Therapeutic Advances in Regulating the Hepcidin/Ferroportin Axis

Abstract
1. Introduction
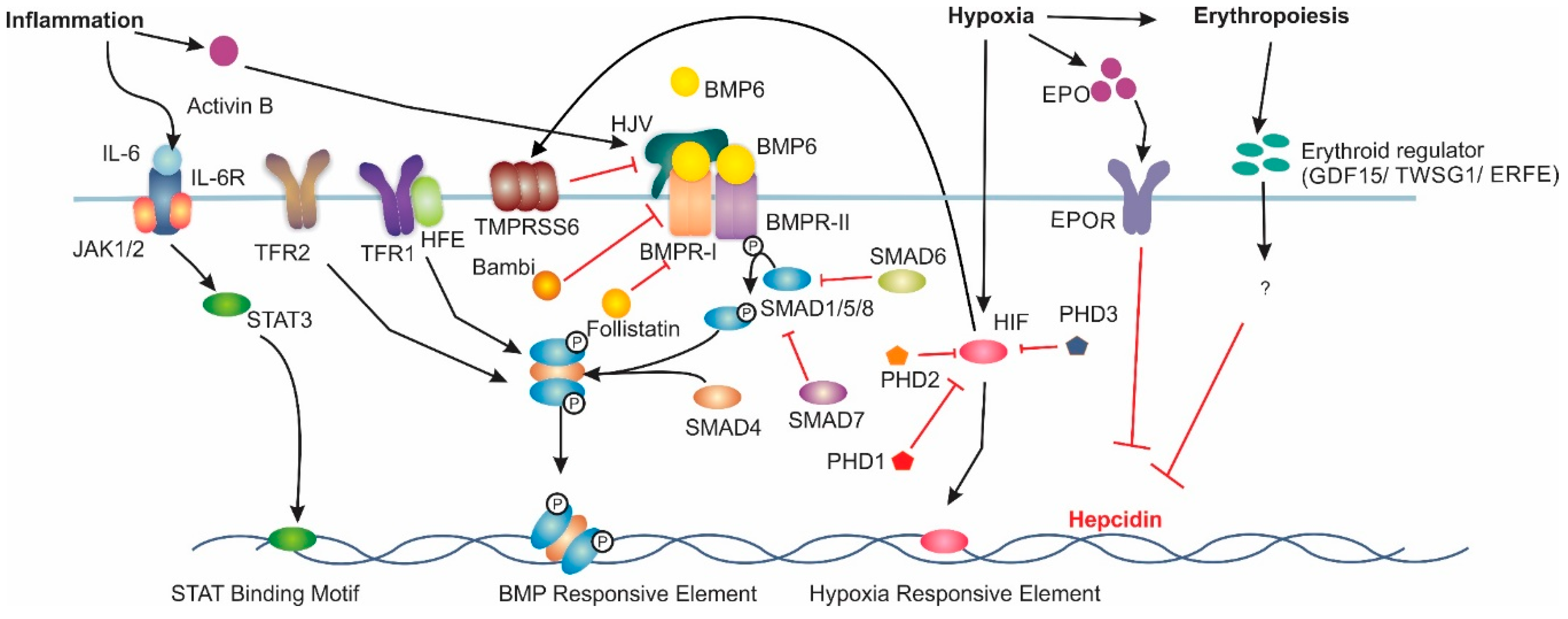
2. Hepcidin Regulation
Extrahepatic Hepcidin
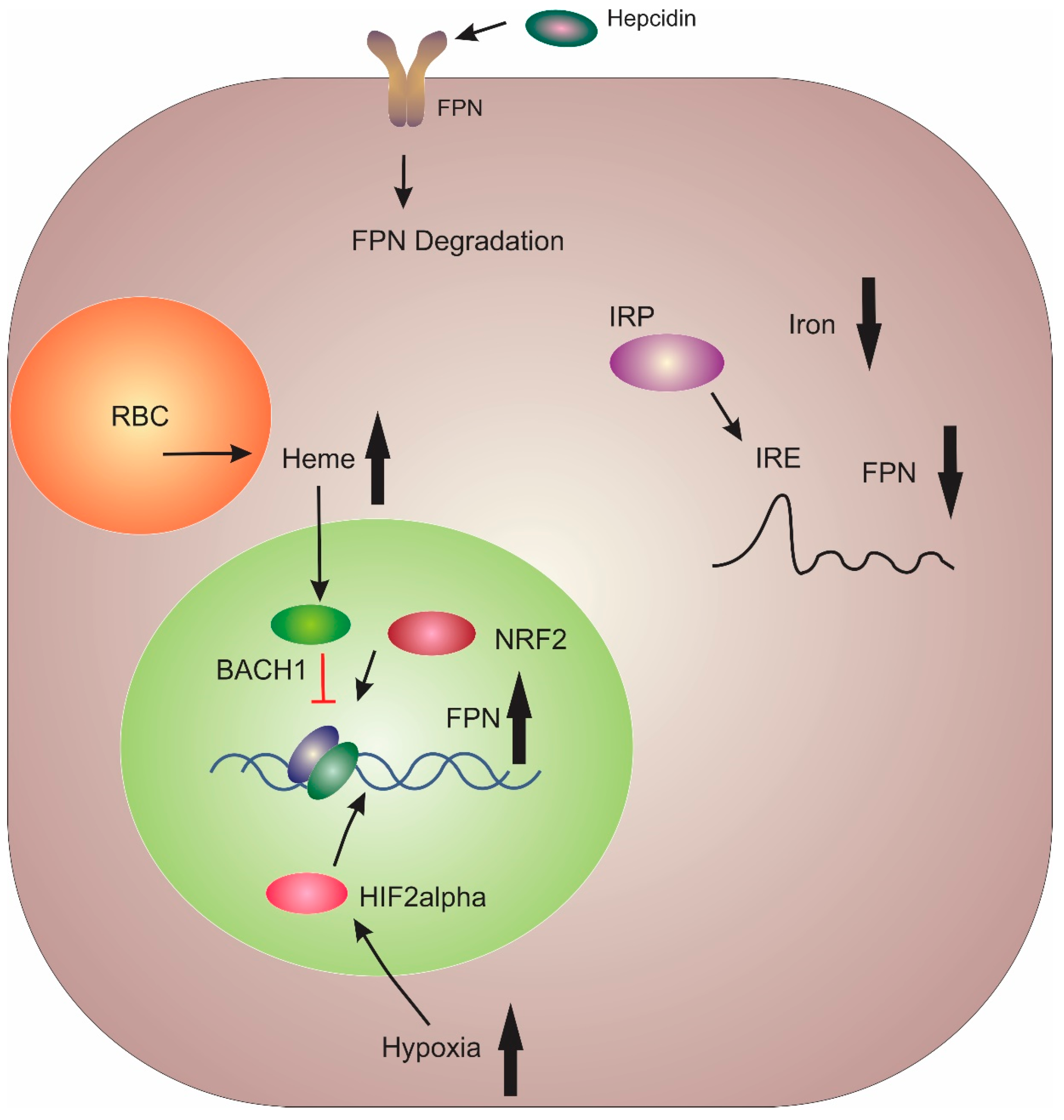
3. Regulation of FPN
4. Hepcidin/FPN Axis Dysregulation
5. Current Therapeutic Treatments for Hepcidin Deficiency
6. Current Therapeutic Treatments for Lowering Hepcidin
6.1. Direct Hepcidin Inhibitors
6.1.1. Anti-Hepcidin Antibodies
6.1.2. Short Interfering and Short Hairpin RNA
6.1.3. Hepcidin-Binding Molecules
6.1.4. Hepcidin-Binding L-RNA Aptamers (Spiegelmers)
6.2. Inhibitors of Hepcidin Production/Synthesis
6.2.1. Heparin-Based Targeting of the BMP/SMAD Pathway
6.2.2. Bone Morphogenetic Protein Receptor (BMPR) Inhibitors
6.2.3. Hemojuvelin (HJV) and Transferrin Receptor 2 (TFR2) Inhibitors
6.2.4. Targeting the IL-6/STAT3 Pathway
6.2.5. Hypoxia-Inducible Factors (HIF) Stabilisers
7. Current Therapeutic Treatments Targeting Ferroportin
Ferroportin Agonists
8. Current Therapeutic Treatments for Increasing Hepcidin
8.1. Hepcidin Agonists
8.2. Hepcidin Analogues
8.3. Small Molecule Hepcidin Agonists
9. Conclusion
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, J.; Sun, B.; Yin, H.; Liu, S. Hepcidin: A promising therapeutic target for iron disorders: A systematic review. Medicine 2016, 95, e3150. [Google Scholar] [CrossRef]
- Sebastiani, G.; Wilkinson, N.; Pantopoulos, K. Pharmacological targeting of the hepcidin/ferroportin axis. Front. Pharmacol. 2016, 7, 160. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, G.; Pantopoulos, K. Systemic iron homeostasis and erythropoiesis. IUBMB Life 2017, 69, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, G.; Pantopoulos, K. Iron metabolism and toxicity. Toxicol. Appl. Pharmacol. 2005, 202, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A red carpet for iron metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef]
- Drakesmith, H.; Nemeth, E.; Ganz, T. Ironing out ferroportin. Cell Metab. 2015, 22, 777–787. [Google Scholar] [CrossRef]
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef]
- Nemeth, E.; Valore, E.V.; Territo, M.; Schiller, G.; Lichtenstein, A.; Ganz, T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003, 101, 2461–2463. [Google Scholar] [CrossRef]
- Krause, A.; Sillard, R.; Kleemeier, B.; Kluver, E.; Maronde, E.; Conejo-Garcia, J.R.; Forssmann, W.G.; Schulz-Knappe, P.; Nehls, M.C.; Wattler, F.; et al. Isolation and biochemical characterization of LEAP-2, a novel blood peptide expressed in the liver. Protein Sci. 2003, 12, 143–152. [Google Scholar] [CrossRef]
- Valore, E.V.; Ganz, T. Posttranslational processing of hepcidin in human hepatocytes is mediated by the prohormone convertase furin. Blood Cells Mol. Dis. 2008, 40, 132–138. [Google Scholar] [CrossRef]
- Nemeth, E.; Preza, G.C.; Jung, C.L.; Kaplan, J.; Waring, A.J.; Ganz, T. The N-terminus of hepcidin is essential for its interaction with ferroportin: Structure-function study. Blood 2006, 107, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Systemic iron homeostasis. Physiol. Rev. 2013, 93, 1721–1741. [Google Scholar] [CrossRef] [PubMed]
- Canali, S.; Zumbrennen-Bullough, K.B.; Core, A.B.; Wang, C.Y.; Nairz, M.; Bouley, R.; Swirski, F.K.; Babitt, J.L. Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood 2017, 129, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Andriopoulos, B.; Corradini, E.; Xia, Y.; Faasse, S.A.; Chen, S.Z.; Grgurevic, L.; Knutson, M.D.; Pietrangelo, A.; Vukicevic, S.; Lin, H.Y.; et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 2009, 41, 482–487. [Google Scholar] [CrossRef]
- Meynard, D.; Kautz, L.; Darnaud, V.; Canonne-Hergaux, F.; Coppin, H.; Roth, M.P. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat. Genet. 2009, 41, 478–481. [Google Scholar] [CrossRef]
- Truksa, J.; Peng, H.; Lee, P.; Beutler, E. Bone morphogenetic proteins 2, 4, and 9 stimulate murine hepcidin 1 expression independently of Hfe, transferrin receptor 2 (Tfr2), and IL-6. Proc. Natl. Acad. Sci. USA 2006, 103, 10289–10293. [Google Scholar] [CrossRef]
- Canali, S.; Wang, C.-Y.; Zumbrennen-Bullough, K.B.; Bayer, A.; Babitt, J.L. Bone morphogenetic protein 2 controls iron homeostasis in mice independent of Bmp6. Am. J. Hematol. 2017, 92, 1204–1213. [Google Scholar] [CrossRef]
- Babitt, J.L.; Huang, F.W.; Wrighting, D.M.; Xia, Y.; Sidis, Y.; Samad, T.A.; Campagna, J.A.; Chung, R.T.; Schneyer, A.L.; Woolf, C.J.; et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet. 2006, 38, 531–539. [Google Scholar] [CrossRef]
- Steinbicker, A.U.; Bartnikas, T.B.; Lohmeyer, L.K.; Leyton, P.; Mayeur, C.; Kao, S.M.; Pappas, A.E.; Peterson, R.T.; Bloch, D.B.; Yu, P.B.; et al. Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood 2011, 118, 4224–4230. [Google Scholar] [CrossRef]
- Mayeur, C.; Leyton, P.A.; Kolodziej, S.A.; Yu, B.L.; Bloch, K.D. BMP type II receptors have redundant roles in the regulation of hepatic hepcidin gene expression and iron metabolism. Blood 2014, 124, 2116–2123. [Google Scholar] [CrossRef]
- Miyazono, K.; Kamiya, Y.; Morikawa, M. Bone morphogenetic protein receptors and signal transduction. J. Biochem. 2010, 147, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.H.; Li, C.; Xu, X.; Zheng, Y.; Xiao, C.; Zerfas, P.; Cooperman, S.; Eckhaus, M.; Rouault, T.; Mishra, L.; et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005, 2, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Truksa, J.; Lee, P.; Beutler, E. Two BMP responsive elements, STAT, and bZIP/HNF4/COUP motifs of the hepcidin promoter are critical for BMP, SMAD1, and HJV responsiveness. Blood 2009, 113, 688–695. [Google Scholar] [CrossRef] [PubMed]
- An, P.; Wang, H.; Wu, Q.; Wang, J.; Xia, Z.; He, X.; Wang, X.; Chen, Y.; Min, J.; Wang, F. Smad7 deficiency decreases iron and haemoglobin through hepcidin up-regulation by multilayer compensatory mechanisms. J. Cell. Mol. Med. 2018, 22, 3035–3044. [Google Scholar] [CrossRef]
- Meynard, D.; Vaja, V.; Sun, C.C.; Corradini, E.; Chen, S.; Lopez-Otin, C.; Grgurevic, L.; Hong, C.C.; Stirnberg, M.; Gutschow, M.; et al. Regulation of TMPRSS6 by BMP6 and iron in human cells and mice. Blood 2011, 118, 747–756. [Google Scholar] [CrossRef]
- Silvestri, L.; Pagani, A.; Nai, A.; De Domenico, I.; Kaplan, J.; Camaschella, C. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008, 8, 502–511. [Google Scholar] [CrossRef]
- Lin, L.; Goldberg, Y.P.; Ganz, T. Competitive regulation of hepcidin mRNA by soluble and cell-associated hemojuvelin. Blood 2005, 106, 2884–2889. [Google Scholar] [CrossRef]
- Verga Falzacappa, M.V.; Vujic Spasic, M.; Kessler, R.; Stolte, J.; Hentze, M.W.; Muckenthaler, M.U. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood 2007, 109, 353–358. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Iron homeostasis in host defence and inflammation. Nat. Rev. Immunol. 2015, 15, 500–510. [Google Scholar] [CrossRef]
- Canali, S.; Core, A.B.; Zumbrennen-Bullough, K.B.; Merkulova, M.; Wang, C.Y.; Schneyer, A.L.; Pietrangelo, A.; Babitt, J.L. Activin B induces noncanonical SMAD1/5/8 signaling via BMP type I receptors in hepatocytes: Evidence for a role in hepcidin induction by inflammation in male mice. Endocrinology 2016, 157, 1146–1162. [Google Scholar] [CrossRef]
- Kanamori, Y.; Sugiyama, M.; Hashimoto, O.; Murakami, M.; Matsui, T.; Funaba, M. Regulation of hepcidin expression by inflammation-induced activin B. Sci. Rep. 2016, 6, 38702. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Kapitsinou, P.P.; Liu, Q.; Unger, T.L.; Rha, J.; Davidoff, O.; Keith, B.; Epstein, J.A.; Moores, S.L.; Erickson-Miller, C.L.; Haase, V.H. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood 2010, 116, 3039–3048. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Ohneda, O.; Yamashita, T.; Takahashi, S.; Suzuki, N.; Nakajima, O.; Kawauchi, S.; Ema, M.; Shibahara, S.; Udono, T.; et al. HLF/HIF-2alpha is a key factor in retinopathy of prematurity in association with erythropoietin. EMBO J. 2003, 22, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Davidoff, O.; Niss, K.; Haase, V.H. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J. Clin. Investig. 2012, 122, 4635–4644. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Zinkernagel, A.S.; Schuepbach, R.A.; Rankin, E.; Vaulont, S.; Haase, V.H.; Nizet, V.; Johnson, R.S. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J. Clin. Investig. 2007, 117, 1926–1932. [Google Scholar] [CrossRef]
- Volke, M.; Gale, D.P.; Maegdefrau, U.; Schley, G.; Klanke, B.; Bosserhoff, A.K.; Maxwell, P.H.; Eckardt, K.U.; Warnecke, C. Evidence for a lack of a direct transcriptional suppression of the iron regulatory peptide hepcidin by hypoxia-inducible factors. PLoS ONE 2009, 4, e7875. [Google Scholar] [CrossRef]
- Silvestri, L.; Pagani, A.; Camaschella, C. Furin-mediated release of soluble hemojuvelin: A new link between hypoxia and iron homeostasis. Blood 2008, 111, 924–931. [Google Scholar] [CrossRef]
- Maurer, E.; Gutschow, M.; Stirnberg, M. Matriptase-2 (TMPRSS6) is directly up-regulated by hypoxia inducible factor-1: Identification of a hypoxia-responsive element in the TMPRSS6 promoter region. Biol. Chem. 2012, 393, 535–540. [Google Scholar] [CrossRef]
- Lakhal, S.; Schodel, J.; Townsend, A.R.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. Regulation of type II transmembrane serine proteinase TMPRSS6 by hypoxia-inducible factors: New link between hypoxia signaling and iron homeostasis. J. Biol. Chem. 2011, 286, 4090–4097. [Google Scholar] [CrossRef]
- Pasricha, S.R.; McHugh, K.; Drakesmith, H. Regulation of hepcidin by erythropoiesis: The story so far. Annu. Rev. Nutr. 2016, 36, 417–434. [Google Scholar] [CrossRef] [PubMed]
- Tanno, T.; Bhanu, N.V.; Oneal, P.A.; Goh, S.H.; Staker, P.; Lee, Y.T.; Moroney, J.W.; Reed, C.H.; Luban, N.L.; Wang, R.H.; et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat. Med. 2007, 13, 1096–1101. [Google Scholar] [CrossRef]
- Tanno, T.; Porayette, P.; Sripichai, O.; Noh, S.J.; Byrnes, C.; Bhupatiraju, A.; Lee, Y.T.; Goodnough, J.B.; Harandi, O.; Ganz, T.; et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood 2009, 114, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Casanovas, G.; Vujic Spasic, M.; Casu, C.; Rivella, S.; Strelau, J.; Unsicker, K.; Muckenthaler, M.U. The murine growth differentiation factor 15 is not essential for systemic iron homeostasis in phlebotomized mice. Haematologica 2013, 98, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Mirciov, C.S.; Wilkins, S.J.; Dunn, L.A.; Anderson, G.J.; Frazer, D.M. Characterization of putative erythroid regulators of hepcidin in mouse models of anemia. PLoS ONE 2017, 12, e0171054. [Google Scholar] [CrossRef] [PubMed]
- Kautz, L.; Jung, G.; Du, X.; Gabayan, V.; Chapman, J.; Nasoff, M.; Nemeth, E.; Ganz, T. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of beta-thalassemia. Blood 2015, 126, 2031–2037. [Google Scholar] [CrossRef] [PubMed]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684. [Google Scholar] [CrossRef]
- Daher, R.; Lefebvre, T.; Puy, H.; Karim, Z. Extrahepatic hepcidin production: The intriguing outcomes of recent years. World J. Clin. Cases 2019, 7, 1926–1936. [Google Scholar] [CrossRef]
- Sow, F.B.; Florence, W.C.; Satoskar, A.R.; Schlesinger, L.S.; Zwilling, B.S.; Lafuse, W.P. Expression and localization of hepcidin in macrophages: A role in host defense against tuberculosis. J. Leukoc. Biol. 2007, 82, 934–945. [Google Scholar] [CrossRef]
- Schwarz, P.; Kübler, J.A.M.; Strnad, P.; Müller, K.; Barth, T.F.E.; Gerloff, A.; Feick, P.; Peyssonnaux, C.; Vaulont, S.; Adler, G.; et al. Hepcidin is localised in gastric parietal cells, regulates acid secretion and is induced by Helicobacter pylori infection. Gut 2012, 61, 193–201. [Google Scholar] [CrossRef]
- Qian, Z.M.; He, X.; Liang, T.; Wu, K.C.; Yan, Y.C.; Lu, L.N.; Yang, G.; Luo, Q.Q.; Yung, W.H.; Ke, Y. Lipopolysaccharides upregulate hepcidin in neuron via microglia and the IL-6/STAT3 signaling pathway. Mol. Neurobiol. 2014, 50, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Urrutia, P.; Aguirre, P.; Esparza, A.; Tapia, V.; Mena, N.P.; Arredondo, M.; Gonzalez-Billault, C.; Nunez, M.T. Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 2013, 126, 541–549. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.T.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 2000, 5, 299–309. [Google Scholar] [CrossRef]
- Donovan, A.; Brownlie, A.; Zhou, Y.; Shepard, J.; Pratt, S.J.; Moynihan, J.; Paw, B.H.; Drejer, A.; Barut, B.; Zapata, A.; et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000, 403, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Abboud, S.; Haile, D.J. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem. 2000, 275, 19906–19912. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Sugianto, P.; Fung, E.; del-Castillo-Rueda, A.; Moran-Jimenez, M.J.; Ganz, T.; Nemeth, E. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012, 15, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Marro, S.; Chiabrando, D.; Messana, E.; Stolte, J.; Turco, E.; Tolosano, E.; Muckenthaler, M.U. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position-7007 of the FPN1 promoter. Haematologica 2010, 95, 1261–1268. [Google Scholar] [CrossRef]
- Sangokoya, C.; Doss, J.F.; Chi, J.T. Iron-responsive miR-485-3p regulates cellular iron homeostasis by targeting ferroportin. PLoS Genet. 2013, 9, e1003408. [Google Scholar] [CrossRef]
- Galy, B.; Ferring-Appel, D.; Becker, C.; Gretz, N.; Grone, H.J.; Schumann, K.; Hentze, M.W. Iron regulatory proteins control a mucosal block to intestinal iron absorption. Cell Rep. 2013, 3, 844–857. [Google Scholar] [CrossRef]
- Nairz, M.; Ferring-Appel, D.; Casarrubea, D.; Sonnweber, T.; Viatte, L.; Schroll, A.; Haschka, D.; Fang, F.C.; Hentze, M.W.; Weiss, G.; et al. Iron regulatory proteins mediate host resistance to salmonella infection. Cell Host Microbe 2015, 18, 254–261. [Google Scholar] [CrossRef]
- Agoro, R.; Mura, C. Inflammation-induced up-regulation of hepcidin and down-regulation of ferroportin transcription are dependent on macrophage polarization. Blood Cells Mol. Dis. 2016, 61, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Liu, X.B.; Quinones, M.; Melby, P.C.; Ghio, A.; Haile, D.J. Regulation of reticuloendothelial iron transporter MTP1 (Slc11a3) by inflammation. J. Biol. Chem. 2002, 277, 39786–39791. [Google Scholar] [CrossRef] [PubMed]
- Vela, D.; Vela-Gaxha, Z. Differential regulation of hepcidin in cancer and non-cancer tissues and its clinical implications. Exp. Mol. Med. 2018, 50, e436. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zhang, S.; Chen, Y.; Zhang, D.; Yuan, L.; Cong, H.; Liu, S. An important role of the hepcidin-ferroportin signaling in affecting tumor growth and metastasis. Acta Biochim. Biophys. Sin. 2015, 47, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Pinnix, Z.K.; Miller, L.D.; Wang, W.; D’Agostino, R., Jr.; Kute, T.; Willingham, M.C.; Hatcher, H.; Tesfay, L.; Sui, G.; Di, X.; et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci. Transl. Med. 2010, 2, 43ra56. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, Y.; Guo, W.; Yuan, L.; Zhang, D.; Xu, Y.; Nemeth, E.; Ganz, T.; Liu, S. Disordered hepcidin-ferroportin signaling promotes breast cancer growth. Cell. Signal. 2014, 26, 2539–2550. [Google Scholar] [CrossRef]
- Jiang, X.P.; Elliott, R.L.; Head, J.F. Manipulation of iron transporter genes results in the suppression of human and mouse mammary adenocarcinomas. Anticancer Res. 2010, 30, 759–765. [Google Scholar]
- Shan, Z.; Wei, Z.; Shaikh, Z.A. Suppression of ferroportin expression by cadmium stimulates proliferation, EMT, and migration in triple-negative breast cancer cells. Toxicol. Appl. Pharmacol. 2018, 356, 36–43. [Google Scholar] [CrossRef]
- Udali, S.; Guarini, P.; Ruzzenente, A.; Ferrarini, A.; Guglielmi, A.; Lotto, V.; Tononi, P.; Pattini, P.; Moruzzi, S.; Campagnaro, T.; et al. DNA methylation and gene expression profiles show novel regulatory pathways in hepatocellular carcinoma. Clin. Epigenet. 2015, 7, 43. [Google Scholar] [CrossRef]
- Tesfay, L.; Clausen, K.A.; Kim, J.W.; Hegde, P.; Wang, X.; Miller, L.D.; Deng, Z.; Blanchette, N.; Arvedson, T.; Miranti, C.K.; et al. Hepcidin regulation in prostate and its disruption in prostate cancer. Cancer Res. 2015, 75, 2254–2263. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, L.; Ma, Y.; Wu, X.; Jin, L.; Yu, F. Increased hepcidin expression in non-small cell lung cancer tissue and serum is associated with clinical stage. Thorac. Cancer 2014, 5, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Tuhkanen, H.; Hartikainen, J.M.; Soini, Y.; Velasco, G.; Sironen, R.; Nykopp, T.K.; Kataja, V.; Eskelinen, M.; Kosma, V.M.; Mannermaa, A. Matriptase-2 gene (TMPRSS6) variants associate with breast cancer survival, and reduced expression is related to triple-negative breast cancer. Int. J. Cancer 2013, 133, 2334–2340. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Randall, J.D.; Cahill, C.M.; Eder, P.S.; Huang, X.; Gunshin, H.; Leiter, L.; McPhee, J.; Sarang, S.S.; Utsuki, T.; et al. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 2002, 277, 45518–45528. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.X.; Tsatsanis, A.; Lim, L.Q.; Adlard, P.A.; Bush, A.I.; Duce, J.A. Beta-amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PLoS ONE 2014, 9, e114174. [Google Scholar] [CrossRef] [PubMed]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2016, 139 (Suppl. 1), 179–197. [Google Scholar] [CrossRef]
- Wang, J.Y.; Zhuang, Q.Q.; Zhu, L.B.; Zhu, H.; Li, T.; Li, R.; Chen, S.F.; Huang, C.P.; Zhang, X.; Zhu, J.H. Meta-analysis of brain iron levels of Parkinson’s disease patients determined by postmortem and MRI measurements. Sci. Rep. 2016, 6, 36669. [Google Scholar] [CrossRef]
- Bacon, B.R.; Adams, P.C.; Kowdley, K.V.; Powell, L.W.; Tavill, A.S.; American Association for the Study of Liver Diseases. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011, 54, 328–343. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL clinical practice guidelines for HFE hemochromatosis. J. Hepatol. 2010, 53, 3–22. [Google Scholar] [CrossRef]
- Katsarou, A.; Pantopoulos, K. Hepcidin therapeutics. Pharmaceuticals 2018, 11, 127. [Google Scholar] [CrossRef]
- Brissot, P. Optimizing the diagnosis and the treatment of iron overload diseases. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 359–370. [Google Scholar] [CrossRef]
- Vadhan-Raj, S.; Abonour, R.; Goldman, J.W.; Smith, D.A.; Slapak, C.A.; Ilaria, R.L., Jr.; Tiu, R.V.; Wang, X.; Callies, S.; Cox, J.; et al. A first-in-human phase 1 study of a hepcidin monoclonal antibody, LY2787106, in cancer-associated anemia. J. Hematol. Oncol. 2017, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Sasu, B.J.; Cooke, K.S.; Arvedson, T.L.; Plewa, C.; Ellison, A.R.; Sheng, J.; Winters, A.; Juan, T.; Li, H.Y.; Begley, C.G.; et al. Antihepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation-induced anemia. Blood 2010, 115, 3616–3624. [Google Scholar] [CrossRef] [PubMed]
- Alnylam Pharmaceuticals. Targeting the Hepcidin Pathway with RNAi Therapeutics for the Treatment of Anemia. Available online: http://www.alnylam.com/web/assets/HPN-ALNY-ASH2011-Anemia.pdf (accessed on 1 November 2019).
- Schlehuber, S.; Skerra, A. Lipocalins in drug discovery: From natural ligand-binding proteins to ‘anticalins’. Drug Discov. Today 2005, 10, 23–33. [Google Scholar] [CrossRef]
- Pieris Pharmaceuticals. PRS-080: Best-in-Class Hepcidin Antagonist for Anemia. Available online: http://www.pieris.com/pipeline/anemia-and-other-disease-areas/prs-080 (accessed on 17 April 2018).
- Hohlbaum, A.M.; Gille, H.; Trentmann, S.; Kolodziejczyk, M.; Rattenstetter, B.; Laarakkers, C.M.; Katzmann, G.; Christian, H.J.; Andersen, N.; Allersdorfer, A.; et al. Sustained plasma hepcidin suppression and iron elevation by Anticalin-derived hepcidin antagonist in cynomolgus monkey. Br. J. Pharmacol. 2018, 175, 1054–1065. [Google Scholar] [CrossRef] [PubMed]
- Moebius, U.; Feuerer, W.; Fenzl, E.; van Swelm, R.; Swinkels, D.W.; Hohlbaum, A. A phase I study investigating the safety, tolerability, pharmacokinetics and pharmacodynamic activity of the hepcidin antagonist PRS-080#022. Results from a randomized, placebo controlled, double-blind study following single administration to healthy subjects. Blood 2015, 126, 536. [Google Scholar]
- Renders, L.; Budde, K.; Rosenberger, C.; van Swelm, R.; Swinkels, D.; Dellanna, F.; Feuerer, W.; Wen, M.; Erley, C.; Bader, B.; et al. First-in-human phase I studies of PRS-080#22, a hepcidin antagonist, in healthy volunteers and patients with chronic kidney disease undergoing hemodialysis. PLoS ONE 2019, 14, e0212023. [Google Scholar] [CrossRef]
- Angmo, S.; Tripathi, N.; Abbat, S.; Sharma, S.; Singh, S.S.; Halder, A.; Yadav, K.; Shukla, G.; Sandhir, R.; Rishi, V.; et al. Identification of guanosine 5′-diphosphate as potential iron mobilizer: Preventing the hepcidin-ferroportin interaction and modulating the interleukin-6/Stat-3 pathway. Sci. Rep. 2017, 7, 40097. [Google Scholar] [CrossRef]
- Pendergrast, P.S.; Marsh, H.N.; Grate, D.; Healy, J.M.; Stanton, M. Nucleic acid aptamers for target validation and therapeutic applications. J. Biomol. Tech. 2005, 16, 224–234. [Google Scholar]
- Schwoebel, F.; van Eijk, L.T.; Zboralski, D.; Sell, S.; Buchner, K.; Maasch, C.; Purschke, W.G.; Humphrey, M.; Zollner, S.; Eulberg, D.; et al. The effects of the anti-hepcidin Spiegelmer NOX-H94 on inflammation-induced anemia in cynomolgus monkeys. Blood 2013, 121, 2311–2315. [Google Scholar] [CrossRef]
- Boyce, M.; Warrington, S.; Cortezi, B.; Zollner, S.; Vauleon, S.; Swinkels, D.W.; Summo, L.; Schwoebel, F.; Riecke, K. Safety, pharmacokinetics and pharmacodynamics of the anti-hepcidin Spiegelmer lexaptepid pegol in healthy subjects. Br. J. Pharmacol. 2016, 173, 1580–1588. [Google Scholar] [CrossRef]
- Poli, M.; Girelli, D.; Campostrini, N.; Maccarinelli, F.; Finazzi, D.; Luscieti, S.; Nai, A.; Arosio, P. Heparin: A potent inhibitor of hepcidin expression in vitro and in vivo. Blood 2011, 117, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Poli, M.; Asperti, M.; Naggi, A.; Campostrini, N.; Girelli, D.; Corbella, M.; Benzi, M.; Besson-Fournier, C.; Coppin, H.; Maccarinelli, F.; et al. Glycol-split nonanticoagulant heparins are inhibitors of hepcidin expression in vitro and in vivo. Blood 2014, 123, 1564–1573. [Google Scholar] [CrossRef] [PubMed]
- Poli, M.; Asperti, M.; Ruzzenenti, P.; Mandelli, L.; Campostrini, N.; Martini, G.; Di Somma, M.; Maccarinelli, F.; Girelli, D.; Naggi, A.; et al. Oversulfated heparins with low anticoagulant activity are strong and fast inhibitors of hepcidin expression in vitro and in vivo. Biochem. Pharmacol. 2014, 92, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.B.; Hong, C.C.; Sachidanandan, C.; Babitt, J.L.; Deng, D.Y.; Hoyng, S.A.; Lin, H.Y.; Bloch, K.D.; Peterson, R.T. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 2008, 4, 33–41. [Google Scholar] [CrossRef]
- Theurl, I.; Schroll, A.; Sonnweber, T.; Nairz, M.; Theurl, M.; Willenbacher, W.; Eller, K.; Wolf, D.; Seifert, M.; Sun, C.C.; et al. Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood 2011, 118, 4977–4984. [Google Scholar] [CrossRef]
- Asshoff, M.; Petzer, V.; Warr, M.R.; Haschka, D.; Tymoszuk, P.; Demetz, E.; Seifert, M.; Posch, W.; Nairz, M.; Maciejewski, P.; et al. Momelotinib inhibits ACVR1/ALK2, decreases hepcidin production, and ameliorates anemia of chronic disease in rodents. Blood 2017, 129, 1823–1830. [Google Scholar] [CrossRef]
- Peterson, P.; Soh, K.K.; Lee, Y.S.; Kim, W.; Whatcott, C.J.; Siddiqui-Jain, A.; Bearss, D.J.; Warner, S.L. ALK2 inhibition via TP-0184 abrogates inflammation-induced hepcidin expression and is a potential therapeutic for anemia of chronic disease. Blood 2015, 126, 273. [Google Scholar] [CrossRef]
- Mleczko-Sanecka, K.; da Silva, A.R.; Call, D.; Neves, J.; Schmeer, N.; Damm, G.; Seehofer, D.; Muckenthaler, M.U. Imatinib and spironolactone suppress hepcidin expression. Haematologica 2017, 102, 1173–1184. [Google Scholar] [CrossRef]
- Boser, P.; Seemann, D.; Liguori, M.J.; Fan, L.; Huang, L.; Hafner, M.; Popp, A.; Mueller, B.K. Anti-repulsive guidance molecule C (RGMc) antibodies increases serum iron in rats and cynomolgus monkeys by hepcidin downregulation. AAPS J. 2015, 17, 930–938. [Google Scholar] [CrossRef]
- Zhang, S.P.; Wang, Z.; Wang, L.X.; Liu, S.J. AG490: An inhibitor of hepcidin expression in vivo. World J. Gastroenterol. 2011, 17, 5032–5034. [Google Scholar] [CrossRef]
- Turkson, J.; Ryan, D.; Kim, J.S.; Zhang, Y.; Chen, Z.; Haura, E.; Laudano, A.; Sebti, S.; Hamilton, A.D.; Jove, R. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J. Biol. Chem. 2001, 276, 45443–45455. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xin, H.; Tang, W.; Li, Y.; Zhang, Z.; Fan, L.; Miao, L.; Tan, B.; Wang, X.; Zhu, Y.Z. AMPK serves as a therapeutic target against anemia of inflammation. Antioxid. Redox Signal. 2017, 27, 251–268. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Ueda, K.; Ishiyama, T.; Goto, R.; Muramatsu, S.; Hashimoto, M.; Watanabe, K.; Tanaka, N. Synthesis and SAR studies of 3,6-disubstituted indazole derivatives as potent hepcidin production inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 2148–2152. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Goto, R.; Kiho, T.; Ueda, K.; Muramatsu, S.; Hashimoto, M.; Aki, A.; Watanabe, K.; Tanaka, N. Discovery of DS79182026: A potent orally active hepcidin production inhibitor. Bioorg. Med. Chem. Lett. 2017, 27, 3716–3722. [Google Scholar] [CrossRef]
- Kurzrock, R.; Voorhees, P.M.; Casper, C.; Furman, R.R.; Fayad, L.; Lonial, S.; Borghaei, H.; Jagannath, S.; Sokol, L.; Usmani, S.Z.; et al. A phase I, open-label study of siltuximab, an anti-IL-6 monoclonal antibody, in patients with B-cell non-Hodgkin lymphoma, multiple myeloma, or Castleman disease. Clin. Cancer Res. 2013, 19, 3659–3670. [Google Scholar] [CrossRef]
- Song, S.N.; Tomosugi, N.; Kawabata, H.; Ishikawa, T.; Nishikawa, T.; Yoshizaki, K. Down-regulation of hepcidin resulting from long-term treatment with an anti-IL-6 receptor antibody (tocilizumab) improves anemia of inflammation in multicentric Castleman disease. Blood 2010, 116, 3627–3634. [Google Scholar] [CrossRef]
- Pergola, P.E.; Spinowitz, B.S.; Hartman, C.S.; Maroni, B.J.; Haase, V.H. Vadadustat, a novel oral HIF stabilizer, provides effective anemia treatment in nondialysis-dependent chronic kidney disease. Kidney Int. 2016, 90, 1115–1122. [Google Scholar] [CrossRef]
- Besarab, A.; Chernyavskaya, E.; Motylev, I.; Shutov, E.; Kumbar, L.M.; Gurevich, K.; Chan, D.T.; Leong, R.; Poole, L.; Zhong, M.; et al. Roxadustat (FG-4592): Correction of anemia in incident dialysis patients. J. Am. Soc. Nephrol. 2016, 27, 1225–1233. [Google Scholar] [CrossRef]
- Brigandi, R.A.; Johnson, B.; Oei, C.; Westerman, M.; Olbina, G.; de Zoysa, J.; Roger, S.D.; Sahay, M.; Cross, N.; McMahon, L.; et al. A novel hypoxia-inducible factor-prolyl hydroxylase inhibitor (GSK1278863) for anemia in CKD: A 28-day, phase 2A randomized trial. Am. J. Kidney Dis. 2016, 67, 861–871. [Google Scholar] [CrossRef]
- Fung, E.; Sugianto, P.; Hsu, J.; Damoiseaux, R.; Ganz, T.; Nemeth, E. High-throughput screening of small molecules identifies hepcidin antagonists. Mol. Pharmacol. 2013, 83, 681–690. [Google Scholar] [CrossRef]
- Leung, D.D.M.; Luan, P.; Manetta, J.V.; Tang, Y.; Witcher, D.R. Anti-Ferroportin 1 Monoclonal Antibodies and Uses Thereof. U.S. Patent 8,183,346B2, 22 May 2012. [Google Scholar]
- Pandur, E.; Fekete, Z.; Tamasi, K.; Grama, L.; Varga, E.; Sipos, K. The C19S substitution enhances the stability of hepcidin while conserving its biological activity. Protein J. 2018, 37, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Mleczko-Sanecka, K.; Roche, F.; da Silva, A.R.; Call, D.; D’Alessio, F.; Ragab, A.; Lapinski, P.E.; Ummanni, R.; Korf, U.; Oakes, C.; et al. Unbiased RNAi screen for hepcidin regulators links hepcidin suppression to proliferative Ras/RAF and nutrient-dependent mTOR signaling. Blood 2014, 123, 1574–1585. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Rhee, D.K.; Malhotra, R.; Mayeur, C.; Hurst, L.A.; Ager, E.; Shelton, G.; Kramer, Y.; McCulloh, D.; Keefe, D.; et al. Progesterone receptor membrane component-1 regulates hepcidin biosynthesis. J. Clin. Investig. 2016, 126, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Arezes, J.; Jung, G.; Gabayan, V.; Valore, E.; Ruchala, P.; Gulig, P.A.; Ganz, T.; Nemeth, E.; Bulut, Y. Hepcidin-induced hypoferremia is a critical host defense mechanism against the siderophilic bacterium vibrio vulnificus. Cell Host Microbe 2015, 17, 47–57. [Google Scholar] [CrossRef]
- Ramos, E.; Ruchala, P.; Goodnough, J.B.; Kautz, L.; Preza, G.C.; Nemeth, E.; Ganz, T. Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood 2012, 120, 3829–3836. [Google Scholar] [CrossRef]
- Chua, K.; Fung, E.; Micewicz, E.D.; Ganz, T.; Nemeth, E.; Ruchala, P. Small cyclic agonists of iron regulatory hormone hepcidin. Bioorg. Med. Chem. Lett. 2015, 25, 4961–4969. [Google Scholar] [CrossRef]
- Casu, C.; Oikonomidou, P.R.; Chen, H.Y.; Nandi, V.; Ginzburg, Y.; Prasad, P.; Fleming, R.E.; Shah, Y.M.; Valore, E.V.; Nemeth, E.; et al. Minihepcidin peptides as disease modifiers in mice affected by beta-thalassemia and polycythemia vera. Blood 2016, 128, 265–276. [Google Scholar] [CrossRef]
- Guo, S.; Aghajan, M.; Casu, C.; Gardenghi, S.; Booten, S.; Rivella, S.; Monia, B.P. Targeting TMPRSS6 using antisense technology for the treatment of beta-thalassemia. Blood 2015, 126, 753. [Google Scholar] [CrossRef]
- Aghajan, M.; Casu, C.; lo Presti, V.; Booten, S.; Monia, B.P.; Rivella, S.; Guo, S. Developing a galnac-conjugated tmprss6 antisense therapy for the treatment of β-thalassemia. Blood 2016, 128, 1013. [Google Scholar] [CrossRef]
- Schmidt, P.J.; Liu, K.; Visner, G.; Fitzgerald, K.; Fishman, S.; Racie, T.; Hettinger, J.L.; Butler, J.S.; Fleming, M.D. RNAi-mediated reduction of hepatic Tmprss6 diminishes anemia and secondary iron overload in a splenectomized mouse model of beta-thalassemia intermedia. Am. J. Hematol. 2018, 93, 745–750. [Google Scholar] [CrossRef]
- Bayele, H.K.; Balesaria, S.; Srai, S.K. Phytoestrogens modulate hepcidin expression by Nrf2: Implications for dietary control of iron absorption. Free Radic. Biol. Med. 2015, 89, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.; Wu, Q.; Wang, H.; Zhao, L.; Wang, X.; Mu, M.; Xie, E.; He, X.; Shao, D.; et al. Adenine alleviates iron overload by cAMP/PKA mediated hepatic hepcidin in mice. J. Cell. Physiol. 2018, 233, 7268–7278. [Google Scholar] [CrossRef] [PubMed]
- Zhen, A.W.; Nguyen, N.H.; Gibert, Y.; Motola, S.; Buckett, P.; Wessling-Resnick, M.; Fraenkel, E.; Fraenkel, P.G. The small molecule, genistein, increases hepcidin expression in human hepatocytes. Hepatology 2013, 58, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Gaun, V.; Patchen, B.; Volovetz, J.; Zhen, A.W.; Andreev, A.; Pollastri, M.P.; Fraenkel, P.G. A chemical screen identifies small molecules that regulate hepcidin expression. Blood Cells Mol. Dis. 2014, 53, 231–240. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, J.; Guo, W.; Liu, X.; Liu, S.; Yin, H. Icariin regulates systemic iron metabolism by increasing hepatic hepcidin expression through Stat3 and Smad1/5/8 signaling. Int. J. Mol. Med. 2016, 37, 1379–1388. [Google Scholar] [CrossRef]
- Yaeger, D.; Piga, A.; Lal, A.; Kattamis, A.; Salman, S.; Byrnes, B.; Tidmarsh, G.; Chawla, L. A Phase 1, Placebo-Controlled Study to Determine the Safety, Tolerability, and Pharmacokinetics of Escalating Subcutaneous Doses of LJPC-401 (Synthetic Human Hepcidin) in Healthy Adults. Available online: http://lajollapharmaceutical.com/wp-content/uploads/2018/06/EHA_Poster.pdf (accessed on 1 November 2019).
- Schmidt, P.J.; Toudjarska, I.; Sendamarai, A.K.; Racie, T.; Milstein, S.; Bettencourt, B.R.; Hettinger, J.; Bumcrot, D.; Fleming, M.D. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(-/-) mice and ameliorates anemia and iron overload in murine beta-thalassemia intermedia. Blood 2013, 121, 1200–1208. [Google Scholar] [CrossRef]
- Silvestri, L.; Nai, A.; Dulja, A.; Pagani, A. Hepcidin and the BMP-SMAD pathway: An unexpected liaison. Vitam. Horm. 2019, 110, 71–99. [Google Scholar] [CrossRef]
- Boergermann, J.H.; Kopf, J.; Yu, P.B.; Knaus, P. Dorsomorphin and LDN-193189 inhibit BMP-mediated Smad, p38 and Akt signalling in C2C12 cells. Int. J. Biochem. Cell Biol. 2010, 42, 1802–1807. [Google Scholar] [CrossRef]
- Wallace, D.F.; Summerville, L.; Subramaniam, V.N. Targeted disruption of the hepatic transferrin receptor 2 gene in mice leads to iron overload. Gastroenterology 2007, 132, 301–310. [Google Scholar] [CrossRef]
- Fleming, R.E.; Feng, Q.; Britton, R.S. Knockout mouse models of iron homeostasis. Annu. Rev. Nutr. 2011, 31, 117–137. [Google Scholar] [CrossRef]
- Camaschella, C.; Roetto, A.; Calì, A.; De Gobbi, M.; Garozzo, G.; Carella, M.; Majorano, N.; Totaro, A.; Gasparini, P. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat. Genet. 2000, 25, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, H. Transferrin and transferrin receptors update. Free Radic. Biol. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Forejtnikovà, H.; Vieillevoye, M.; Zermati, Y.; Lambert, M.; Pellegrino, R.M.; Guihard, S.; Gaudry, M.; Camaschella, C.; Lacombe, C.; Roetto, A.; et al. Transferrin receptor 2 is a component of the erythropoietin receptor complex and is required for efficient erythropoiesis. Blood 2010, 116, 5357–5367. [Google Scholar] [CrossRef] [PubMed]
- De Vos, J.; Jourdan, M.; Tarte, K.; Jasmin, C.; Klein, B. JAK2 tyrosine kinase inhibitor tyrphostin AG490 downregulates the mitogen-activated protein kinase (MAPK) and signal transducer and activator of transcription (STAT) pathways and induces apoptosis in myeloma cells. Br. J. Haematol. 2000, 109, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Wish, J.B. Hypoxia-inducible factor prolyl hydroxylase inhibitors: A potential new treatment for anemia in patients with CKD. Am. J. Kidney Dis. 2017, 69, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Hao, C.; Liu, B.C.; Lin, H.; Wang, C.; Xing, C.; Liang, X.; Jiang, G.; Liu, Z.; Li, X.; et al. Roxadustat treatment for anemia in patients undergoing long-term dialysis. N. Engl. J. Med. 2019, 381, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Hao, C.; Peng, X.; Lin, H.; Yin, A.; Hao, L.; Tao, Y.; Liang, X.; Liu, Z.; Xing, C.; et al. Roxadustat for anemia in patients with kidney disease not receiving dialysis. N. Engl. J. Med. 2019, 381, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Ariazi, J.L.; Duffy, K.J.; Adams, D.F.; Fitch, D.M.; Luo, L.; Pappalardi, M.; Biju, M.; DiFilippo, E.H.; Shaw, T.; Wiggall, K.; et al. Discovery and preclinical characterization of GSK1278863 (daprodustat), a small molecule hypoxia inducible factor-prolyl hydroxylase inhibitor for anemia. J. Pharmacol. Exp. Ther. 2017, 363, 336–347. [Google Scholar] [CrossRef]
- Ross, S.L.; Biswas, K.; Rottman, J.; Allen, J.R.; Long, J.; Miranda, L.P.; Winters, A.; Arvedson, T.L. Identification of antibody and small molecule antagonists of ferroportin-hepcidin interaction. Front. Pharmacol. 2017, 8, 838. [Google Scholar] [CrossRef]
- Preza, G.C.; Ruchala, P.; Pinon, R.; Ramos, E.; Qiao, B.; Peralta, M.A.; Sharma, S.; Waring, A.; Ganz, T.; Nemeth, E. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J. Clin. Investig. 2011, 121, 4880–4888. [Google Scholar] [CrossRef]
- Guo, S.; Casu, C.; Gardenghi, S.; Booten, S.; Aghajan, M.; Peralta, R.; Watt, A.; Freier, S.; Monia, B.P.; Rivella, S. Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J. Clin. Investig. 2013, 123, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Casu, C.; Aghajan, M.; Oikonomidou, P.R.; Guo, S.; Monia, B.P.; Rivella, S. Combination of Tmprss6-ASO and the iron chelator deferiprone improves erythropoiesis and reduces iron overload in a mouse model of beta-thalassemia intermedia. Haematologica 2016, 101, e8–e11. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.J.; Racie, T.; Westerman, M.; Fitzgerald, K.; Butler, J.S.; Fleming, M.D. Combination therapy with a Tmprss6 RNAi-therapeutic and the oral iron chelator deferiprone additively diminishes secondary iron overload in a mouse model of beta-thalassemia intermedia. Am. J. Hematol. 2015, 90, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, A.M.; Maurer, E.; Lulsdorff, V.; Wilms, A.; Furtmann, N.; Bajorath, J.; Gutschow, M.; Stirnberg, M. En route to new therapeutic options for iron overload diseases: Matriptase-2 as a target for kunitz-type inhibitors. Chembiochem 2016, 17, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesh, S.; Kyo, S.; Banerjee, P.P. Genistein represses telomerase activity via both transcriptional and posttranslational mechanisms in human prostate cancer cells. Cancer Res. 2006, 66, 2107–2115. [Google Scholar] [CrossRef] [PubMed]
- Chau, M.N.; El Touny, L.H.; Jagadeesh, S.; Banerjee, P.P. Physiologically achievable concentrations of genistein enhance telomerase activity in prostate cancer cells via the activation of STAT3. Carcinogenesis 2007, 28, 2282–2290. [Google Scholar] [CrossRef] [PubMed]
- Patchen, B.; Koppe, T.; Cheng, A.; Seo, Y.A.; Wessling-Resnick, M.; Fraenkel, P.G. Dietary supplementation with ipriflavone decreases hepatic iron stores in wild type mice. Blood Cells Mol. Dis. 2016, 60, 36–43. [Google Scholar] [CrossRef]
- Akhtar, S.; Benter, I.F. Nonviral delivery of synthetic siRNAs in vivo. J. Clin. Investig. 2007, 117, 3623–3632. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Model | Drug | Target |
|---|---|---|
| Direct Hepcidin Inhibitors | ||
| In vitro | Guanosine 5, -diphosphate (GDP) [89] | Hepcidin |
| In vivo | Ab2.7, H6 and H10 [82], ALN-HPN [83] | |
| Clinical trial | LY2787106 [81], PRS-080 [87], Nox-H94 [92] | |
| Inhibitors of Hepcidin Production/Synthesis | ||
| In vitro | Unfractionated heparin | BMP6 |
| Enoxaparin, Fondaparinux [93] | ||
| RO-68 and RO-82 [94] | ||
| SSLMWH-19 [95] | ||
| In vivo | Dorsomorphin [96], LDN-1913189 [97] | Type 1 BMPRs |
| Momelotinib [98], TP-0184 [99] | ALK2 | |
| Imatinib, spironolactone [100] | BMP/SMAD Pathway | |
| sHJV.Fc [97] | BMP6 | |
| ABT-207, h5F9-AM8 [101] | HJV | |
| RNAi [83] | TFR2 | |
| AG490 [102] | JAK | |
| PpYLKTK [103] | STAT3 | |
| Metformin [104] | AMPK | |
| Indazole [105], DS79182026 [106] | ALK2 and ALK3 | |
| Clinical Trail | Siltuximab [107], Tocilizumab [108] | IL-6 |
| Hypoxia-Inducible Factors (HIF) Stabilisers | ||
| Clinical trial | Vadadustat [109], Roxadustat [110], Daproustat [111] | PHD |
| Ferroportin Agonists | ||
| In vitro | Fursultiamine [112], Anti-FPN mouse antibody [113] | FPN |
| Hepcidin Agonists | ||
| In vitro | Hepcidin Cys19Ser [114] | FPN |
| Sorafenib, Wortmannin, Rapamycin [115] | RAS/RAF and PI3 | |
| Epitiostanol, Progesterone, Mifepristone [116] | PGRMC1 | |
| In vivo | PR73 [117], PR65 [118], mHS17, mHS26 [119], M004, M009 [120] | FPN |
| TMPRSS6-ASO#1 [121], TMPRSS6-ASO#2 [121], GalNAc-ASO [122], RNAi-GalNAc [123], | TMPRSS6 | |
| Naringenin, Quercetin, Resveratrol [124] | NRF2 | |
| Adenine [125] | BMP6/SMAD pathway | |
| Genistein [126] | BMP-RE and STAT | |
| Ipriflavone, Vorinostat [127] | BMP6 and STAT3 | |
| Icariin, Epimedin [128] | STAT3 and Smad1/5/8 | |
| Clinical Trial | LJPC-401 [129] | FPN |
| RNAi LNP [130] | TMPRSS6 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hawula, Z.J.; Wallace, D.F.; Subramaniam, V.N.; Rishi, G. Therapeutic Advances in Regulating the Hepcidin/Ferroportin Axis. Pharmaceuticals 2019, 12, 170. https://doi.org/10.3390/ph12040170
Hawula ZJ, Wallace DF, Subramaniam VN, Rishi G. Therapeutic Advances in Regulating the Hepcidin/Ferroportin Axis. Pharmaceuticals. 2019; 12(4):170. https://doi.org/10.3390/ph12040170
Chicago/Turabian StyleHawula, Zachary J., Daniel F. Wallace, V. Nathan Subramaniam, and Gautam Rishi. 2019. "Therapeutic Advances in Regulating the Hepcidin/Ferroportin Axis" Pharmaceuticals 12, no. 4: 170. https://doi.org/10.3390/ph12040170
APA StyleHawula, Z. J., Wallace, D. F., Subramaniam, V. N., & Rishi, G. (2019). Therapeutic Advances in Regulating the Hepcidin/Ferroportin Axis. Pharmaceuticals, 12(4), 170. https://doi.org/10.3390/ph12040170