Non-Nucleoside Agonists of the Adenosine Receptors: An Overview

 ,
, 
 and
and 
Abstract
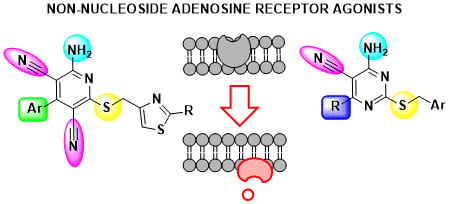
1. Introduction
2. Non-Nucleoside Agonists of the ARs
2.1. Structural Features and Biological Activity
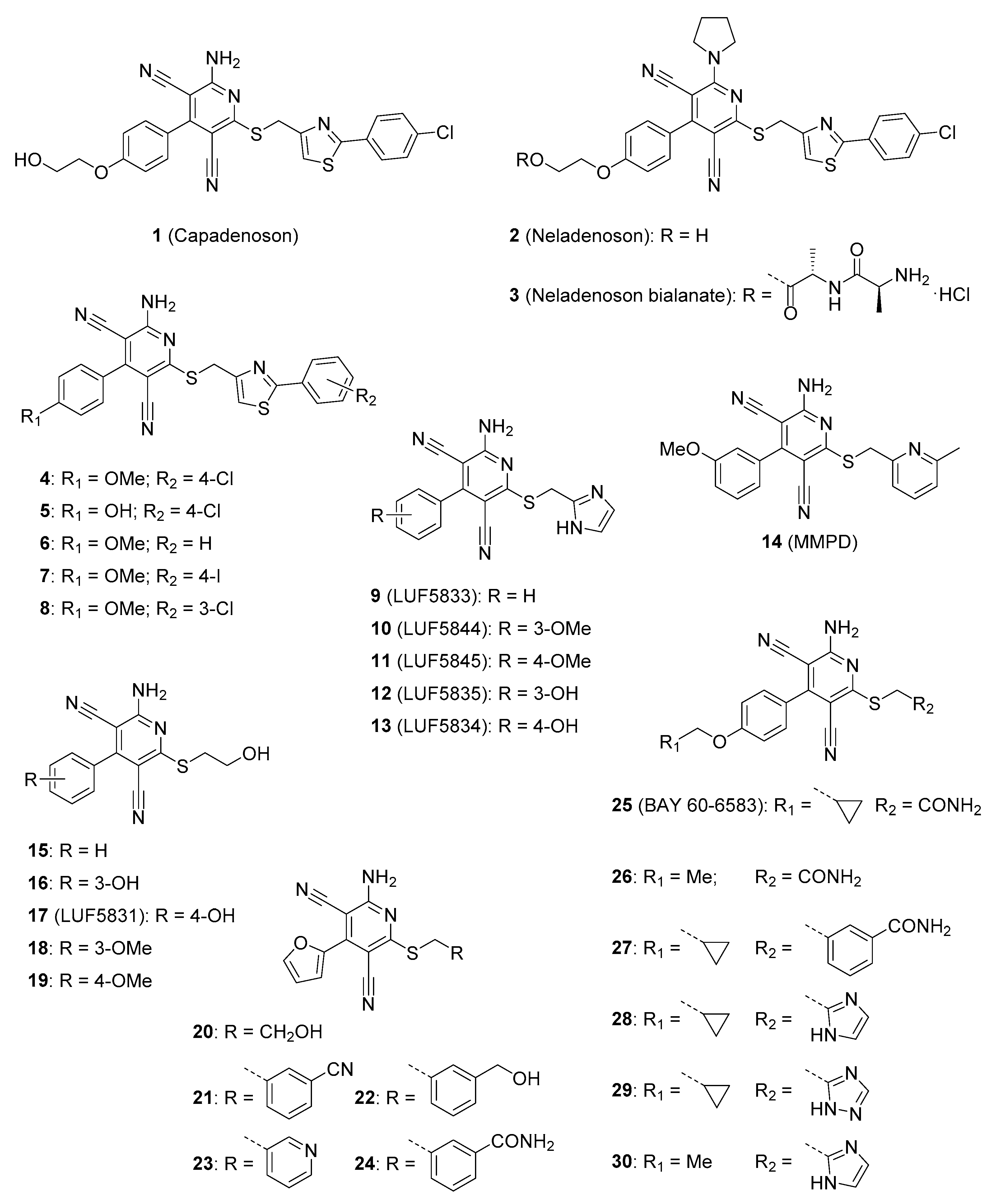
2.1.1. Pyridine Derivatives
2.1.2. Pyrimidine Derivatives
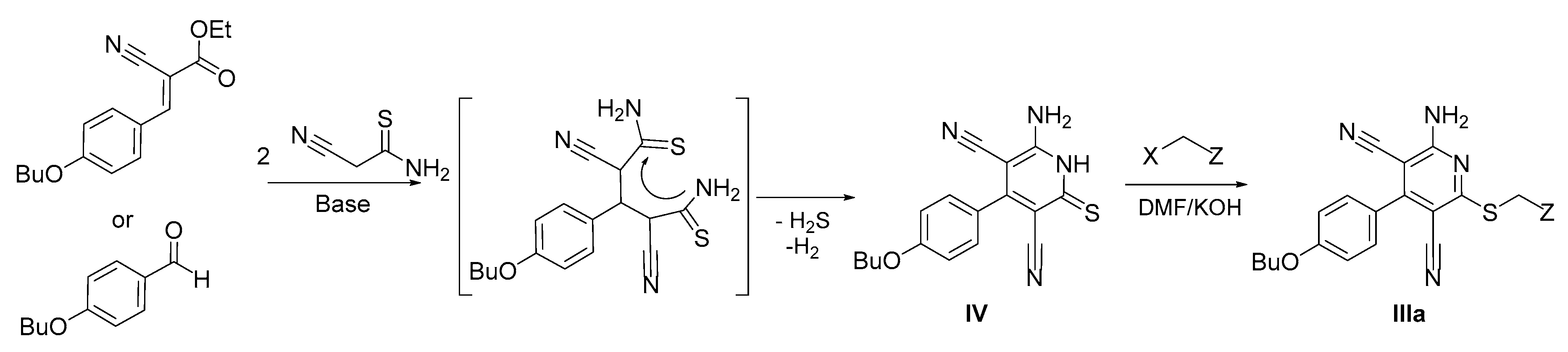
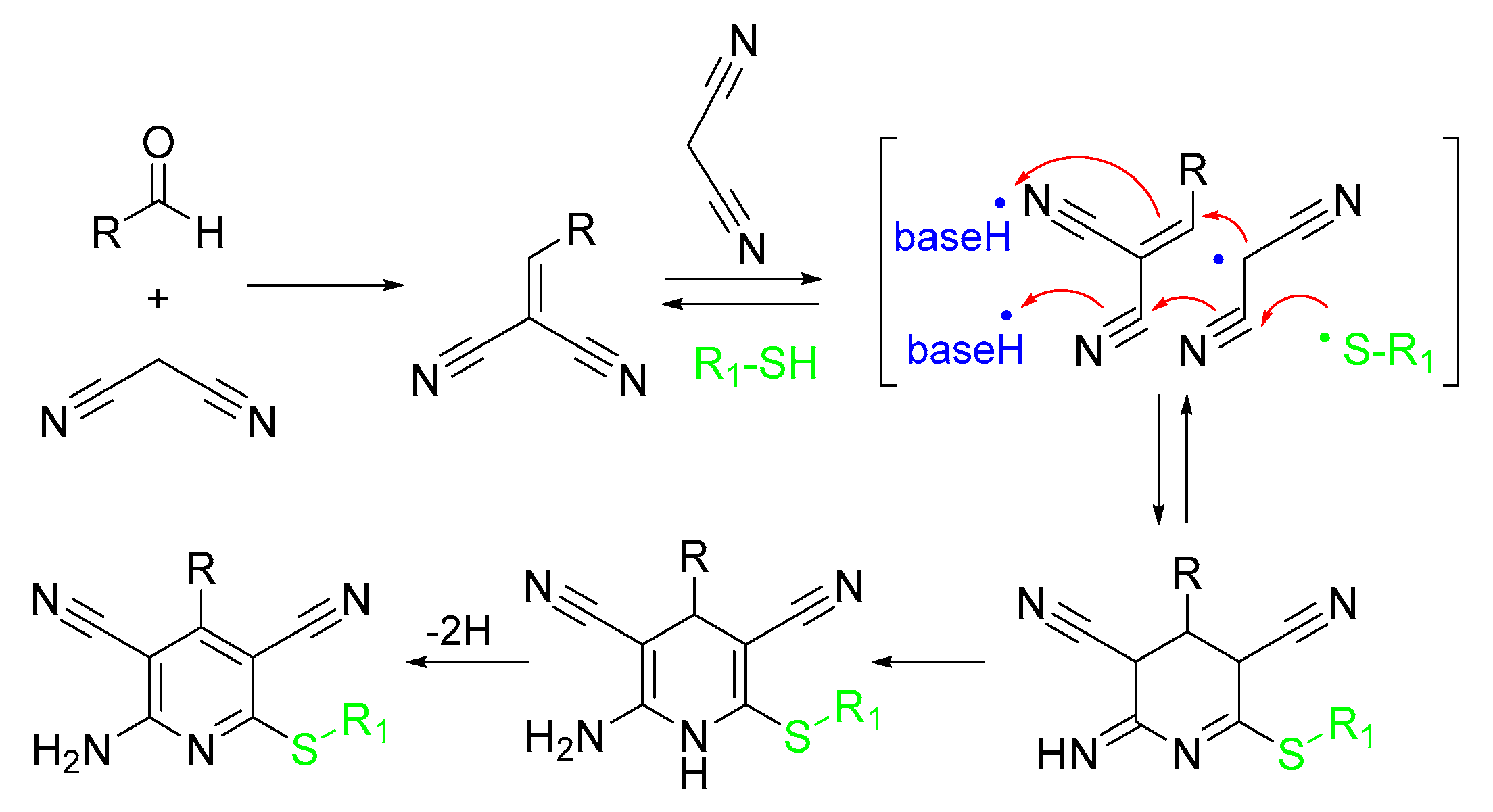
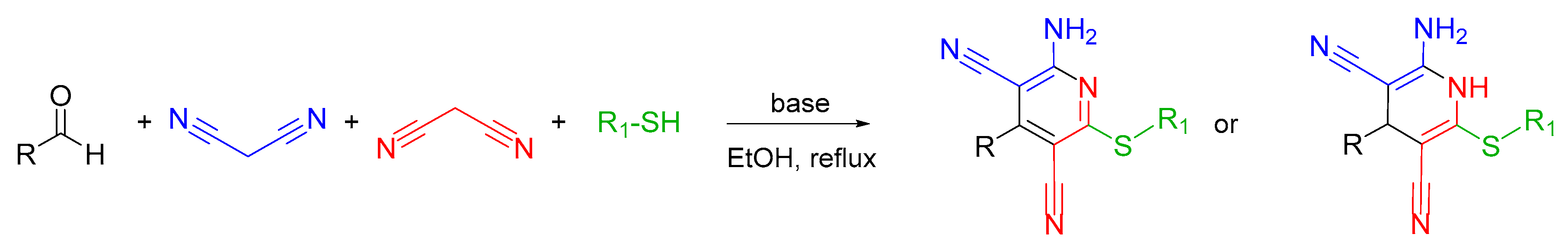
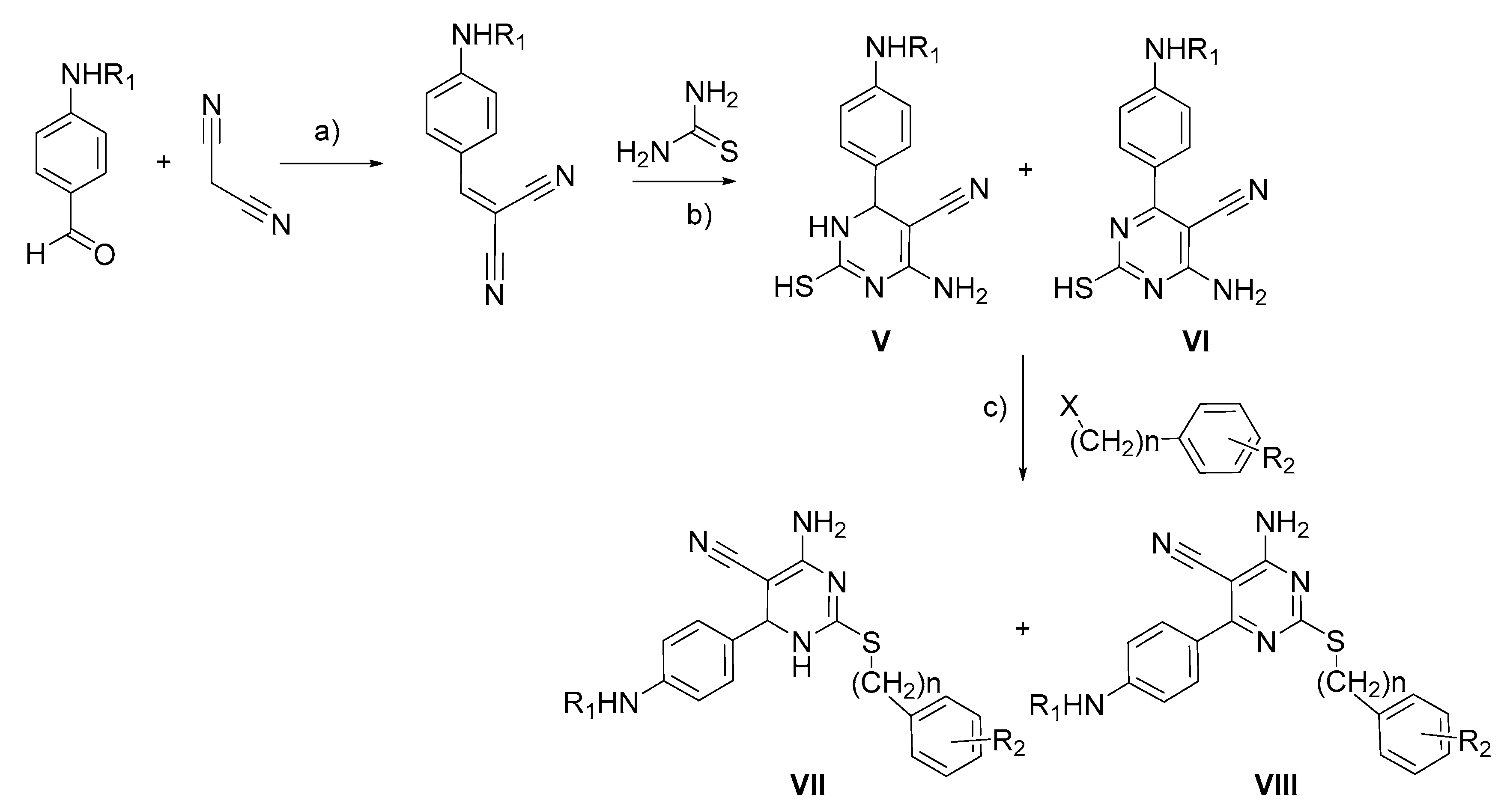
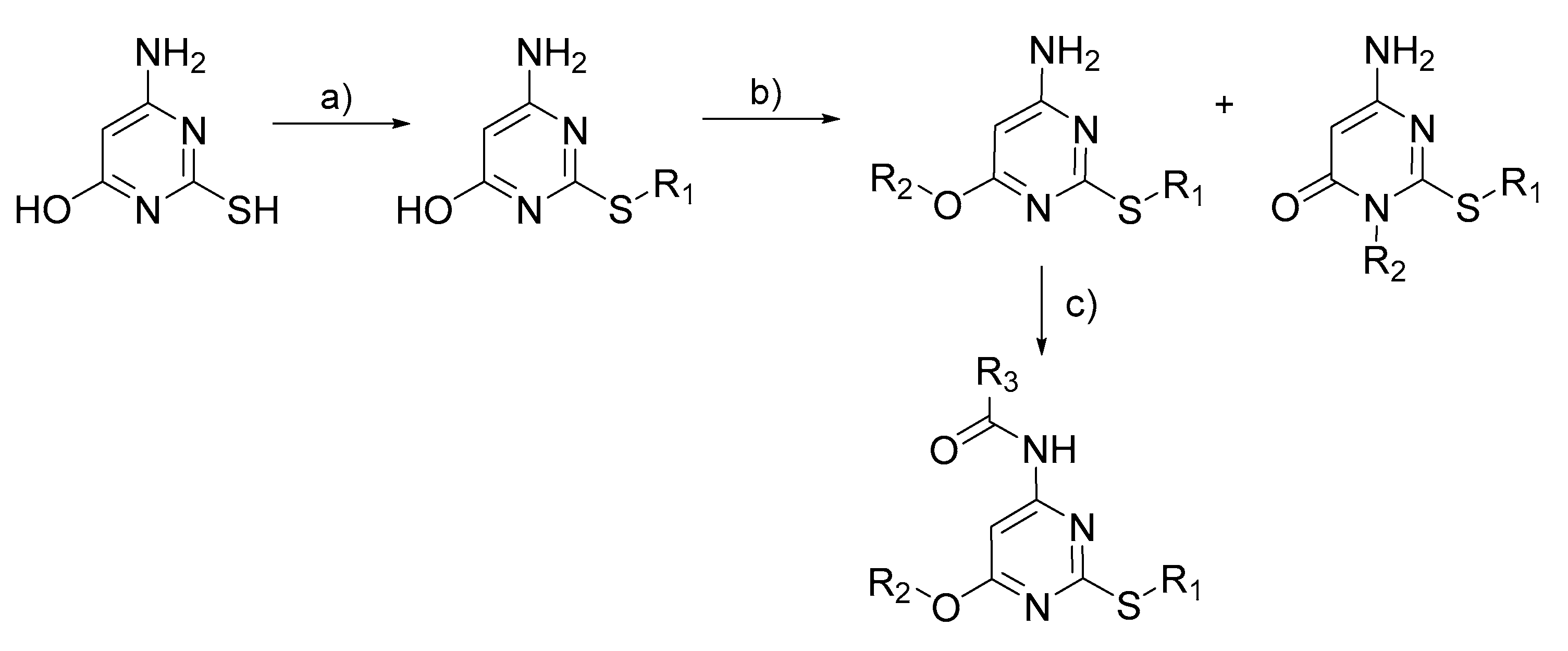
2.2. Synthetic Approaches
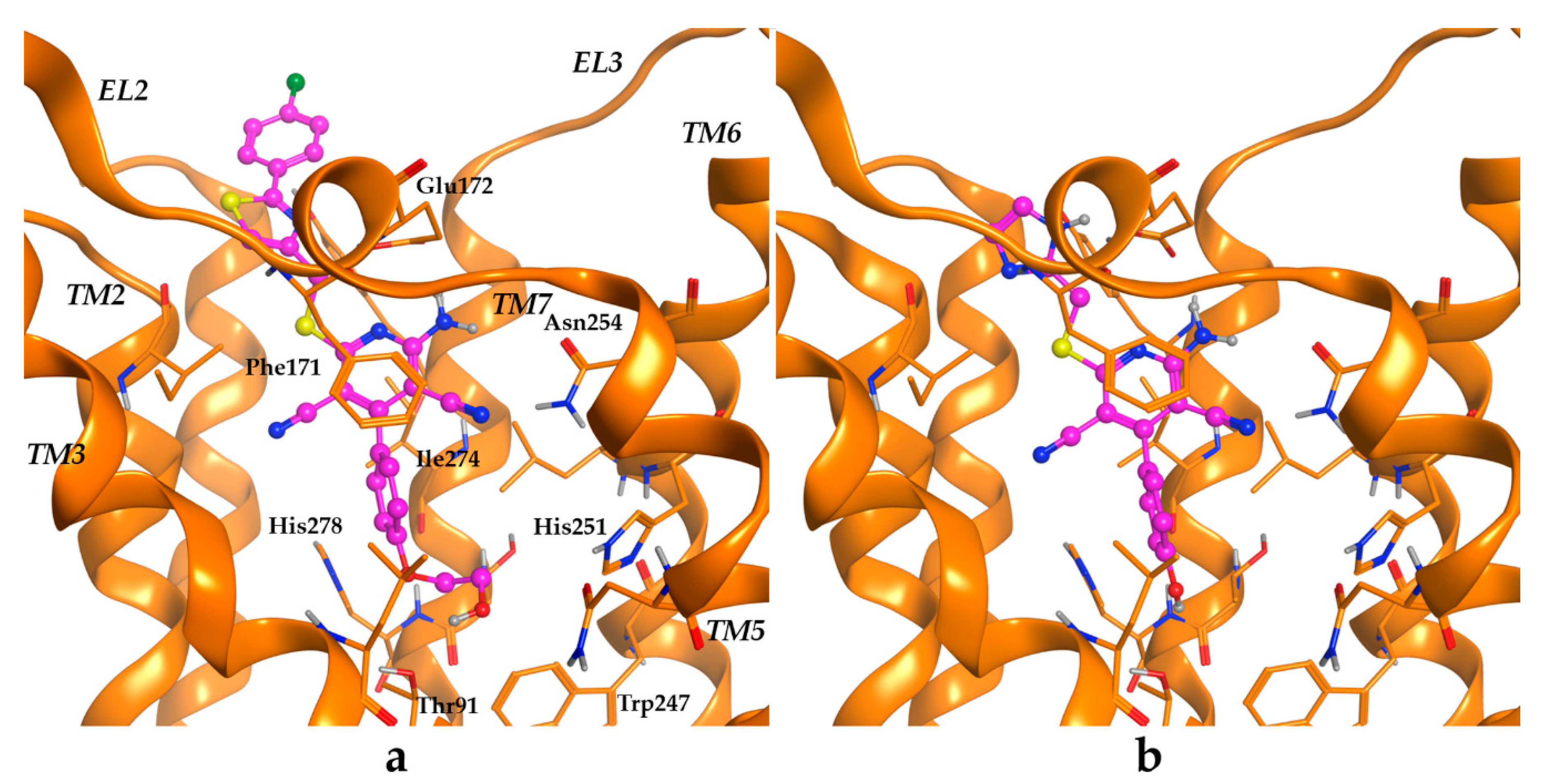
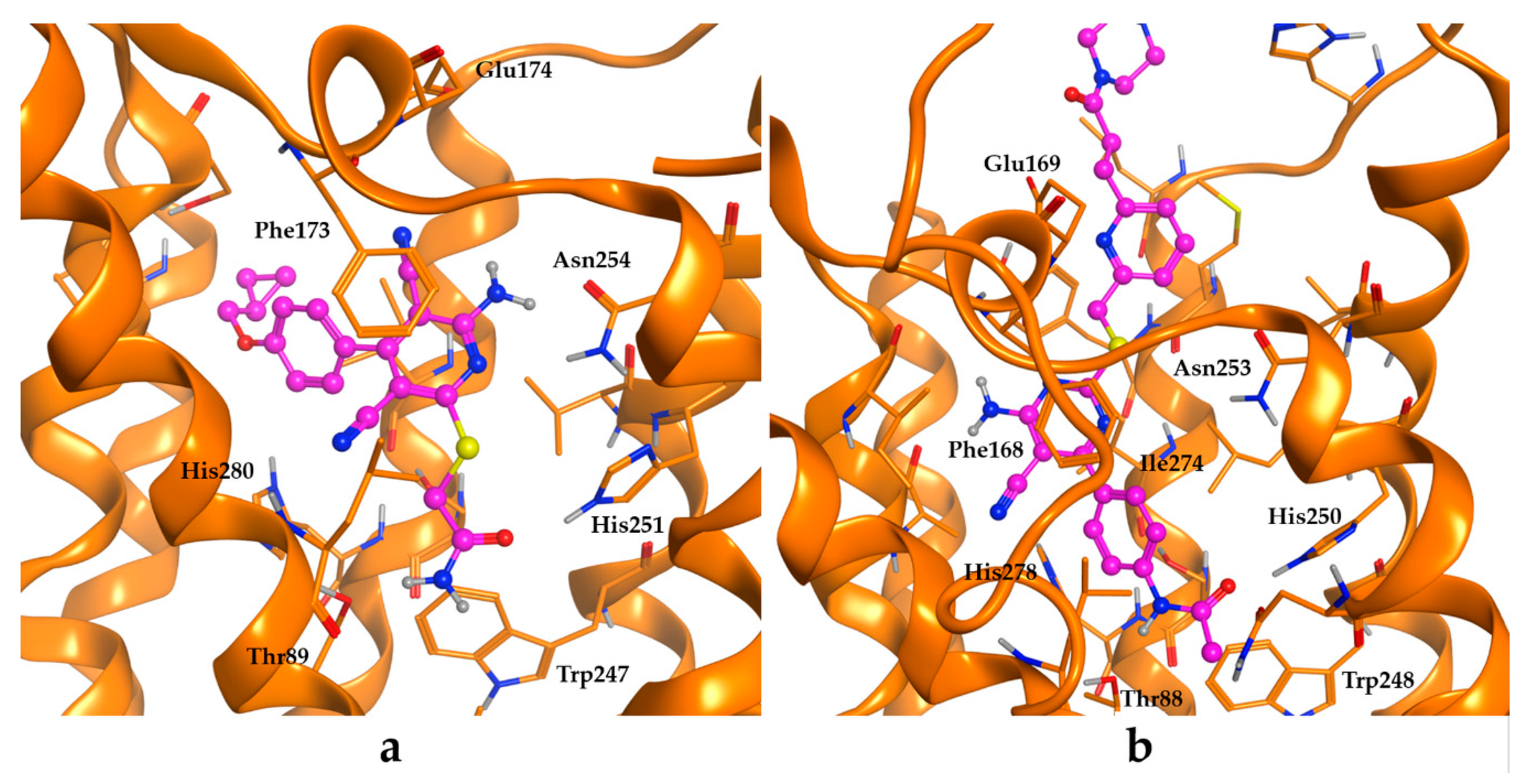
2.3. Molecular Modelling
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Klotz, K.-N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharm. Rev. 2001, 53, 527–552. [Google Scholar] [PubMed]
- Robeva, A.S.; Woodard, R.L.; Jin, X.; Gao, Z.; Bhattacharya, S.; Taylor, H.E.; Rosin, D.L.; Linden, J. Molecular characterization of recombinant human adenosine receptors. Drug Dev. Res. 1996, 39, 243–252. [Google Scholar] [CrossRef]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Muller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors--an update. Pharm. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of adenosine receptors: The state of the art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef] [PubMed]
- Cristalli, G.; Volpini, R. Adenosine receptors: Chemistry and pharmacology. Curr. Top. Med. Chem. 2003, 3, 355–469. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Gao, Z.G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug. Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef] [PubMed]
- Sperlagh, B.; Vizi, E.S. The role of extracellular adenosine in chemical neurotransmission in the hippocampus and basal ganglia: Pharmacological and clinical aspects. Curr. Top. Med. Chem. 2011, 11, 1034–1046. [Google Scholar] [CrossRef]
- Headrick, J.P.; Ashton, K.J.; Rose’meyer, R.B.; Peart, J.N. Cardiovascular adenosine receptors: Expression, actions and interactions. Pharm. Ther. 2013, 140, 92–111. [Google Scholar] [CrossRef] [PubMed]
- Dal Ben, D.; Antonioli, L.; Lambertucci, C.; Fornai, M.; Blandizzi, C.; Volpini, R. Purinergic ligands as potential therapeutic Tools for the treatment of inflammation-related intestinal diseases. Front. Pharm. 2018, 9, 212. [Google Scholar] [CrossRef]
- Antonioli, L.; Colucci, R.; Pellegrini, C.; Giustarini, G.; Tuccori, M.; Blandizzi, C.; Fornai, M. The role of purinergic pathways in the pathophysiology of gut diseases: Pharmacological modulation and potential therapeutic applications. Pharm. Ther. 2013, 139, 157–188. [Google Scholar] [CrossRef]
- Vallon, V.; Osswald, H. Adenosine receptors and the kidney. In Adenosine Receptors in Health and Disease; Springer: Berlin/Heidelberg, Germany, 2009; pp. 443–470. [Google Scholar]
- Johnston-Cox, H.A.; Yang, D.; Ravid, K. Physiological implications of adenosine receptor-mediated platelet aggregation. J. Cell. Physiol. 2011, 226, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Linden, J. Regulation of leukocyte function by adenosine receptors. Adv. Pharm. 2011, 61, 95–114. [Google Scholar]
- Chen, J.-F.; Eltzschig, H.K.; Fredholm, B.B. Adenosine receptors as drug targets–What are the challenges? Nat. Rev. Drug Discov. 2013, 12, 265–286. [Google Scholar] [CrossRef] [PubMed]
- CVT. CV Therapeutics and Astellas Announce FDA Approval for Lexiscan(TM) (regadenoson) Injection. Available online: http://www.cvt.com/PressRelease.aspx?releaseID=1128317 (accessed on 10 April 2008).
- Kyowa Hakko Kirin. Approval for Manufacturing and Marketing of NOURIAST® Tablets 20 mg, a Novel Antiparkinsonian Agent. Available online: http://www.kyowa-kirin.com/news_releases/2013/e20130325_04.html (accessed on 7 October 2019).
- FDA. Drug Trials Snapshots: NOURIANZ. Available online: https://www.fda.gov/drugs/drug-trials-snapshots-nourianz (accessed on 7 October 2019).
- Muller, C.E.; Jacobson, K.A. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Biophys. Acta 2011, 1808, 1290–1308. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Cacciari, B.; Pineda de Las Infantas, M.J.; Romagnoli, R.; Spalluto, G.; Volpini, R.; Costanzi, S.; Vittori, S.; Cristalli, G.; Melman, N.; et al. Synthesis and biological activity of a new series of N6-arylcarbamoyl, 2-(Ar)alkynyl-N6-arylcarbamoyl, and N6-carboxamido derivatives of adenosine-5’-N-ethyluronamide as A1 and A3 adenosine receptor agonists. J. Med. Chem. 1998, 41, 3174–3185. [Google Scholar] [CrossRef] [PubMed]
- Cristalli, G.; Volpini, R.; Vittori, S.; Camaioni, E.; Monopoli, A.; Conti, A.; Dionisotti, S.; Zocchi, C.; Ongini, E. 2-Alkynyl derivatives of adenosine-5′-N-ethyluronamide: Selective A2 adenosine receptor agonists with potent inhibitory activity on platelet aggregation. J. Med. Chem. 1994, 37, 1720–1726. [Google Scholar] [CrossRef] [PubMed]
- Cristalli, G.; Camaioni, E.; Vittori, S.; Volpini, R.; Borea, P.A.; Conti, A.; Dionisotti, S.; Ongini, E.; Monopoli, A. 2-Aralkynyl and 2-heteroalkynyl derivatives of adenosine-5′-N-ethyluronamide as selective A2a adenosine receptor agonists. J. Med. Chem. 1995, 38, 1462–1472. [Google Scholar] [CrossRef] [PubMed]
- Volpini, R.; Buccioni, M.; Dal Ben, D.; Lambertucci, C.; Lammi, C.; Marucci, G.; Ramadori, A.T.; Klotz, K.-N.; Cristalli, G. Synthesis and biological evaluation of 2-alkynyl-N6-methyl-5′-N-methylcarboxamidoadenosine derivatives as potent and highly selective agonists for the human adenosine A3 receptor. J. Med. Chem. 2009, 52, 7897–7900. [Google Scholar] [CrossRef] [PubMed]
- Lambertucci, C.; Antonini, I.; Buccioni, M.; Dal Ben, D.; Kachare, D.D.; Volpini, R.; Klotz, K.-N.; Cristalli, G. 8-Bromo-9-alkyl adenine derivatives as tools for developing new adenosine A2A and A2B receptors ligands. Bioorg. Med. Chem. 2009, 17, 2812–2822. [Google Scholar] [CrossRef] [PubMed]
- Lambertucci, C.; Buccioni, M.; Dal Ben, D.; Kachler, S.; Marucci, G.; Spinaci, A.; Thomas, A.; Klotz, K.-N.; Volpini, R. New substituted 9-propyladenine derivatives as A2A adenosine receptor antagonists. Medchemcomm 2015, 6, 963–970. [Google Scholar] [CrossRef]
- Lambertucci, C.; Spinaci, A.; Buccioni, M.; Dal Ben, D.; Ngouadjeu Ngnintedem, M.A.; Kachler, S.; Marucci, G.; Klotz, K.-N.; Volpini, R. New A2A adenosine receptor antagonists: A structure-based upside-down interaction in the receptor cavity. Bioorg. Chem. 2019, 92. [Google Scholar] [CrossRef] [PubMed]
- Rosentreter, U.; Henning, R.; Bauser, M.; Krämer, T.; Vaupel, A.; Hübsch, W.; Dembowsky, K.; Salcher-Schraufstätter, O.; Stasch, J.P.; Krahn, T.; et al. Substituted 2-Thio-3,5-Dicyano-4-Aryl-6-Aminopyridines and the Use Thereof as Adenosine Receptor Ligands. U.S. Patent 7,135,486, 14 November 2006. [Google Scholar]
- Rosentreter, U.; Krämer, T.; Shimada, M.; Hübsch, W.; Diedrichs, N.; Krahn, T.; Henninger, K.; Stasch, J.P. Substituted 2-Thio-3,5-Dicyano-4-Phenyl-6-Aminopyridines and their Use as Adenosine Receptor-Selective Ligands. U.S. Patent 7,045,631, 16 May 2006. [Google Scholar]
- Rosentreter, U.; Kramer, T.; Vaupel, A.; Hubsch, W.; Diedrichs, N.; Krahn, T.; Dembowsky, K.; Stasch, J.P.; Shimada, M. Substituted 2-Thio-3,5-Dicyano-4-Phenyl-6-Aminopyridines with Adenosine Receptor-Binding Activity and their Use Cardiovascular Preparations. U.S. Patent 7,078,417, 18 July 2006. [Google Scholar]
- Nell, P.; Albrecht-Kupper, B.; Hübsch, W.; Wuttke, M.; Krahn, T.; Diedrichs, N.; Bischoff, H. Use of Adenosine A1 and/or Dual A1/A2B Agonists for Production of Medicaments for Treating Diseases. U.S. Patent Application No. 12/224,417, 25 February 2010. [Google Scholar]
- Kato, M.; Sato, N.; Okada, M.; Uno, T.; Ito, N.; Takeji, Y.; Shinohara, H.; Fuwa, M. 4-Amino-5-Cyanopyrimidine Derivatives. Patent Application KR20070008715, 17 January 2007. [Google Scholar]
- Beukers, M.W.; Chang, L.C.; von Frijtag Drabbe Kunzel, J.K.; Mulder-Krieger, T.; Spanjersberg, R.F.; Brussee, J.; IJzerman, A.P. New, non-adenosine, high-potency agonists for the human adenosine A2B receptor with an improved selectivity profile compared to the reference agonist N-ethylcarboxamidoadenosine. J. Med. Chem. 2004, 47, 3707–3709. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.C.; von Frijtag Drabbe Kunzel, J.K.; Mulder-Krieger, T.; Spanjersberg, R.F.; Roerink, S.F.; van den Hout, G.; Beukers, M.W.; Brussee, J.; IJzerman, A.P. A series of ligands displaying a remarkable agonistic-antagonistic profile at the adenosine A1 receptor. J. Med. Chem. 2005, 48, 2045–2053. [Google Scholar] [CrossRef] [PubMed]
- Heitman, L.H.; Mulder-Krieger, T.; Spanjersberg, R.F.; von Frijtag Drabbe Kunzel, J.K.; Dalpiaz, A.; IJzerman, A.P. Allosteric modulation, thermodynamics and binding to wild-type and mutant (T277A) adenosine A1 receptors of LUF5831, a novel nonadenosine-like agonist. Br. J. Pharm. 2006, 147, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.R.; Klaasse, E.; Lin, J.; van Bruchem, J.; Beukers, M.W.; IJzerman, A.P. Characterization of [3H] LUF5834: A novel non-ribose high-affinity agonist radioligand for the adenosine A1 receptor. Biochem. Pharm. 2010, 80, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Louvel, J.; Guo, D.; Agliardi, M.; Mocking, T.A.; Kars, R.; Pham, T.P.; Xia, L.; de Vries, H.; Brussee, J.; Heitman, L.H.; et al. Agonists for the adenosine A1 receptor with tunable residence time. A Case for nonribose 4-amino-6-aryl-5-cyano-2-thiopyrimidines. J. Med. Chem. 2014, 57, 3213–3222. [Google Scholar] [CrossRef] [PubMed]
- Louvel, J.; Guo, D.; Soethoudt, M.; Mocking, T.A.; Lenselink, E.B.; Mulder-Krieger, T.; Heitman, L.H.; IJzerman, A.P. Structure-kinetics relationships of Capadenoson derivatives as adenosine A1 receptor agonists. Eur. J. Med. Chem. 2015, 101, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.R.; Klein Herenbrink, C.; van Westen, G.J.; Spoorendonk, J.A.; Hoffmann, C.; IJzerman, A.P. A novel nonribose agonist, LUF5834, engages residues that are distinct from those of adenosine-like ligands to activate the adenosine A2A receptor. Mol. Pharm. 2012, 81, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Yuki, Y.; Shinohara, H.; Takeji, Y.; Ito, K.; Michikami, D.; Hino, K.; Yamazaki, H. A Novel Cyanopyrimidine Derivative. Patent Application KR20100111687, 15 October 2010. [Google Scholar]
- Nell, P.G.; Albrecht-Kupper, B. The Adenosine A1 Receptor and its Ligands. Prog. Med. Chem. 2009, 47, 163–201. [Google Scholar] [PubMed]
- Kiesman, W.F.; Elzein, E.; Zablocki, J. A1 adenosine receptor antagonists, agonists, and allosteric enhancers. In Adenosine Receptors in Health and Disease; Springer: Berlin/Heidelberg, Germany, 2009; pp. 25–58. [Google Scholar]
- Baltos, J.A.; Vecchio, E.A.; Harris, M.A.; Qin, C.X.; Ritchie, R.H.; Christopoulos, A.; White, P.J.; May, L.T. Capadenoson, a clinically trialed partial adenosine A1 receptor agonist, can stimulate adenosine A2B receptor biased agonism. Biochem. Pharm. 2017, 135, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Bott-Flugel, L.; Bernshausen, A.; Schneider, H.; Luppa, P.; Zimmermann, K.; Albrecht-Kupper, B.; Kast, R.; Laugwitz, K.L.; Ehmke, H.; Knorr, A.; et al. Selective attenuation of norepinephrine release and stress-induced heart rate increase by partial adenosine A1 agonism. PLoS ONE 2011, 6, e18048. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, H.N.; Gupta, R.C.; Kohli, S.; Wang, M.; Rastogi, S.; Zhang, K.; Zimmermann, K.; Diedrichs, N.; Albrecht-Kupper, B.E. Chronic therapy with a partial adenosine A1-receptor agonist improves left ventricular function and remodeling in dogs with advanced heart failure. Circ. Heart Fail. 2013, 6, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Tendera, M.; Gaszewska-Zurek, E.; Parma, Z.; Ponikowski, P.; Jankowska, E.; Kawecka-Jaszcz, K.; Czarnecka, D.; Krzeminska-Pakula, M.; Bednarkiewicz, Z.; Sosnowski, M.; et al. The new oral adenosine A1 receptor agonist capadenoson in male patients with stable angina. Clin. Res. Cardiol. 2012, 101, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Clinicaltrials.gov. Capadenoson in Angina Pectoris. Available online: https://clinicaltrials.gov/ct2/show/NCT00518921 (accessed on 7 October 2019).
- Albrecht-Kupper, B.E.; Leineweber, K.; Nell, P.G. Partial adenosine A1 receptor agonists for cardiovascular therapies. Purinergic Signal 2012, 8, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00568945 (accessed on 7 October 2019).
- Bailey, I.R.; Laughlin, B.; Moore, L.A.; Bogren, L.K.; Barati, Z.; Drew, K.L. Optimization of Thermolytic Response to A1 Adenosine Receptor Agonists in Rats. J. Pharm. Exp. Ther. 2017, 362, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Baltos, J.A.; Gregory, K.J.; White, P.J.; Sexton, P.M.; Christopoulos, A.; May, L.T. Quantification of adenosine A1 receptor biased agonism: Implications for drug discovery. Biochem. Pharm. 2016, 99, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Meibom, D.; Albrecht-Kupper, B.; Diedrichs, N.; Hubsch, W.; Kast, R.; Kramer, T.; Krenz, U.; Lerchen, H.G.; Mittendorf, J.; Nell, P.G.; et al. Neladenoson bialanate hydrochloride: A prodrug of a partial adenosine A1 receptor agonist for the chronic treatment of heart diseases. Chem. Med. Chem. 2017, 12, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Voors, A.A.; Dungen, H.D.; Senni, M.; Nodari, S.; Agostoni, P.; Ponikowski, P.; Bax, J.J.; Butler, J.; Kim, R.J.; Dorhout, B.; et al. Safety and tolerability of neladenoson bialanate, a novel oral partial adenosine A1 receptor agonist, in patients with chronic heart failure. J. Clin. Pharm. 2017, 57, 440–451. [Google Scholar] [CrossRef]
- Dinh, W.; Albrecht-Kupper, B.; Gheorghiade, M.; Voors, A.A.; van der Laan, M.; Sabbah, H.N. Partial adenosine A1 agonist in heart failure. Handb. Exp. Pharm. 2017, 243, 177–203. [Google Scholar]
- Tamargo, J.; Caballero, R.; Delpon, E. New drugs in preclinical and early stage clinical development in the treatment of heart failure. Expert Opin. Investig. Drugs 2019, 28, 51–71. [Google Scholar] [CrossRef]
- Rossignol, P.; Hernandez, A.F.; Solomon, S.D.; Zannad, F. Heart failure drug treatment. Lancet 2019, 393, 1034–1044. [Google Scholar] [CrossRef]
- Deb, P.K.; Deka, S.; Borah, P.; Abed, S.N.; Klotz, K.-N. Medicinal chemistry and therapeutic potential of agonists, antagonists and allosteric modulators of A1 adenosine receptor: Current status and perspectives. Curr. Pharm. Des. 2019. [Google Scholar] [CrossRef] [PubMed]
- Clinicaltrials.gov. A Trial to Study Neladenoson Bialanate Over 20 Weeks in Patients with Chronic Heart Failure with Preserved Ejection Fraction (PANACHE). Available online: https://www.clinicaltrials.gov/ct2/show/NCT03098979 (accessed on 7 October 2019).
- Clinicaltrials.gov. A Trial to Study Neladenoson Bialanate Over 20 Weeks in Patients with Chronic Heart Failure with Reduced Ejection Fraction (PANTHEON). Available online: https://www.clinicaltrials.gov/ct2/show/NCT02992288 (accessed on 7 October 2019).
- Voors, A.A.; Bax, J.J.; Hernandez, A.F.; Wirtz, A.B.; Pap, A.F.; Ferreira, A.C.; Senni, M.; van der Laan, M.; Butler, J. Safety and efficacy of the partial adenosine A1 receptor agonist neladenoson bialanate in patients with chronic heart failure with reduced ejection fraction: a phase IIb, randomized, double-blind, placebo-controlled trial. Eur. J. Heart Fail. 2019. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.J.; Voors, A.A.; McMurray, J.J.V.; Kitzman, D.W.; Viethen, T.; Bomfim Wirtz, A.; Huang, E.; Pap, A.F.; Solomon, S.D. Effect of neladenoson bialanate on exercise capacity among patients with heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 2019, 321, 2101–2112. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Gao, Z.G.; Tyler, R.; Stodden, T.; Li, Y.; Ramsey, J.; Zhao, W.J.; Wang, G.J.; Wiers, C.E.; Fowler, J.S.; et al. Preclinical Evaluation of the First Adenosine A1 Receptor Partial Agonist Radioligand for Positron Emission Tomography Imaging. J. Med. Chem. 2018, 61, 9966–9975. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Tosh, D.K.; Jain, S.; Gao, Z.G. Historical and Current Adenosine Receptor Agonists in Preclinical and Clinical Development. Front. Cell Neurosci. 2019, 13, 124. [Google Scholar] [CrossRef] [PubMed]
- Betti, M.; Catarzi, D.; Varano, F.; Falsini, M.; Varani, K.; Vincenzi, F.; Pasquini, S.; di Cesare Mannelli, L.; Ghelardini, C.; Lucarini, E.; et al. Modifications on the Amino-3,5-dicyanopyridine Core to Obtain Multifaceted Adenosine Receptor Ligands with Antineuropathic Activity. J. Med. Chem. 2019, 62, 6894–6912. [Google Scholar] [CrossRef] [PubMed]
- Seibt, B.F.; Schiedel, A.C.; Thimm, D.; Hinz, S.; Sherbiny, F.F.; Muller, C.E. The second extracellular loop of GPCRs determines subtype-selectivity and controls efficacy as evidenced by loop exchange study at A2 adenosine receptors. Biochem. Pharmacol. 2013, 85, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Betti, M.; Catarzi, D.; Varano, F.; Falsini, M.; Varani, K.; Vincenzi, F.; Dal Ben, D.; Lambertucci, C.; Colotta, V. The aminopyridine-3,5-dicarbonitrile core for the design of new non-nucleoside-like agonists of the human adenosine A2B receptor. Eur. J. Med. Chem. 2018, 150, 127–139. [Google Scholar] [CrossRef]
- Eckle, T.; Krahn, T.; Grenz, A.; Kohler, D.; Mittelbronn, M.; Ledent, C.; Jacobson, M.A.; Osswald, H.; Thompson, L.F.; Unertl, K.; et al. Cardioprotection by ecto-5’-nucleotidase (CD73) and A2B adenosine receptors. Circulation 2007, 115, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Baraldi, P.G.; Tabrizi, M.A.; Fruttarolo, F.; Romagnoli, R.; Preti, D. Recent improvements in the development of A2B adenosine receptor agonists. Purinergic Signal. 2009, 5, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, A.C.; Hinz, S.; Thimm, D.; Sherbiny, F.; Borrmann, T.; Maass, A.; Muller, C.E. The four cysteine residues in the second extracellular loop of the human adenosine A2B receptor: role in ligand binding and receptor function. Biochem. Pharm. 2011, 82, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Thimm, D.; Schiedel, A.C.; Sherbiny, F.F.; Hinz, S.; Hochheiser, K.; Bertarelli, D.C.G.; Maass, A.; Müller, C.E. Ligand-Specific binding and activation of the human adenosine A2B receptor. Biochemistry 2013, 52, 726–740. [Google Scholar] [CrossRef] [PubMed]
- Hinz, S.; Lacher, S.K.; Seibt, B.F.; Muller, C.E. BAY60-6583 acts as a partial agonist at adenosine A2B receptors. J. Pharm. Exp. Ther. 2014, 349, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Piras, B.A.; Kron, I.L.; French, B.A.; Yang, Z. Adenosine 2B receptor activation reduces myocardial reperfusion injury by promoting anti-inflammatory macrophages differentiation via PI3K/Akt pathway. Oxid. Med. Cell. Longev. 2015, 2015, 585297. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Liang, D.; Tian, Y.; Kron, I.L.; French, B.A.; Yang, Z. Infarct-Sparing effect of adenosine A2B receptor agonist is primarily due to its action on splenic leukocytes via a PI3K/Akt/IL-10 pathway. J. Surg. Res. 2018, 232, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Johnston-Cox, H.; Koupenova, M.; Yang, D.; Corkey, B.; Gokce, N.; Farb, M.G.; LeBrasseur, N.; Ravid, K. The A2B adenosine receptor modulates glucose homeostasis and obesity. PLoS ONE 2012, 7, e40584. [Google Scholar] [CrossRef] [PubMed]
- Hoegl, S.; Brodsky, K.S.; Blackburn, M.R.; Karmouty-Quintana, H.; Zwissler, B.; Eltzschig, H.K. Alveolar epithelial A2B adenosine receptors in pulmonary protection during acute lung injury. J. Immunol. 2015, 195, 1815–1824. [Google Scholar] [CrossRef]
- Johnston-Cox, H.; Eisenstein, A.S.; Koupenova, M.; Carroll, S.; Ravid, K. The macrophage A2B adenosine receptor regulates tissue insulin sensitivity. PLoS ONE 2014, 9, e98775. [Google Scholar] [CrossRef]
- Csoka, B.; Koscso, B.; Toro, G.; Kokai, E.; Virag, L.; Nemeth, Z.H.; Pacher, P.; Bai, P.; Hasko, G. A2B adenosine receptors prevent insulin resistance by inhibiting adipose tissue inflammation via maintaining alternative macrophage activation. Diabetes 2014, 63, 850–866. [Google Scholar] [CrossRef]
- Chang, L.C.; Spanjersberg, R.F.; von Frijtag Drabbe Kunzel, J.K.; Mulder-Krieger, T.; van den Hout, G.; Beukers, M.W.; Brussee, J.; IJzerman, A.P. 2,4,6-trisubstituted pyrimidines as a new class of selective adenosine A1 receptor antagonists. J. Med. Chem. 2004, 47, 6529–6540. [Google Scholar] [CrossRef] [PubMed]
- van Veldhoven, J.P.; Chang, L.C.; von Frijtag Drabbe Kunzel, J.K.; Mulder-Krieger, T.; Struensee-Link, R.; Beukers, M.W.; Brussee, J.; IJzerman, A.P. A new generation of adenosine receptor antagonists: from di- to trisubstituted aminopyrimidines. Bioorg. Med. Chem. 2008, 16, 2741–2752. [Google Scholar] [CrossRef] [PubMed]
- Vidal, B.; Nueda, A.; Esteve, C.; Domenech, T.; Benito, S.; Reinoso, R.F.; Pont, M.; Calbet, M.; Lopez, R.; Cadavid, M.I.; et al. Discovery and characterization of 4’-(2-furyl)-N-pyridin-3-yl-4,5’-bipyrimidin-2’-amine (LAS38096), a potent, selective, and efficacious A2B adenosine receptor antagonist. J. Med. Chem. 2007, 50, 2732–2736. [Google Scholar] [CrossRef] [PubMed]
- Cosimelli, B.; Greco, G.; Laneri, S.; Novellino, E.; Sacchi, A.; Trincavelli, M.L.; Giacomelli, C.; Taliani, S.; Da Settimo, F.; Martini, C. 4-amino-6-alkyloxy-2-alkylthiopyrimidine derivatives as novel non-nucleoside agonists for the adenosine A1 receptor. Chem. Biol. Drug Des. 2016, 88, 724–729. [Google Scholar] [CrossRef] [PubMed]
- Bharate, S.B.; Singh, B.; Kachler, S.; Oliveira, A.; Kumar, V.; Bharate, S.S.; Vishwakarma, R.A.; Klotz, K.-N.; Gutierrez de Teran, H. Discovery of 7-(Prolinol-N-yl)-2-phenylamino-thiazolo[5,4-d]pyrimidines as novel non-nucleoside partial agonists for the A2A adenosine receptor: Prediction from molecular modeling. J. Med. Chem. 2016, 59, 5922–5928. [Google Scholar] [CrossRef] [PubMed]
- Cosimelli, B.; Greco, G.; Ehlardo, M.; Novellino, E.; Da Settimo, F.; Taliani, S.; La Motta, C.; Bellandi, M.; Tuccinardi, T.; Martinelli, A.; et al. Derivatives of 4-amino-6-hydroxy-2-mercaptopyrimidine as novel, potent, and selective A3 adenosine receptor antagonists. J. Med. Chem. 2008, 51, 1764–1770. [Google Scholar] [CrossRef]
- Dyachenko, V.D.; Krivokolysko, S.; Sharanin, Y.; Litvinov, V.P. New route to 6-amino-4-aryl-3,5-dicyano-pyridine-2(1H)-thiones. Russ. J. Org. Chem. 1997, 33, 1014–1017. [Google Scholar]
- Dyachenko, V.D.; Krivokolysko, S.; Sharanin, Y.; Litvinov, V.P. Synthesis and recyclization of 4-aryl-2,6-diamino-3,5-dicyano-4H-thiopyrans. Russ. J. Org. Chem. 1998, 34, 557–563. [Google Scholar]
- Quintela, J.M.; Peinador, C.; Veiga, M.C.; Botana, L.M.; Alfonso, A.; Riguera, R. Synthesis, antihistaminic and cytotoxic activity of pyridothieno- and pyridodithienotriazines. Eur. J. Med. Chem. 1998, 33, 887–897. [Google Scholar] [CrossRef]
- Dyachenko, V.D.; Litvinov, V.P. Michael reaction in synthesis of 6-amino-4-(4-butoxyphenyl)-3,5-dicyanopyridine-2(1H)-thione. Chem. Heterocycl. Compd. 1998, 34, 188–194. [Google Scholar] [CrossRef]
- Evdokimov, N.M.; Magedov, I.V.; Kireev, A.S.; Kornienko, A. One-step, three-component synthesis of pyridines and 1,4-dihydropyridines with manifold medicinal utility. Org. Lett. 2006, 8, 899–902. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, M.; Ramanaiah, B.C.; Narsaiah, C.; Mahesh, B.; Kumaraswamy, M.; Mallu, K.K.R.; Ankathi, V.M.; Shanthan Rao, P. Novel ZnCl2-catalyzed one-pot multicomponent synthesis of 2-amino-3,5-dicarbonitrile-6-thio-pyridines. Tetrahedron Lett. 2009, 50, 3897–3900. [Google Scholar] [CrossRef]
- Kottawar, S.S.; Siddiqui, S.A.; Bhusare, S.R. Scandium triflate-catalyzed one-pot multi-component synthesis of 2-amino-6-thiopyridine-3,5-dicarbonitriles. Heterocycl. Commun. 2012, 18, 249. [Google Scholar] [CrossRef]
- Takale, S.; Patil, J.; Padalkar, V.; Pisal, R.; Chaskar, A. O-iodoxybenzoic acid in water: Optimized green alternative for multicomponent one-pot synthesis of 2-amino-3,5-dicarbonitrile-6-thiopyridines. J. Braz. Chem. Soc. 2012, 23, 966–969. [Google Scholar] [CrossRef]
- Thimmaiah, M.; Li, P.; Regati, S.; Chen, B.; Zhao, J.C.-G. Multi-component synthesis of 2-amino-6-(alkylthio)pyridine-3,5-dicarbonitriles using Zn(II) and Cd(II) metal–organic frameworks (MOFs) under solvent-free conditions. Tetrahedron Lett. 2012, 53, 4870–4872. [Google Scholar] [CrossRef] [PubMed]
- Safaei-Ghomi, J.; Ghasemzadeh, M.A. CuI nanoparticles: A highly active and easily recyclable catalyst for the synthesis of 2-amino-3,5-dicyano-6-sulfanyl pyridines. J. Sulfur Chem. 2013, 34, 233–241. [Google Scholar] [CrossRef]
- Tian, J.; Guo, H. One-Pot Synthesis of 2-Amino-4-phenyl-6-(phenylsulfanyl)-3,5-dicyanopyridines in Ionic Liquids. Chin. J. Org. Chem. 2012, 32, 193–196. [Google Scholar] [CrossRef]
- Sobhani, S.; Honarmand, M. 2-Hydroxyethylammonium acetate: A reusable task-specific ionic liquid promoting one-pot, three-component synthesis of 2-amino-3,5-dicarbonitrile-6-thio-pyridines. C. R. Chim. 2013, 16, 279–286. [Google Scholar] [CrossRef]
- Emerson, W.S.; Patrick, T.M. The preparation of 2-thiophenealdehyde and some of its derivatives. J. Org. Chem. 1949, 14, 790–797. [Google Scholar] [CrossRef]
- Daboun, H.A.; El-Reedy, A.M. A one step synthesis of new 4-aminopyrimidine derivatives: Preparation of tetrazolo- and s-triazolopyrimidines. Z. Nat. B 1983, 38, 1686. [Google Scholar] [CrossRef]
- Urquhart, G.G.; Gates, J.W.J.; Connor, R. n-Dodecyl (lauryl) mercaptan. Org. Synth. 1941, 21, 36. [Google Scholar]
- El-Sharabsy, S.A.; Gawad, S.M.A.; Hussain, S.M. Reactions with substituted acrylonitriles: A novel synthesis of polysubstituted pyrimidines. J. Prakt. Chem. 1989, 331, 207–211. [Google Scholar] [CrossRef]
- Cosimelli, B.; Greco, G.; Laneri, S.; Novellino, E.; Sacchi, A.; Collina, S.; Rossi, D.; Cosconati, S.; Barresi, E.; Taliani, S.; et al. Studies on enantioselectivity of chiral 4-acetylamino-6-alkyloxy-2-alkylthiopyrimidines acting as antagonists of the human A3 adenosine receptor. Medchemcomm 2018, 9, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Cristalli, G.; Eleuteri, A.; Vittori, S.; Volpini, R.; Lohse, M.J.; Klotz, K.-N. 2-Alkynyl derivatives of adenosine and adenosine-5’-N-ethyluronamide as selective agonists at A2 adenosine receptors. J. Med. Chem. 1992, 35, 2363–2368. [Google Scholar] [CrossRef]
- Cristalli, G.; Costanzi, S.; Lambertucci, C.; Taffi, S.; Vittori, S.; Volpini, R. Purine and deazapurine nucleosides: Synthetic approaches, molecular modelling and biological activity. IL Farm. 2003, 58, 193–204. [Google Scholar] [CrossRef]
- Volpini, R.; Camaioni, E.; Vittori, S.; Barboni, L.; Lambertucci, C.; Cristalli, G. Synthesis of New Nucleosides by coupling of chloropurines with 2- and 3-deoxy derivatives of N-methyl-D-ribofuranuronamide. Helv. Chim. Acta 1998, 81, 145–152. [Google Scholar] [CrossRef]
- Lebon, G.; Warne, T.; Edwards, P.C.; Bennett, K.; Langmead, C.J.; Leslie, A.G.; Tate, C.G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. [Google Scholar] [CrossRef]
- Xu, F.; Wu, H.; Katritch, V.; Han, G.W.; Jacobson, K.A.; Gao, Z.G.; Cherezov, V.; Stevens, R.C. Structure of an agonist-bound human A2A adenosine receptor. Science 2011, 332, 322–327. [Google Scholar] [CrossRef]
- Lebon, G.; Edwards, P.C.; Leslie, A.G.; Tate, C.G. Molecular determinants of CGS21680 binding to the human adenosine A2A receptor. Mol. Pharm. 2015, 87, 907–915. [Google Scholar] [CrossRef]
- Draper-Joyce, C.J.; Khoshouei, M.; Thal, D.M.; Liang, Y.L.; Nguyen, A.T.N.; Furness, S.G.B.; Venugopal, H.; Baltos, J.A.; Plitzko, J.M.; Danev, R.; et al. Structure of the adenosine-bound human adenosine A1 receptor-Gi complex. Nature 2018, 558, 559–563. [Google Scholar] [CrossRef]
- Dal Ben, D.; Buccioni, M.; Lambertucci, C.; Thomas, A.; Volpini, R. Simulation and comparative analysis of binding modes of nucleoside and non-nucleoside agonists at the A2B adenosine receptor. Silico Pharm. 2013, 1, 24. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jonsson, A.L.; Beuming, T.; Shelley, J.C.; Voth, G.A. Ligand-dependent activation and deactivation of the human adenosine A2A receptor. J. Am. Chem. Soc. 2013, 135, 8749–8759. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.; Gao, Z.G.; Moss, S.M.; Jacobson, K.A.; Carlsson, J. Molecular docking screening using agonist-bound GPCR structures: Probing the A2A adenosine receptor. J. Chem. Inf. Model. 2015, 55, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Dal Ben, D.; Buccioni, M.; Lambertucci, C.; Marucci, G.; Santinelli, C.; Spinaci, A.; Thomas, A.; Volpini, R. Simulation and comparative analysis of different binding modes of Non-nucleoside agonists at the A2A adenosine receptor. Mol. Inf. 2016, 35, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- Dal Ben, D.; Lambertucci, C.; Marucci, G.; Volpini, R.; Cristalli, G. Adenosine receptor modeling: What does the A2A crystal structure tell us? Curr. Top. Med. Chem. 2010, 10, 993–1018. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Affinity Data (Ki nM, or % Radioligand Displ.) | Potency Data (EC50 nM) | |||||
|---|---|---|---|---|---|---|---|
| A1AR | A2AAR | A2BAR | A3AR | A1AR | A2AAR | A2BAR | |
| 1 [36,41] | 1.4 a,d | 0% b,e | 2.5% a,f | 1.2% a,g | 0.66 c | 1400 c | 1.1 c |
| 2 [50] | - | - | - | - | 0.1 a | 670 a | 80 a |
| 4 [36] | 5.0 a,d | 5.8% b,e | 20% a,f | 19% a,g | 2.9 a | - | - |
| 5 [36] | 1.5 a,d | - | - | - | - | - | - |
| 6 [36] | 8.4 a,d | 0.0% b,e | 63% a,f | 27% a,g | 1.9 a | - | - |
| 7 [36] | 3.9 a,d | - | - | - | - | - | - |
| 8 [36] | 12 a,d | - | - | - | - | - | - |
| 9 [31] | 2.4 a,d | 28 b,e | - | 171 b,h | - | - | 19 a |
| 10 [31] | 2.0 a,d | 105 b,e | - | 74 b,h | - | - | 34 a |
| 11 [31] | 7.0 a,d | 214 b,e | - | 24 b,h | - | - | 9 a |
| 12 [31] | 4.4 a,d | 21 b,e | - | 104 b,h | - | - | 10 a |
| 13 [31,37] | 2.6 a,d | 28 b,e | - | 538 b,h | 3.29 a | 16.2 b | 12 a |
| 14 [60] | 0.49 b,d | 71 b,e | 75 b,i | 42% b,h | 1.0 b | - | - |
| 15 [32,39] | 15 a,d | 23% a,e | - | 26% b,h | 0.7 a | > 3000 a | 670 a |
| 16 [32] | 12 a,d | 25% a,e | - | 16% b,h | - | - | - |
| 17 [32,39] | 23 a,d | 37% a,e | - | 0% b,h | 0.5 a | > 3000 a | 248 a |
| 18 [32] | 4.3 a,d | 21% a,e | - | 18% b,h | - | - | - |
| 19 [32,39] | 41 a,d | 8% a,e | - | 21% b,h | 2.7 a | > 3000 a | > 3000 a |
| 20 [62] | 57 a,d | 27% a,e | - | 29% a,h | - | - | > 1000 a |
| 21 [62] | 1.02 a,d | 93 a,e | - | 668 a,h | - | - | > 1000 a |
| 22 [62] | 0.98 a,d | 31 a,e | - | 25% a,h | - | - | > 1000 a |
| 23 [62] | 1.42 a,d | 24 a,e | - | 948 a,h | - | - | > 1000 a |
| 24 [62] | 1.32 a,d | 67 a,e | - | 326 a,h | - | - | > 1000 a |
| 25 [39,63,64] | 31% a,d | 2% a,e | 114 a,f | 8% a,h | - | - | 10 a |
| 26 [64] | 345 a,d | 1% a,e | - | 20% a,h | - | - | 38 a |
| 27 [64] | 83 a,d | 25% a,e | - | 1% a,h | - | - | 12.7 a |
| 28 [64] | 235 a,d | 764 a,e | - | 474 a,h | - | - | 9.5 a |
| 29 [64] | 338 a,d | 1% a,e | - | 1% a,h | - | - | 51 a |
| 30 [64] | 8.2 a,d | 221 a,e | - | 85 a,h | - | - | 11.7 a |
| Cpd | Affinity Data (Ki nM, or % Radioligand Displacement) | Potency Data (EC50 nM) | |||||
|---|---|---|---|---|---|---|---|
| A1AR | A2AAR | A2BAR | A3AR | A1AR | A2AAR | A2BAR | |
| 31 [30,38] | - | - | - | - | - | 2.8 b | - |
| 32 [30,38] | - | - | - | - | - | 2.5 b | - |
| 33 [30,38] | - | - | - | - | - | 3.0 b | - |
| 34 [30,38] | - | - | - | - | - | 3.3 b | - |
| 35 [30,38] | - | - | - | - | - | 2.9 b | - |
| 36 [30,38] | - | - | - | - | - | 1.8 b | - |
| 37 [35] | 4.8 a,c | 10% b,d | 3.5% a,e | 3.0% a,f | 12 a | - | - |
| 38 [35] | 14 a,c | 44% b,d | 2.5% a,e | 5.5% a,f | 3.9 a | - | - |
| 39 [35] | 2.4 a,c | −2.0% b,d | 20% a,e | 24% a,f | 4.6 a | - | - |
| 40 [35] | 14 a,c | 4.5% b,d | 0% a,e | 4.5% a,f | 4.9 a | - | - |
| 41 [35] | 5.2 a,c | 2.9% b,d | 1.7% a,e | −1.8% a,f | 4.6 a | - | - |
| 42 [35] | 1.8 a,c | 3.2 b,d | 3.5 a,e | 11 a,f | 1.9 a | - | - |
| 43 [80] | 1240 a,c | >10,000 a,g | - | >10,000 a,g | 490 a | - | Inactive a |
| 44 [80] | 1945 a,c | >10,000 a,g | - | >10,000 a,g | 870 a | - | Inactive a |
| 45 [81] | 555 a,h | 200 a,g | - | 978 a,i | - | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dal Ben, D.; Lambertucci, C.; Buccioni, M.; Martí Navia, A.; Marucci, G.; Spinaci, A.; Volpini, R. Non-Nucleoside Agonists of the Adenosine Receptors: An Overview. Pharmaceuticals 2019, 12, 150. https://doi.org/10.3390/ph12040150
Dal Ben D, Lambertucci C, Buccioni M, Martí Navia A, Marucci G, Spinaci A, Volpini R. Non-Nucleoside Agonists of the Adenosine Receptors: An Overview. Pharmaceuticals. 2019; 12(4):150. https://doi.org/10.3390/ph12040150
Chicago/Turabian StyleDal Ben, Diego, Catia Lambertucci, Michela Buccioni, Aleix Martí Navia, Gabriella Marucci, Andrea Spinaci, and Rosaria Volpini. 2019. "Non-Nucleoside Agonists of the Adenosine Receptors: An Overview" Pharmaceuticals 12, no. 4: 150. https://doi.org/10.3390/ph12040150
APA StyleDal Ben, D., Lambertucci, C., Buccioni, M., Martí Navia, A., Marucci, G., Spinaci, A., & Volpini, R. (2019). Non-Nucleoside Agonists of the Adenosine Receptors: An Overview. Pharmaceuticals, 12(4), 150. https://doi.org/10.3390/ph12040150