Neural Predictors of the Antidepressant Placebo Response


Abstract
:1. Introduction
2. Imaging Data Captured in Clinical Trials Primarily Focused on Antidepressant Efficacy or Predicting Antidepressant Response
3. Imaging Experiments Specifically Interrogating Mechanism of Antidepressant Placebo Response
4. Discussion
4.1. Endogenous Opioids and the Antidepressant Placebo Response
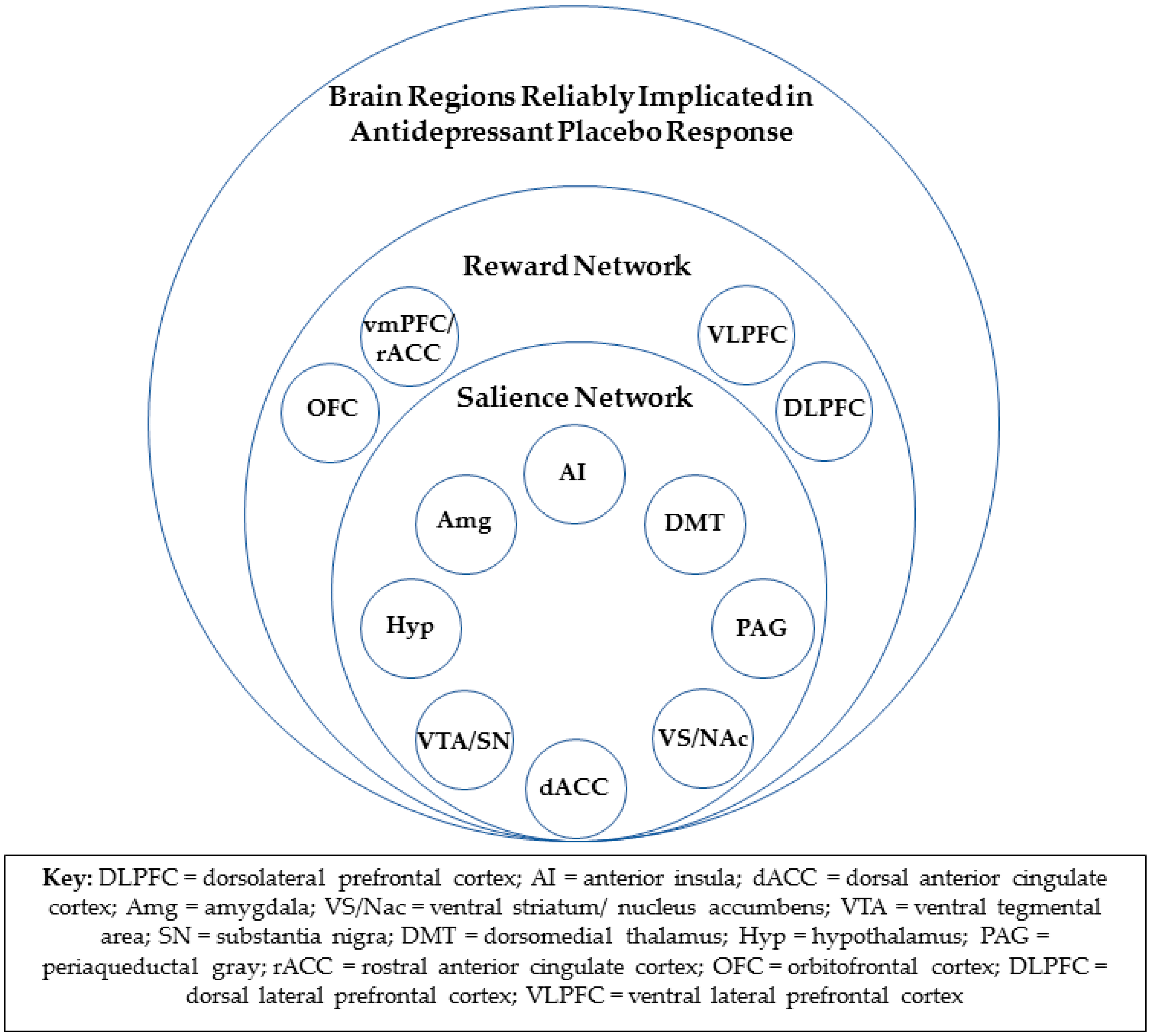
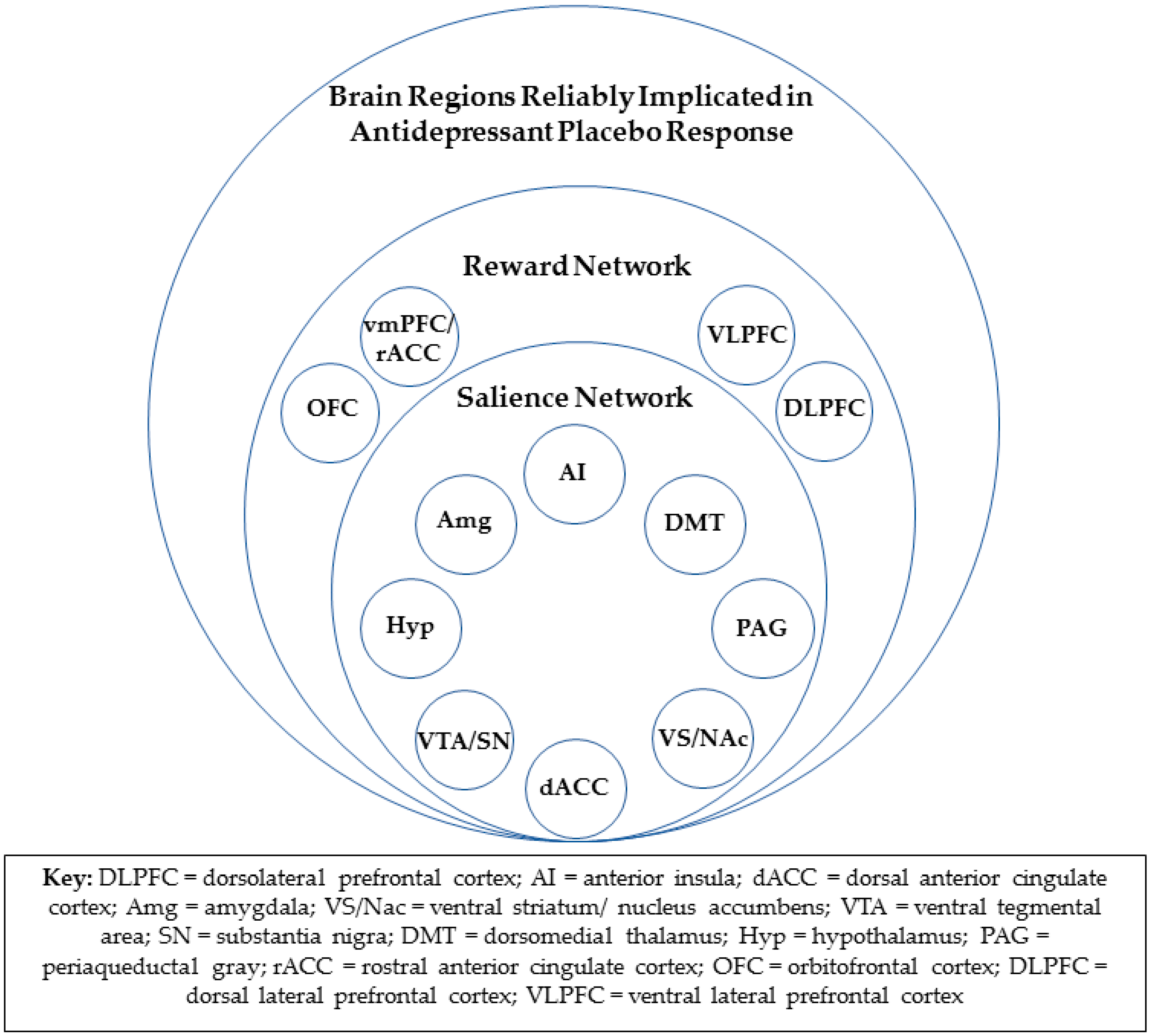
4.2. The Reward Network and Antidepressant Placebo Response
4.3. Rostral Anterior Cingulate Cortex and Antidepressant Placebo Response
4.4. Consistent Overlap in Neural Substrates of Antidepressant Placebo and Medication Response
4.5. Salience/Reward Network Functional Connectivity and Other Tools for Future Brain-Based Prospective Identification of Likely Placebo Responders
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Depression [Fact Sheet]. 2018. Available online: https://www.who.int/en/news-room/fact-sheets/detail/depression (accessed on 18 October 2019).
- Rush, J.A.; Trivedi, M.H.; Wisniewski, S.R.; Nierenberg, A.A.; Stewart, J.W.; Warden, D.; Fava, M. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: A STAR*D report. Am. J. Psychiatry 2006, 163, 1905–1917. [Google Scholar] [CrossRef] [PubMed]
- Walsh, B.T.; Seidman, S.N.; Sysko, R.; Gould, M. Placebo response in studies of major depression: Variable, substantial, and growing. JAMA Psychiatry 2002, 287, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Stolk, P.; ten Berg, M.J.; Hemels, M.E.; Einarson, T.R. Meta-analysis of placebo rates in major depressive disorder trials. Ann. Pharmacother. 2003, 37, 1891–1899. [Google Scholar] [CrossRef] [PubMed]
- Iovieno, N.; Papakostas, G.I. Correlation between different levels of placebo response rate. J. Clin. Psychiatry 2012, 73, 1300–1306. [Google Scholar] [CrossRef]
- Fava, M.; Evins, E.A.; Dorer, D.J.; Schoenfeld, D.A. The problem of the placebo response in clinical trials for psychiatric disorders: Culprits, possible remedies, and a novel study design approach. Psychother. Psychosom. 2003, 72, 115–127. [Google Scholar] [CrossRef]
- Hills, M.; Armitage, P. The two-period cross-over clinical trial. Br. J. Clin. Pharmacol. 1979, 8, 7–20. [Google Scholar] [CrossRef]
- Merlo-Pich, E.; Alexander, R.C.; Fava, M.; Gomeni, R. A new population-enrichment strategy to improve efficiency of placebo-controlled clinical trials of antidepressant drugs. Clin. Pharmacol. Ther. 2010, 88, 634–642. [Google Scholar] [CrossRef]
- Mallinckrodt, C.H.; Tamura, R.N.; Tanaka, Y. Recent developments in improving signal detection and reducing placebo response in psychiatric clinical trials. J. Psychiatr. Res. 2011, 45, 1202–1207. [Google Scholar] [CrossRef]
- Faries, D.; Herrera, J.; Rayamajhi, J.; Debrota, D.; Demitrack, M.; Potter, W.Z. The responsiveness of the Hamilton Depression Rating Scale. J. Psychiatr. Res. 2000, 34, 3–10. [Google Scholar] [CrossRef]
- Bech, P.; Kajdasz, D.K.; Porsdal, V. Dose-response relationship of duloxetine in placebo-controlled clinical trials in patients with major depressive disorder. Psychopharmacology 2006, 188, 273–280. [Google Scholar] [CrossRef]
- McIntyre, R.S.; Konarski, J.Z.; Mancini, D.A.; Fulton, K.A.; Parikh, S.V.; Grigoriadis, S.; Kennedy, S.H. Measuring the severity of depression and remission in primary care: Validation of the HAMD-7 scale. Can. Med. Assoc. J. 2005, 173, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Fava, M.; Mischoulon, D.; Iosifescu, D.; Witte, J.; Pecina, M.; Flynn, M.; Mark, P. Adouble-blind, placebo-controlled study of aripiprazole adjunctive to antidepressant therapy among depressed outpatients with inadequate response to prior antidepressant therapy (ADAPT-A study). Psychother. Psychosom. 2012, 81, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M.P.; Pooley, J.; Flynn, M.J.; Baer, L.; Mischoulon, D.; Mou, D.; Fava, M. Guarding the gate: Remote structured assessments to enhance enrollment precision in depression trials. J. Clin. Psychopharmacol. 2017, 37, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Mayberg, H.S.; Arturo Silva, J.; Brannan, S.K.; Tekell, J.L.; Mahurin, R.K.; McGinnis, S.; Jerabek, P.A. The functional neuroanatomy of the placebo effect. Am. J. Psychiatry 2002, 159, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Leuchter, A.F.; Cook, I.A.; Witte, E.A.; Morgan, M.; Abrams, M. Changes in brain function of depressed subjects during treatment with placebo. Am. J. Psychiatry 2002, 159, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Leuchter, A.F.; Morgan, M.; Cook, I.A.; Dunkin, J.; Abrams, M.; Witte, E. Pretreatment neurophysiological and clinical characteristics of placebo responders in treatment trials for major depression. Psychopharmacology 2004, 177, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Korb, A.S.; Aimee, H.M.; Cook, I.A.; Leuchter, A.F. Rostral anterior cingulate cortex theta current density and response to Antidepressants and Placebo in Major Depression. Clin. Neurophysiol. 2009, 120, 1313–1319. [Google Scholar] [CrossRef]
- Trivedi, M.H.; South, C.; Jha, M.K.; Rush, A.J.; Cao, J.; Kurian, B.; Fava, M. A novel strategy to identify placebo responders: Prediction index of clinical and biological markers in the EMBARC trial. Psychother. Psychosom. 2018, 87, 285–295. [Google Scholar] [CrossRef]
- Pizzagalli, D.A.; Webb, C.A.; Dillon, D.G.; Tenke, C.E.; Kayser, J.; Goer, F.; Trivedi, M.H. Pretreatment rostral anterior cingulate cortex theta activity in relation to symptom improvement in depression: A randomized clinical trial. JAMA Psychiatry 2018, 75, 547–554. [Google Scholar] [CrossRef]
- Whitton, A.E.; Webb, C.A.; Dillon, D.G.; Kayser, J.; Rutherford, A.; Goer, F.; Pizzagalli, D.A. Pretreatment rostral anterior cingulate cortex connectivity with salience network predicts depression recovery: Findings from the EMBARC randomized clinical trial. Biol. Psychiatry 2019, 85, 872–880. [Google Scholar] [CrossRef]
- Peciña, M.; Bohnert, A.S.B.; Sikora, M.; Avery, E.T.; Langenecker, S.A.; Mickey, B.J.; Zubieta, J.-K. Association between placebo-activated neural systems are linked to antidepressant responses: Neurochemistry of placebo effects in major depression. JAMA Psychiatry 2015, 72, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Sikora, M.; Heffernan, J.; Avery, E.T.; Mickey, B.J.; Zubieta, J.-K.; Peciña, M. Salience network functional connectivity predicts placebo effects in major depression. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2016, 1, 68–76. [Google Scholar] [CrossRef]
- Seeley, W.W.; Menon, V.; Schatzberg, A.F.; Keller, J.; Glover, G.H.; Kenna, H.; Greicius, M.D. Dissociable intrinsic connectivity networks for salience processing and executive control. J. Neurosci. 2007, 27, 2349–2356. [Google Scholar] [CrossRef]
- Peciña, M.; Heffernan, J.; Wilson, J.; Zubieta, J.K.; Dombrovski, A.Y. Prefrontal expectancy and reinforcement-driven antidepressant placebo effects. Transl. Psychiatry 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Schultz, W. Predictive reward signal of dopamine neurons. J. Neurophysiol. 1998, 80, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Laurent, V.; Morse, A.K.; Balleine, B.W. The role of opioid processes in reward and decision-making. Br. J. Pharmacol. 2015, 172, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Haber, S.N.; Knutson, B. The reward circuit: Linking primate anatomy and human imaging. Neuropsychopharmacology 2010, 35, 4–26. [Google Scholar] [CrossRef]
- Wilson, R.P.; Colizzi, M.; Bossong, M.G.; Allen, P.; Kempton, M.; Bhattacharyya, S. The neural substrate of reward anticipation in health: A meta-analysis of fMRI findings in the monetary incentive delay task. Neuropsychol. Rev. 2018, 28, 496–506. [Google Scholar] [CrossRef]
- Papakostas, G.I.; Fava, M. Does the probability of receiving placebo influence clinical trial outcome? A meta-regression of double-blind, randomized clinical trials in MDD. Eur. Neuropsychopharmacol. 2009, 19, 34–40. [Google Scholar] [CrossRef]
- Rutherford, B.R.; Wall, M.M.; Brown, P.J.; Choo, T.-H.; Wager, T.D.; Peterson, B.S.; Roose, S.P. Patient expectancy as a mediator of placebo effects in antidepressant clinical trials. Am. J. Psychiatry 2017, 174, 135–142. [Google Scholar] [CrossRef]
- Rutherford, B.R.; Roose, S.P. A model of placebo response in antidepressant clinical trials. Am. J. Psychiatry 2013, 170, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Lidstone, S.C.; Stoessl, J.A. Understanding the placebo effect: Contributions from neuroimaging. Mol. Imaging Biol. 2007, 9, 176–185. [Google Scholar] [CrossRef] [PubMed]
- De La Fuente-Fernández, R. The placebo-reward hypothesis: Dopamine and the placebo effect. Parkinsonism Relat. Disord. 2009, 15 (Suppl. 3), S72–S74. [Google Scholar] [CrossRef]
- Borges, S.; Chen, Y.F.; Laughren, T.P.; Temple, R.; Patel, H.D.; David, P.A.; Khin, N.A. Review of maintenance trials for major depressive disorder: A 25-year perspective from the US Food and Drug Administration. J. Clin. Psychiatry 2014, 75, 205–214. [Google Scholar] [CrossRef]

{kind=link}
| Reference | N | Method | Brain Regions Associated with Antidepressant Placebo Response |
|---|---|---|---|
| [15] | 17 | FDG PET after 1 and 6 wks PBO or ADT | rACC, pACC, NAc, OFC, dlPFC, PMC, IPC, Hyp, Tha, SSA, pIns, pHipp; also predict ADT response. |
| [16,17] | 51 | EEG at baseline, after 1 week single-blind PBO lead-in, and 2, 4, 6, and 8 wks ADT | Frontocentral cortex |
| [18] | 72 | EEG at baseline | None |
| [19] | 144 | EEG at baseline, after 1 week PBO | rACC; |
| [20] | 248 | EEG at baseline, after 1 week PBO or ADT | rACC; also predicts ADT response |
| [21] | 238 | EEG at baseline, after 1 week PBO or ADT, functional connectivity analysis. | rACC, rAI; also predicts ADT response |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rette, D.; McDonald, E.; Iosifescu, D.V.; Collins, K.A. Neural Predictors of the Antidepressant Placebo Response. Pharmaceuticals 2019, 12, 158. https://doi.org/10.3390/ph12040158
Rette D, McDonald E, Iosifescu DV, Collins KA. Neural Predictors of the Antidepressant Placebo Response. Pharmaceuticals. 2019; 12(4):158. https://doi.org/10.3390/ph12040158
Chicago/Turabian StyleRette, Danielle, Erin McDonald, Dan V. Iosifescu, and Katherine A. Collins. 2019. "Neural Predictors of the Antidepressant Placebo Response" Pharmaceuticals 12, no. 4: 158. https://doi.org/10.3390/ph12040158
APA StyleRette, D., McDonald, E., Iosifescu, D. V., & Collins, K. A. (2019). Neural Predictors of the Antidepressant Placebo Response. Pharmaceuticals, 12(4), 158. https://doi.org/10.3390/ph12040158