Multilevel Impacts of Iron in the Brain: The Cross Talk between Neurophysiological Mechanisms, Cognition, and Social Behavior


{kind=link}
{kind=link}
{kind=link}
Abstract
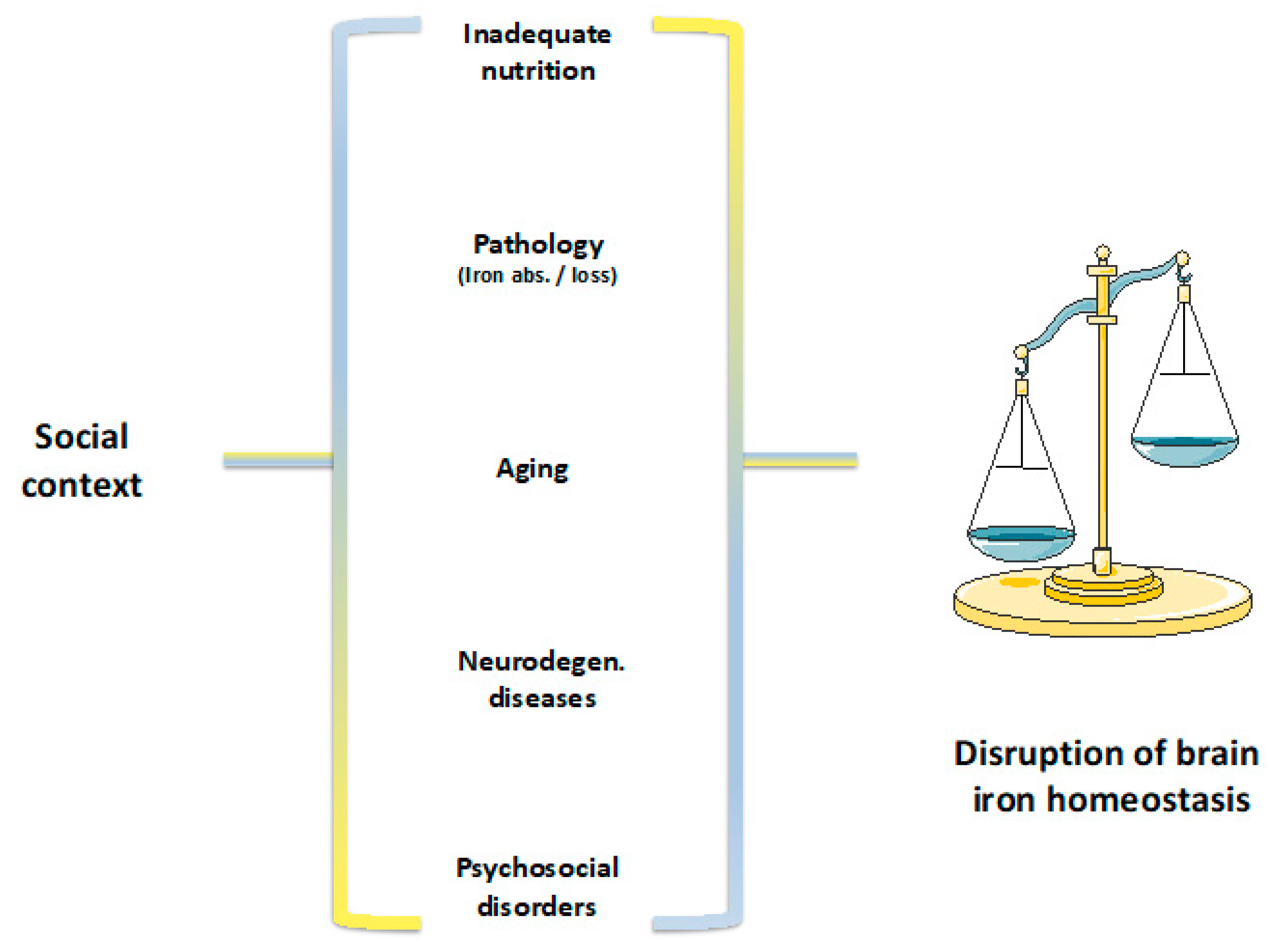
1. Introduction
2. Iron in the Brain
3. Iron Metabolism
4. The Cytotoxicity of Iron in the Brain
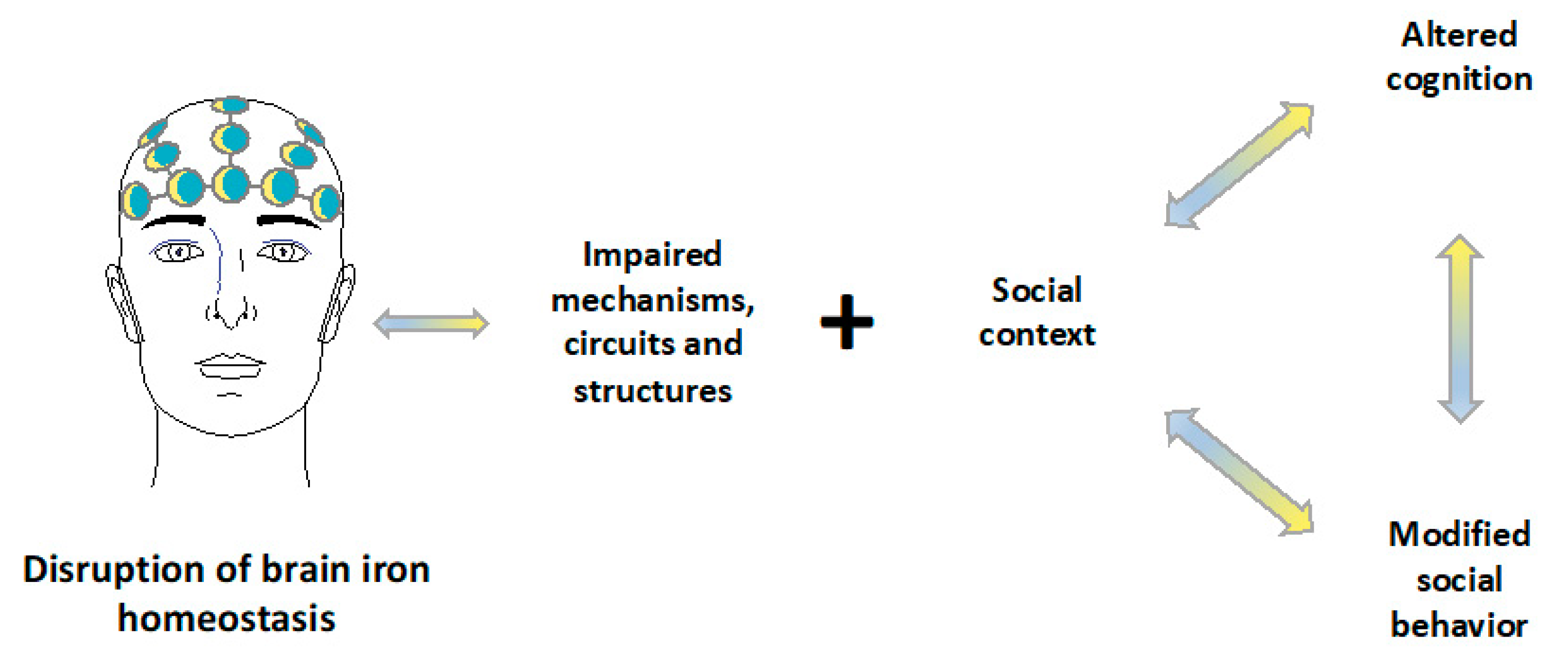
5. Neural Circuits and Social Behavior
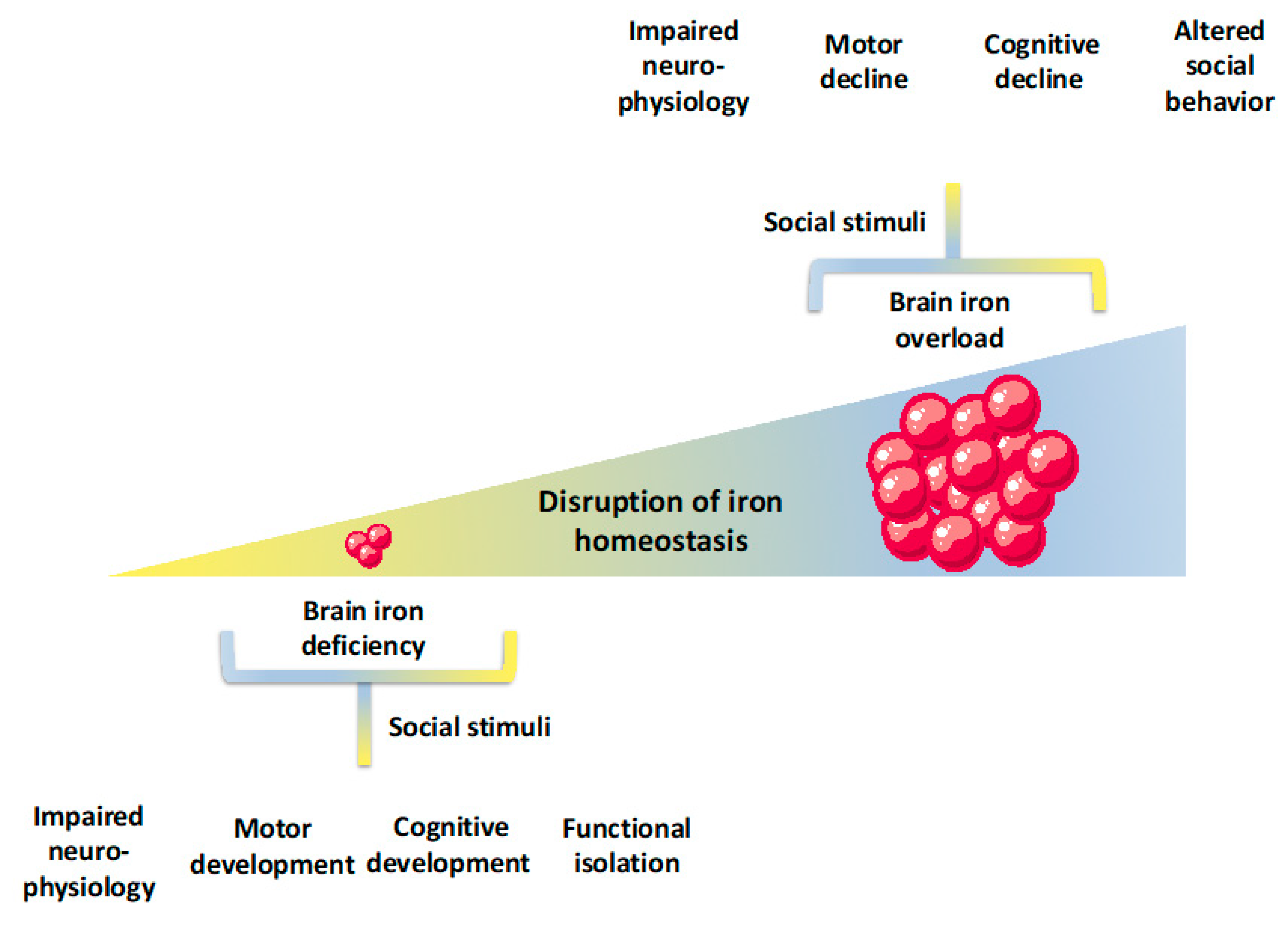
6. Linking Iron Deficiency to Cognition and Social Behavior in Human Subjects
7. Increased Iron Levels, Cognition, and Social Behavior in Human Subjects
8. Linking Iron with Chronic Stress, Anxiety, and Depression
9. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Gozzelino, R.; Arosio, P. The importance of iron in pathophysiologic conditions. Front. Pharmacol. 2015, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Gozzelino, R. The Pathophysiology of Heme in the Brain. Curr. Alzheimer Res. 2016, 13, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A. Iron metabolism in the CNS: Implications for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Beard, J.L.; Connor, J.R. IRON STATUS AND NEURAL FUNCTIONING. Annu. Rev. Nutr. 2003, 23, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Nnah, I.C.; Wessling-Resnick, M. Brain Iron Homeostasis: A Focus on Microglial Iron. Pharmaceuticals 2018, 11, 129. [Google Scholar] [CrossRef]
- World Health Organization. Iron deficiency anaemia: Assessment, prevention and control. A Guide for Programme Managers; WHO Press: Geneva, Switzerland, 2001. [Google Scholar]
- Petry, N.; Olofin, I.; Hurrell, R.F.; Boy, E.; Wirth, J.P.; Moursi, M.; Donahue Angel, M.; Rohner, F. The Proportion of Anemia Associated with Iron Deficiency in Low, Medium, and High Human Development Index Countries: A Systematic Analysis of National Surveys. Nutrients 2016, 8, 693. [Google Scholar] [CrossRef] [PubMed]
- Low, M.S.; Grigoriadis, G. Iron deficiency and new insights into therapy. Med. J. Aust. 2017, 207, 81–87. [Google Scholar] [CrossRef]
- James, P.W.; Nelson, M.; Ralph, A.; Leather, S. Socioeconomic determinants of health: The contribution of nutrition to inequalities in health. BMJ 1997, 314, 1545. [Google Scholar] [CrossRef]
- Watt, R.G.; Dykes, J.; Sheiham, A. Socio-economic determinants of selected dietary indicators in British pre-school children. Public Health Nutr. 2001, 4, 1229–1233. [Google Scholar] [CrossRef]
- Keskin, Y.; Moschonis, G.; mitriou Sur, H.; Kocaoglu, B.; Hayran, O.; Manios, Y. Prevalence of iron deficiency among schoolchildren of different socio-economic status in urban Turkey. Eur. J. Clin. Nutr. 2005, 59, 1602035. [Google Scholar] [CrossRef]
- Brotanek, J.M.; Gosz, J.; Weitzman, M.; Flores, G. Secular Trends in the Prevalence of Iron Deficiency Among US Toddlers, 1976–2002. Arch. Pediat. Adol. Med. 2008, 162, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Wieser, S.; Plessow, R.; Eichler, K.; Malek, O.; Capanzana, M.V.; Agdeppa, I.; Bruegger, U. Burden of micronutrient deficiencies by socio-economic strata in children aged 6 months to 5 years in the Philippines. BMC Public Health 2013, 13, 1167. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, M.; Bertocco, F.; Ricciardi, A.; Ferrara, D.; Incarnato, L.; Capozzi, L. Iron deficiency screening in the first three years of life: A three-decade-long retrospective case study. Hematology 2014, 19, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Plessow, R.; Arora, N.; Brunner, B.; Tzogiou, C.; Eichler, K.; Brügger, U.; Wieser, S. Social Costs of Iron Deficiency Anemia in 6–59-Month-Old Children in India. PLoS ONE 2015, 10, e0136581. [Google Scholar] [CrossRef] [PubMed]
- Lang, T.; Caraher, M. Access to healthy foods: Part II. Food poverty and shopping deserts: What are the implications for health promotion policy and practice? Health Educ. J. 1998, 57, 202–211. [Google Scholar] [CrossRef]
- World Health Organization. Guideline: Daily Iron Supplementation in Adult Women and Adolescent Girls; WHO Press: Geneva, Switzerland, 2016. [Google Scholar]
- Jáuregui-Lobera, I. Iron deficiency and cognitive functions. Neuropsychiatr. Dis. Treat. 2014, 10, 2087–2095. [Google Scholar] [CrossRef] [PubMed]
- Larson, L.M.; Phiri, K.S.; Pasricha, S.R. Iron and Cognitive Development: What Is the Evidence? Ann. Nutr. Metab. 2017, 71, 25–38. [Google Scholar] [CrossRef]
- Armitage, A.E.; Moretti, D. The Importance of Iron Status for Young Children in Low- and Middle-Income Countries: A Narrative Review. Pharmaceuticals 2019, 12, 59. [Google Scholar] [CrossRef]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2016, 139, 179–197. [Google Scholar] [CrossRef]
- Cahn-Weiner, D.A.; Grace, J.; Ott, B.R.; Fernandez, H.H.; Friedman, J.H. Cognitive and behavioral features discriminate between Alzheimer’s and Parkinson’s disease. Cogn. Behav. Neurol. 2002, 15, 79–87. [Google Scholar]
- Meireles, J.; Massano, J. Cognitive Impairment and Dementia in Parkinson’s Disease: Clinical Features, Diagnosis, and Management. Front. Neurol. 2012, 3, 88. [Google Scholar] [CrossRef] [PubMed]
- Miyasaki, J.; Shannon, K.; Voon, V.; Ravina, B.; Kleiner-Fisman, G.; Anderson, K.; Shulman, L.M.; Gronseth, G.; Weiner, W.J. Practice Parameter: Evaluation and treatment of depression, psychosis, and dementia in Parkinson disease (an evidence-based review) Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2006, 66, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Lanctôt, K.L.; Amatniek, J.; Ancoli-Israel, S.; Arnold, S.E.; Ballard, C.; Cohen-Mansfield, J.; Ismail, Z.; Lyketsos, C.; Miller, D.S.; Musiek, E.; et al. Neuropsychiatric signs and symptoms of Alzheimer’s disease: New treatment paradigms. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Wessling-Resnick, M. Iron and mechanisms of emotional behavior. J. Nutritional. Biochem. 2014, 25, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.L.; Weigel, T.K.; Elkahloun, A.G.; Herkenham, M. Chronic social defeat reduces myelination in the mouse medial prefrontal cortex. Sci. Rep. UK 2017, 7, 46548. [Google Scholar] [CrossRef]
- Gozzelino, R.; Arosio, P. Iron Homeostasis in Health and Disease. Int. J. Mol. Sci. 2016, 17, 130. [Google Scholar] [CrossRef] [PubMed]
- Sankiewicz, A.; Goscik, J.; Swiergiel, A.H.; Majewska, A.; Wieczorek, M.; Juszczak, G.R.; Lisowski, P. Social stress increases expression of hemoglobin genes in mouse prefrontal cortex. BMC Neurosci. 2014, 15, 130. [Google Scholar] [CrossRef]
- Mucha, M.; Skrzypiec, A.E.; Schiavon, E.; Attwood, B.K.; Kucerova, E.; Pawlak, R. Lipocalin-2 controls neuronal excitability and anxiety by regulating dendritic spine formation and maturation. Proc. Natl. Acad. Sci. USA 2011, 108, 18436–18441. [Google Scholar] [CrossRef]
- Jha, M.; Lee, S.; Park, D.; Kook, H.; Park, K.G.; Lee, I.K.; Suk, K. Diverse functional roles of lipocalin-2 in the central nervous system. Neurosci. Biobehav. Rev. 2015, 49, 135–156. [Google Scholar] [CrossRef]
- Agrawal, S.; Berggren, K.L.; Marks, E.; Fox, J.H. Impact of high iron intake on cognition and neurodegeneration in humans and in animal models: A systematic review. Nutr. Rev. 2017, 75, 456–470. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, M.T.; Chana-Cuevas, P. New Perspectives in Iron Chelation Therapy for the Treatment of Neurodegenerative Diseases. Pharmaceuticals 2018, 11, 109. [Google Scholar] [CrossRef] [PubMed]
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.; Moreau, C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic. Biol. Med. 2019, 133, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Crichton, R.R. Essential Metals in Medicine: Therapeutic Use and Toxicity of Metal Ions in the Clinic. Metal Ions Life Sci. 2019, 19, 87–122. [Google Scholar] [CrossRef]
- Andraca, I.; Castillo, M.; Walter, T. Psychomotor Development and Behavior in Iron-deficient Anemic Infants. Nutr. Rev. 1997, 55, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Crichton, R.R.; Ward, R.J. Metal-Based Neurodegeneration: From Molecular Mechanisms to Therapeutic Strategies, 2nd ed.; Wiley: Chichester, UK, 2013; ISBN 978-1-119-97714-8. [Google Scholar]
- Sobotka, T.J.; Whittaker, P.; Sobotka, J.M.; Brodie, R.E.; Wander, D.Y.; Robl, M.; Bryant, M.; Barton, C. Neurobehavioral dysfunctions associated with dietary iron overload. Physiol. Behav. 1996, 59, 213–219. [Google Scholar] [CrossRef]
- Vinceti, M.; Mandrioli, J.; Borella, P.; Michalke, B.; Tsatsakis, A.; Finkelstein, Y. Selenium neurotoxicity in humans: Bridging laboratory and epidemiologic studies. Toxicol. Lett. 2014, 230, 295–303. [Google Scholar] [CrossRef]
- Youdim, M.B. Brain iron deficiency and excess; cognitive impairment and neurodegenration with involvement of striatum and hippocampus. Neurotox. Res. 2008, 14, 45–56. [Google Scholar] [CrossRef]
- Lozoff, B. Iron Deficiency and Child Development. Food Nutr. Bull. 2007, 28, S560–S571. [Google Scholar] [CrossRef]
- Rudisill, S.S.; Martin, B.R.; Mankowski, K.M.; Tessier, C.R. Iron Deficiency Reduces Synapse Formation in the Drosophila Clock Circuit. Biol. Trace Elem. Res. 2019, 189, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Beard, J. Recent Evidence from Human and Animal Studies Regarding Iron Status and Infant Development. J. Nutr. 2007, 137, 524S–530S. [Google Scholar] [CrossRef] [PubMed]
- Lozoff, B.; Beard, J.; Connor, J.; Felt, B.; Georgieff, M.; Schallert, T. Long-Lasting Neural and Behavioral Effects of Iron Deficiency in Infancy. Nutr. Rev. 2006, 64, S34–S43. [Google Scholar] [CrossRef] [PubMed]
- Lozoff, B. Early Iron Deficiency Has Brain and Behavior Effects Consistent with Dopaminergic Dysfunction. J. Nutr. 2011, 141, 740S–746S. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R.L.; Gahche, J.J.; Lentino, C.V.; Dwyer, J.T.; Engel, J.S.; Thomas, P.R.; Betz, J.M.; Sempos, C.T.; Picciano, M.F. Dietary Supplement Use in the United States, 2003–2006. J. Nutr. 2011, 141, 261–266. [Google Scholar] [CrossRef]
- Eid, R.; Arab, N.; Greenwood, M.T. Iron mediated toxicity and programmed cell death: A review and a re-examination of existing paradigms. Biochim. Biophys. Acta BBA Mol. Cell Res. 2017, 1864, 399–430. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Goldin, E.; Stahl, S.; Falardeau, J.L.; Kennedy, J.C.; Acierno, J.S., Jr.; Bove, C.; Kaneski, C.R.; Nagle, J.; Bromley, M.C.; et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum. Mol. Genet. 2000, 9, 2471–2478. [Google Scholar] [CrossRef]
- Dong, X.P.; Cheng, X.; Mills, E.; Delling, M.; Wang, F.; Kurz, T.; Xu, H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 2008, 455, 992. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Campagna, D.R.; McDonald, A.; Fleming, M.D. The Steap proteins are metalloreductases. Blood 2006, 108, 1388–1394. [Google Scholar] [CrossRef]
- Vargas, J.D.; Herpers, B.; McKie, A.T.; Gledhill, S.; nnell, J.; van den Heuvel, M.; Davies, K.E.; Ponting, C.P. Stromal cell-derived receptor 2 and cytochrome b561 are functional ferric reductases. Biochim. Biophys. Acta BBA Proteins Proteom. 2003, 1651, 116–123. [Google Scholar] [CrossRef]
- Fishman, J.; Rubin, J.; Handrahan, J.; Connor, J.; Fine, R. Receptor-mediated transcytosis of transferrin across the blood-brain barrier. J. Neurosci. Res. 1987, 18, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Beard, J.L.; Wiesinger, J.A.; Li, N.; Connor, J.R. Brain iron uptake in hypotransferrinemic mice: Influence of systemic iron status. J. Neurosci. Res. 2005, 79, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Carmona, F.; Gozzelino, R.; Maccarinelli, F.; Poli, M. The importance of eukaryotic ferritins in iron handling and cytoprotection. Biochem. J. 2015, 472, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin Regulates Cellular Iron Efflux by Binding to Ferroportin and Inducing Its Internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, J.M. Ferrous ions detected in cerebrospinal fluid by using bleomycin and DNA damage. Clin. Sci. 1992, 82, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.; Dong, X.; Wang, F.; Xu, H. Mechanisms of brain iron transport: Insight into neurodegeneration and CNS disorders. Future Med. Chem. 2010, 2, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Chiou, B.; Lucassen, E.; Sather, M.; Kallianpur, A.; Connor, J. Semaphorin4A and H-ferritin utilize Tim-1 on human oligodendrocytes: A novel neuro-immune axis. Glia 2018, 66, 1317–1330. [Google Scholar] [CrossRef]
- Todorich, B.; Zhang, X.; Slagle-Webb, B.; Seaman, W.E.; Connor, J.R. Tim-2 is the receptor for H-ferritin on oligodendrocytes. J. Neurochem. 2008, 107, 1495–1505. [Google Scholar] [CrossRef]
- McCarthy, R.C.; Sosa, J.; Gardeck, A.M.; Baez, A.S.; Lee, C.H.; Wessling-Resnick, M. Inflammation-induced iron transport and metabolism by brain microglia. J. Biol. Chem. 2018, 293, 7853–7863. [Google Scholar] [CrossRef]
- Hubert, V.; Chauveau, F.; Dumot, C.; Ong, E.; Berner, L.P.; Canet-Soulas, E.; Ghersi-Egea, J.F.; Wiart, M. Clinical Imaging of Choroid Plexus in Health and in Brain Disorders: A Mini-Review. Front. Mol. Neurosci. 2019, 12, 34. [Google Scholar] [CrossRef]
- Moos, T.; Nielsen, T.; Skjørringe, T.; Morgan, E.H. Iron trafficking inside the brain. J. Neurochem. 2007, 103, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Vela, D. The Dual Role of Hepcidin in Brain Iron Load and Inflammation. Front. Neurosci. 2018, 12, 740. [Google Scholar] [CrossRef] [PubMed]
- McAuley, G.; Schrag, M.; Barnes, S.; Obenaus, A.; Dickson, A.; Holshouser, B.; Kirsch, W. Iron quantification of microbleeds in postmortem brain. Magnet. Reson. Med. 2011, 65, 1592–1601. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ramirez, S.; Greenberg, S.M.; Viswanathan, A. Cerebral microbleeds: Overview and implications in cognitive impairment. Alzheimer’s Res. Ther. 2014, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Yip, S.; Sastry, B.R. Effects of hemoglobin and its breakdown products on synaptic transmission in rat hippocampal CA1 neurons. Brain Res. 2000, 864, 1–12. [Google Scholar] [CrossRef]
- Noorbakhsh-Sabet, N.; Pulakanti, V.; Zand, R. Uncommon Causes of Cerebral Microbleeds. J. Stroke Cerebrovasc. Dis. 2017, 26, 2043–2049. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, H.; Khawar, N.; Kulpa, J.; Bellin, A.; Proteasa, S.; Sundaram, R. Neurological Complications following Blood Transfusions in Sickle Cell Anemia. Case Rep. Hematol. 2017, 2017, 1–3. [Google Scholar] [CrossRef]
- Lee, V.H.; Wijdicks, E.F.; Manno, E.M.; Rabinstein, A.A. Clinical Spectrum of Reversible Posterior Leukoencephalopathy Syndrome. Arch. Neurol. 2008, 65, 205–210. [Google Scholar] [CrossRef]
- Carmona-Gutierrez, D.; Hughes, A.L.; Madeo, F.; Ruckenstuhl, C. The crucial impact of lysosomes in aging and longevity. Ageing Res. Rev. 2016, 32, 2–12. [Google Scholar] [CrossRef]
- Asano, T.; Komatsu, M.; Yamaguchi-Iwai, Y.; Ishikawa, F.; Mizushima, N.; Iwai, K. Distinct Mechanisms of Ferritin Delivery to Lysosomes in Iron-Depleted and Iron-Replete Cells. Mol. Cell Biol. 2011, 31, 2040–2052. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, W.J.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105. [Google Scholar] [CrossRef] [PubMed]
- Del Rey, M.; Mancias, J.D. NCOA4-Mediated Ferritinophagy: A Potential Link to Neurodegeneration. Front. Neurosci. 2019, 13, 238. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Tiranti, V. Neurodegeneration with Brain Iron Accumulation Disorders: Valuable Models Aimed at Understanding the Pathogenesis of Iron Deposition. Pharmaceuticals 2019, 12, 27. [Google Scholar] [CrossRef] [PubMed]
- Dusek, P.; Schneider, S.A.; Aaseth, J. Iron chelation in the treatment of neurodegenerative diseases. J. Trace Elem. Med. Bio. 2016, 38, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Andersen, H.; Johnsen, K.; Moos, T. Iron deposits in the chronically inflamed central nervous system and contributes to neurodegeneration. Cell Mol. Life Sci. 2014, 71, 1607–1622. [Google Scholar] [CrossRef]
- Zhou, D.R.; Eid, R.; Miller, K.A.; Boucher, E.; Mandato, C.A.; Greenwood, M.T. Intracellular second messengers mediate stress inducible hormesis and Programmed Cell Death: A review. Biochim. Biophys. Acta BBA Mol. Cell Res. 2019, 1866, 773–792. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Moreau, C.; Devedjian, J.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garçon, G.; Rouaix, N.; et al. Targeting Chelatable Iron as a Therapeutic Modality in Parkinson’s Disease. Antioxid. Redox Sign. 2014, 21, 195–210. [Google Scholar] [CrossRef]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. UK 2017, 7, 1398. [Google Scholar] [CrossRef]
- Fenton, H.J.; Jackson, H.J.I. The oxidation of polyhydric alcohols in presence of iron. J. Chem. Soc. Trans. 1899, 75, 1–11. [Google Scholar] [CrossRef]
- Weiland, A.; Wang, Y.; Wu, W.; Lan, X.; Han, X.; Li, Q.; Wang, J. Ferroptosis and Its Role in Diverse Brain Diseases. Mol. Neurobiol. 2019, 56, 4880–4893. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Y.; Li, T.; Wang, X.; Zhu, C. Iron Metabolism and Brain Development in Premature Infants. Front. Physiol. 2019, 10, 463. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Role of Free Radicals in the Neurodegenerative Diseases. Drug Aging 2001, 18, 685–716. [Google Scholar] [CrossRef] [PubMed]
- Black, C.N.; Bot, M.; Scheffer, P.G.; Cuijpers, P.; Penninx, B. Is depression associated with increased oxidative stress? A systematic review and meta-analysis. Psychoneuroendocrinology 2015, 51, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.P.; Greenamyre, T.J. Mitochondrial Iron Metabolism and Its Role in Neurodegeneration. J. Alzheimer’s Dis. 2010, 20, S551–S568. [Google Scholar] [CrossRef] [PubMed]
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Muñoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog. Neurobiol. 2017, 155, 96–119. [Google Scholar] [CrossRef] [PubMed]
- Fearnhead, H.O.; Vandenabeele, P.; Berghe, T. How do we fit ferroptosis in the family of regulated cell death? Cell Death Differ. 2017, 24, 1991–1998. [Google Scholar] [CrossRef] [PubMed]
- Reimers, A.; Ljung, H. The emerging role of omega-3 fatty acids as a therapeutic option in neuropsychiatric disorders. Ther. Adv. Psychopharmacol. 2019, 9, 2045125319858901. [Google Scholar] [CrossRef]
- Kennedy, D.P.; Adolphs, R. The social brain in psychiatric and neurological disorders. Trends Cogn. Sci. 2012, 16, 559–572. [Google Scholar] [CrossRef]
- Adolphs, R.; Damasio, H.; Tranel, D.; Cooper, G.; Damasio, A.R. A Role for Somatosensory Cortices in the Visual Recognition of Emotion as Revealed by Three-Dimensional Lesion Mapping. J. Neurosci. 2000, 20, 2683–2690. [Google Scholar] [CrossRef]
- Pessoa, L.; Adolphs, R. Emotion processing and the amygdala: From a “low road” to “many roads” of evaluating biological significance. Nat. Rev. Neurosci. 2010, 11, 773. [Google Scholar] [CrossRef]
- Nelson, E.E.; Guyer, A.E. The development of the ventral prefrontal cortex and social flexibility. Dev. Cogn. Neurosci. 2011, 1, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Barrash, J.; Tranel, D.; Anderson, S.W. Acquired Personality Disturbances Associated with Bilateral Damage to the Ventromedial Prefrontal Region. Dev. Neuropsychol. 2000, 18, 355–381. [Google Scholar] [CrossRef] [PubMed]
- Meiser, J.; Weindl, D.; Hiller, K. Complexity of dopamine metabolism. Cell Commun. Signal. 2013, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Murray-Kolb, L.E. Iron status and neuropsychological consequences in women of reproductive age: What do we know and where are we headed? J. Nutr. 2011, 141, 747S–755S. [Google Scholar] [CrossRef] [PubMed]
- McClung, J.P.; Murray-Kolb, L.E. Iron Nutrition and Premenopausal Women: Effects of Poor Iron Status on Physical and Neuropsychological Performance. Annu. Rev. Nutr. 2013, 33, 271–288. [Google Scholar] [CrossRef] [PubMed]
- Low, M.; Speedy, J.; Styles, C.E.; De-Regil, L.; Pasricha, S. Daily iron supplementation for improving anaemia, iron status and health in menstruating women. Cochrane Database Syst. Rev. 2016, 4, CD009747. [Google Scholar] [CrossRef] [PubMed]
- Breymann, C. Iron Deficiency Anemia in Pregnancy. Semin. Hematol. 2015, 52, 339–347. [Google Scholar] [CrossRef]
- Algarín, C.; Peirano, P.; Garrido, M.; Pizarro, F.; Lozoff, B. Iron Deficiency Anemia in Infancy. Pediatr. Res. 2003, 53, 217–223. [Google Scholar] [CrossRef]
- Walter, T. Effect of Iron-Deficiency Anemia on Cognitive Skills and Neuromaturation in Infancy and Childhood. Food Nutr. Bull. 2003, 24, S104–S110. [Google Scholar] [CrossRef]
- Algarín, C.; Nelson, C.A.; Peirano, P.; Westerlund, A.; Reyes, S.; Lozoff, B. Iron-deficiency anemia in infancy and poorer cognitive inhibitory control at age 10 years. Dev. Med. Child Neurol. 2013, 55, 453–458. [Google Scholar] [CrossRef]
- McCann, J.C.; Ames, B.N. An overview of evidence for a causal relation between iron deficiency during development and deficits in cognitive or behavioral function. Am. J. Clin. Nutr. 2007, 85, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Bressan, R.; Crippa, J. The role of dopamine in reward and pleasure behavior—Review of data from preclinical research. Acta Psychiat. Scand. 2005, 111, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Wild, B.; Rodden, F.A.; Grodd, W.; Ruch, W. Neural correlates of laughter and humour. Brain 2003, 126, 2121–2138. [Google Scholar] [CrossRef] [PubMed]
- Oski, F.; Honig, A.; Helu, B.; Howanitz, P. Effect of iron therapy on behavior performance in nonanemic, iron-deficient infants. Pediatrics 1983, 71, 877–880. [Google Scholar] [PubMed]
- Walter, T.; Kovalskys, J.; Stekel, A. Effect of mild iron deficiency on infant mentaldevelopment scores. J. Pediatrics 1983, 102, 519–522. [Google Scholar] [CrossRef]
- Lozoff, B.; Wolf, A.W.; Urrutia, J.J.; Viteri, F.E. Abnormal Behavior and Low Developmental Test Scores in Iron-Deficient Anemic Infants. J. Dev. Behav. Pediatrics 1985, 6, 69–75. [Google Scholar] [CrossRef]
- Deinard, A.S.; List, A.; Lindgren, B.; Hunt, J.V.; Chang, P.N. Cognitive deficits in iron-deficient and iron-deficient anemic children. J. Pediatrics 1986, 108, 681–689. [Google Scholar] [CrossRef]
- Lozoff, B.; Klein, N.K.; Prabucki, K.M. Iron-Deficient Anemic Infants at Play. J. Dev. Behav. Pediatrics 1986, 7, 152–158. [Google Scholar] [CrossRef]
- Lozoff, B.; Klein, N.K.; Nelson, E.C.; McClish, D.K.; Manuel, M.; Chacon, M. Behavior of Infants with Iron-Deficiency Anemia. Child Dev. 1998, 69, 24–36. [Google Scholar] [CrossRef]
- Lozoff, B.; Clark, K.M.; Jing, Y.; Armony-Sivan, R.; Angelilli, M.; Jacobson, S.W. Dose-Response Relationships between Iron Deficiency with or without Anemia and Infant Social-Emotional Behavior. J. Pediatrics 2008, 152, 696–702. [Google Scholar] [CrossRef]
- Honig, A.; Oski, F.A. Solemnity: A clinical risk index for iron deficient infants. Early Child Dev. Care 2010, 16, 69–83. [Google Scholar] [CrossRef]
- Shafir, T.; Angulo-Barroso, R.; Jing, Y.; Angelilli, M.; Jacobson, S.W.; Lozoff, B. Iron deficiency and infant motor development. Early Hum. Dev. 2008, 84, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Hokama, T.; Ken, G.M.; Nosoko, N. Iron Deficiency Anaemia and Child Development. Asia Pac. J. Public Health 2005, 17, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Lozoff, B.; Armony-Sivan, R.; Kaciroti, N.; Jing, Y.; Golub, M.; Jacobson, S.W. Eye-Blinking Rates Are Slower in Infants with Iron-Deficiency Anemia than in Nonanemic Iron-Deficient or Iron-Sufficient Infants. J. Nutr. 2010, 140, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Lozoff, B.; Felt, B.T.; Nelson, E.C.; Wolf, A.W.; Meltzer, H.W.; Jimenez, E. Serum prolactin levels and behavior in infants. Biol. Psychiat. 1995, 37, 4–12. [Google Scholar] [CrossRef]
- Hensch, T.K. Critical period regulation. Annu. Rev. Neurosci. 2004, 27, 549–579. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Hanson, M.A. Living with the Past: Evolution, Development, and Patterns of Disease. Science 2004, 305, 1733–1736. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.R.; Jacobson, J.L.; Burden, M.J.; Armony-Sivan, R.; Dodge, N.C.; Angelilli, M.; Lozoff, B.; Jacobson, S.W. Iron Deficiency Anemia and Cognitive Function in Infancy. Pediatrics 2010, 126, e427–e434. [Google Scholar] [CrossRef] [PubMed]
- Kivisto, P. Social Theory: Roots and Branches, 5th ed.; Oxford University Press: Oxford, UK; New York, NY, USA, 2013; ISBN 978-0199937127. [Google Scholar]
- Collins, R. Four Sociological Traditions, Revised & Enlarged edition; Oxford University Press: Oxford, UK; New York, NY, USA, 1994; ISBN 978-0195082081. [Google Scholar]
- Archer, M. Structure, Agency and the Internal Conversation; Cambridge University Press: Cambridge, UK, 2003; ISBN 978-1139087315. [Google Scholar]
- Caetano, A. Coping with Life: A Typology of Personal Reflexivity. Sociol. Q. 2016, 58, 32–50. [Google Scholar] [CrossRef]
- Caetano, A. Designing social action: The impact of reflexivity on practice. J. Theor. Soc. Behav. 2019, 49, 146–160. [Google Scholar] [CrossRef]
- Alloway, T.; Bibile, V.; Lau, G. Computerized working memory training: Can it lead to gains in cognitive skills in students? Comput. Hum. Behav. 2013, 29, 632–638. [Google Scholar] [CrossRef]
- Buschkuehl, M.; Jaeggi, S.M.; Jonides, J. Neuronal effects following working memory training. Dev. Cogn. Neurosci. 2012, 2, S167–S179. [Google Scholar] [CrossRef] [PubMed]
- Jaeggi, S.M.; Buschkuehl, M.; Jonides, J.; Shah, P. Short- and long-term benefits of cognitive training. Proc. Natl. Acad. Sci. USA 2011, 108, 10081–10086. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.B.; Chein, J.M. Does working memory training work? The promise and challenges of enhancing cognition by training working memory. Psychon. Bull. Rev. 2011, 18, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Oyarzún, D.I.; Aliaga, S.I.; de la Cieciwa, P.A.; López, G.B. Mother-child interaction and child behavior in preschool children with a history of iron-deficiency anemia in infancy. Arch. Latinoam. Nutr. 1993, 43, 191–198. [Google Scholar]
- Brown, L.J.; Pollitt, E. Malnutrition, Poverty and Intellectual Development. Sci. Am. 1996, 274, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Levitsky, D.A.; Strupp, B.J. Malnutrition and the Brain: Changing Concepts, Changing Concerns. J. Nutr. 1995, 125, 2212S–2220S. [Google Scholar] [CrossRef]
- Corapci, F.; Radan, A.E.; Lozoff, B. Iron Deficiency in Infancy and Mother-Child Interaction at 5 Years. J. Dev. Behav. Pediatrics 2006, 27, 371–378. [Google Scholar] [CrossRef]
- Armony-Sivan, R.; Kaplan-Estrin, M.; Jacobson, S.W.; Lozoff, B. Iron-Deficiency Anemia in Infancy and Mother-Infant Interaction During Feeding. J. Dev. Behav. Pediatrics 2010, 31, 326–332. [Google Scholar] [CrossRef] [PubMed]
- East, P.; Lozoff, B.; Blanco, E.; Delker, E.; Delva, J.; Encina, P.; Gahagan, S. Infant Iron Deficiency, Child Affect, and Maternal Unresponsiveness: Testing the Long-Term Effects of Functional Isolation. Dev. Psychol. 2017, 53, 2233–2244. [Google Scholar] [CrossRef]
- Lozoff, B.; Smith, J.B.; Clark, K.M.; Perales, C.; Rivera, F.; Castillo, M. Home Intervention Improves Cognitive and Social-Emotional Scores in Iron-Deficient Anemic Infants. Pediatrics 2010, 126, e884–e894. [Google Scholar] [CrossRef] [PubMed]
- Alloway, T.P. What do we know about the long-term cognitive effects of iron-deficiency anemia in infancy? Dev. Med. Child Neurol. 2013, 55, 401–402. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Jin, Y.; Unverzagt, F.W.; Ma, F.; Hall, K.S.; Murrell, J.R.; Cheng, Y.; Shen, J.; Ying, B.; Ji, R.; et al. Trace Element Levels and Cognitive Function in Rural Elderly Chinese. J. Gerontol. Ser. 2008, 63, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Milward, E.A.; Bruce, D.G.; Knuiman, M.W.; Divitini, M.L.; Cole, M.; Inderjeeth, C.A.; Clarnette, R.M.; Maier, G.; Jablensky, A.; Olynyk, J.K. A Cross-Sectional Community Study of Serum Iron Measures and Cognitive Status in Older Adults. J. Alzheimer’s Dis. 2010, 20, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Schiepers, O.J.; van Boxtel, M.P.; de Groot, R.H.; Jolles, J.; de Kort, W.L.; Swinkels, D.W.; Kok, F.J.; Verhoef, P.; Durga, J. Serum Iron Parameters, HFE C282Y Genotype, and Cognitive Performance in Older Adults: Results from the FACIT Study. J. Gerontol. Ser. 2010, 65, 1312–1321. [Google Scholar] [CrossRef]
- Lam, P.; Kritz-Silverstein, D.; Barrett-Connor, E.; Milne, D.; Nielsen, F.; Gamst, A.; Morton, D.; Wingard, D. Plasma trace elements and cognitive function in older men and women: The Rancho Bernardo study. J. Nutr. Health Aging 2008, 12, 22–27. [Google Scholar] [CrossRef]
- Umur, E.E.; Oktenli, C.; Celik, S.; Tangi, F.; Sayan, O.; Sanisoglu, Y.S.; Ipcioglu, O.; Terekeci, H.M.; Top, C.; Nalbant, S.; et al. Increased iron and oxidative stress are separately related to cognitive decline in elderly. Geriatr. Gerontol. Int. 2011, 11, 504–509. [Google Scholar] [CrossRef]
- Andreeva, V.A.; Galan, P.; Arnaud, J.; Julia, C.; Hercberg, S.; Kesse-Guyot, E. Midlife Iron Status Is Inversely Associated with Subsequent Cognitive Performance, Particularly in Perimenopausal Women. J. Nutr. 2013, 143, 1974–1981. [Google Scholar] [CrossRef]
- McCarthy, R.C.; Kosman, D.J. Glial Cell Ceruloplasmin and Hepcidin Differentially Regulate Iron Efflux from Brain Microvascular Endothelial Cells. PLoS ONE 2014, 9, e89003. [Google Scholar] [CrossRef]
- Kumar, N. Neuroimaging in Superficial Siderosis: An In-Depth Look. Am. J. Neuroradiol. 2010, 31, 5–14. [Google Scholar] [CrossRef]
- Van Es, A.; van der Grond, J.; de Craen, A.; Admiraal-Behloul, F.; Blauw, G.J.; van Buchem, M.A. Caudate nucleus hypointensity in the elderly is associated with markers of neurodegeneration on MRI. Neurobiol. Aging 2008, 29, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, E.V.; Adalsteinsson, E.; Rohlfing, T.; Pfefferbaum, A. Relevance of Iron Deposition in Deep Gray Matter Brain Structures to Cognitive and Motor Performance in Healthy Elderly Men and Women: Exploratory Findings. Brain Imaging Behav. 2009, 3, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Penke, L.; Hernandéz, M.C.; Maniega, S.; Gow, A.J.; Murray, C.; Starr, J.M.; Bastin, M.E.; Deary, I.J.; Wardlaw, J.M. Brain iron deposits are associated with general cognitive ability and cognitive aging. Neurobiol. Aging 2012, 33, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, K.M.; Daugherty, A.M.; Haacke, M.E.; Raz, N. The Role of Hippocampal Iron Concentration and Hippocampal Volume in Age-Related Differences in Memory. Cereb. Cortex. 2013, 23, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, A.M.; Haacke, M.E.; Raz, N. Striatal Iron Content Predicts Its Shrinkage and Changes in Verbal Working Memory after Two Years in Healthy Adults. J. Neurosci. 2015, 35, 6731–6743. [Google Scholar] [CrossRef] [PubMed]
- Del Hernández, M.C.; Ritchie, S.; Glatz, A.; Allerhand, M.; Maniega, S.; Gow, A.J.; Royle, N.A.; Bastin, M.E.; Starr, J.M.; Deary, I.J.; et al. Brain iron deposits and lifespan cognitive ability. Age 2015, 37, 100. [Google Scholar] [CrossRef] [PubMed]
- Hernández, V.M.; Allerhand, M.; Glatz, A.; Clayson, L.; Maniega, M.S.; Gow, A.; Royle, N.; Bastin, M.; Starr, J.; Deary, I.; et al. Do white matter hyperintensities mediate the association between brain iron deposition and cognitive abilities in older people? Eur. J. Neurol. 2016, 23, 1202–1209. [Google Scholar] [CrossRef]
- Scarpellini, F.; Sia, M.; Scarpellini, L. Psychological Stress and Lipoperoxidation in Miscarriage. Ann. N. Y. Acad. Sci. 1994, 709, 210–213. [Google Scholar] [CrossRef]
- Miyashita, T.; Yamaguchi, T.; Motoyama, K.; Unno, K.; Nakano, Y.; Shimoi, K. Social stress increases biopyrrins, oxidative metabolites of bilirubin, in mouse urine. Biochem. Biophys. Res. Commun. 2006, 349, 775–780. [Google Scholar] [CrossRef]
- Wang, L.; Wang, W.; Zhao, M.; Ma, L.; Li, M. Psychological stress induces dysregulation of iron metabolism in rat brain. Neuroscience 2008, 155, 24–30. [Google Scholar] [CrossRef]
- Yu, S.; Feng, Y.; Shen, Z.; Li, M. Diet supplementation with iron augments brain oxidative stress status in a rat model of psychological stress. Nutrition 2011, 27, 1048–1052. [Google Scholar] [CrossRef] [PubMed]
- Fleming, D.J.; Tucker, K.L.; Jacques, P.F.; Dallal, G.E.; Wilson, P.W.; Wood, R.J. Dietary factors associated with the risk of high iron stores in the elderly Framingham Heart Study cohort. Am. J. Clin. Nutr. 2002, 76, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Milman, N.; Byg, K.; Ovesen, L.; Kirchhoff, M.; Jürgensen, K. Iron status in Danish men 1984–94: A cohort comparison of changes in iron stores and the prevalence of iron deficiency and iron overload. Eur. J. Haematol. 2002, 68, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Starcke, K.; Brand, M. Decision making under stress: A selective review. Neurosci. Biobehav. Rev. 2012, 36, 1228–1248. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Teixeira, A. Intra- and extra-organisational foundations of innovation process—The information and communication technology sector under the crisis in Portugal. Int. J. Innov. Manag. 2016, 20, 1650056. [Google Scholar] [CrossRef]
- Ferreira, A. Reconnecting Nature and Culture: A Model of Social Decision-making in Innovation. Int. J. Interdiscip. Organ. Study 2014, 8, 13–18. [Google Scholar] [CrossRef]
- Ferreira, A. Reasoning on Emotions: Drawing an Integrative Approach. In Complexity Sciences: Theoretical and Empirical Approaches to Social Action; Cambridge Scholars Publishing: Newcastle upon Tyne, UK, 2018; pp. 125–139. [Google Scholar]
- Steptoe, A.; Hamer, M.; Chida, Y. The effects of acute psychological stress on circulating inflammatory factors in humans: A review and meta-analysis. Brain Behav. Immun. 2007, 21, 901–912. [Google Scholar] [CrossRef]
- Weik, U.; Herforth, A.; Kolb-Bachofen, V.; Deinzer, R. Acute Stress Induces Proinflammatory Signaling at Chronic Inflammation Sites. Psychosom. Med. 2008, 70, 906–912. [Google Scholar] [CrossRef]
- Morey, J.N.; Boggero, I.A.; Scott, A.B.; Segerstrom, S.C. Current directions in stress and human immune function. Curr. Opin. Psychol. 2015, 5, 13–17. [Google Scholar] [CrossRef]
- Slavich, G.M.; Way, B.M.; Eisenberger, N.I.; Taylor, S.E. Neural sensitivity to social rejection is associated with inflammatory responses to social stress. Proc. Natl. Acad. Sci. USA 2010, 107, 14817–14822. [Google Scholar] [CrossRef]
- Yu, R. Stress potentiates decision biases: A stress induced deliberation-to-intuition (SIDI) model. Neurobiol. Stress 2016, 3, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Felger, J.C.; Treadway, M.T. Inflammation Effects on Motivation and Motor Activity: Role of Dopamine. Neuropsychopharmacol 2016, 42, 216–241. [Google Scholar] [CrossRef] [PubMed]
- Slavich, G.M.; Irwin, M.R. From stress to inflammation and major depressive disorder: A social signal transduction theory of depression. Psychol. Bull. 2014, 140, 774. [Google Scholar] [CrossRef] [PubMed]
- Starcke, K.; Brand, M. Effects of Stress on Decisions Under Uncertainty: A Meta-Analysis. Psychol. Bull. 2016, 142, 909–933. [Google Scholar] [CrossRef] [PubMed]
- González-Morales, G.M.; Neves, P. When stressors make you work: Mechanisms linking challenge stressors to performance. Work Stress 2015, 29, 213–229. [Google Scholar] [CrossRef]
- Beery, A.K.; Kaufer, D. Stress, social behavior, and resilience: Insights from rodents. Neurobiol. Stress 2015, 1, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Sapolsky, R.M. The Influence of Social Hierarchy on Primate Health. Science 2005, 308, 648–652. [Google Scholar] [CrossRef]
- American Psychology Association. APA Dictionary of Psychology: Chronic Stress. Available online: https://dictionary.apa.org/chronic-stress (accessed on 30 July 2019).
- Ford, M.T.; Cerasoli, C.P.; Higgins, J.A.; Decesare, A.L. Relationships between psychological, physical, and behavioural health and work performance: A review and meta-analysis. Work Stress 2011, 25, 185–204. [Google Scholar] [CrossRef]
- Porcelli, A.J.; Delgado, M.R. Stress and decision making: Effects on valuation, learning, and risk-taking. Curr. Opin. Behav. Sci. 2017, 14, 33–39. [Google Scholar] [CrossRef]
- McEwen, B.S.; Magarinos, A. Stress and hippocampal plasticity: Implications for the pathophysiology of affective disorders. Hum. Psychopharmacol. Clin. Exp. 2001, 16, S7–S19. [Google Scholar] [CrossRef]
- Pesarico Extracortical Regions. Front. Cell Neurosci. 2019, 13, 197. [CrossRef]
- Drevets, W.C.; Price, J.L.; Furey, M.L. Brain structural and functional abnormalities in mood disorders: Implications for neurocircuitry models of depression. Brain Struct. Funct. 2008, 213, 93–118. [Google Scholar] [CrossRef] [PubMed]
- Felt, B.T.; Peirano, P.; Algarín, C.; Chamorro, R.; Sir, T.; Kaciroti, N.; Lozoff, B. Long-term neuroendocrine effects of iron-deficiency anemia in infancy. Pediatr. Res. 2012, 71, 707. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.; Morais, S.L.; Saad, S. Reduced Cortisol Secretion in Patients with Iron Deficiency. Ann. Nutr. Metab. 1991, 35, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Kalhan, S.C.; Ghosh, A. Dietary Iron, Circadian Clock, and Hepatic Gluconeogenesis. Diabetes 2015, 64, 1091–1093. [Google Scholar] [CrossRef][Green Version]
- Debono, M.; Ghobadi, C.; Rostami-Hodjegan, A.; Huatan, H.; Campbell, M.J.; Newell-Price, J.; Darzy, K.; Merke, D.P.; Arlt, W.; Ross, R.J. Modified-release hydrocortisone to provide circadian cortisol profiles. J. Clin. Endocrinol. Metabolism 2009, 94, 1548–1554. [Google Scholar] [CrossRef]
- Ridefelt, P.; Larsson, A.; Rehman, J.; Axelsson, J. Influences of sleep and the circadian rhythm on iron-status indices. Clin. Biochem. 2010, 43, 1323–1328. [Google Scholar] [CrossRef]
- Reinke, H.; Asher, G. Crosstalk between metabolism and circadian clocks. Nat. Rev. Mol. Cell Biol. 2019, 20, 1. [Google Scholar] [CrossRef]
- American Psychiatric Association. APA Diagnostic and Statistical Manual of Mental Disorders; DSM Library, American Psychiatric Association: Philadelphia, PA, USA, 2013. [Google Scholar] [CrossRef]
- Beck, A.T.; Brown, G.; Steer, R.A.; Eidelson, J.I.; Riskind, J.H. Differentiating anxiety and depression: A test of the cognitive content-specificity hypothesis. J. Abnorm. Psychol. 1987, 96, 179. [Google Scholar] [CrossRef]
- Twenge, J.M. The age of anxiety? The birth cohort change in anxiety and neuroticism, 1952–1993. J. Personal. Soc. Psychol. 2000, 79, 1007. [Google Scholar] [CrossRef]
- Lozoff, B.; Jimenez, E.; Hagen, J.; Mollen, E.; Wolf, A.W. Poorer Behavioral and Developmental Outcome More Than 10 Years After Treatment for Iron Deficiency in Infancy. Pediatrics 2000, 105, e51. [Google Scholar] [CrossRef] [PubMed]
- Shelton, R.; Brown, L. Mechanisms of action in the treatment of anxiety. J. Clin. Psychiatry 2001, 62, 10–15. [Google Scholar] [PubMed]
- Eizadi-Mood, N.; Ahmadi, R.; Babazadeh, S.; Yaraghi, A.; Sadeghi, M.; Peymani, P.; Sabzghabaee, A.M. Anemia, depression, and suicidal attempts in women: Is there a relationship? J. Res. Pharm. Pract. 2018, 7, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Hidese, S.; Saito, K.; Asano, S.; Kunugi, H. Association between iron-deficiency anemia and depression: A web-based Japanese investigation. Psychiatry Clin. Neurosci. 2018, 72, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Dryman, T.M.; Heimberg, R.G. Emotion regulation in social anxiety and depression: A systematic review of expressive suppression and cognitive reappraisal. Clin. Psychol. Rev. 2018, 65, 17–42. [Google Scholar] [CrossRef]
- Howren, B.M.; Lamkin, D.M.; Suls, J. Associations of Depression With C-Reactive Protein, IL-1, and IL-6: A Meta-Analysis. Psychosom. Med. 2009, 71, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A Meta-Analysis of Cytokines in Major Depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Martins, A.C.; Almeida, J.I.; Lima, I.S.; Kapitão, A.S.; Gozzelino, R. Iron Metabolism and the Inflammatory Response. IUBMB Life 2017, 69, 442–450. [Google Scholar] [CrossRef]
- Mehrpouya, S.; Nahavandi, A.; Khojasteh, F.; Soleimani, M.; Ahmadi, M.; Barati, M. Iron administration prevents BDNF decrease and depressive-like behavior following chronic stress. Brain Res. 2015, 1596, 79–87. [Google Scholar] [CrossRef]
- Mizuno, Y.; Ozeki, M.; Iwata, H.; Takeuchi, T.; Ishihara, R.; Hashimoto, N.; Kobayashi, H.; Iwai, K.; Ogasawara, S.; Ukai, K.; et al. A case of clinically and neuropathologically atypical corticobasal degeneration with widespread iron deposition. Acta Neuropathol. 2002, 103, 288–294. [Google Scholar] [CrossRef]
- Farajdokht, F.; Soleimani, M.; Mehrpouya, S.; Barati, M.; Nahavandi, A. The role of hepcidin in chronic mild stress-induced depression. Neurosci. Lett. 2015, 588, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.M.; Connor, T.J.; Harkin, A. Stress-Related Immune Markers in Depression: Implications for Treatment. Int. J. Neuropsychoph. 2016, 19, pyw001. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, A.; Neves, P.; Gozzelino, R. Multilevel Impacts of Iron in the Brain: The Cross Talk between Neurophysiological Mechanisms, Cognition, and Social Behavior. Pharmaceuticals 2019, 12, 126. https://doi.org/10.3390/ph12030126
Ferreira A, Neves P, Gozzelino R. Multilevel Impacts of Iron in the Brain: The Cross Talk between Neurophysiological Mechanisms, Cognition, and Social Behavior. Pharmaceuticals. 2019; 12(3):126. https://doi.org/10.3390/ph12030126
Chicago/Turabian StyleFerreira, Ana, Pedro Neves, and Raffaella Gozzelino. 2019. "Multilevel Impacts of Iron in the Brain: The Cross Talk between Neurophysiological Mechanisms, Cognition, and Social Behavior" Pharmaceuticals 12, no. 3: 126. https://doi.org/10.3390/ph12030126
APA StyleFerreira, A., Neves, P., & Gozzelino, R. (2019). Multilevel Impacts of Iron in the Brain: The Cross Talk between Neurophysiological Mechanisms, Cognition, and Social Behavior. Pharmaceuticals, 12(3), 126. https://doi.org/10.3390/ph12030126