Recent Developments in Peptidyl Diaryl Phoshonates as Inhibitors and Activity-Based Probes for Serine Proteases

Abstract
1. Introduction
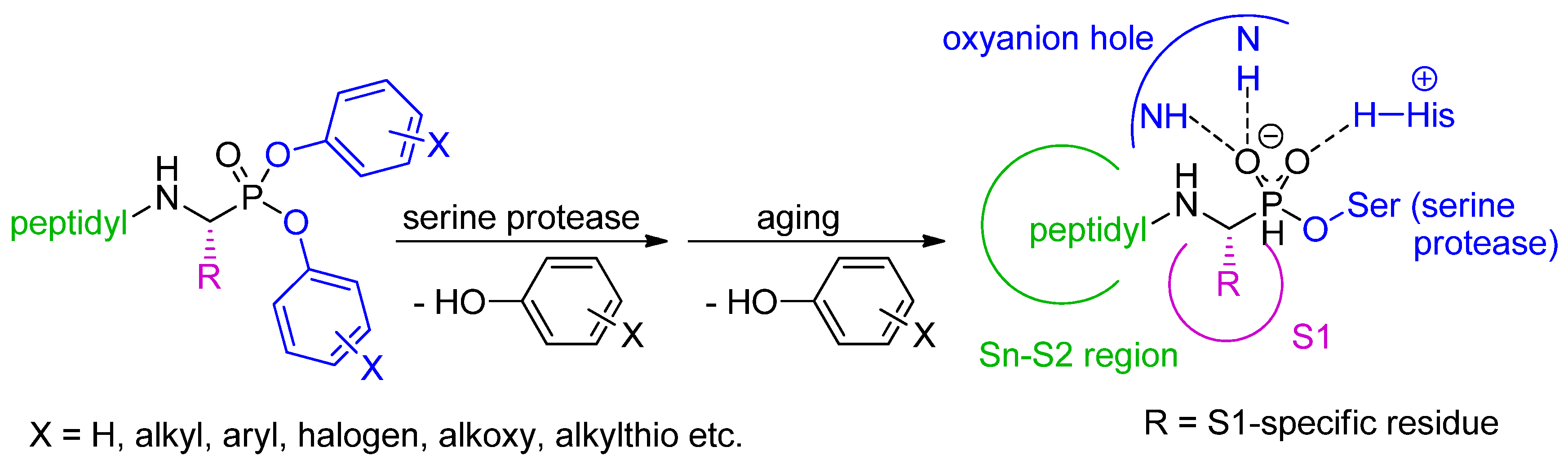
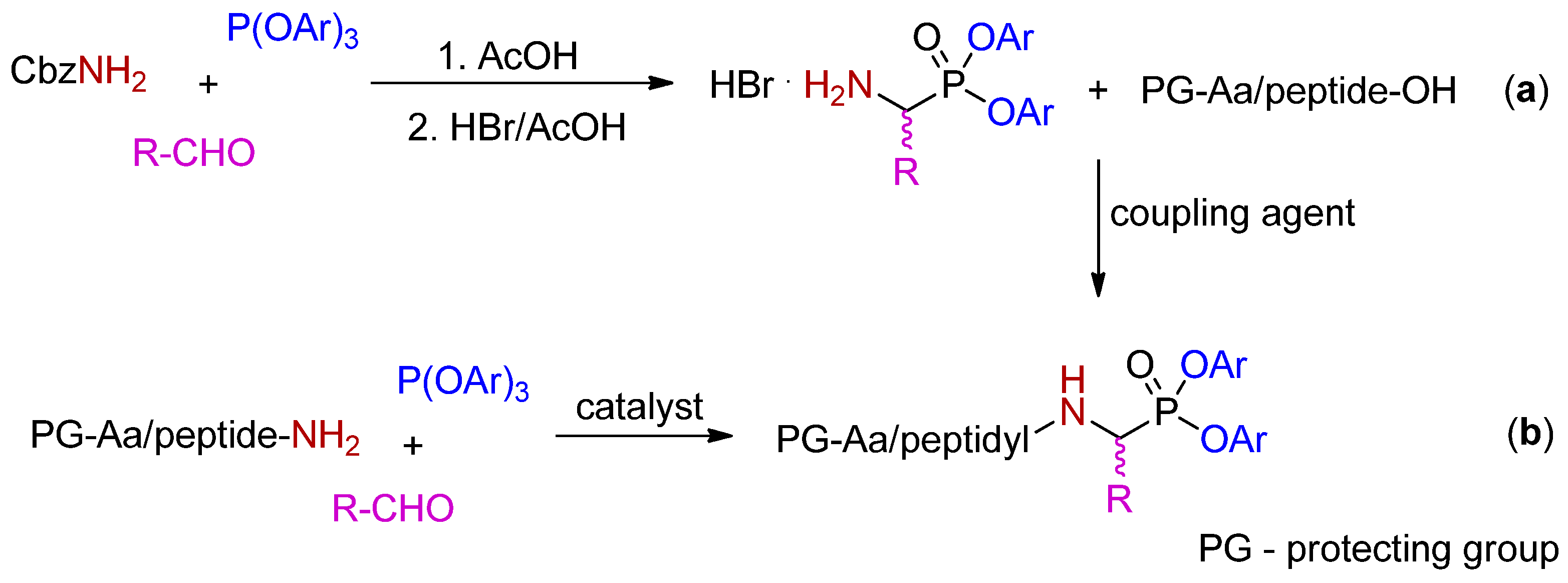
2. Synthetic and Mechanistic Considerations
3. Inhibition of Serine Proteases
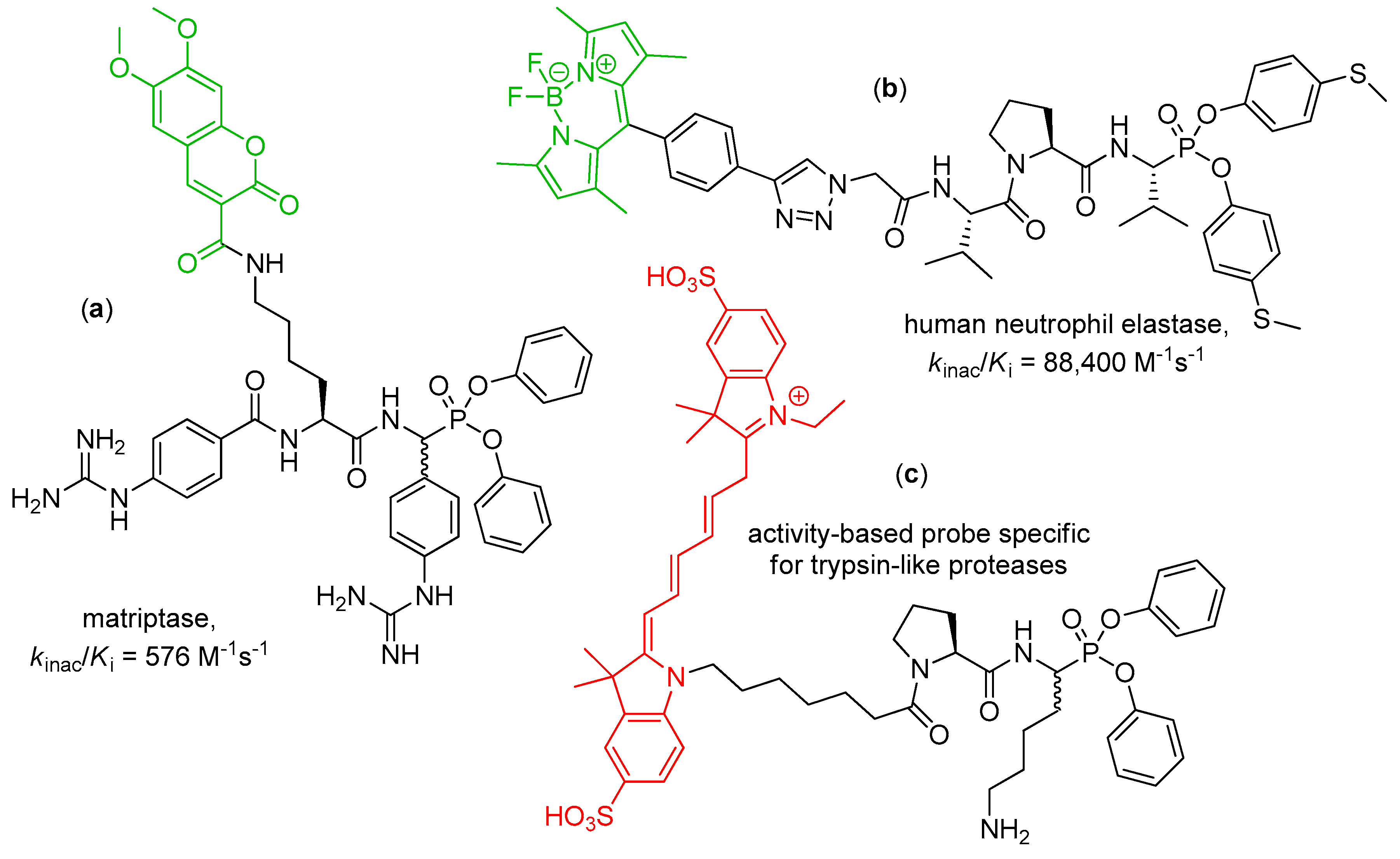
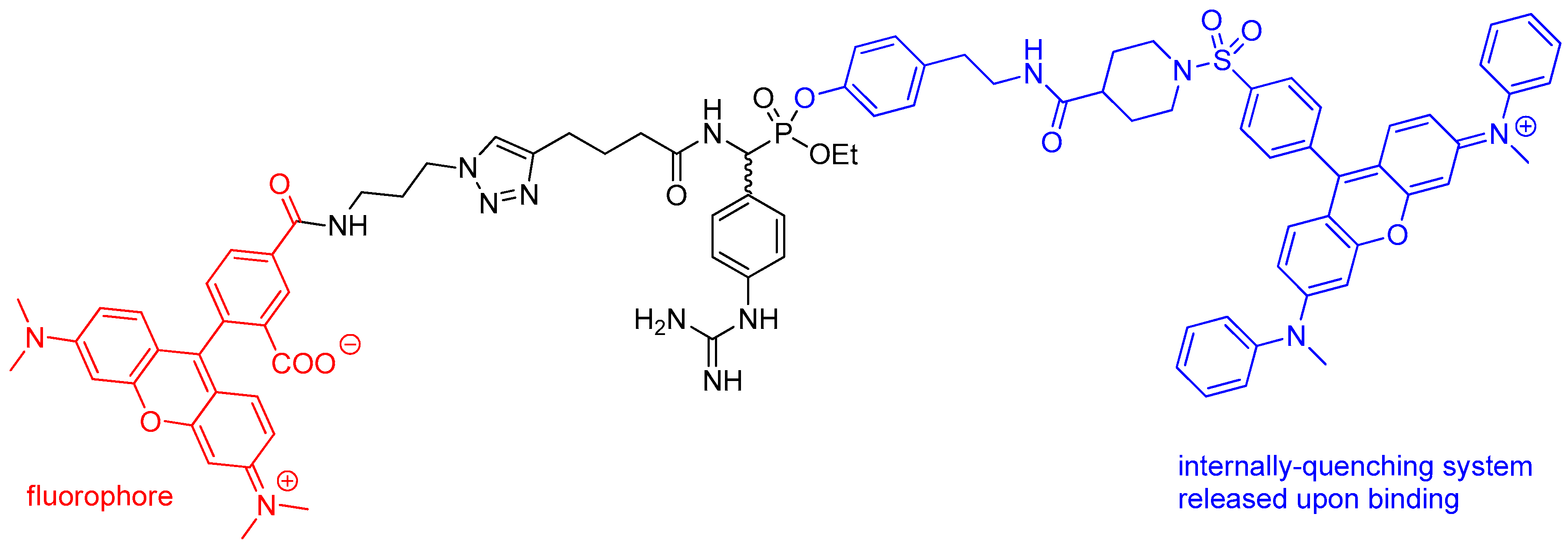
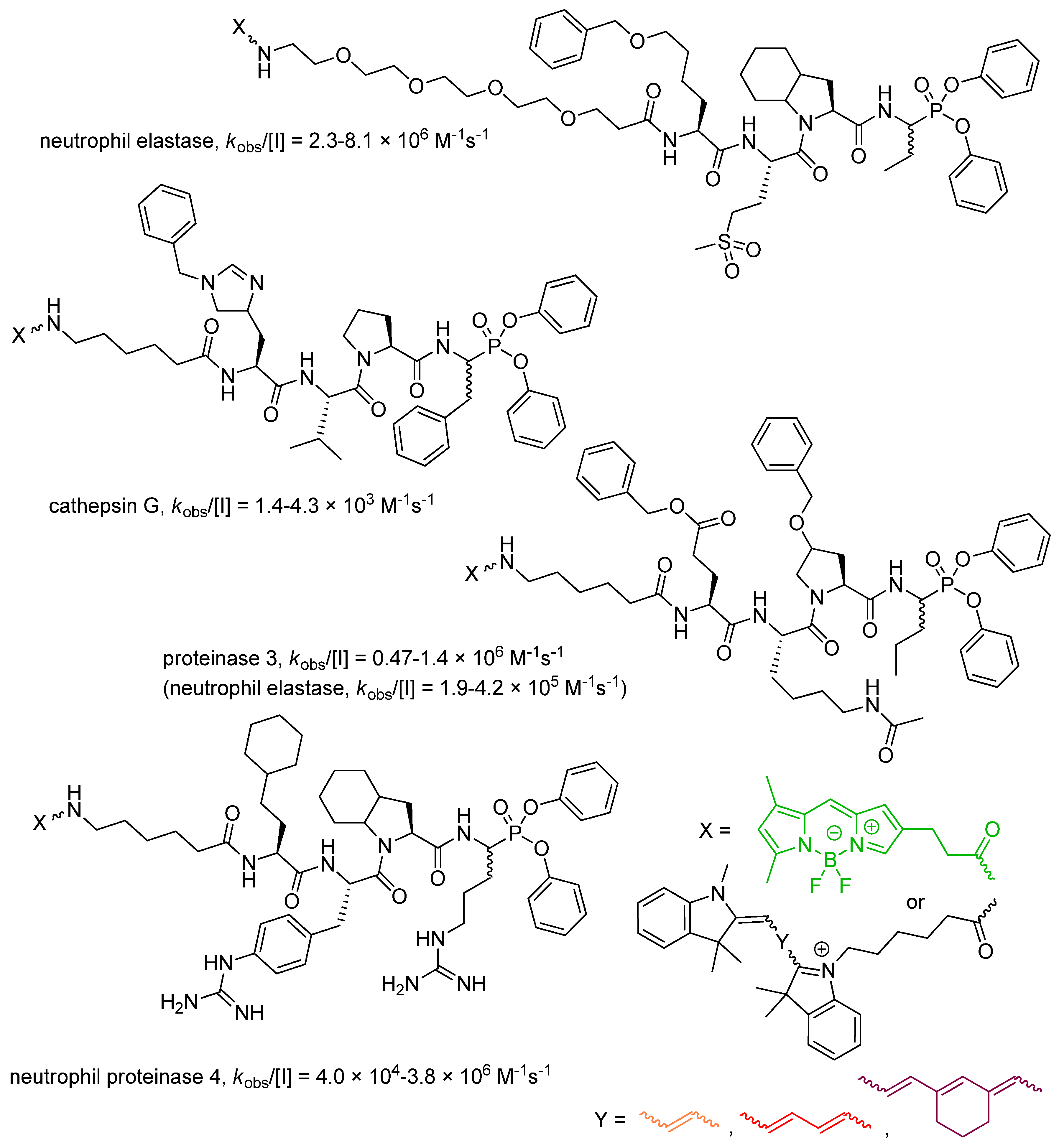
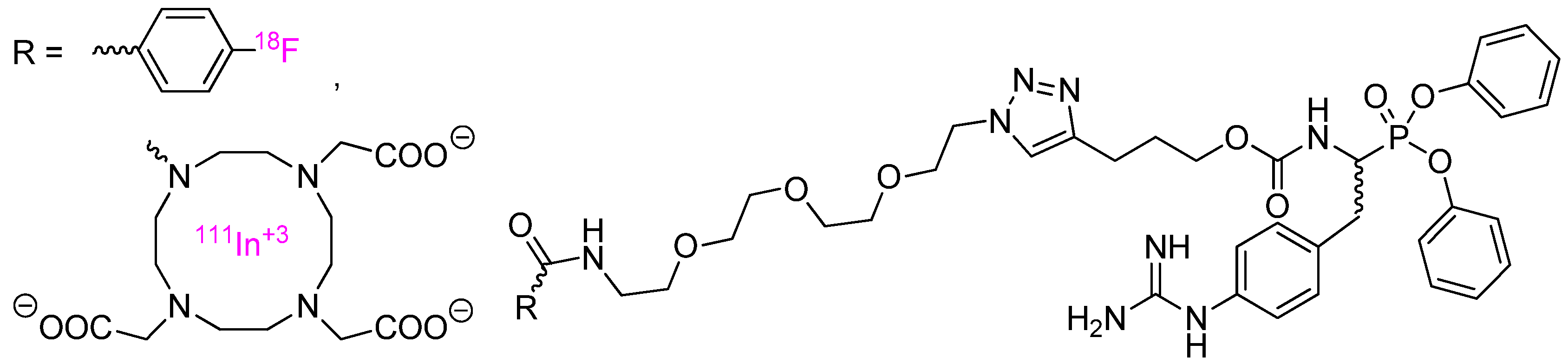
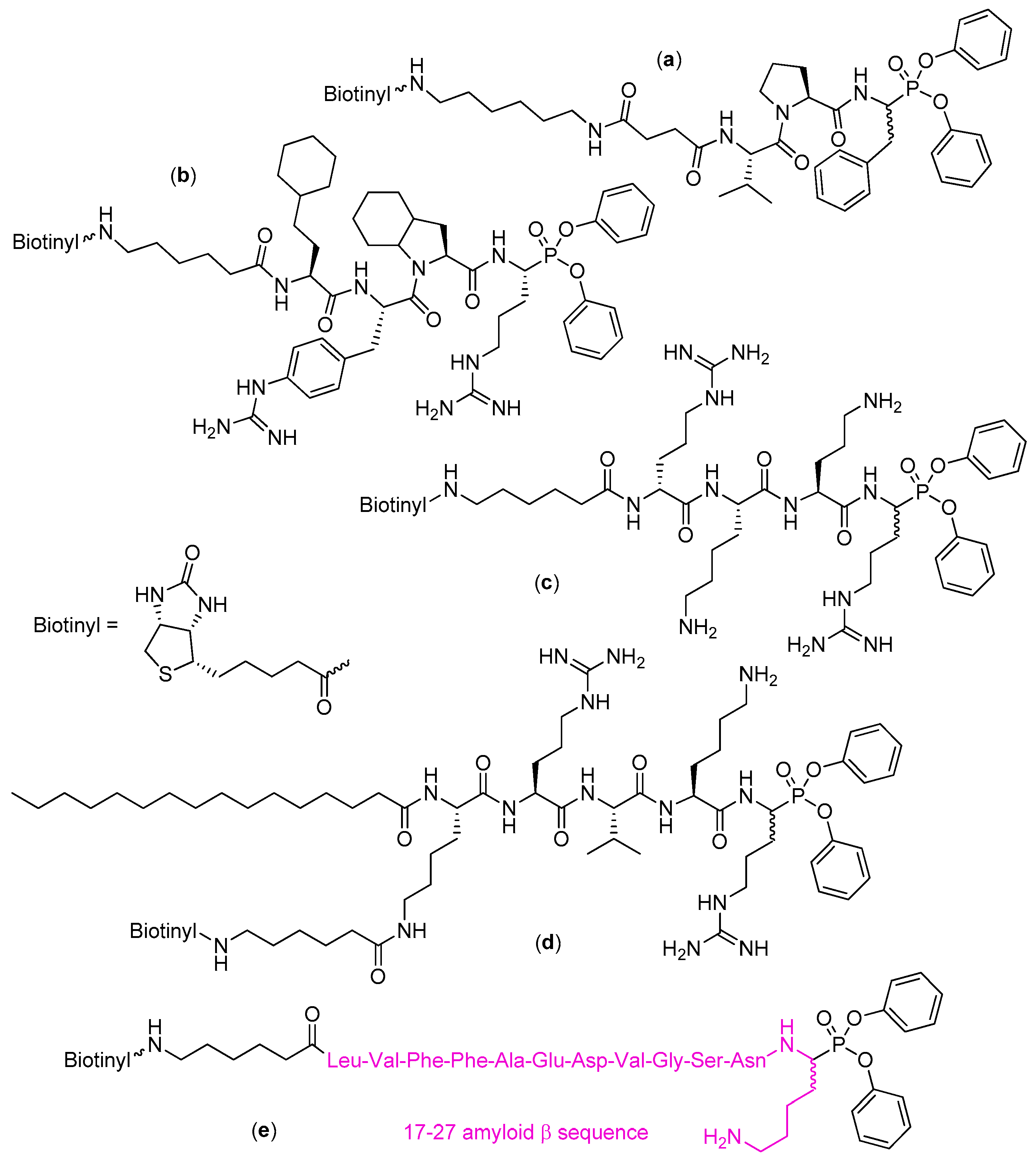
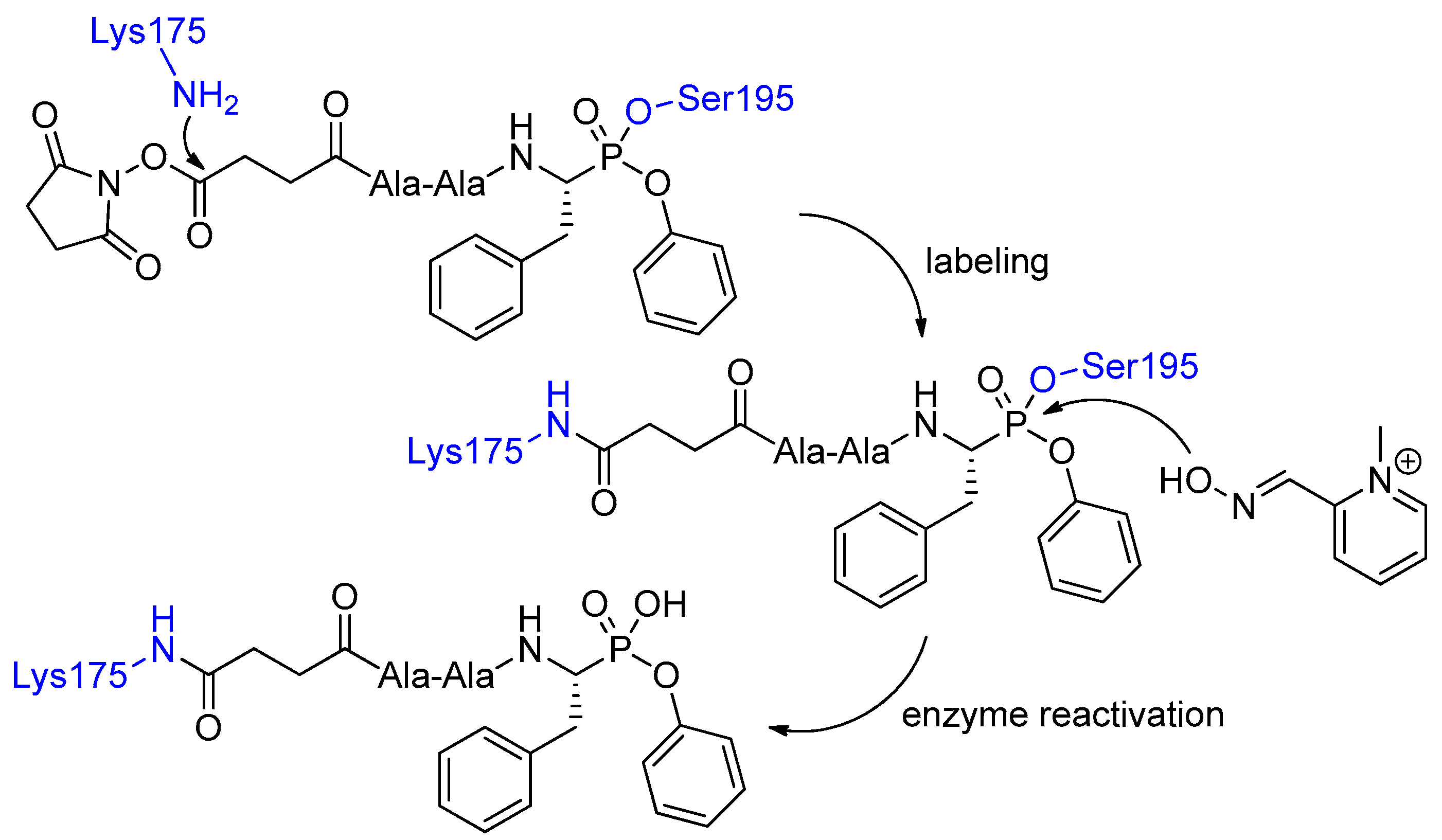
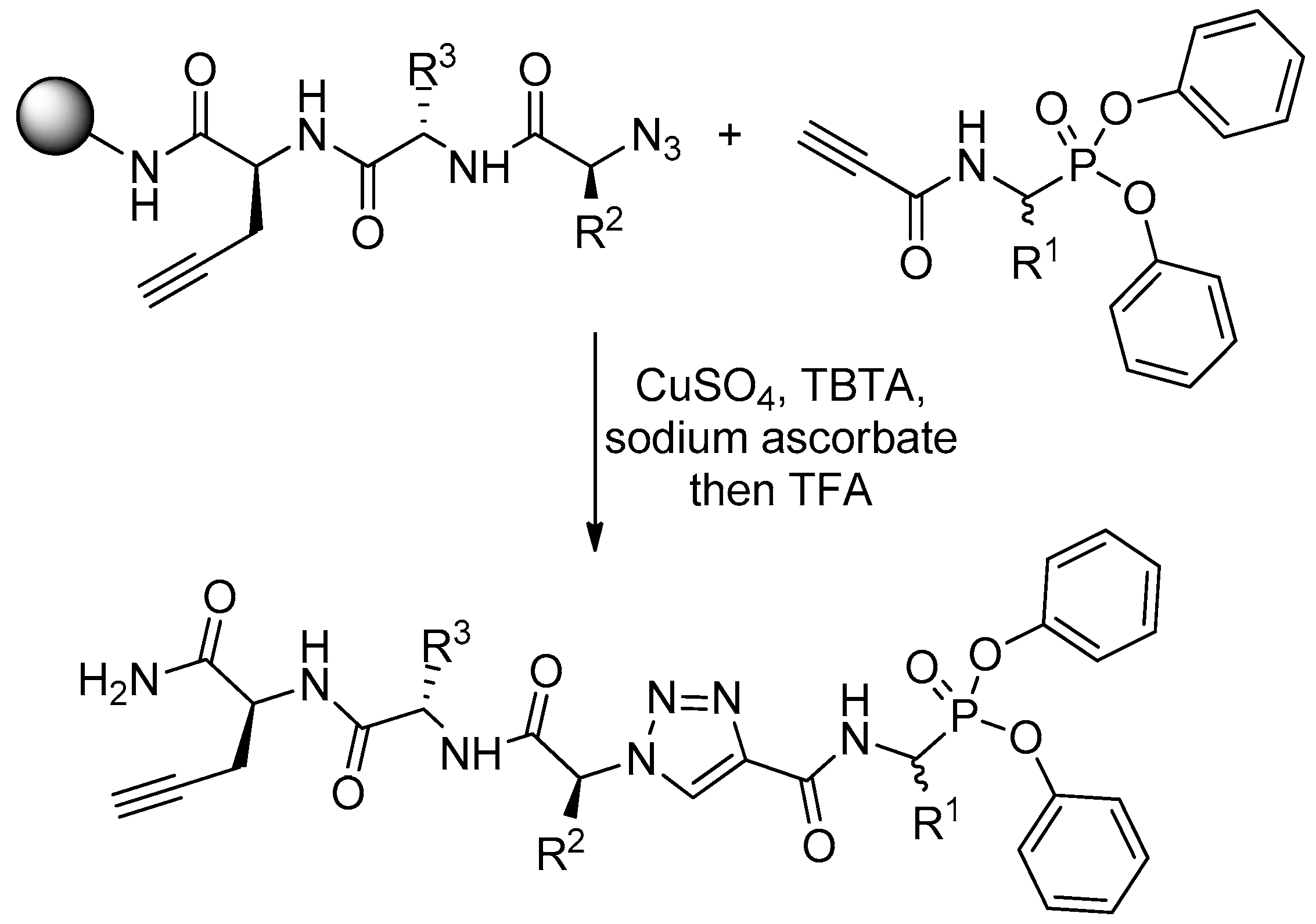
4. Activity-Based Probes
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Oleksyszyn, J.; Powers, J.C. Amino acid and peptide phosphonate derivatives as specific inhibitors of serine peptidases. Methods Enzymol. 1994, 244, 423–441. [Google Scholar] [CrossRef] [PubMed]
- Oleksyszyn, J.; Powers, J.C. Irreversible inhibition of serine proteases by peptide derivatives of α-aminoalkyl)phosphonate diphenyl esters. Biochemistry 1991, 30, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Grzywa, R.; Sieńczyk, M. Phosphonic esters and their application of protease control. Curr. Pharm. Des. 2013, 19, 1154–1178. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, J.A.; Oleksyszyn, J.; Kam, C.M.; Boduszek, B.; Presnell, S.; Plaskon, R.R.; Suddath, F.L.; Powers, J.C.; Williams, L.D. Inhibition of trypsin and thrombin by amino(4-amidinophenyl)methanephosphonate diphenyl ester derivatives: X-ray structures and molecular models. Biochemistry 1996, 35, 3147–3155. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.; Wharry, S.; Hamilton, R.J.; Martin, S.L.; Healy, A.; Walker, B.J. Asymmetric preference of serine proteases toward phosphonate and phosphinate esters. Biochem. Biophys. Res. Commun. 2000, 276, 1235–1239. [Google Scholar] [CrossRef] [PubMed]
- Boduszek, B.; Brown, A.D.; Powers, J.C. α-Aminoalkylphosphonate di(chlorophenyl) esters as inhibitors of serine proteases. J. Enzyme Inhib. 1994, 8, 147–158. [Google Scholar] [CrossRef]
- Sieńczyk, M.; Oleksyszyn, J. Inhibition of trypsin and urokinase by Cbz-amino(4-guanidinophenyl)methanephosphonate aromatic ester derivatives: The influence of the ester group on their biological activity. Bioorg. Med. Chem. Lett. 2006, 16, 2886–2890. [Google Scholar] [CrossRef]
- Winiarski, Ł.; Oleksyszyn, J.; Sieńczyk, M. Human neutrophil elastase phosphonic inhibitors with improved potency of action. J. Med. Chem. 2012, 55, 6541–6553. [Google Scholar] [CrossRef]
- Schulz-Fincke, A.C.; Blaut, M.; Braune, A.; Gütschow, M. A BODIPY-tagged phosphono peptide as activity-based probe for human leukocyte elastase. ACS Med. Chem. Lett. 2018, 9, 345–350. [Google Scholar] [CrossRef]
- Ono, S.; Murai, J.; Furuta, S.; Doike, K.; Manzaki, F.; Yoshimura, T.; Kuroda, H.; Umezaki, M.; Oyama, H. Covalent chromatography for chymotrypsin-like proteases using a diphenyl 1-amino-2-phenylethylphosphonate derivative. J. Biol. Macromol. 2013, 13, 78–85. [Google Scholar] [CrossRef]
- Ono, S.; Murai, J.; Nakai, T.; Kuroda, H.; Horino, Y.; Yoshimura, T.; Oyama, H.; Umezaki, M. Site-selective chemical modification of chymotrypsin using a peptidyl diphenyl 1-amino-2-phenylethylphosphonate derivative. Chem. Lett. 2013, 42, 860–862. [Google Scholar] [CrossRef]
- Ono, S.; Nakai, T.; Kuroda, H.; Miyatake, R.; Horino, Y.; Abe, H.; Umezaki, M.; Oyama, H. Site-selective chemical modification of chymotrypsin using peptidyl derivatives bearing optically active diphenyl 1-amino-2-phenylethylphosphonate: Stereochemical effect of the diphenyl phosphonate moiety. Biopolymers 2016, 106, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Oleksyszyn, J.; Subotkowska, L.; Mastalerz, P. Diphenyl 1-aminoalkanephosphonates. Synthesis 1979, 985–986. [Google Scholar] [CrossRef]
- Skoreński, M.; Oleksyszyn, J.; Sieńczyk, M. A convenient method for the one step synthesis of phosphonic peptides. Tetrahedron Lett. 2013, 54, 4975–4977. [Google Scholar] [CrossRef]
- Serim, S.; Mayer, S.V.; Verhelst, S.H.L. Tuning activity-based probe selectivity for serine proteases by on-resin ‘click’ construction of peptide diphenyl phosphonates. Org. Biomol. Chem. 2013, 11, 5714–5721. [Google Scholar] [CrossRef] [PubMed]
- Stapels, D.A.C.; Geisbrecht, B.V.; Rooijakkers, S.H.M. Neutrophil serine proteases in antibacterial defense. Curr. Opin. Microbiol. 2015, 23, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef]
- Loison, F.; Zhu, H.; Karatepe, K.; Kasorn, A.; Liu, P.; Ye, K.; Zhou, J.; Cao, S.; Gong, H.; Jenne, D.E.; et al. Proteinase 3-dependent caspase-3 cleavage modulates neutrophil death and inflammation. J. Clin. Investig. 2014, 124, 4445–4458. [Google Scholar] [CrossRef]
- Guarino, C.; Legowska, M.; Epinette, C.; Kellenberger, C.; Dallet-Choisy, S.; Sieńczyk, M.; Gabant, G.; Cadene, M.; Zoidakis, J.; Vlahou, A.; et al. New selective peptidyl di(chlorophenyl) phosphonate esters for visualizing and blocking neutrophil proteinase 3 in human diseases. J. Biol. Chem. 2014, 289, 31777–31791. [Google Scholar] [CrossRef]
- Guarino, C.; Gruba, N.; Grzywa, R.; Dyguda-Kazimierowicz, E.; Hamon, Y.; Łȩgowska, M.; Skoreński, M.; Dallet-Choisy, S.; Marchand-Adam, S.; Kellenberger, C.; et al. Exploiting the S4-S5 specificity of human neutrophil proteinase 3 to improve the potency of peptidyl di(chlorophenyl)-phosphonate ester inhibitors: A kinetic and molecular modeling analysis. J. Med. Chem. 2018, 61, 1858–1870. [Google Scholar] [CrossRef]
- Häußler, D.; Scheidt, T.; Stirnberg, M.; Steinmetzer, T.; Gütschow, M. A bisbenzamidine phosphonate as a Janus-faced inhibitor for trypsin-like serine proteases. ChemMedChem 2015, 10, 1641–1646. [Google Scholar] [CrossRef] [PubMed]
- Häußler, D.; Mangold, M.; Furtmann, N.; Braune, A.; Blaut, M.; Bajorath, J.; Stirnberg, M.; Gütschow, M. Phosphono bisbenzguanidines as irreversible dipeptidomimetic inhibitors and activity-based probes of matriptase-2. Chem. Eur. J. 2016, 22, 8525–8535. [Google Scholar] [CrossRef] [PubMed]
- Wiemhoefer, A.; Stargardt, A.; van der Linden, W.A.; Renner, M.C.; van Kesteren, R.E.; Stap, J.; Raspe, M.A.; Tomkinson, B.; Kessels, H.W.; Ovaa, H.; et al. Tripeptidyl peptidase II mediates levels of nuclear phosphorylated ERK1 and ERK2. Mol. Cell. Proteomics 2015, 14, 2177–2193. [Google Scholar] [CrossRef] [PubMed]
- Skoreński, M.; Pachota, M.; Pyrć, K.; Sieńczyk, M.; Oleksyszyn, J. Novel peptidyl α-aminoalkylphosphonates as inhibitors of hepatitis C virus NS3/4A protease. Antivir. Res. 2017, 144, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Burchacka, E.; Skoreński, M.; Sieńczyk, M.; Oleksyszyn, J. Phosphonic analogues of glutamic acid as irreversible inhibitors of Staphylococcus aureus endoproteinase GluC: An efficient synthesis and inhibition of the human IgG degradation. Bioorg. Med. Chem. Lett. 2013, 23, 1412–1415. [Google Scholar] [CrossRef]
- Burchacka, E.; Zdżalik, M.; Niemczyk, J.S.; Pustelny, K.; Popowicz, G.; Wladyka, B.; Dubin, A.; Potempa, J.; Sieńczyk, M.; Dubin, G.; et al. Development and binding characteristics of phosphonate inhibitors of SplA protease from Staphylococcus aureus. Protein Sci. 2014, 23, 179–189. [Google Scholar] [CrossRef]
- Burchacka, E.; Walczak, M.; Sieńczyk, M.; Dubin, G.; Zdżalik, M.; Potempa, J.; Oleksyszyn, J. The development of first Staphylococcus aureus SplB protease inhibitors: Phosphonic analogues of glutamine. Bioorg. Med. Chem. Lett. 2012, 22, 5574–5578. [Google Scholar] [CrossRef]
- Raju, R.M.; Goldberg, A.L.; Rubin, E.J. Bacterial proteolytic complexes as therapeutic targets. Nat. Rev. Drug Discov. 2012, 11, 777–789. [Google Scholar] [CrossRef]
- Skoreński, M.; Sieńczyk, M. Viral proteases as targets for drug design. Cur. Pharm. Design 2013, 19, 1126–1153. [Google Scholar] [CrossRef]
- Morikawa, K.; Lange, C.M.; Gouttenoire, J.; Meylan, E.; Brass, V.; Penin, F.; Moradpour, D. Nonstructural protein 3-4A: The Swiss army knife of hepatitis C virus. J. Viral Hepat. 2011, 18, 305–315. [Google Scholar] [CrossRef]
- McCauley, J.A.; Rudd, M.T. Hepatitis C virus NS3/4a protease inhibitors. Curr. Opin. Pharmacol. 2016, 30, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, S.; Bogen, S.L.; Arasappan, A.; Bennett, F.; Chen, K.; Jao, E.; Liu, Y.-T.; Lovey, R.; Hendrata, S.; Huang, Y.; et al. Discovery of (1R,5S)-N-[3-Amino-1-(cyclobutylmethyl)-2,3-dioxopropyl]-3-[2(S)-[[[(1,1-dimethylethyl)amino]carbonyl]amino]-3,3-dimethyl-1-oxobutyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexan-2(S)-carboxamide (SCH 503034), a selective, potent, orally bioavailable hepatitis C virus NS3 protease inhibitor: A potential therapeutic agent for the treatment of hepatitis C infection. J. Med. Chem. 2006, 49, 6074–6086. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Kwong, A.D.; Perni, R.B. Discovery and development of VX-950, a novel, covalent, and reversible inhibitor of hepatitis C virus NS3-4A serine protease. Infect. Disord. Drug Targets 2006, 6, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Skoreński, M.; Milewska, A.; Pyrć, K.; Sieńczyk, M.; Oleksyszyn, J. Phosphonate inhibitors of West Nile virus NS2B/NS3 protease. J. Enzyme Inhib. Med. Chem. 2019, 34, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Agbowuro, A.A.; Mazraani, R.; McCaughey, L.C.; Huston, W.M.; Gamble, A.B.; Tyndall, J.D.A. Stereochemical basis for the anti-chlamydial activity of the phosphonate protease inhibitor JO146. Tetrahedron 2018, 74, 1184–1190. [Google Scholar] [CrossRef]
- Moreno-Cinos, C.; Sassetti, E.; Salado, I.G.; Witt, G.; Benramdane, S.; Reinhardt, L.; Cruz, C.D.; Joossens, J.; Van der Veken, P.; Brötz-Oesterhelt, H.; et al. α‑Amino diphenyl phosphonates as novel inhibitors of Escherichia coli ClpP protease. J. Med. Chem. 2019, 62, 774–797. [Google Scholar] [CrossRef]
- Brotz-Oesterhelt, H.; Sass, P. Bacterial caseinolytic proteases as novel targets for antibacterial treatment. Int. J. Med. Microbiol. 2014, 304, 23–30. [Google Scholar] [CrossRef]
- Sanman, L.E.; Bogyo, M. Activity-based profiling of proteases. Annu. Rev. Biochem. 2014, 83, 249–273. [Google Scholar] [CrossRef]
- Kasperkiewicz, P.; Poreba, M.; Groborz, K.; Drag, M. Emerging challenges in the design of selective substrates, inhibitors and activity-based probes for indistinguishable proteases. FEBS J. 2017, 284, 1518–1539. [Google Scholar] [CrossRef]
- Abuelyaman, A.S.; Hudig, D.; Woodard, S.L.; Powers, J.C. Fluorescent derivatives of diphenyl [1-(N-peptidylamino)alkyl]phosphonate esters: Synthesis and use in the inhibition and cellular localization of serine proteases. Bioconjug. Chem. 1994, 5, 400–405. [Google Scholar] [CrossRef]
- Pan, Z.; Jeffery, D.A.; Chehade, K.; Beltman, J.; Clark, J.M.; Grothaus, P.; Bogyo, M.; Baruch, A. Development of activity-based probes for trypsin-family serine proteases. Bioorg. Med. Chem. Lett. 2006, 16, 2882–2885. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, B.F.; Quinn, D.J.; Duff, T.; Cathcart, G.R.; Scott, C.J.; Walker, B. Expedited solid-phase synthesis of fluorescently labeled and biotinylated aminoalkane diphenyl phosphonate affinity probes for chymotrypsin- and elastase-like serine proteases. Bioconjug. Chem. 2009, 20, 2098–2105. [Google Scholar] [CrossRef] [PubMed]
- Sabidó, E.; Tarragó, T.; Giralt, E. Towards the identification of unknown neuropeptide precursor-processing enzymes: Design and synthesis of a new family of dipeptidyl phosphonate activity probes for substrate-based protease identification. Bioorg. Med. Chem. 2010, 18, 8350–8355. [Google Scholar] [CrossRef] [PubMed]
- Häußler, D.; Schulz-Fincke, A.C.; Beckmann, A.M.; Keils, A.; Gilberg, E.; Mangold, M.; Bajorath, J.; Stirnberg, M.; Steinmetzer, T.; Gütschow, M. A fluorescent-labeled phosphono bisbenzguanidine as an activity-based probe for matriptase. Chem. Eur. J. 2017, 23, 5205–5209. [Google Scholar] [CrossRef] [PubMed]
- Edgington-Mitchell, L.E.; Barlow, N.; Aurelio, L.; Samha, A.; Szabo, M.; Graham, B.; Bunnett, N. Fluorescent diphenylphosphonate-based probes for detection of serine protease activity during inflammation. Bioorg. Med. Chem. Lett. 2017, 27, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Serim, S.; Baer, P.; Verhelst, S.H.L. Mixed alkyl aryl phosphonate esters as quenched fluorescent activity-based probes for serine proteases. Org. Biomol. Chem. 2015, 13, 2293–2299. [Google Scholar] [CrossRef] [PubMed]
- Kasperkiewicz, P.; Altman, Y.; D’Angelo, M.; Salvesen, G.S.; Drag, M. Toolbox of fluorescent probes for parallel imaging reveals uneven location of serine proteases in neutrophils. J. Am. Chem. Soc. 2017, 139, 10115–10125. [Google Scholar] [CrossRef] [PubMed]
- Kasperkiewicz, P.; Poreba, M.; Snipas, S.J.; Parker, H.; Winterbourn, C.C.; Salvesen, G.S.; Drag, M. Design of ultrasensitive probes for human neutrophil elastase through hybrid combinatorial substrate library profiling. Proc. Natl. Acad. Sci. USA 2014, 111, 2518–2523. [Google Scholar] [CrossRef] [PubMed]
- Ides, J.; Thomae, D.; Wyffels, L.; Vangestel, C.; Messagie, J.; Joossens, J.; Lardon, F.; Van der Veken, P.; Agustyns, K.; Stroobants, S.; et al. Synthesis and in vivo preclinical evaluation of an 18F labeled uPA inhibitor as a potential PET imaging agent. Nucl. Med. Biol. 2014, 41, 477–484. [Google Scholar] [CrossRef]
- Vangestel, C.; Thomae, D.; Van Soom, J.; Ides, J.; Wyffels, L.; Pauwels, P.; Stroobants, S.; Van der Veken, P.; Magdolen, V.; Joossens, J.; et al. Preclinical evaluation of [111 In]MICA-401, an activity-based probe for SPECT imaging of in vivo uPA activity. Contrast Media Mol. Imaging 2016, 11, 448–458. [Google Scholar] [CrossRef]
- Zou, F.; Schmon, M.; Sieńczyk, M.; Grzywa, R.; Palesch, D.; Boehm, B.O.; Sun, Z.L.; Watts, C.; Schirmbeck, R.; Burster, T. Application of a novel highly sensitive activity-based probe for detection of cathepsin G. Anal. Biochem. 2012, 421, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Grzywa, R.; Burchacka, E.; Łęcka, M.; Winiarski, Ł.; Walczak, M.; Łupicka-Słowik, A.; Wysocka, M.; Burster, T.; Bobrek, K.; Csencsits-Smith, K.; et al. Synthesis of novel phosphonic-type activity-based probes for neutrophil serine proteases and their application in spleen lysates of different organisms. ChemBioChem 2014, 15, 2605–2612. [Google Scholar] [CrossRef] [PubMed]
- Kasperkiewicz, P.; Poreba, M.; Snipas, S.J.; Lin, S.J.; Kirchhofer, D.; Salvesen, G.S.; Drag, M. Design of a selective substrate and activity based probe for human neutrophil serine protease 4. PLoS ONE 2015, 10, e0132818. [Google Scholar] [CrossRef] [PubMed]
- Perera, N.C.; Schilling, O.; Kittel, H.; Back, W.; Kremmer, E.; Jenne, D.E. NSP4, an elastase-related protease in human neutrophils with arginine specificity. Proc. Natl. Acad. Sci. USA 2012, 109, 6229–6234. [Google Scholar] [CrossRef] [PubMed]
- Rut, W.; Zhang, L.; Kasperkiewicz, P.; Poreba, M.; Hilgenfeld, R.; Drąg, M. Extended substrate specificity and first potent irreversible inhibitor/activity-based probe design for Zika virus NS2B-NS3 protease. Antivir. Res. 2017, 139, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, T.E.; Reihill, J.A.; Walker, B.; Hamilton, R.A.; Martin, S.L. A selective irreversible inhibitor of furin does not prevent Pseudomonas aeruginosa exotoxin A-induced airway epithelial cytotoxicity. PLoS ONE 2016, 11, e0159868. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Sadr, M.S.; Chrétien, M.; Mbikay, M. The multifaceted proprotein convertases: Their unique, redundant, complementary, and opposite functions. J. Biol. Chem. 2013, 288, 21473–21481. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, H.; Fujita, Y.; Tsuda, Y. Development of an activity-based probe for amyloid β-hydrolyzing antibodies. Bioorg. Med. Chem. Lett. 2016, 26, 2210–2213. [Google Scholar] [CrossRef]
- Augustyns, K.; Van der Veken, P.; Senten, K.; Haemers, A. The therapeutic potential of inhibitors of dipeptidyl peptidase IV (DPP IV) and related proline-specific dipeptidyl aminopeptidases. Curr. Med. Chem. 2005, 12, 971–998. [Google Scholar] [CrossRef] [PubMed]
- Sienczyk, M.; Oleksyszyn, J. Irreversible inhibition of serine proteases—Design and in vivo activity of diaryl α-aminophosphonate derivatives. Curr. Med. Chem. 2009, 16, 1673–1687. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Inhibitor Structure | Targeted Enzyme, Activity and Reference |
|---|---|---|
| 1 |  | Human neutrophil elastase, kinact/KI = 2,353,000 M−1s−1 [8] |
| 2 |  | Human neutrophil protease 3, kobs/[I] = 4418 M−1s−1 [19], |
| 3 |  | Human matriptase-2 kinact/Ki = 2790 M−1s−1 [22] |
| 4 |  | Tripeptidyl peptidase II, IC50 = 0.46 nM [23] |
| 5 |  | Hepatitis C virus NS3/4A protease, k2/Ki = 79,850 M−1s−1 (genotype A), k2/Ki = 60,850 M−1s−1 (genotype B) [24] |
| 6 |  | Staphylococcus aureus endoproteinase GluC k2/Ki = 8540 M−1s−1 [25] |
| 7 |  | Staphylococcus aureus SpIA protease k2/Ki > 8000 M−1s−1 [26] |
| 8 |  | Staphylococcus aureus SpIB protease k2/Ki = 1400 M−1s−1 [27] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maślanka, M.; Mucha, A. Recent Developments in Peptidyl Diaryl Phoshonates as Inhibitors and Activity-Based Probes for Serine Proteases. Pharmaceuticals 2019, 12, 86. https://doi.org/10.3390/ph12020086
Maślanka M, Mucha A. Recent Developments in Peptidyl Diaryl Phoshonates as Inhibitors and Activity-Based Probes for Serine Proteases. Pharmaceuticals. 2019; 12(2):86. https://doi.org/10.3390/ph12020086
Chicago/Turabian StyleMaślanka, Marta, and Artur Mucha. 2019. "Recent Developments in Peptidyl Diaryl Phoshonates as Inhibitors and Activity-Based Probes for Serine Proteases" Pharmaceuticals 12, no. 2: 86. https://doi.org/10.3390/ph12020086
APA StyleMaślanka, M., & Mucha, A. (2019). Recent Developments in Peptidyl Diaryl Phoshonates as Inhibitors and Activity-Based Probes for Serine Proteases. Pharmaceuticals, 12(2), 86. https://doi.org/10.3390/ph12020086