Bucillamine Prevents Afatinib-Mediated Inhibition of Epidermal Growth Factor Receptor Signaling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
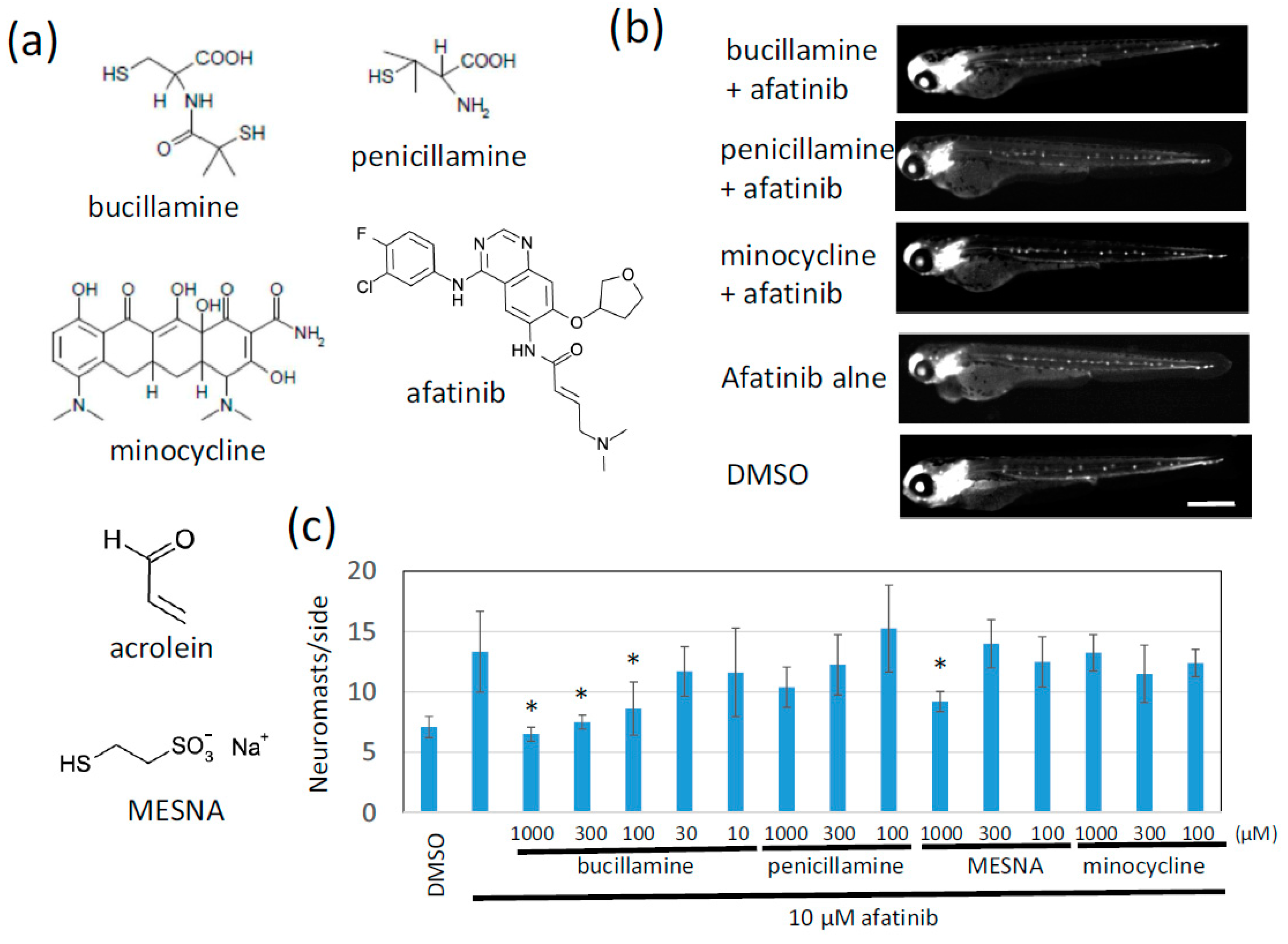
2.1. Afatinib Induces Extra Formation of Lateral Line Neuromasts in Zebrafish Larvae
2.2. Bucillamine Is an Irreversible EGFR-TKI Quencher
2.3. Bucillamine Forms Adducts With Afatinib and Prevents In Vitro Kinase Inhibition of Afatinib
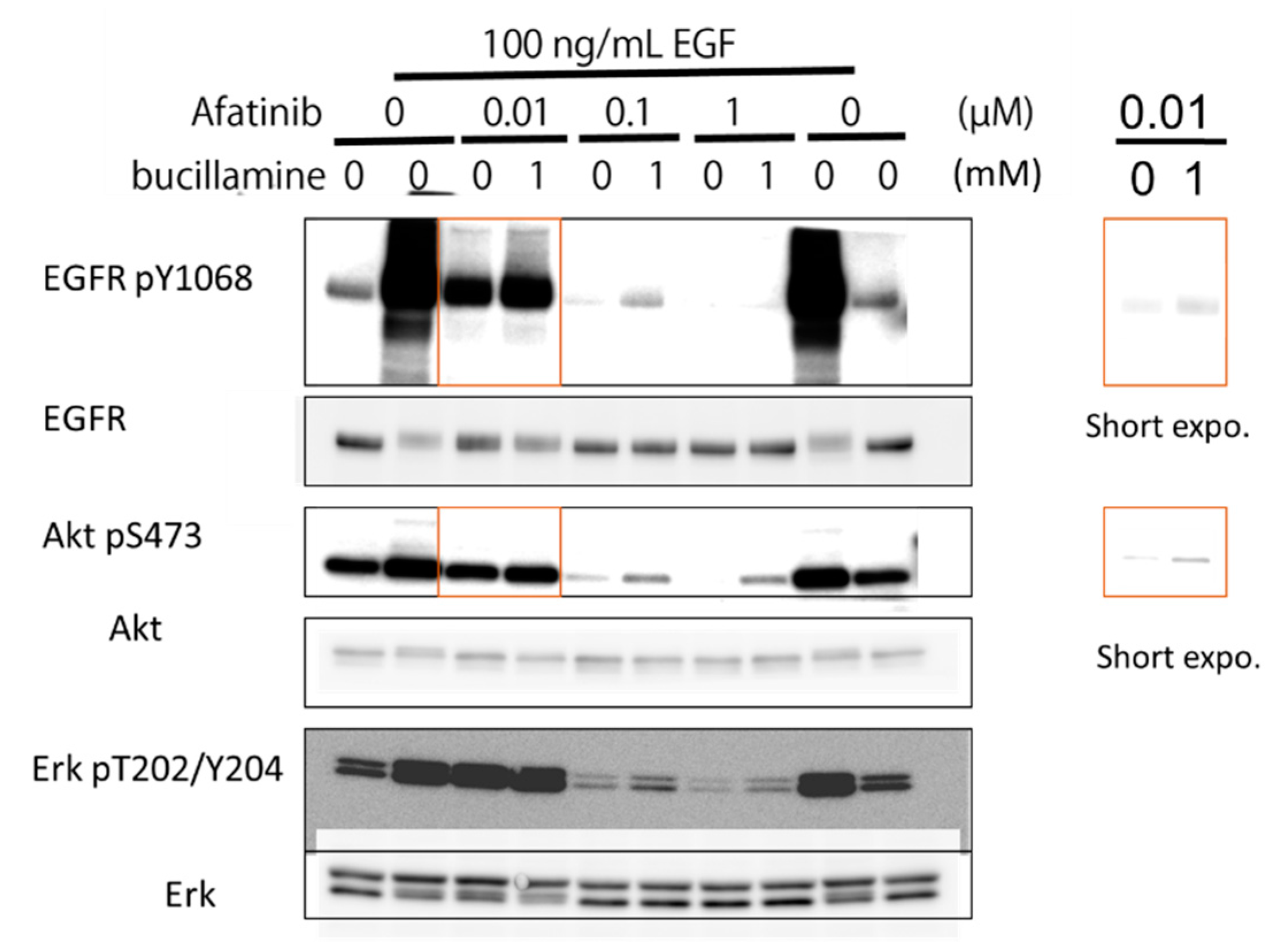
2.4. Bucillamine Restores EGFR Autophosphorylation and Its Downstream Signaling in Afatinib-Treated A431 Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Cell Lines
4.2. Zebrafish
4.3. Alkaline Phosphatase (ALP) Staining
4.4. Thin-layer Chromatography (TLC)
4.5. In Vitro Kinase Assay
4.6. Phosphorylation Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ciardiello, F.; Tortora, G. EGFR antagonists in cancer treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef] [PubMed]
- Eilers, R.E., Jr.; Gandhi, M.; Patel, J.D.; Mulcahy, M.F.; Agulnik, M.; Hensing, T.; Lacouture, M.E. Dermatologic infections in cancer patients treated with epidermal growth factor receptor inhibitor therapy. J. Nation Cancer Inst. 2010, 102, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Lacouture, M.E. Mechanisms of cutaneous toxicities to EGFR inhibitors. Nat. Rev. Cancer 2006, 6, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Perez-Soler, R.; Saltz, L. Cutaneous adverse effects with HER1/EGFR-targeted agents: Is there a silver lining? J. Clin. Oncol. 2005, 23, 5235–5246. [Google Scholar] [CrossRef] [PubMed]
- Wacker, B.; Nagrani, T.; Weinberg, J.; Witt, K.; Clark, G.; Cagnoni, P.J. Correlation between development of rash and efficacy in patients treated with the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib in two large phase III studies. Clin. Cancer Res. 2007, 13, 3913–3921. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Mita, A.; De Jonge, M.; Matthews, L.; McCarthy, S.; Iwata, K.K.; Verweij, J.; Rowinsky, E.K.; Krueger, J.G. Characterisation of the cutaneous pathology in non-small cell lung cancer (NSCLC) patients treated with the EGFR tyrosine kinase inhibitor erlotinib. Eur. J. Cancer 2010, 46, 2010–2019. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.A.; Alexander, N.; Hogan, M.E.; Sundberg, J.P.; Dlugosz, A.; Threadgill, D.W.; Magnuson, T.; Yuspa, S.H. Genetically null mice reveal a central role for epidermal growth factor receptor in the differentiation of the hair follicle and normal hair development. Am. J. Pathol. 1997, 150, 1959–1975. [Google Scholar]
- Murillas, R.; Larcher, F.; Conti, C.J.; Santos, M.; Ullrich, A.; Jorcano, J.L. Expression of a dominant negative mutant of epidermal growth factor receptor in the epidermis of transgenic mice elicits striking alterations in hair follicle development and skin structure. EMBO J. 1995, 14, 5216–5223. [Google Scholar] [CrossRef]
- Mascia, F.; Mariani, V.; Girolomoni, G.; Pastore, S. Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation. Am. J. Pathol. 2003, 163, 303–312. [Google Scholar] [CrossRef]
- Pastore, S.; Mascia, F.; Mariotti, F.; Dattilo, C.; Mariani, V.; Girolomoni, G. ERK1/2 regulates epidermal chemokine expression and skin inflammation. J. Immunol. 2005, 174, 5047–5056. [Google Scholar] [CrossRef]
- Mascia, F.; Lam, G.; Keith, C.; Garber, C.; Steinberg, S.M.; Kohn, E.; Yuspa, S.H. Genetic ablation of epidermal EGFR reveals the dynamic origin of adverse effects of anti-EGFR therapy. Sci. Transl. Med. 2013, 5, 199ra110. [Google Scholar] [CrossRef]
- Schrumpf, H.; Boelke, E.; Ansari, P.; Mackenzie, C.; Lichtenberger, B.M.; Gerber, P.A.; Holcmann, M.; Buhren, B.A.; Amberg, N.; Smolle, V.; et al. Epidermal EGFR controls cutaneous host defense and prevents inflammation. Sci. Transl. Med. 2013, 5, 199ra111. [Google Scholar] [CrossRef]
- Fry, D.W.; Bridges, A.J.; Denny, W.A.; Doherty, A.; Greis, K.D.; Hicks, J.L.; Hook, K.E.; Keller, P.R.; Leopold, W.R.; Loo, J.A.; et al. Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor. Proc. Natl. Acad. Sci. USA 1998, 95, 12022–12027. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Munoz, A.; Rajadhyksha, S.; Gilmour, D.; van Bebber, F.; Antos, C.; Rodriguez Esteban, C.; Nusslein-Volhard, C.; Izpisua Belmonte, J.C. ErbB2 and ErbB3 regulate amputation-induced proliferation and migration during vertebrate regeneration. Dev. Biol. 2009, 327, 177–190. [Google Scholar] [CrossRef]
- Park, K.; Tan, E.H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.C.; Lee, K.H.; Lu, S.; et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016, 17, 577–589. [Google Scholar] [CrossRef]
- Wada, H.; Ghysen, A.; Satou, C.; Higashijima, S.; Kawakami, K.; Hamaguchi, S.; Sakaizumi, M. Dermal morphogenesis controls lateral line patterning during postembryonic development of teleost fish. Dev. Biol. 2010, 340, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Brock, N.; Stekar, J.; Pohl, J.; Niemeyer, U.; Scheffler, G. Acrolein, the causative factor of urotoxic side-effects of cyclophosphamide, ifosfamide, trofosfamide and sufosfamide. Arzneimittelforschung 1979, 29, 659–661. [Google Scholar]
- Brock, N. The development of mesna for the inhibition of urotoxic side effects of cyclophosphamide, ifosfamide, and other oxazaphosphorine cytostatics. Recent Results Cancer Res. 1980, 74, 270–278. [Google Scholar] [CrossRef]
- Van Doorn, R.; Kirtschig, G.; Scheffer, E.; Stoof, T.J.; Giaccone, G. Follicular and epidermal alterations in patients treated with ZD1839 (Iressa), an inhibitor of the epidermal growth factor receptor. Br. J. Dermatol. 2002, 147, 598–601. [Google Scholar] [CrossRef]
- Nishiya, N. Chemical Modifier Screenings as Methods for Identifying Pathway-Targeting Compounds and for Predicting Drug-Drug Interactions. Org. Chem. Curr. Res. 2014, 3. [Google Scholar] [CrossRef]
- Whitfield, T.T.; Granato, M.; van Eeden, F.J.; Schach, U.; Brand, M.; Furutani-Seiki, M.; Haffter, P.; Hammerschmidt, M.; Heisenberg, C.P.; Jiang, Y.J.; et al. Mutations affecting development of the zebrafish inner ear and lateral line. Development 1996, 123, 241–254. [Google Scholar] [PubMed]
- Kraus, M.H.; Issing, W.; Miki, T.; Popescu, N.C.; Aaronson, S.A. Isolation and characterization of ERBB3, a third member of the ERBB/epidermal growth factor receptor family: Evidence for overexpression in a subset of human mammary tumors. Proc. Natl. Acad. Sci. USA 1989, 86, 9193–9197. [Google Scholar] [CrossRef] [PubMed]
- Lush, M.E.; Piotrowski, T. ErbB expressing Schwann cells control lateral line progenitor cells via non-cell-autonomous regulation of Wnt/beta-catenin. Elife 2014, 3, e01832. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, I.A. Intra-articular dissociation of the rheumatoid factor. J. Lab. Clin. Med. 1962, 60, 409–421. [Google Scholar] [PubMed]
- Thong, Y.H.; Ferrante, A. Inhibition of mitogen-induced human lymphocyte proliferative responses by tetracycline analogues. Clin. Exp. Immunol. 1979, 35, 443–446. [Google Scholar] [PubMed]
- Ingham, E.; Turnbull, L.; Kearney, J.N. The effects of minocycline and tetracycline on the mitotic response of human peripheral blood-lymphocytes. J. Antimicrob. Chemother. 1991, 27, 607–617. [Google Scholar] [CrossRef]
- Gonzales, A.J.; Hook, K.E.; Althaus, I.W.; Ellis, P.A.; Trachet, E.; Delaney, A.M.; Harvey, P.J.; Ellis, T.A.; Amato, D.M.; Nelson, J.M.; et al. Antitumor activity and pharmacokinetic properties of PF-00299804, a second-generation irreversible pan-erbB receptor tyrosine kinase inhibitor. Mol. Cancer Ther. 2008, 7, 1880–1889. [Google Scholar] [CrossRef]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef]
- Goto, M.; Sasano, M.; Nishioka, K. Decrease in disease activity and concomitant increase in the percentage of peripheral blood suppressor T-cells in rheumatoid arthritis by a newly synthesized slow-acting anti-rheumatic drug (Bucillamine). Int. J. Immunopharmacol. 1989, 11, 327–331. [Google Scholar] [CrossRef]
- Eguchi, K.; Kawakami, A.; Ida, H.; Nakashima, M.; Yamashita, I.; Sakai, M.; Shimada, H.; Terada, K.; Fukuda, T.; Ishimaru, T.; et al. Bucillamine inhibits T cell adhesion to human endothelial cells. J. Rheumatol. 1992, 19, 1045–1050. [Google Scholar]
- Hashimoto, K.; Lipsky, P.E. Immunosuppression by the disease modifying antirheumatic drug bucillamine: Inhibition of human T lymphocyte function by bucillamine and its metabolites. J. Rheumatol. 1993, 20, 953–957. [Google Scholar] [PubMed]
- Hirohata, S.; Lipsky, P.E. Regulation of B cell function by bucillamine, a novel disease-modifying antirheumatic drug. Clin. Immunol. Immunopathol. 1993, 66, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Segaert, S.; Van Cutsem, E. Clinical signs, pathophysiology and management of skin toxicity during therapy with epidermal growth factor receptor inhibitors. Ann. Oncol. 2005, 16, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Gibson, T.B.; Ranganathan, A.; Grothey, A. Randomized phase III trial results of panitumumab, a fully human anti-epidermal growth factor receptor monoclonal antibody, in metastatic colorectal cancer. Clin. Colorectal Cancer 2006, 6, 29–31. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishiya, N.; Murai, M.; Hosoda, A.; Yonezawa, H.; Omori, N. Bucillamine Prevents Afatinib-Mediated Inhibition of Epidermal Growth Factor Receptor Signaling. Pharmaceuticals 2019, 12, 165. https://doi.org/10.3390/ph12040165
Nishiya N, Murai M, Hosoda A, Yonezawa H, Omori N. Bucillamine Prevents Afatinib-Mediated Inhibition of Epidermal Growth Factor Receptor Signaling. Pharmaceuticals. 2019; 12(4):165. https://doi.org/10.3390/ph12040165
Chicago/Turabian StyleNishiya, Naoyuki, Moeka Murai, Ayumi Hosoda, Honami Yonezawa, and Norikazu Omori. 2019. "Bucillamine Prevents Afatinib-Mediated Inhibition of Epidermal Growth Factor Receptor Signaling" Pharmaceuticals 12, no. 4: 165. https://doi.org/10.3390/ph12040165
APA StyleNishiya, N., Murai, M., Hosoda, A., Yonezawa, H., & Omori, N. (2019). Bucillamine Prevents Afatinib-Mediated Inhibition of Epidermal Growth Factor Receptor Signaling. Pharmaceuticals, 12(4), 165. https://doi.org/10.3390/ph12040165