100 | −486 ± 0.07 | 13.86 ± 2.20 | YES |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fruit, C.; Couly, F.; Bhansali, R.; Rammohan, M.; Lindberg, M.F.; Crispino, J.D.; Meijer, L.; Besson, T. Biological Characterization of 8-Cyclopropyl-2-(pyridin-3-yl)thiazolo[5,4-f]quinazolin-9(8H)-one, a Promising Inhibitor of DYRK1A. Pharmaceuticals 2019, 12, 185. https://doi.org/10.3390/ph12040185
Fruit C, Couly F, Bhansali R, Rammohan M, Lindberg MF, Crispino JD, Meijer L, Besson T. Biological Characterization of 8-Cyclopropyl-2-(pyridin-3-yl)thiazolo[5,4-f]quinazolin-9(8H)-one, a Promising Inhibitor of DYRK1A. Pharmaceuticals. 2019; 12(4):185. https://doi.org/10.3390/ph12040185
Chicago/Turabian StyleFruit, Corinne, Florence Couly, Rahul Bhansali, Malini Rammohan, Mattias F. Lindberg, John D. Crispino, Laurent Meijer, and Thierry Besson. 2019. "Biological Characterization of 8-Cyclopropyl-2-(pyridin-3-yl)thiazolo[5,4-f]quinazolin-9(8H)-one, a Promising Inhibitor of DYRK1A" Pharmaceuticals 12, no. 4: 185. https://doi.org/10.3390/ph12040185
APA StyleFruit, C., Couly, F., Bhansali, R., Rammohan, M., Lindberg, M. F., Crispino, J. D., Meijer, L., & Besson, T. (2019). Biological Characterization of 8-Cyclopropyl-2-(pyridin-3-yl)thiazolo[5,4-f]quinazolin-9(8H)-one, a Promising Inhibitor of DYRK1A. Pharmaceuticals, 12(4), 185. https://doi.org/10.3390/ph12040185