Targeting the Serine Pathway: A Promising Approach against Tuberculosis? †


Abstract
1. Introduction
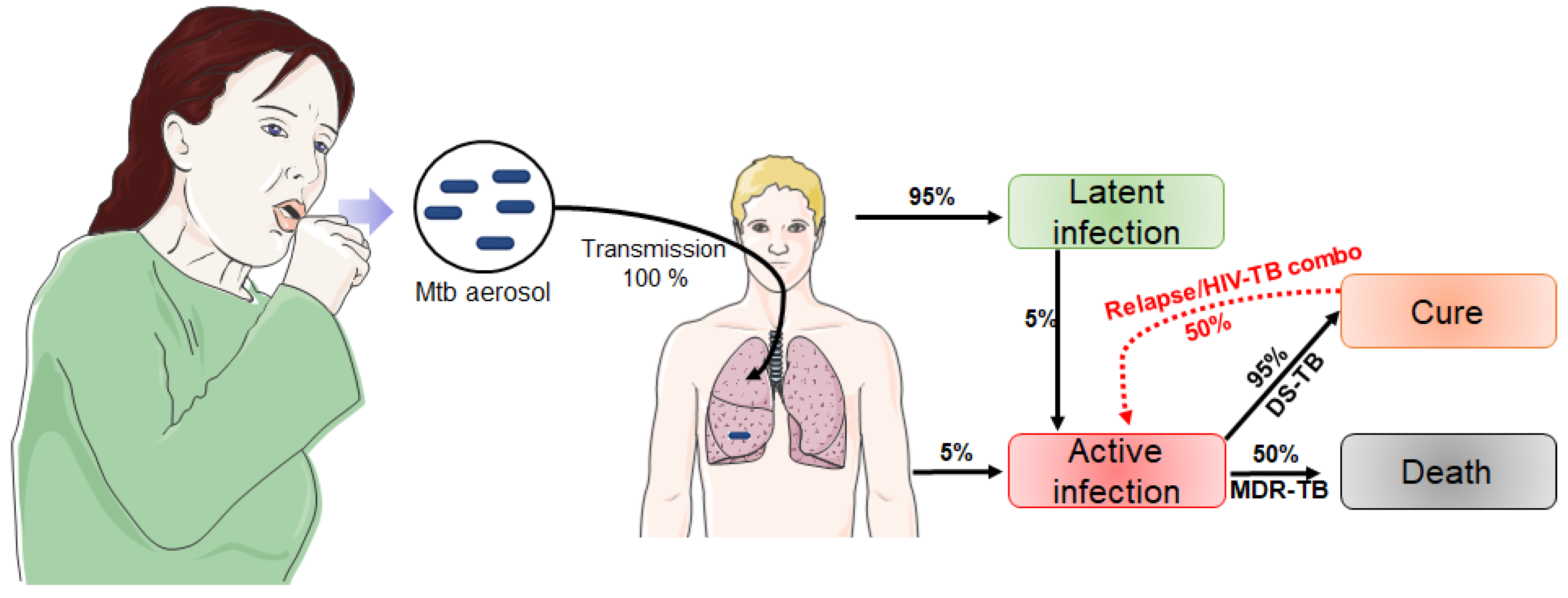
2. Tuberculosis: Overview
- integrated, patient-centred care and prevention
- bold policies and supportive systems
- intensified research and innovation
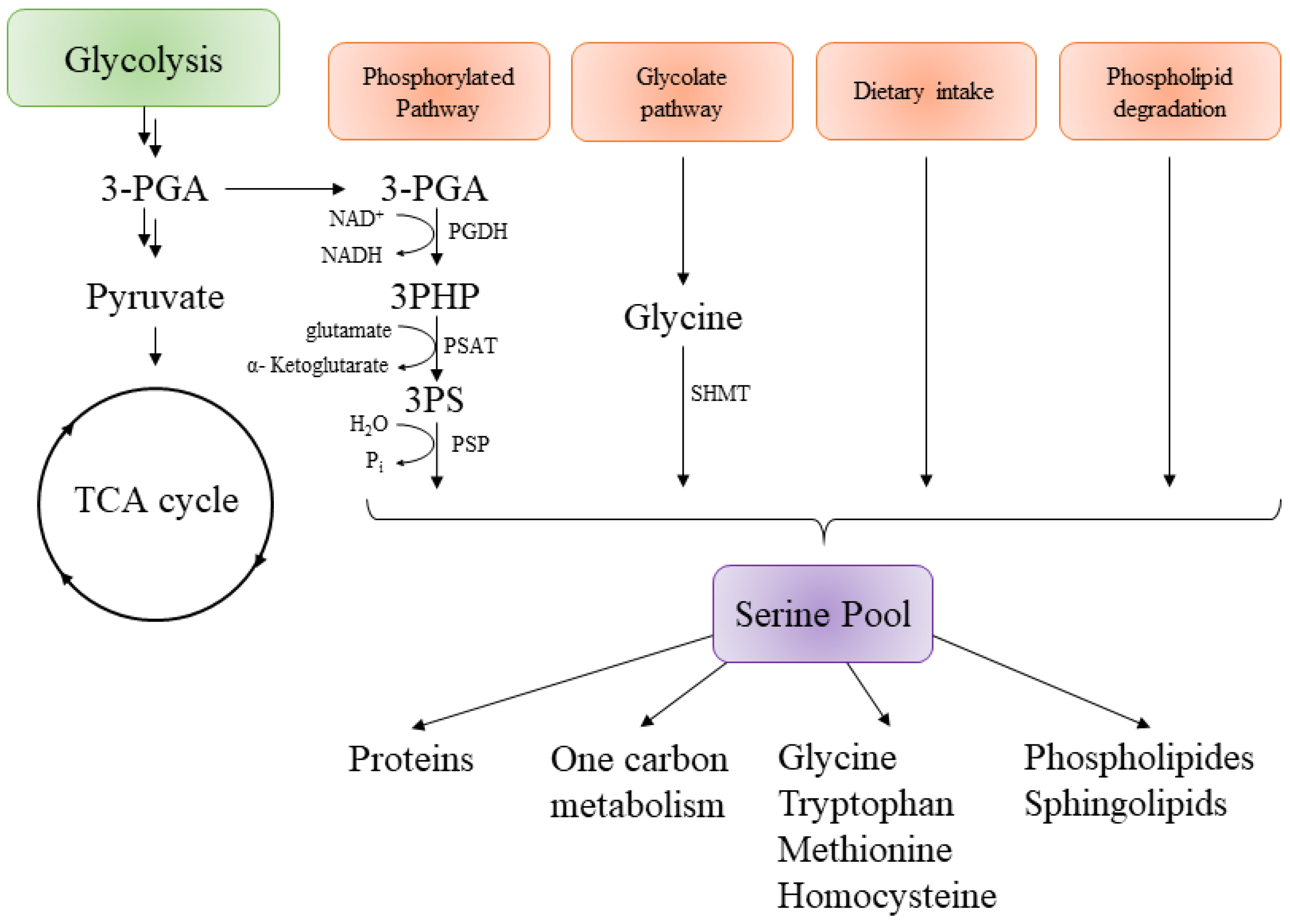
3. Targeting the Serine Pathway
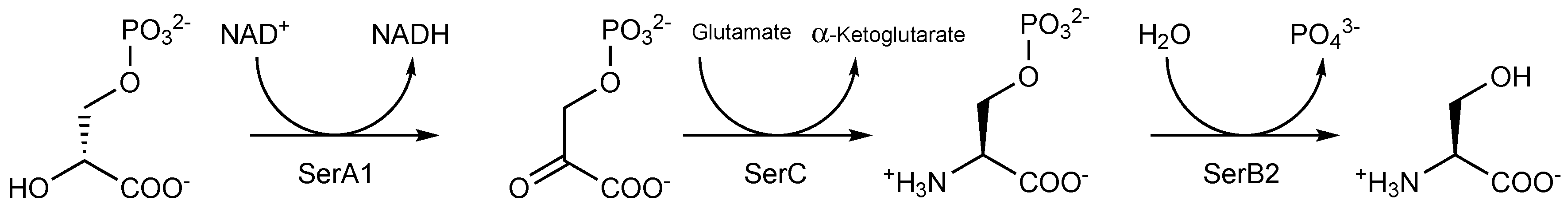
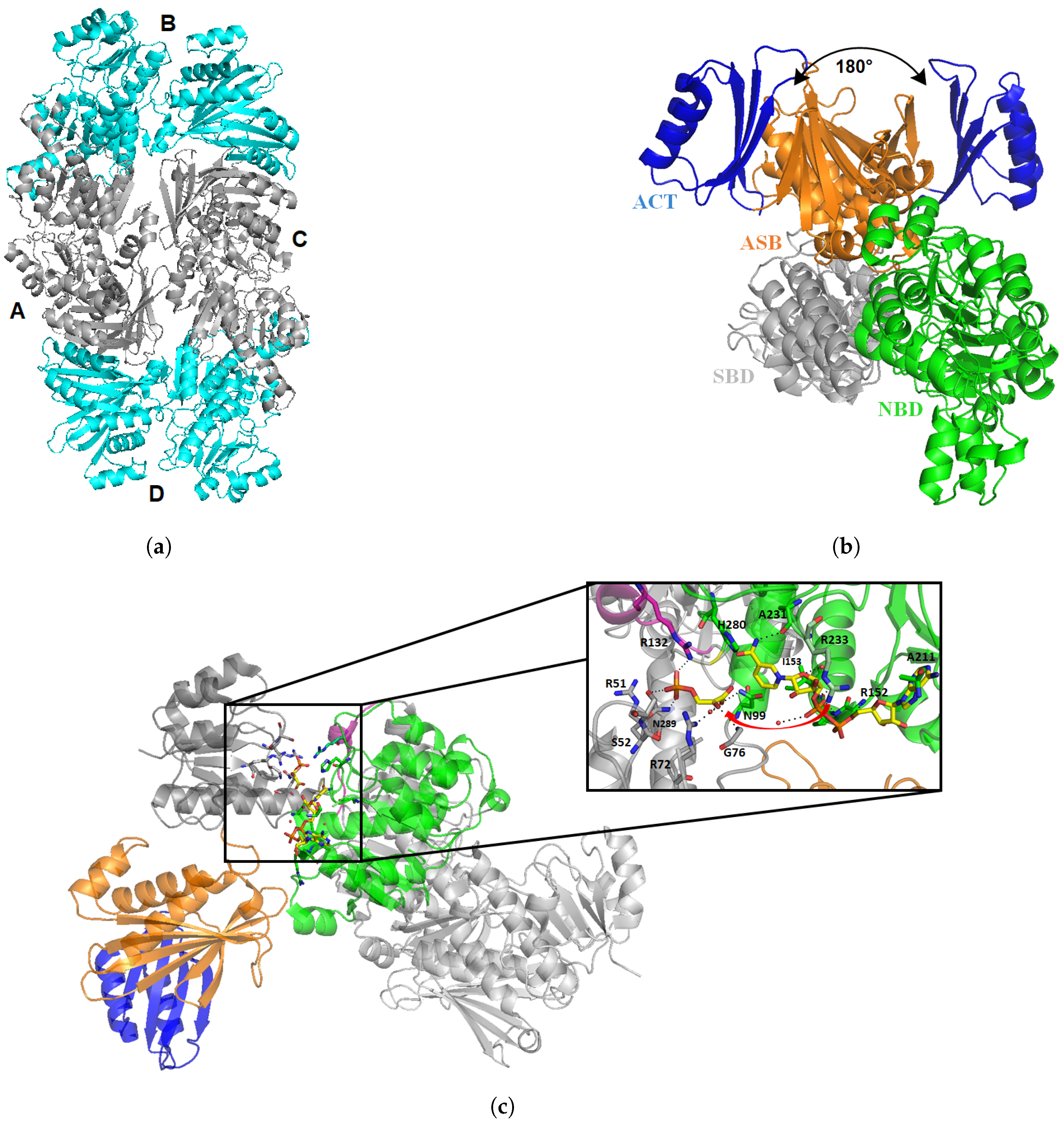
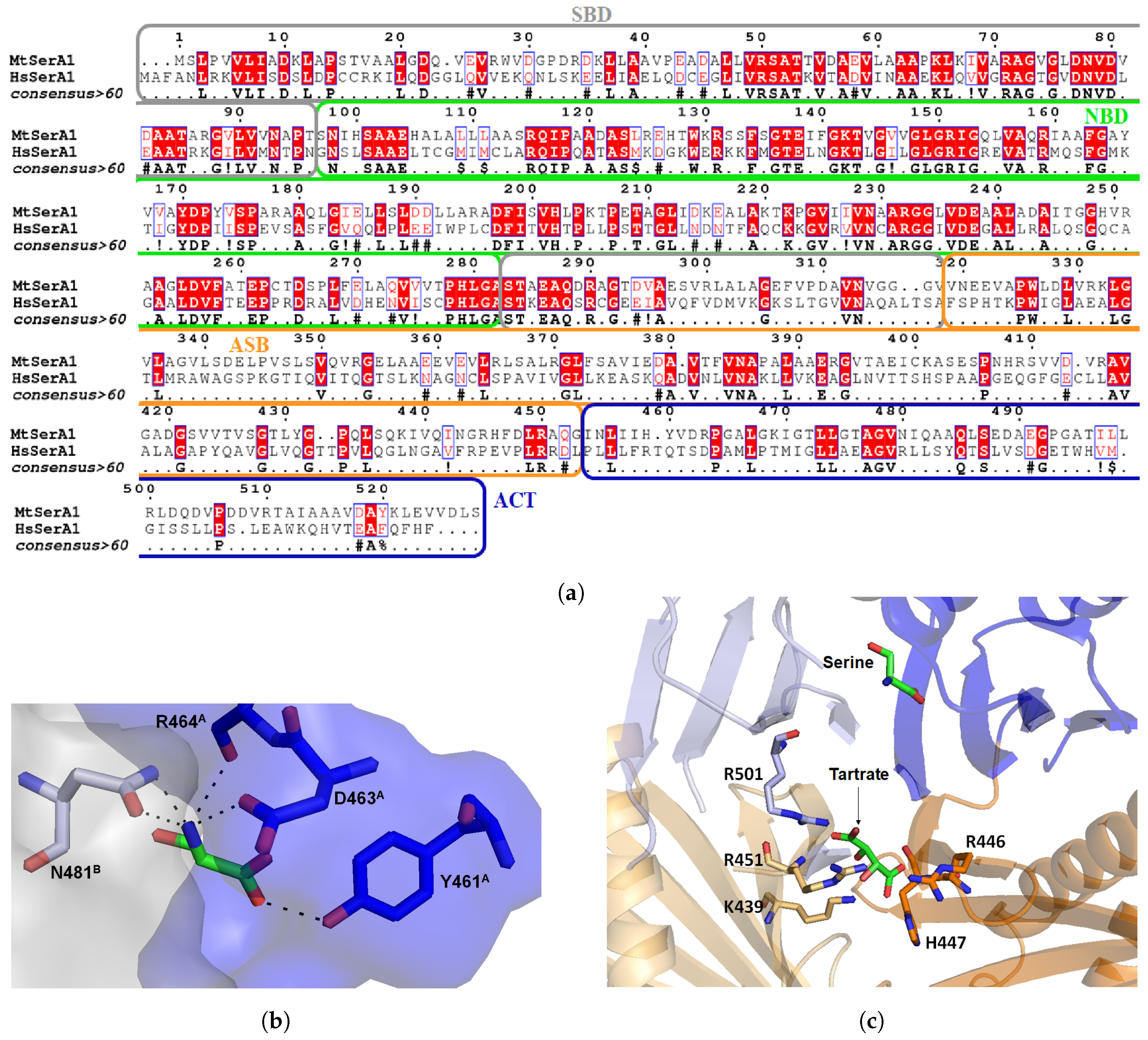
3.1. Mycobacterium tuberculosis Phosphoglycerate Dehydrogenase SerA1: Old But Gold?
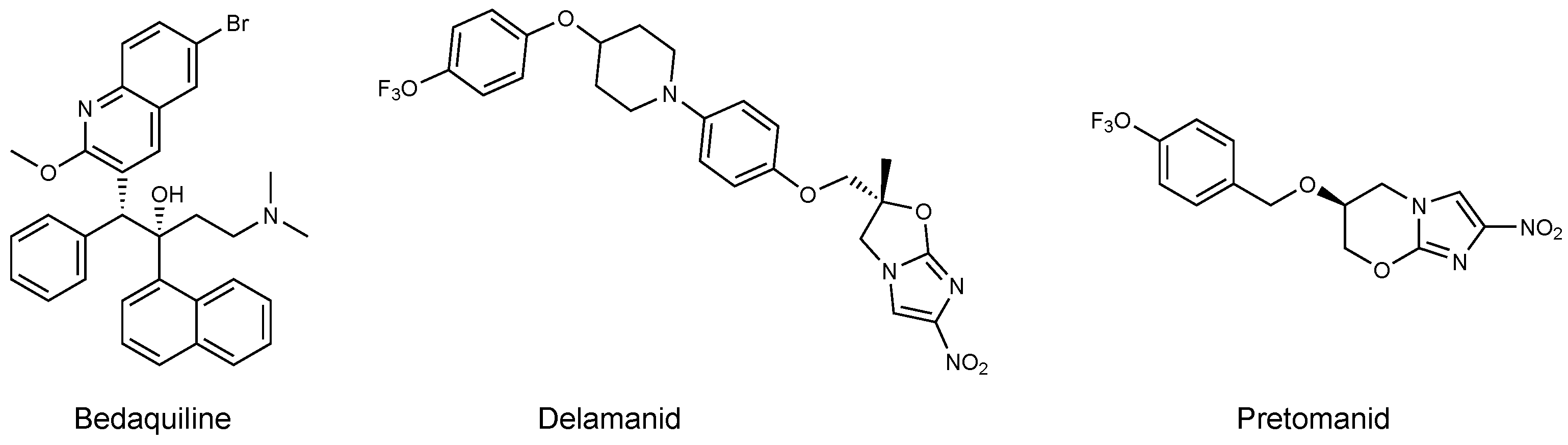
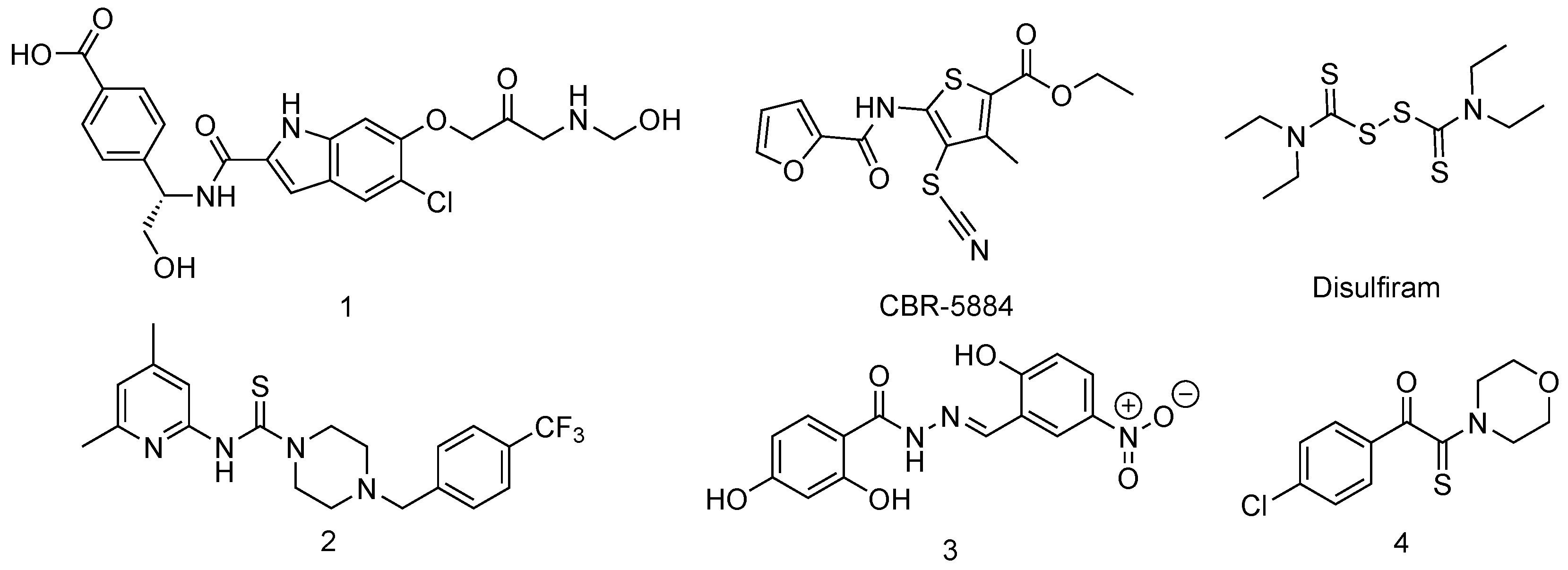
Inhibitors of Phosphoglycerate Dehydrogenases
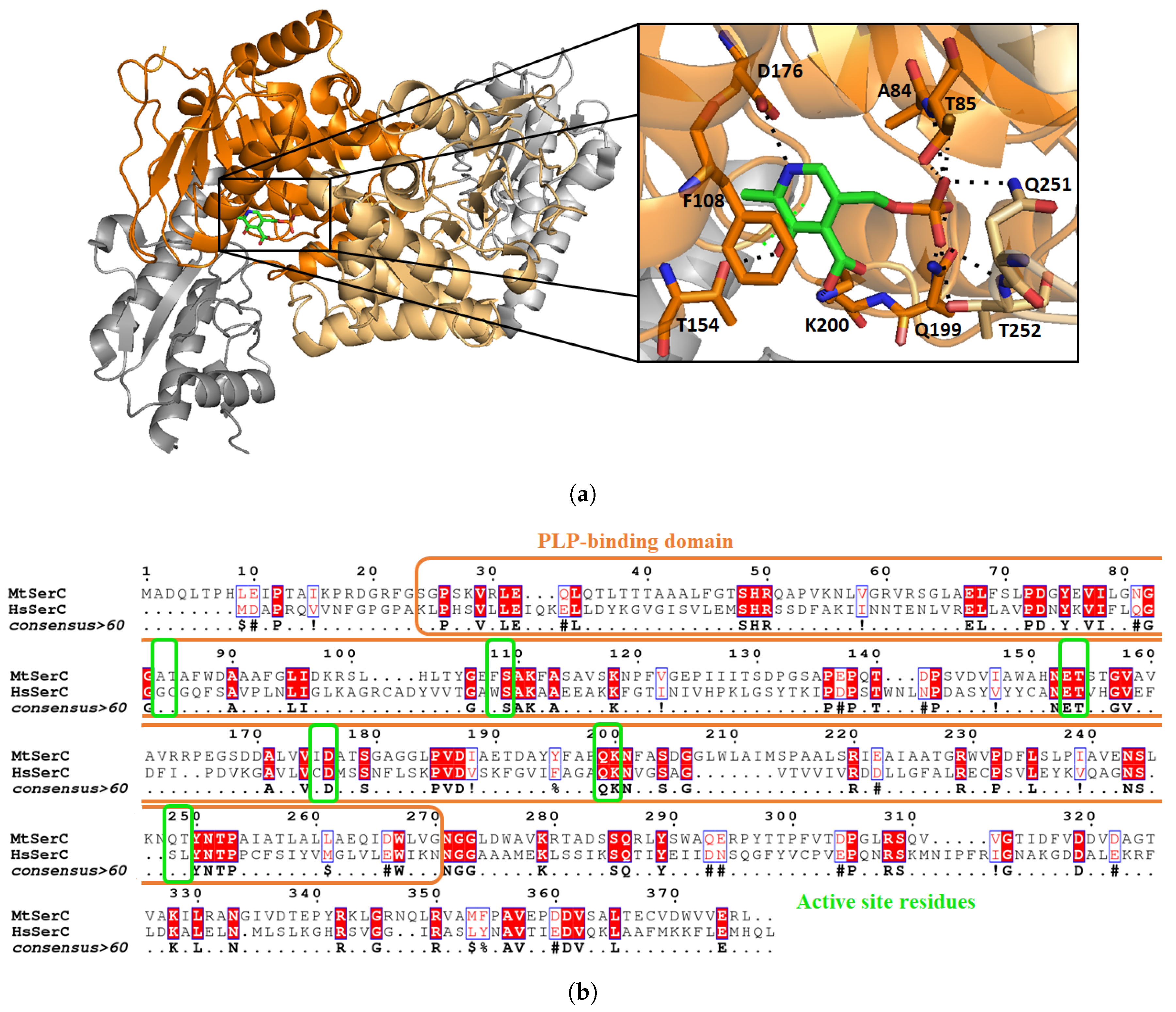
3.2. Mycobacterium tuberculosis Phosphoserine Aminotransferase SerC
3.3. Mycobacterium tuberculosis Phosphoserine Phosphatase SerB2: A Promising Therapeutic Target?
Inhibitors of Phosphatases
4. Summary
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- O’Neil, J. Tackling a Crisis for the Health and Wealth of Nations. 2014. Available online: https://amr-review.org/sites/default/files/AMRReviewPaper-Tacklingacrisisforthehealthandwealthofnations_1.pdf (accessed on 25 June 2018).
- Donadio, S.; Maffioli, S.; Monciardini, P.; Sosio, M.; Jabes, D. Antibiotic discovery in the twenty-first century: Current trends and future perspectives. J. Antibiot. 2010, 63, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention, Office of Infectious Disease. Antibiotic Resistance Threats in the United States. Available online: http://www.cdc.gov/drugresistance/threat-report-2013 (accessed on 25 October 2017).
- Masi, M.; Réfregiers, M.; Pos, K.M.; Pagès, J.M. Mechanisms of envelope permeability and antibiotic influx and efflux in Gram-negative bacteria. Nat. Microbiol. 2017, 2, 17001. [Google Scholar] [CrossRef]
- Silver, L.; Bostian, K. Discovery and development of new antibiotics: The problem of antibiotic resistance. Antimicrob. Agents Chemother. 1993, 37, 377. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.A.; Walsh, C.T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Dorman, S.E.; Chaisson, R.E. From magic bullets back to the magic mountain: The rise of extensively drug-resistant tuberculosis. Nat. Med. 2007, 13, 295–298. [Google Scholar] [CrossRef]
- Gillespie, S.H. Evolution of drug resistance in Mycobacterium tuberculosis: Clinical and molecular perspective. Antimicrob. Agents Chemother. 2002, 46, 267–274. [Google Scholar] [CrossRef]
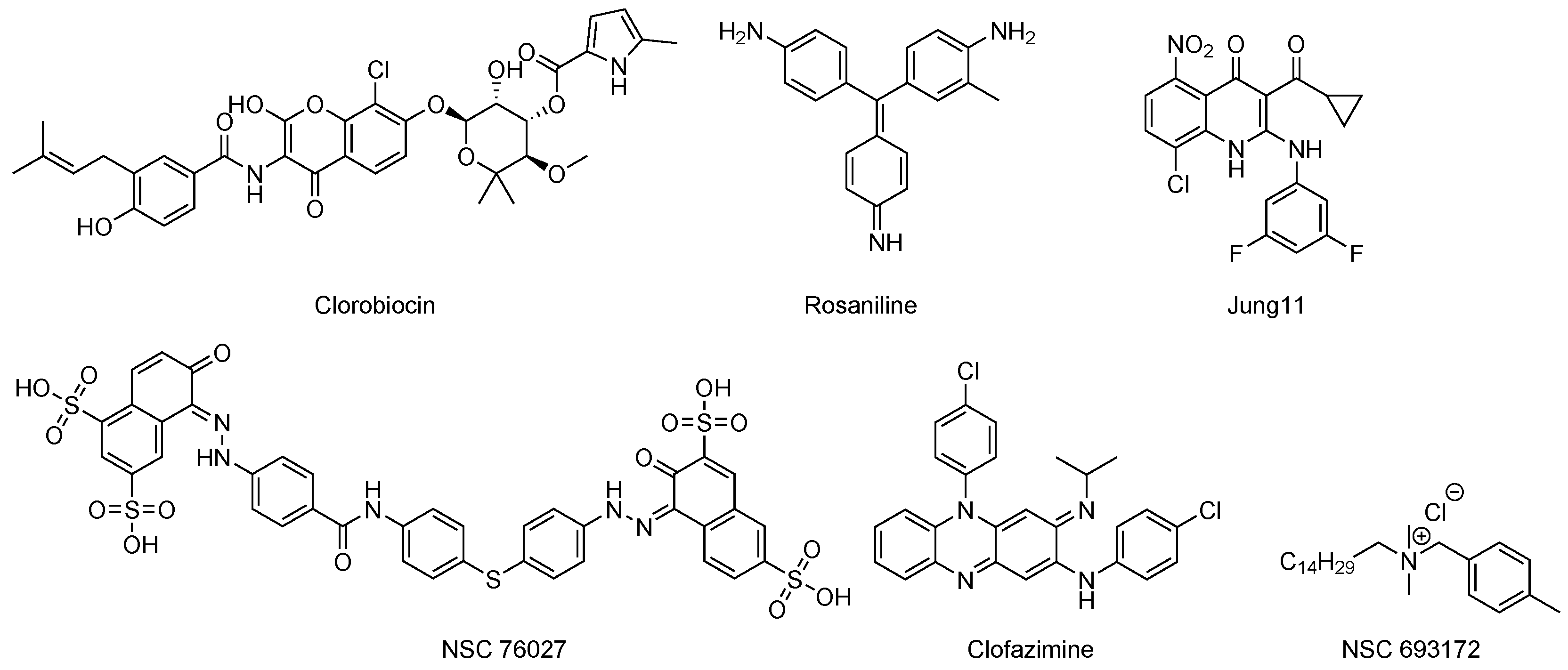
- Shree, S.; Singh, A.K.; Saxena, R.; Kumar, H.; Agarwal, A.; Sharma, V.K.; Srivastava, K.; Srivastava, K.K.; Sanyal, S.; Ramachandran, R. The M. tuberculosis HAD phosphatase (Rv3042c) interacts with host proteins and is inhibited by Clofazimine. Cell. Mol. Life Sci. 2016, 37, 3401–3417. [Google Scholar] [CrossRef]
- Koul, A.; Arnoult, E.; Lounis, N.; Guillemont, J.; Andries, K. The challenge of new drug discovery for tuberculosis. Nature 2011, 469, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Millard, J.; Ugarte-Gil, C.; Moore, D. Multidrug resistant tuberculosis. BMJ 2015, 350, h882. [Google Scholar] [CrossRef]
- World Health Organization. World Health Statistics 2016: Monitoring Health for the SDGs. Available online: http://www.who.int/ (accessed on 25 September 2017).
- World Health Organization. Global Tuberculosis Report 2015. Available online: http://www.who.int/ (accessed on 25 September 2017).
- The Global Alliance for TB Drug Development. Available online: https://www.tballiance.org/ (accessed on 25 November 2017).
- Butler, D. New fronts in an old war. Nature 2000, 406, 670–672. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Suresh, M.R. An overview of tuberculosis chemotherapy—A literature review. J. Pharm. Pharm. Sci. 2011, 14, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Cambier, C.; Falkow, S.; Ramakrishnan, L. Host evasion and exploitation schemes of Mycobacterium tuberculosis. Cell 2014, 159, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Treatment of Tuberculosis: Guidelines, 2010. Available online: http://www.who.int/ (accessed on 25 September 2017).
- Falzon, D.; Schünemann, H.J.; Harausz, E.; González-Angulo, L.; Lienhardt, C.; Jaramillo, E.; Weyer, K. World Health Organization treatment guidelines for drug-resistant tuberculosis, 2016 update. Eur. Respir. J. 2017, 49, 1602308. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report 2017. Available online: http://www.who.int/ (accessed on 25 September 2017).
- Dutt, A.K.; Stead, W.W. Present chemotherapy for tuberculosis. J. Infect. Dis. 1982, 146, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Pablos-Méndez, A.; Raviglione, M.C.; Laszlo, A.; Binkin, N.; Rieder, H.L.; Bustreo, F.; Cohn, D.L.; Lambregts-van Weezenbeek, C.S.; Kim, S.J.; Chaulet, P.; et al. Global surveillance for antituberculosis-drug resistance, 1994–1997. N. Engl. J. Med. 1998, 338, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. The magic bullets and tuberculosis drug targets. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 529–564. [Google Scholar] [CrossRef] [PubMed]
- National Institutes of Health. Available online: https://clinicaltrials.gov/ (accessed on 25 November 2017).
- Hards, K.; Robson, J.R.; Berney, M.; Shaw, L.; Bald, D.; Koul, A.; Andries, K.; Cook, G.M. Bactericidal mode of action of bedaquiline. J. Antimicrob. Chemother. 2015, 70, 2028–2037. [Google Scholar] [CrossRef]
- Cox, E.; Laessig, K. FDA approval of bedaquiline—The benefit–risk balance for drug-resistant tuberculosis. N. Engl. J. Med. 2014, 371, 689–691. [Google Scholar] [CrossRef]
- Aung, H.L.; Nyunt, W.W.; Fong, Y.; Cook, G.M.; Aung, S.T. First 2 Extensively Drug-Resistant Tuberculosis Cases From Myanmar Treated with Bedaquiline. Clin. Infect. Dis. 2017, 65, 531–532. [Google Scholar] [CrossRef]
- Blair, H.A.; Scott, L.J. Delamanid: A review of its use in patients with multidrug-resistant tuberculosis. Drugs 2015, 75, 91–100. [Google Scholar] [CrossRef]
- Xavier, A.S.; Lakshmanan, M. Delamanid: A new armor in combating drug-resistant tuberculosis. J. Pharmacol. Pharmacother. 2014, 5, 222. [Google Scholar] [CrossRef] [PubMed]
- Hartkoorn, R.C.; Uplekar, S.; Cole, S.T. Cross-resistance between clofazimine and bedaquiline through upregulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 2979–2981. [Google Scholar]
- Hoffmann, H.; Kohl, T.A.; Hofmann-Thiel, S.; Merker, M.; Beckert, P.; Jaton, K.; Nedialkova, L.; Sahalchyk, E.; Rothe, T.; Keller, P.M.; et al. Delamanid and bedaquiline resistance in Mycobacterium tuberculosis ancestral Beijing genotype causing extensively drug-resistant tuberculosis in a Tibetan refugee. Am. J. Respir. Crit. Care Med. 2016, 193, 337–340. [Google Scholar] [CrossRef]
- Manjunatha, U.; Boshoff, H.I.; Barry, C.E. The mechanism of action of PA-824: Novel insights from transcriptional profiling. Commun. Integr. Biol. 2009, 2, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Macielag, M.J. Chemical Properties of Antimicrobials and Their Uniqueness. In Antibiotic Discovery and Development; Dougherty, T.J., Pucci, M.J., Eds.; Springer: Boston, MA, USA, 2012; pp. 793–820. [Google Scholar] [CrossRef]
- Cole, S.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.; Eiglmeier, K.; Gas, S.; Barry, C.R.; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [Google Scholar] [CrossRef]
- El-Hattab, A.W. Serine biosynthesis and transport defects. Mol. Genet. Metabol. 2016, 118, 153–159. [Google Scholar] [CrossRef]
- McCoy, T.A.; Maxwell, M.; Neuman, R.E. The amino acid requirements of the Walker carcinosarcoma 256 in vitro. Cancer Res. 1956, 16, 979–984. [Google Scholar]
- Eagle, H. Amino acid metabolism in mammalian cell cultures. Science 1959, 130, 432–437. [Google Scholar] [CrossRef] [PubMed]
- De Koning, T.J.; Snell, K.; Duran, M.; Berger, R.; Surtees, R. L-serine in disease and development. Biochem. J. 2003, 371, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Lowry, M.; Hall, D.E.; Hall, M.S.; Brosnan, J.T. Renal metabolism of amino acids in vivo: Studies on serine and glycine fluxes. Am. J. Physiol.-Ren. Physiol. 1987, 252, F304–F309. [Google Scholar] [CrossRef] [PubMed]
- Snell, K. The duality of pathways for serine biosynthesis is a fallacy. Trends Biochem. Sci. 1986, 11, 241–243. [Google Scholar] [CrossRef]
- Snell, K. Enzymes of serine metabolism in normal, developing and neoplastic rat tissues. Adv. Enzyme Regul. 1984, 22, 325–400. [Google Scholar] [CrossRef]
- Snell, K.; Natsumeda, Y.; Eble, J.; Glover, J.; Weber, G. Enzymic imbalance in serine metabolism in human colon carcinoma and rat sarcoma. Br. J. Cancer 1988, 57, 87–90. [Google Scholar] [CrossRef]
- Ros, R.; Muñoz-Bertomeu, J.; Krueger, S. Serine in plants: Biosynthesis, metabolism, and functions. Trends Plant Sci. 2014, 19, 564–569. [Google Scholar] [CrossRef]
- Umbarger, H.E.; Umbarger, M.A. The biosynthetic pathway of serine in Salmonella typhimurium. Biochim. Biophys. Acta 1962, 62, 193–195. [Google Scholar] [CrossRef]
- Bai, G.; Schaak, D.D.; Smith, E.A.; McDonough, K.A. Dysregulation of serine biosynthesis contributes to the growth defect of a Mycobacterium tuberculosis crp mutant. Mol. Microbiol. 2011, 82, 180–198. [Google Scholar] [CrossRef]
- Subramanyam, V.; Pal, B.; Mohanty, K.K. Inducibility and stability of auxotrophic mutations in Mycobacterium fortuitum, M. smegmatis and M. vaccae. Lett. Appl. Microbiol. 1989, 8, 161–164. [Google Scholar] [CrossRef]
- Kalpana, G.V.; Bloom, B.R.; Jacobs, W.R. Insertional mutagenesis and illegitimate recombination in mycobacteria. Proc. Natl. Acad. Sci. USA 1991, 88, 5433–5437. [Google Scholar] [CrossRef]
- Hinds, J.; Mahenthiralingam, E.; Kempsell, K.E.; Duncan, K.; Stokes, R.W.; Parish, T.; Stoker, N.G. Enhanced gene replacement in mycobacteria. Microbiology 1999, 145, 519–527. [Google Scholar] [CrossRef]
- Parish, T.; Stoker, N.G. Electroporation of mycobacteria. In Mycobacteria Protocols; Humana Press: New York, NY, USA, 1998; pp. 129–144. [Google Scholar]
- Bardarov, S.; Bardarov, S., Jr.; Pavelka, M.S., Jr.; Sambandamurthy, V.; Larsen, M.; Tufariello, J.; Chan, J.; Hatfull, G.; Jacobs, W.R., Jr. Specialized transduction: An efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 2002, 148, 3007–3017. [Google Scholar] [CrossRef]
- Tufariello, J.M.; Malek, A.A.; Vilchèze, C.; Cole, L.E.; Ratner, H.K.; González, P.A.; Jain, P.; Hatfull, G.F.; Larsen, M.H.; Jacobs, W.R. Enhanced specialized transduction using recombineering in Mycobacterium tuberculosis. mBio 2014, 5, e01179-14. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Hu, Z.; Xu, X.L.; Sacchettini, J.C.; Grant, G.A. D-3-Phosphoglycerate dehydrogenase from Mycobacterium tuberculosis is a link between the Escherichia coli and mammalian enzymes. J. Biol. Chem. 2005, 280, 14884–14891. [Google Scholar] [CrossRef] [PubMed]
- Coulibaly, F.; Lassalle, E.; Baker, H.M.; Baker, E.N. Structure of phosphoserine aminotransferase from Mycobacterium tuberculosis. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Arora, G.; Tiwari, P.; Mandal, R.S.; Gupta, A.; Sharma, D.; Saha, S.; Singh, R. High throughput screen identifies small molecule inhibitors specific for Mycobacterium tuberculosis phosphoserine phosphatase. J. Biol. Chem. 2014, 289, 25149–25165. [Google Scholar] [CrossRef] [PubMed]
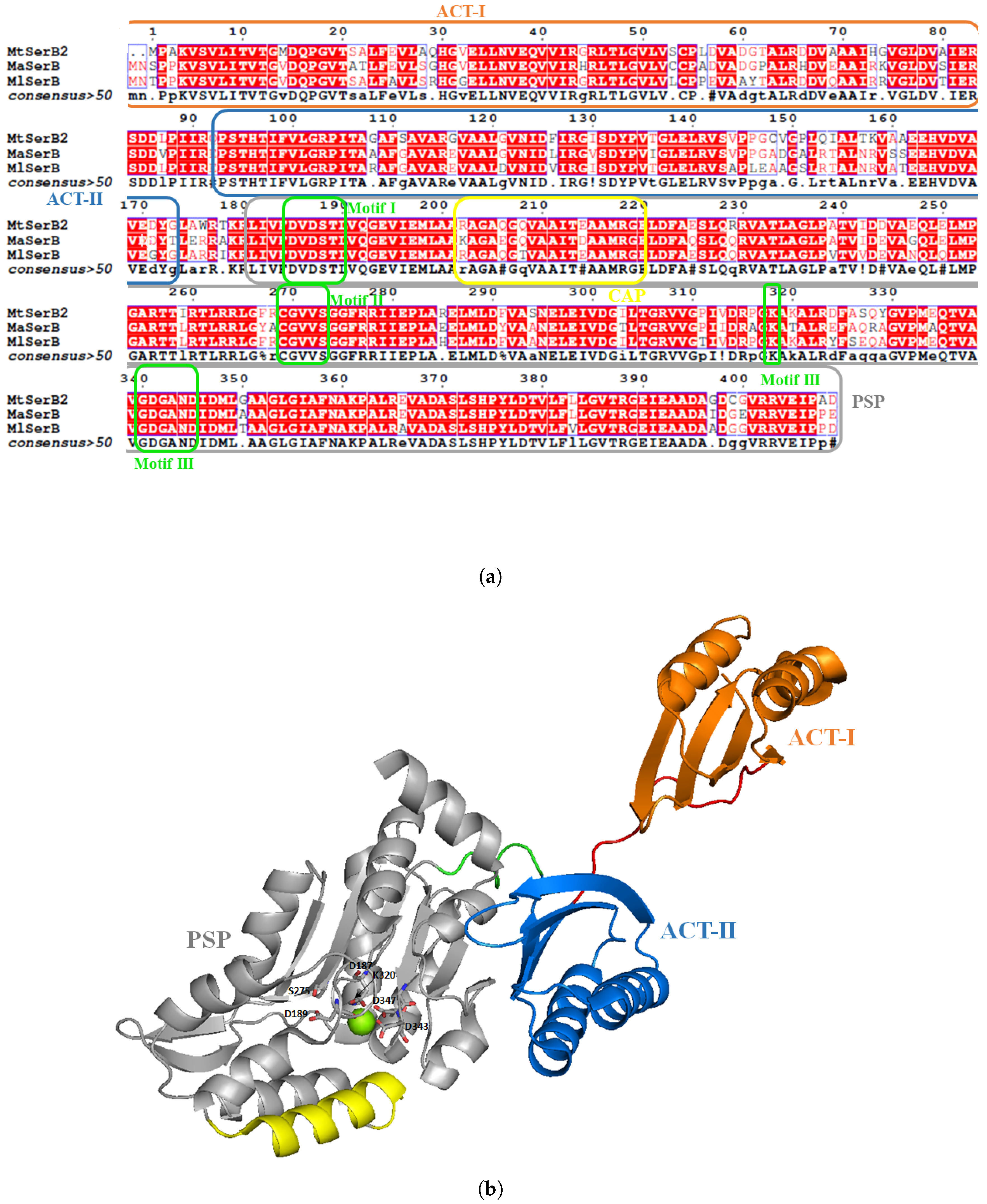
- Yadav, G.P.; Shree, S.; Maurya, R.; Rai, N.; Singh, D.K.; Srivastava, K.K.; Ramachandran, R. Characterization of M. tuberculosis SerB2, an essential HAD-family phosphatase, reveals novel properties. PLoS ONE 2014, 9, e115409. [Google Scholar] [CrossRef]
- El-Sharoud, W.; Delorme, C.; Darwish, M.; Renault, P. Genotyping of Streptococcus thermophilus strains isolated from traditional Egyptian dairy products by sequence analysis of the phosphoserine phosphatase (serB) gene with phenotypic characterizations of the strains. J. Appl. Microbiol. 2012, 112, 329–337. [Google Scholar] [CrossRef]
- Garnant, M.; Stauffer, G. Construction and analysis of plasmids containing the Escherichia coli serB gene. Mol. Gen. Genet. MGG 1984, 193, 72–75. [Google Scholar] [CrossRef]
- Tobey, K.L.; Grant, G.A. The nucleotide sequence of the serA gene of Escherichia coli and the amino acid sequence of the encoded protein, D-3-phosphoglycerate dehydrogenase. J. Biol. Chem. 1986, 261, 12179–12183. [Google Scholar]
- Acuna-Hidalgo, R.; Schanze, D.; Kariminejad, A.; Nordgren, A.; Kariminejad, M.H.; Conner, P.; Grigelioniene, G.; Nilsson, D.; Nordenskjöld, M.; Wedell, A.; et al. Neu-Laxova syndrome is a heterogeneous metabolic disorder caused by defects in enzymes of the L-serine biosynthesis pathway. Am. J. Hum. Genet. 2014, 95, 285–293. [Google Scholar] [CrossRef]
- Grant, G.A. Contrasting catalytic and allosteric mechanisms for phosphoglycerate dehydrogenases. Arch. Biochem. Biophys. 2012, 519, 175–185. [Google Scholar] [CrossRef]
- Grant, G.A. A new family of 2-hydroxyacid dehydrogenases. Biochem. Biophys. Res. Commun. 1989, 165, 1371–1374. [Google Scholar] [CrossRef]
- Dey, S.; Grant, G.A.; Sacchettini, J.C. Crystal Structure of Mycobacterium tuberculosis D-3-Phosphoglycerate Dehydrogenase EXTREME ASYMMETRY IN A TETRAMER OF IDENTICAL SUBUNITS. J. Biol. Chem. 2005, 280, 14892–14899. [Google Scholar] [CrossRef]
- Dey, S.; Burton, R.L.; Grant, G.A.; Sacchettini, J.C. Structural analysis of substrate and effector binding in Mycobacterium tuberculosis D-3-phosphoglycerate dehydrogenase. Biochemistry 2008, 47, 8271–8282. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Burton, R.L.; Chen, S.; Xu, X.L.; Grant, G.A. Role of the anion-binding site in catalysis and regulation of Mycobacterium tuberculosis D-3-phosphoglycerate dehydrogenase. Biochemistry 2009, 48, 4808–4815. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Burton, R.L.; Chen, S.; Xu, X.L.; Grant, G.A. A novel mechanism for substrate inhibition in Mycobacterium tuberculosis D-3-phosphoglycerate dehydrogenase. J. Biol. Chem. 2007, 282, 31517–31524. [Google Scholar]
- Ravez, S.; Spillier, Q.; Marteau, R.; Feron, O.; Frédérick, R. Challenges and Opportunities in the Development of Serine Synthetic Pathway Inhibitors for Cancer Therapy: Miniperspective. J. Med. Chem. 2016, 60, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Fuller, N.; Spadola, L.; Cowen, S.; Patel, J.; Schönherr, H.; Cao, Q.; McKenzie, A.; Edfeldt, F.; Rabow, A.; Goodnow, R. An improved model for fragment-based lead generation at AstraZeneca. Drug Discov. Today 2016, 21, 1272–1283. [Google Scholar] [PubMed]
- Fuller, N. Fragment-based discovery of the first known inhibitors of PHGDH. Am. Chem. Soc. 2015, 250, MEDI-366. [Google Scholar]
- Mullarky, E.; Lucki, N.C.; Zavareh, R.B.; Anglin, J.L.; Gomes, A.P.; Nicolay, B.N.; Wong, J.C.; Christen, S.; Takahashi, H.; Singh, P.K.; et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc. Natl. Acad. Sci. USA 2016, 113, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Pacold, M.E.; Brimacombe, K.R.; Chan, S.H.; Rohde, J.M.; Lewis, C.A.; Swier, L.J.; Possemato, R.; Chen, W.W.; Sullivan, L.B.; Fiske, B.P.; et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol. 2016, 12, 452. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liberti, M.V.; Liu, P.; Deng, X.; Liu, Y.; Locasale, J.W.; Lai, L. Rational design of selective allosteric inhibitors of PHGDH and serine synthesis with anti-tumor activity. Cell Chem. Biol. 2017, 24, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Ravez, S.; Corbet, C.; Spillier, Q.; Dutu, A.; Robin, A.D.; Mullarky, E.; Cantley, L.C.; Feron, O.; Frédérick, R. α-Ketothioamide derivatives: A promising tool to interrogate phosphoglycerate dehydrogenase (PHGDH). J. Med. Chem. 2017, 60, 1591–1597. [Google Scholar] [CrossRef]
- Kennelly, P.J. Protein kinases and protein phosphatases in prokaryotes: A genomic perspective. FEMS Microbiol. Lett. 2002, 206, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Barford, D. The effects of phosphorylation on the structure and function of proteins. Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 199–232. [Google Scholar] [CrossRef] [PubMed]
- Koul, A.; Choidas, A.; Treder, M.; Tyagi, A.K.; Drlica, K.; Singh, Y.; Ullrich, A. Cloning and characterization of secretory tyrosine phosphatases of Mycobacterium tuberculosis. J. Bacteriol. 2000, 182, 5425–5432. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.; Chao, J.D.; Av-Gay, Y. Mycobacterium tuberculosis-secreted phosphatases: From pathogenesis to targets for TB drug development. Trends Microbiol. 2013, 21, 100–109. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, A.; Sim, A.T.; Sakoff, J.A. Serine- threonine protein phosphatase inhibitors: Development of potential therapeutic strategies. J. Med. Chem. 2002, 45, 1151–1175. [Google Scholar] [CrossRef] [PubMed]
- Pieters, J. Mycobacterium tuberculosis and the macrophage: Maintaining a balance. Cell Host Microbe 2008, 3, 399–407. [Google Scholar] [CrossRef]
- Beresford, N.J.; Mulhearn, D.; Szczepankiewicz, B.; Liu, G.; Johnson, M.E.; Fordham-Skelton, A.; Abad-Zapatero, C.; Cavet, J.S.; Tabernero, L. Inhibition of MptpB phosphatase from Mycobacterium tuberculosis impairs mycobacterial survival in macrophages. J. Antimicrob. Chemother. 2009, 63, 928–936. [Google Scholar] [CrossRef]
- Wong, D.; Bach, H.; Sun, J.; Hmama, Z.; Av-Gay, Y. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+–ATPase to inhibit phagosome acidification. Proc. Natl. Acad. Sci. USA 2011, 108, 19371–19376. [Google Scholar] [CrossRef] [PubMed]
- Hmama, Z.; Peña-Díaz, S.; Joseph, S.; Av-Gay, Y. Immunoevasion and immunosuppression of the macrophage by Mycobacterium tuberculosis. Immunol. Rev. 2015, 264, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Anishetty, S.; Pulimi, M.; Pennathur, G. Potential drug targets in Mycobacterium tuberculosis through metabolic pathway analysis. Comput. Biol. Chem. 2005, 29, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Tribble, G.D.; Mao, S.; James, C.E.; Lamont, R.J. A Porphyromonas gingivalis haloacid dehalogenase family phosphatase interacts with human phosphoproteins and is important for invasion. Proc. Natl. Acad. Sci. USA 2006, 103, 11027–11032. [Google Scholar] [CrossRef]
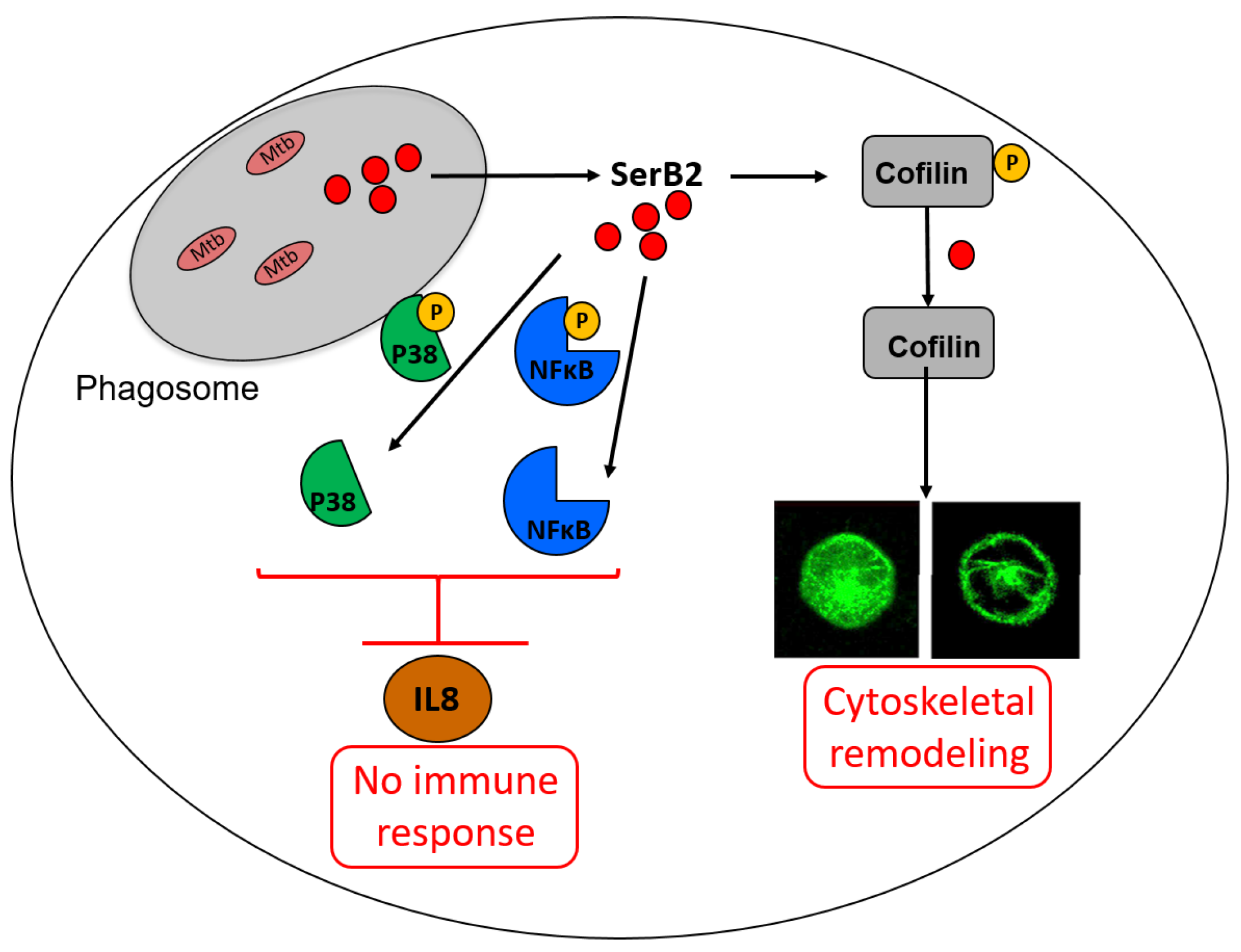
- Hasegawa, Y.; Tribble, G.D.; Baker, H.V.; Mans, J.J.; Handfield, M.; Lamont, R.J. Role of Porphyromonas gingivalis SerB in gingival epithelial cell cytoskeletal remodeling and cytokine production. Infect. Immun. 2008, 76, 2420–2427. [Google Scholar] [CrossRef]
- Bainbridge, B.; Verma, R.K.; Eastman, C.; Yehia, B.; Rivera, M.; Moffatt, C.; Bhattacharyya, I.; Lamont, R.J.; Kesavalu, L. Role of Porphyromonas gingivalis phosphoserine phosphatase enzyme SerB in inflammation, immune response, and induction of alveolar bone resorption in rats. Infect. Immun. 2010, 78, 4560–4569. [Google Scholar] [CrossRef]
- Moffatt, C.E.; Inaba, H.; Hirano, T.; Lamont, R.J. Porphyromonas gingivalis SerB-mediated dephosphorylation of host cell cofilin modulates invasion efficiency. Cell. Microbiol. 2012, 14, 577–588. [Google Scholar] [CrossRef][Green Version]
- Takeuchi, H.; Hirano, T.; Whitmore, S.E.; Morisaki, I.; Amano, A.; Lamont, R.J. The serine phosphatase SerB of Porphyromonas gingivalis suppresses IL-8 production by dephosphorylation of NF-κB RelA/p65. PLoS Pathog. 2013, 9, e1003326. [Google Scholar] [CrossRef]
- Olsen, I.; Hajishengallis, G. Major neutrophil functions subverted by Porphyromonas gingivalis. J. Oral Microbiol. 2016, 8, 30936. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Dhasmana, N.; Dubey, N.; Kumar, N.; Gangwal, A.; Gupta, M.; Singh, Y. Bacterial Virulence Factors: Secreted for Survival. Indian J. Microbiol. 2016, 57, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Dunaway-Mariano, D.; Allen, K.N. HAD superfamily phosphotransferase substrate diversification: structure and function analysis of HAD subclass IIB sugar phosphatase BT4131. Biochemistry 2005, 44, 8684–8696. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.N.; Dunaway-Mariano, D. Markers of fitness in a successful enzyme superfamily. Curr. Opin. Struct. Biol. 2009, 19, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.F.; Stroobant, V.; Pirard, M.; Delpierre, G.; Van Schaftingen, E. A new class of phosphotransferases phosphorylated on an aspartate residue in an amino-terminal DXDX (T/V) motif. J. Biol. Chem. 1998, 273, 14107–14112. [Google Scholar] [CrossRef]
- Aravind, L.; Galperin, M.Y.; Koonin, E.V. The catalytic domain of the P-type ATPase has the haloacid dehalogenase fold. Trends Biochem. Sci. 1998, 23, 127–129. [Google Scholar] [CrossRef]
- Aravind, L.; Koonin, E.V. The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. Sci. 1998, 23, 469–472. [Google Scholar] [CrossRef]
- Koonin, E.V.; Tatusov, R.L. Computer analysis of bacterial haloacid dehalogenases defines a large superfamily of hydrolases with diverse specificity: Application of an iterative approach to database search. J. Mol. Biol. 1994, 244, 125–132. [Google Scholar] [CrossRef]
- Seifried, A.; Schultz, J.; Gohla, A. Human HAD phosphatases: Structure, mechanism, and roles in health and disease. FEBS J. 2013, 280, 549–571. [Google Scholar] [CrossRef]
- Shi, Y. Serine/threonine phosphatases: Mechanism through structure. Cell 2009, 139, 468–484. [Google Scholar] [CrossRef]
- Glasner, M.E.; Gerlt, J.A.; Babbitt, P.C. Evolution of enzyme superfamilies. Curr. Opin. Chem. Biol. 2006, 10, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Abendroth, J.; Gardberg, A.S.; Robinson, J.I.; Christensen, J.S.; Staker, B.L.; Myler, P.J.; Stewart, L.J.; Edwards, T.E. SAD phasing using iodide ions in a high-throughput structural genomics environment. J. Struct. Funct. Genom. 2011, 12, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Bitto, E.; Kim, D.J.; Bingman, C.A.; Kim, H.J.; Han, B.W.; Phillips, G.N. Crystal structure of tandem ACT domain-containing protein ACTP from Galdieria sulphuraria. Proteins Struct. Funct. Bioinform. 2012, 80, 2105–2109. [Google Scholar] [CrossRef] [PubMed]
- Grant, G.A. Regulatory Mechanism of Mycobacterium tuberculosis Phosphoserine Phosphatase SerB2. Biochemistry 2017, 56, 6481–6490. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.K.; Ko, Y.; Yu, K.R.; Kim, J.H.; Lee, J.Y.; Chae, C.H.; Ji, S.; Kim, C.H.; Lee, H.K.; Choi, E.B.; et al. Identification of 3-acyl-2-phenylamino-1, 4-dihydroquinolin-4-one derivatives as inhibitors of the phosphatase SerB653 in Porphyromonas gingivalis, implicated in periodontitis. Bioorg. Med. Chem. Lett. 2012, 22, 2084–2088. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.C.; Sharma, S. Molecular modeling studies of 3-acyl-2-phenylamino-1, 4-dihydroquinolin-4-one derivatives as phosphatase SerB653 inhibitors. Med. Chem. Res. 2016, 25, 2119–2126. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, N.; Shum, D.; Huddar, S.; Park, C.M.; Jang, S. Identification of Antipneumococcal Molecules Effective Against Different Streptococcus pneumoniae Serotypes Using a Resazurin-Based High-Throughput Screen. ASSAY Drug Dev. Technol. 2017, 15, 198–209. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haufroid, M.; Wouters, J. Targeting the Serine Pathway: A Promising Approach against Tuberculosis? Pharmaceuticals 2019, 12, 66. https://doi.org/10.3390/ph12020066
Haufroid M, Wouters J. Targeting the Serine Pathway: A Promising Approach against Tuberculosis? Pharmaceuticals. 2019; 12(2):66. https://doi.org/10.3390/ph12020066
Chicago/Turabian StyleHaufroid, Marie, and Johan Wouters. 2019. "Targeting the Serine Pathway: A Promising Approach against Tuberculosis?" Pharmaceuticals 12, no. 2: 66. https://doi.org/10.3390/ph12020066
APA StyleHaufroid, M., & Wouters, J. (2019). Targeting the Serine Pathway: A Promising Approach against Tuberculosis? Pharmaceuticals, 12(2), 66. https://doi.org/10.3390/ph12020066