Juvenile Arthritis Patients Suffering from Chronic Inflammation Have Increased Activity of Both IDO and GTP-CH1 Pathways But Decreased BH4 Efficacy: Implications for Well-Being, Including Fatigue, Cognitive Impairment, Anxiety, and Depression


Abstract
1. Introduction
2. Results
2.1. Disease Activity and MTX Treatment
2.2. GTP–CH1 Activity
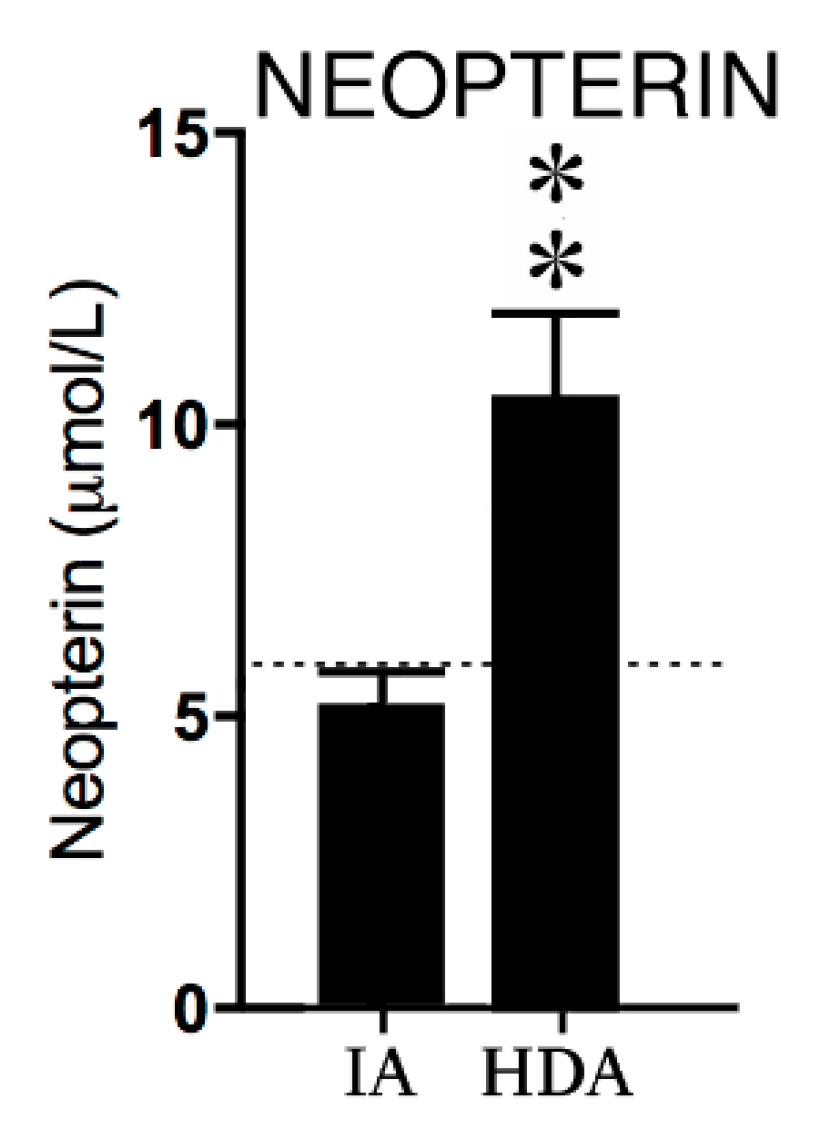
2.2.1. Neopterin
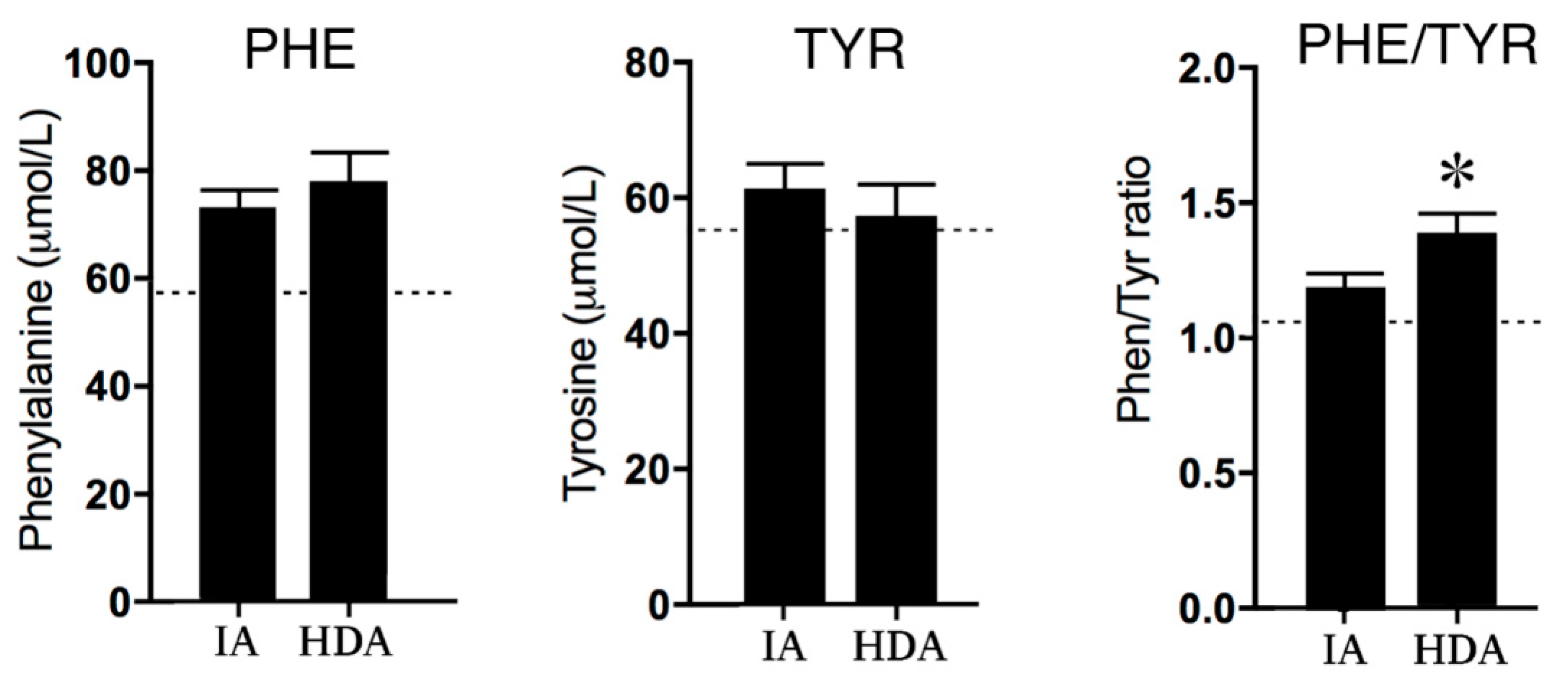
2.2.2. Phenylalanine and Tyrosine
2.3. IDO Activity
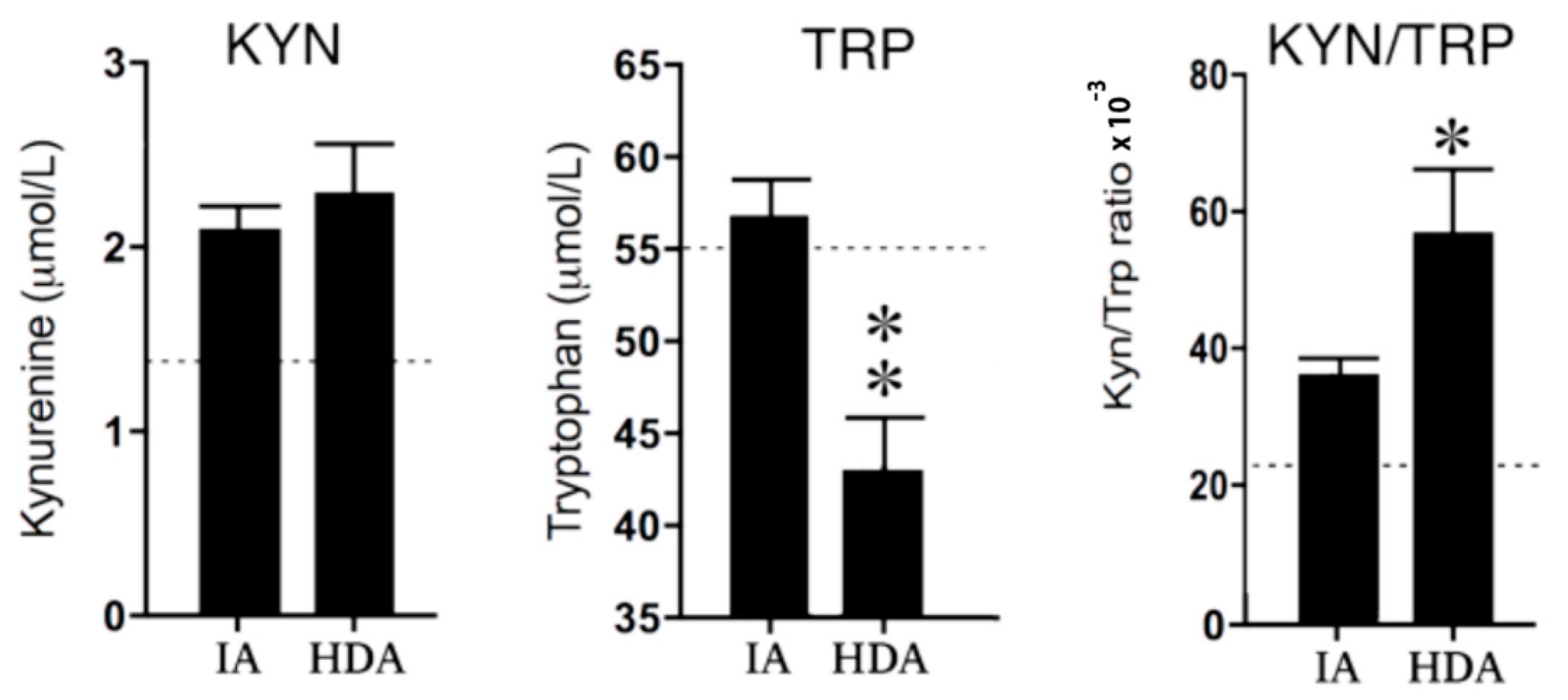
2.3.1. Kynurenine and Tryptophan
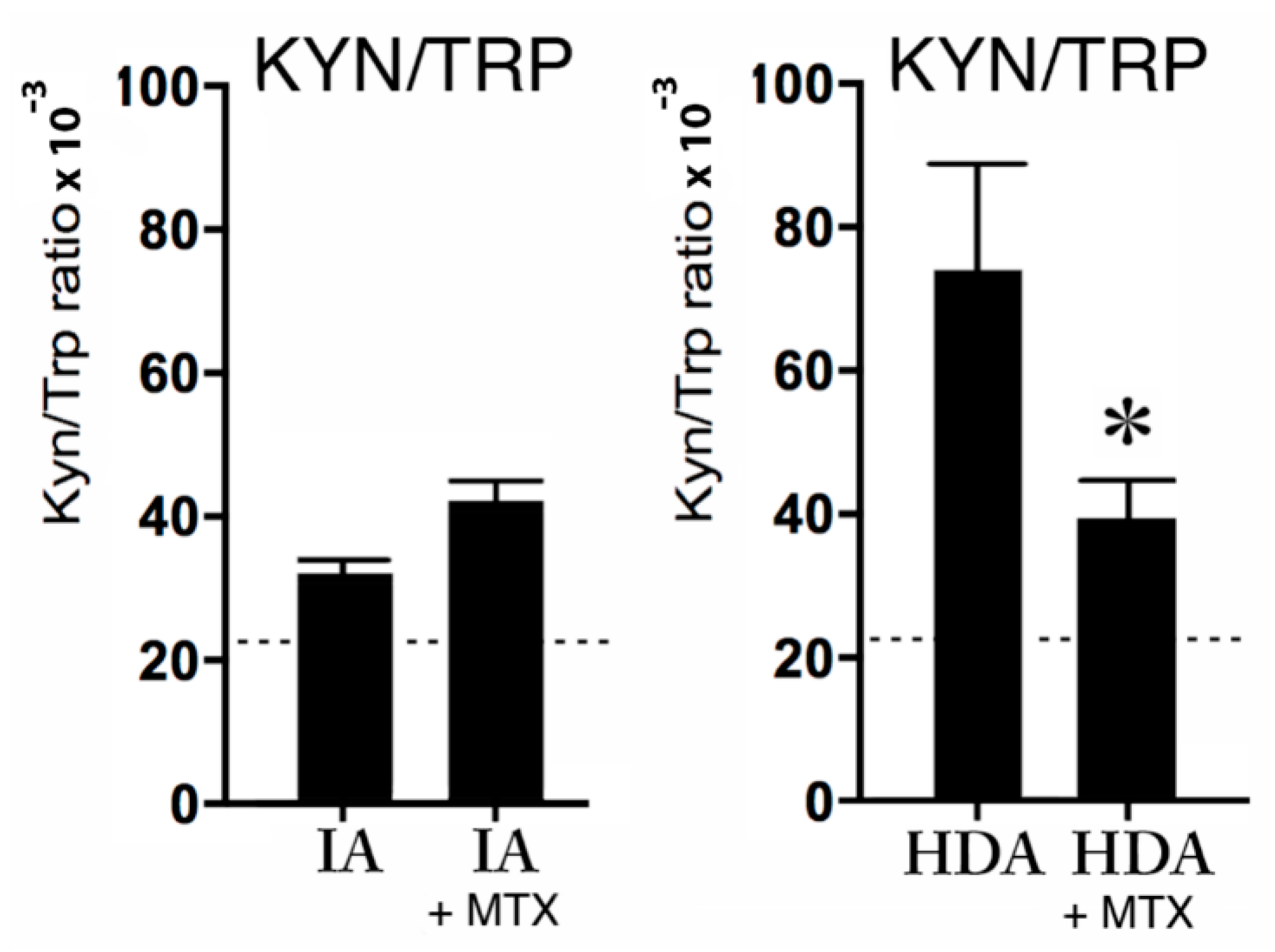
2.3.2. Kynurenine, Tryptophan, and MTX Treatment
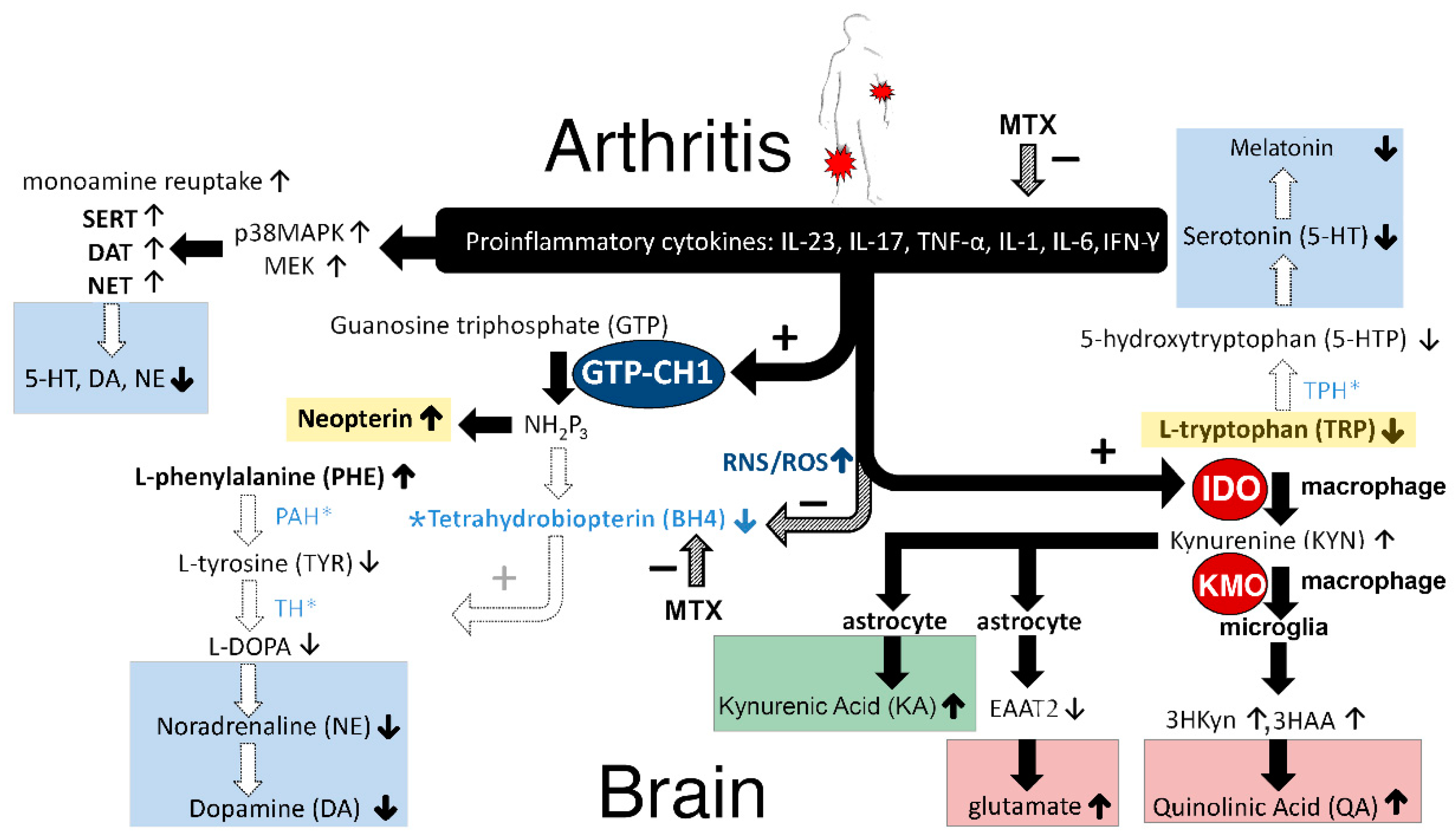
3. Discussion
3.1. GTP–CH1 Pathway
3.1.1. Neopterin
3.1.2. Tetrahydrobiopterin (BH4)
3.2. IDO Pathway
3.2.1. Tryptophan, Serotonin, and Melatonin
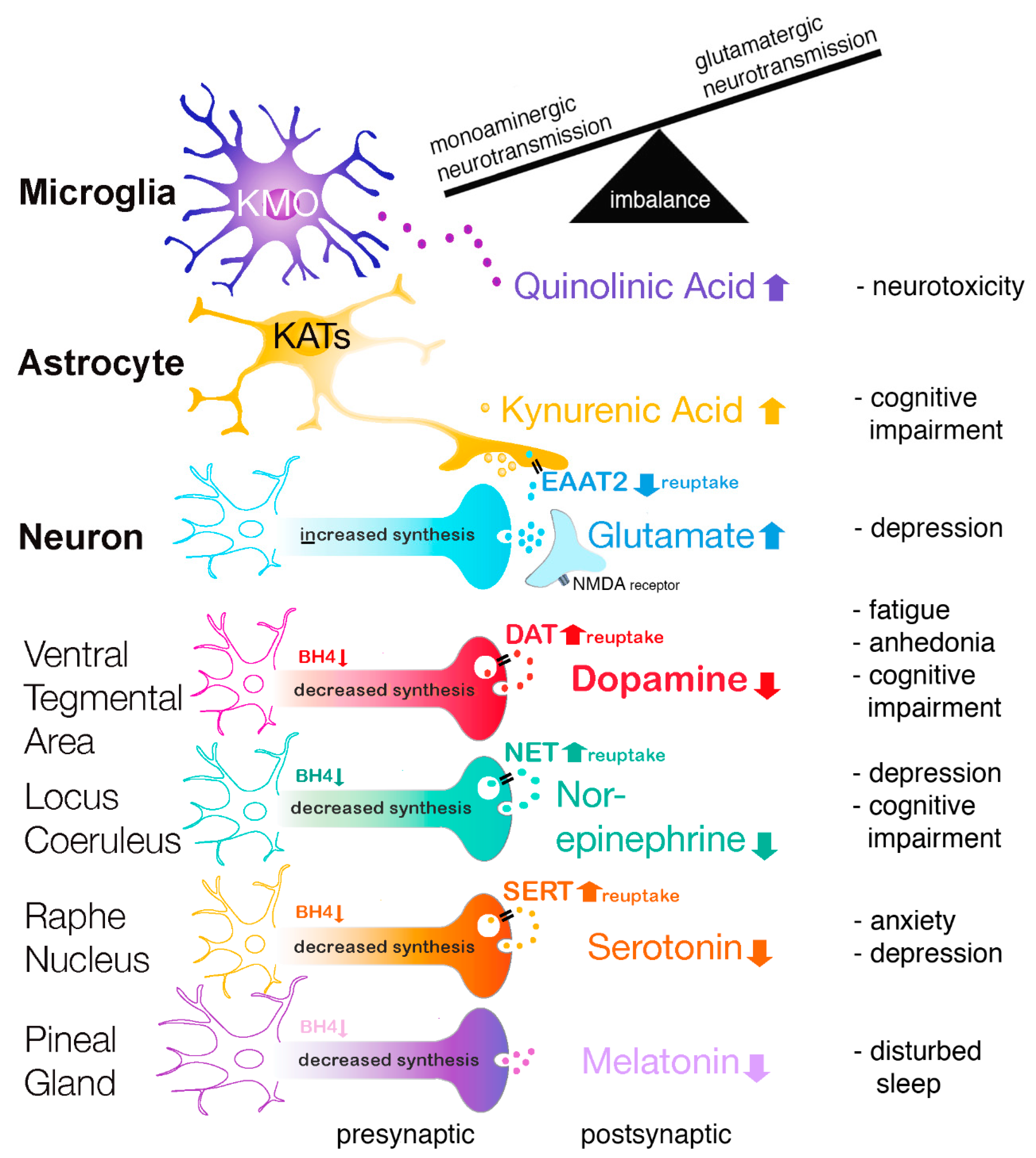
3.2.2. Kynurenine, Quinolinic Acid, and Glutamate
3.3. Increased Monoamine Reuptake
3.4. Pharma-Food Interventions
3.4.1. Inflammation and Immunosuppressants
3.4.2. Monoamines, Transporters, and Monoamine Reuptake Inhibitors
3.4.3. Monoamines, BH4, and Nutritional Interventions
3.4.4. Glutamate and Nutritional Interventions
4. Materials and Methods
4.1. Study Population
4.2. Blood Samples
4.3. Statistics
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ravelli, A.; Martini, A. Juvenile idiopathic arthritis. Lancet 2007, 369, 767–778. [Google Scholar] [CrossRef]
- Laxer, R.M.; Sherry, D.D.; Hashkes, P.J. Juvenile Idiopathic Arthritis (JIA). In Pediatric Rheumatology in Clinical Practice; Springer: Cham, Switzerland, 2016; pp. 31–62. [Google Scholar]
- Nijhof, L.N.; van de Putte, E.M.; Wulffraat, N.M.; Nijhof, S.L. Prevalence of severe fatigue among adolescents with pediatric rheumatic diseases. Arthritis Care Res. 2016, 68, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Norheim, K.B.; Jonsson, G.; Omdal, R. Biological mechanisms of chronic fatigue. Rheumatology 2011, 50, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, S.; Cockshott, Z.; Byron, M.; Kitchen, K.; Tipler, S.; Pope, D.; Hehir, M. Patients’ perceptions of fatigue in rheumatoid arthritis, Overwhelming, uncontrollable, ignored. Arthritis Rheum.-Arthritis Care Res. 2005, 53, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Repping-Wuts, H.; Uitterhoeve, R.; van Riel, P.; van Achterberg, T. Fatigue as experienced by patients with rheumatoid arthritis (RA), A qualitative study. Int. J. Nurs. Stud. 2008, 45, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, S.; Chalder, T.; Choy, E.; Cramp, F.; Davis, B.; Dures, E.; Nicholls, C.; Kirwan, J. Fatigue in rheumatoid arthritis, time for a conceptual model. Rheumatology 2011, 50, 1004–1006. [Google Scholar] [CrossRef] [PubMed]
- Karshikoff, B.; Sundelin, T.; Lasselin, J. Role of inflammation in human fatigue: Relevance of multidimensional assessments and potential neuronal mechanisms. Front. Immunol. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Petersen, L.E.; Baptista, T.S.A.; Molina, J.K.; Motta, J.G.; do Prado, A.; Piovesan, D.M.; de Nardi, T.; Viola, T.W.; Vieira, É.L.M.; Teixeira, A.L.; et al. Cognitive impairment in rheumatoid arthritis: Role of lymphocyte subsets, cytokines and neurotrophic factors. Clin. Rheumatol. 2018, 37, 1171–1181. [Google Scholar] [CrossRef]
- Memari, A.H.; Chamanara, E.; Ziaee, V.; Kordi, R.; Raeeskarami, S.R. Behavioral Problems in Juvenile Idiopathic Arthritis: A Controlled Study to Examine the Risk of Psychopathology in a Chronic Pediatric Disorder. Int. J. Chronic Dis. 2016, 2016, 5726236. [Google Scholar] [CrossRef]
- Schrepf, A.; Kaplan, C.M.; Ichesco, E.; Larkin, T.; Harte, S.E.; Harris, R.E.; Murray, A.D.; Waiter, G.D.; Clauw, D.J.; Basu, N. A multi-modal MRI study of the central response to inflammation in rheumatoid arthritis. Nat. Commun. 2018, 9, 2243. [Google Scholar] [CrossRef]
- Nerurkar, L.; Siebert, S.; McInnes, I.B.; Cavanagh, J. Rheumatoid arthritis and depression: An inflammatory perspective. Lancet Psychiatry 2018. [Google Scholar] [CrossRef]
- Felger, J.C.; Li, L.; Marvar, P.J.; Woolwine, B.J.; Harrison, D.G.; Raison, C.L.; Miller, A.H. Tyrosine metabolism during interferon-alpha administration: Association with fatigue and CSF dopamine concentrations. Brain Behav. Immun. 2013, 31, 153–160. [Google Scholar] [CrossRef]
- Dantzer, R.; Wollman, E.E.; Vitkovic, L.; Yirmiya, R. Cytokines, stress, and depression—Conclusions and perspectives. In Cytokines, Stress, and Depression; Dantzer, R., Wollman, E.E., Yirmiya, R., Eds.; Kluwer Academic/Plenum Publ: New York, NY, USA, 1999; pp. 317–329. [Google Scholar]
- Felger, J.C.; Miller, A.H. Cytokine effects on the basal ganglia and dopamine function: The subcortical source of inflammatory malaise. Front. Neuroendocrinol. 2012, 33, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Bower, J.E. Cancer-related fatigue-mechanisms, risk factors, and treatments. Nat. Rev. Clin. Oncol. 2014, 11, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Anderson, G.; Galecki, P.; Berk, M.; Maes, M.A. Narrative review on the similarities and dissimilarities between myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and sickness behavior. BMC Med. 2013, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Berk, M.; Goehler, L.; Song, C.; Anderson, G.; Gałecki, P.; Leonard, B. Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med. 2012, 10, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Haroon, E.; Raison, C.L.; Miller, A.H. Psychoneuroimmunology Meets Neuropsychopharmacology: Translational implications of the impact of inflammation on Behavior. Neuropsychopharmacology 2012, 37, 137–162. [Google Scholar] [CrossRef]
- Pollard, L.C.; Choy, E.H.; Gonzalez, J.; Khoshaba, B.; Scott, D.L. Fatigue in rheumatoid arthritis reflects pain, not disease activity. Rheumatology 2006, 45, 885–889. [Google Scholar] [CrossRef]
- Van Hoogmoed, D.; Fransen, J.; Bleijenberg, G.; van Riel, P. Physical and psychosocial correlates of severe fatigue in rheumatoid arthritis. Rheumatology 2010, 49, 1294–1302. [Google Scholar] [CrossRef]
- Butbul Aviel, Y.; Stremler, R.; Benseler, S.M.; Cameron, B.; Laxer, R.M.; Ota, S.; Schneider, R.; Spiegel, L.; Stinson, J.N.; Tse, S.M.; et al. Sleep and fatigue and the relationship to pain, disease activity and quality of life in juvenile idiopathic arthritis and juvenile dermatomyositis. Rheumatology 2011, 50, 2051–2060. [Google Scholar] [CrossRef]
- Capuron, L.; Schroecksnadel, S.; Féart, C.; Aubert, A.; Higueret, D.; Barberger-Gateau, P.; Layé, S.; Fuchs, D. Chronic low-grade inflammation in elderly persons is associated with altered tryptophan and tyrosine metabolism: Role in neuropsychiatric symptoms. Biol. Psychiatry 2011, 70, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Brown, L.M.; Blattner, W.A.; Maloney, E.M.; Kurman, C.C.; Nelson, D.L.; Fuchs, D.; Wachter, H.; Tollerud, D.J. Serum neopterin, beta(2)-microglobulin, soluble interleukin-2 receptors, and immunoglobulin levels in healthy adolescents. Clin. Immunol. Immunopathol. 1998, 88, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Sweeten, T.; Milton, M.W.; Posey, D.J.; McDougle, C.J. Plasma Kynurenine Levels in Autistic Disorder. J. Dev. Phys. Dis. 2006, 18, 419–426. [Google Scholar] [CrossRef]
- Bergwerff, C.E.; Luman, M.; Blom, H.J.; Oosterlaan, J. Paediatric reference values for total homocysteine, tryptophan, tyrosine and phenylalanine in blood spots. Scand. J. Clin. Lab. Investig. 2017, 77, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Shady, M.M.; Fathy, H.A.; Ali, A.; Youness, E.R.; Fathy, G.A. Association of neopterin as a marker of immune system activation and juvenile rheumatoid arthritis activity. J. Pediatr. 2015, 91, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Reibnegger, G.; Egg, D.; Fuchs, D.; Günther, R.; Hausen, A.; Werner, E.R.; Wachter, H. Urinary neopterin reflects clinical activity in patients with rheumatoid arthritis. Arthritis Rheum. 1986, 29, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Prior, C.; Bollbach, R.; Fuchs, D.; Hausen, A.; Judmaier, G.; Niederwieser, D.; Reibnegger, G.; Rotthauwe, H.W.; Werner, E.R.; Wachter, H. Urinary neopterin, a marker of clinical activity in patients with Crohn’s disease. Clin. Chim. Acta 1986, 155, 11–21. [Google Scholar] [CrossRef]
- Niederwieser, D.; Fuchs, D.; Hausen, A.; Judmaier, G.; Reibnegger, G.; Wachter, H.; Huber, C. Neopterin as a new biochemical marker in the clinical assessment of ulcerative colitis. Immunobiology 1985, 170, 320–326. [Google Scholar] [CrossRef]
- Murr, C.; Widner, B.; Wirleitner, B.; Fuchs, D. Neopterin as a marker for immune system activation. Curr. Drug Metab. 2002, 3, 175–187. [Google Scholar] [CrossRef]
- Schoenborn, J.R.; Wilson, C.B. Regulation of Interferon-γ During Innate and Adaptive Immune Responses. Adv. Immunol. 2007, 96, 41–101. [Google Scholar]
- Schoedon, G.; Troppmair, J.; Adolf, G.; Huber, C.; Niederwieser, A. Interferon-gamma enhances biosynthesis of pterins in peripheral blood mononuclear cells by induction of GTP-cyclohydrolase I activity. J. Interferon Res. 1986, 6, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Zhang, Y.-Y.; Chen, K.; Hatakeyama, K.; Keaney, J.F. Cytokine-Stimulated GTP Cyclohydrolase I Expression in Endothelial Cells Requires Coordinated Activation of Nuclear Factor-B and Stat1/Stat3. Circ. Res. 2005, 96, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Werner-Felmayer, G.; Werner, E.R.; Fuchs, D.; Hausen, A.; Reibnegger, G.; Wachter, H. Tetrahydrobiopterin-dependent Formation of Nitrite and Nitrate in Murine Fibroblasts. J. Exp. Med. 1990, 172, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Werner, E.R.; Werner-Felmayer, G.; Fuchs, D.; Hausen, A.; Reibnegger, G.; Yim, J.J.; Pfleiderer, W.; Wachter, H. Tetrahydrobiopterin biosynthetic activities in human macrophages, fibroblasts, THP-1, and T 24 cells. GTP-cyclohydrolase I is stimulated by interferon-gamma, and 6-pyruvoyl tetrahydropterin synthase and sepiapterin reductase are constitutively present. J. Biol. Chem. 1990, 265, 3189–3192. [Google Scholar] [PubMed]
- Werner, E.R.; Werner-Felmayer, G.; Wachter, H. Tetrahydrobiopterin and cytokines. Proc. Soc. Exp. Biol. Med. 1993, 203, 1–12. [Google Scholar] [CrossRef]
- Hoffmann, G.; Wirleitner, B.; Fuchs, D. Potential role of immune system activation-associated production of neopterin derivatives in humans. Inflamm. Res. 2003, 52, 313–321. [Google Scholar] [CrossRef]
- Neurauter, G.; Schröcksnadel, K.; Scholl-Bürgi, S.; Sperner-Unterweger, B.; Schubert, C.; Ledochowski, M.; Fuchs, D. Chronic immune stimulation correlates with reduced phenylalanine turnover. Curr. Drug Metab. 2008, 9, 622–627. [Google Scholar] [CrossRef]
- Lipińska, J.; Lipińska, S.; Stańczyk, J.; Sarniak, A.; Przymińska vel Prymont, A.; Kasielski, M.; Smolewska, E. Reactive oxygen species and serum antioxidant defense in juvenile idiopathic arthritis. Clin. Rheumatol. 2015, 34, 451–456. [Google Scholar] [CrossRef]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef]
- Kitagami, T.; Yamada, K.; Miura, H.; Hashimoto, R.; Nabeshima, T.; Ohta, T. Mechanism of systemically injected interferon-alpha impeding monoamine biosynthesis in rats: Role of nitric oxide as a signal crossing the blood-brain barrier. Brain Res. 2003, 978, 104–114. [Google Scholar] [CrossRef]
- Zielasek, J.; Hartung, H.P. Molecular mechanisms of microglial activation. Adv. Neuroimmunol. 1996, 6, 191–222. [Google Scholar] [CrossRef]
- Cunnington, C.; Channon, K.M. Tetrahydrobiopterin: Pleiotropic roles in cardiovascular pathophysiology. Heart 2010, 96, 1872–1877. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Haroon, E.; Raison, C.L.; Felger, J.C. Cytokine targets in the brain: Impact on Neurotransmitters and Neurocircuits. Depress. Anxiety 2013, 30, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Dobryakova, E.; DeLuca, J.; Genova, H.M.; Wylie, G.R. Neural correlates of cognitive fatigue: Cortico-striatal circuitry and effort-reward imbalance. J. Int. Neuropsychol. Soc. 2013, 19, 849–853. [Google Scholar] [CrossRef]
- Yang, S.; Jan, Y.H.; Mishin, V.; Richardson, J.R.; Hossain, M.M.; Heindel, N.D.; Heck, D.E.; Laskin, D.L.; Laskin, J.D. Sulfa drugs inhibit sepiapterin reduction and chemical redox cycling by sepiapterin reductase. J. Pharmacol. Exp. Ther. 2015, 352, 529–540. [Google Scholar] [CrossRef]
- Kim, H.; Chen, L.; Lim, G.; Sung, B.; Wang, S.; McCabe, M.F.; Rusanescu, G.; Yang, L.; Tian, Y.; Mao, J. Brain indoleamine 2,3-dioxygenase contributes to the comorbidity of pain and depression. J. Clin. Investig. 2012, 122, 2940–2954. [Google Scholar] [CrossRef]
- Kwidzinski, E.; Bunse, J.; Aktas, O.; Richter, D.; Mutlu, L.; Zipp, F.; Nitsch, R.; Bechmann, I. Indolamine 2,3-dioxygenase is expressed in the CNS and down-regulates autoimmune inflammation. FASEB J. 2005, 19, 1347–1349. [Google Scholar] [CrossRef]
- Corona, A.W.; Norden, D.M.; Skendelas, J.P.; Huang, Y.; O’Connor, J.C.; Lawson, M.; Dantzer, R.; Kelley, K.W.; Godbout, J.P. Indoleamine 2,3-dioxygenase inhibition attenuates lipopolysaccharide induced persistent microglial activation and depressive-like complications in fractalkine receptor (CX(3)CR1)-deficient mice. Brain Behav. Immun. 2013, 31, 134–142. [Google Scholar] [CrossRef]
- Salazar, A.; Gonzalez-Rivera, B.L.; Redus, L.; Parrott, J.M.; O’Connor, J.C. Indoleamine 2,3-dioxygenase mediates anhedonia and anxiety-like behaviors caused by peripheral lipopolysaccharide immune challenge. Horm. Behav. 2012, 62, 202–209. [Google Scholar] [CrossRef]
- Dantzer, R.; O’Connor, J.C.; Lawson, M.A.; Kelley, K.W. Inflammation-associated depression: From serotonin to kynurenine. Psychoneuroendocrinology 2011, 36, 426–436. [Google Scholar] [CrossRef]
- Leonard, B.; Maes, M. Mechanistic explanations how cell-mediated immune activation, inflammation and oxidative and nitrosative stress pathways and their sequels and concomitants play a role in the pathophysiology of unipolar depression. Neurosci. Biobehav. Rev. 2012, 36, 764–785. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Klasen, M.; Eisner, P.; Zepf, F.D.; Zvyagintsev, M.; Palomero-Gallagher, N.; Weber, R.; Eisert, A.; Mathiak, K. Central serotonin modulates neural responses to virtual violent actions in emotion regulation networks. Brain Struct. Funct. 2018, 223, 3327–3345. [Google Scholar] [CrossRef] [PubMed]
- Korte, S.M.; Prins, J.; Van den Bergh, F.S.; Oosting, R.S.; Dupree, R.; Korte-Bouws, G.A.H.; Westphal, K.G.; Olivier, B.; Denys, D.A.; Garland, A.; et al. The 5-HT(1A/1B)-receptor agonist eltoprazine increases both catecholamine release in the prefrontal cortex and dopamine release in the nucleus accumbens and decreases for reward and “waiting” impulsivity, but increases “stopping” impulsivity. Eur. J. Pharmacol. 2017, 794, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, P.B.; Huitron-Resendiz, S.; Henriksen, S.J.; Sutcliffe, J.G. 5-HT7 Receptor Inhibition and Inactivation Induce Antidepressant-like Behavior and Sleep Pattern. Biol. Psychiatry 2005, 58, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Barf, T.; Korte, S.M.; Korte-Bouws, G.A.H.; Sonesson, C.; Damsma, G.; Bohus, B.; Wikström, H. Potential anxiolytic properties of R-(+)-8-OSO2CF3-PAT, a 5-HT 1A receptor agonist. Eur. J. Pharmacol. 1996, 297, 205–211. [Google Scholar] [CrossRef]
- Korte, S.M.; Prins, J.; Krajnc, A.M.; Hendriksen, H.; Oosting, R.S.; Westphal, K.G.; Korte-Bouws, G.A.H.; Olivier, B. The many different faces of major depression: It is time for personalized medicine. Eur. J. Pharmacol. 2015, 753, 88–104. [Google Scholar] [CrossRef] [PubMed]
- Stinson, J.N.; Hayden, J.A.; Ahola Kohut, S.; Soobiah, C.; Cartwright, J.; Weiss, S.K.; Witmans, M.B. Sleep problems and associated factors in children with juvenile idiopathic arthritis: A systematic review. Pediatr. Rheumatol. Online J. 2014, 12, 1–12. [Google Scholar] [CrossRef]
- Chen, Y.; Brew, B.J.; Guillemin, G.J. Characterization of the kynurenine pathway in NSC-34 cell line: Implications for amyotrophic lateral sclerosis. J. Neurochem. 2011, 118, 816–825. [Google Scholar] [CrossRef]
- Du, F.; Schmidt, W.; Okuno, E.; Kido, R.; Köhler, C.; Schwarcz, R. Localization of kynurenine aminotransferase immunoreactivity in the rat hippocampus. J. Comp. Neurol. 1992, 321, 477–487. [Google Scholar] [CrossRef]
- Foster, A.C.; Collins, J.F.; Schwarcz, R. On the excitotoxic properties of quinolinic acid, 2,3-piperidine dicarboxylic acids and structurally related compounds. Neuropharmacology 1983, 22, 1331–1342. [Google Scholar] [CrossRef]
- Guidetti, P.; Schwarcz, R. 3-Hydroxykynurenine potentiates quinolinate but not NMDA toxicity in the rat striatum. Eur. J. Neurosci. 1999, 11, 3857–6383. [Google Scholar] [CrossRef]
- Lin, C.L.; Kong, Q.; Cuny, G.D.; Glicksman, M.A. Glutamate transporter EAAT2: A new target for the treatment of neurodegenerative diseases. Future Med. Chem. 2012, 4, 1689–1700. [Google Scholar] [CrossRef] [PubMed]
- Haroon, E.; Miller, A.H.; Sanacora, G. Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacology 2017, 42, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 100–105. [Google Scholar] [CrossRef]
- Vercellino, M.; Merola, A.; Piacentino, C.; Votta, B.; Capello, E.; Mancardi, G.L.; Mutani, R.; Giordana, M.T.; Cavalla, P. Altered glutamate reuptake in relapsing-remitting and secondary progressive multiple sclerosis cortex: Correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. J. Neuropathol. Exp. Neurol. 2007, 66, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Swartz, K.J.; During, M.J.; Freese, A.; Beal, M.F. Cerebral synthesis and release of kynurenic acid: An endogenous antagonist of excitatory amino acid receptors. J. Neurosci. 1990, 10, 2965–2973. [Google Scholar] [CrossRef] [PubMed]
- Hilmas, C.; Pereira, E.F.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: Physiopathological implications. J. Neurosci. 2001, 21, 7463–7473. [Google Scholar] [CrossRef]
- Flores-Barrera, E.; Thomases, D.R.; Cass, D.K.; Bhandari, A.; Schwarcz, R.; Bruno, J.P.; Tseng, K.Y. Preferential disruption of prefrontal GABAergic function by nanomolar concentrations of the α7nACh negative modulator Kynurenic Acid. J. Neurosci. 2017, 37, 7921–7929. [Google Scholar] [CrossRef]
- Linderholm, K.R.; Andersson, A.; Olsson, S.; Olsson, E.; Snodgrass, R.; Engberg, G.; Erhardt, S. Activation of rat ventral tegmental area dopamine neurons by endogenous kynurenic acid: A pharmacological analysis. Neuropharmacology 2007, 53, 918–924. [Google Scholar] [CrossRef]
- Wichers, M.C.; Koek, G.H.; Robaeys, G.; Verkerk, R.; Scharpé, S.; Maes, M. IDO and interferon-alpha-induced depressive symptoms: A shift in hypothesis from tryptophan depletion to neurotoxicity. Mol. Psychiatry 2005, 10, 538–544. [Google Scholar] [CrossRef]
- Raison, C.L.; Dantzer, R.; Kelley, K.W.; Lawson, M.A.; Woolwine, B.J.; Vogt, G.; Spivey, J.R.; Saito, K.; Miller, A.H. CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-alpha: Relationship to CNS immune responses and depression. Mol. Psychiatry 2010, 15, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Schwamborn, R.; Brown, E.; Haase, J. Elevation of cortical serotonin transporter activity upon peripheral immune challenge is regulated independently of p38 mitogen-activated protein kinase activation and transporter phosphorylation. J. Neurochem. 2016, 137, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Wang, S.; Huang, Z.; Zhang, L.; Yang, X.; Bai, X.; Zhou, D.; Qin, Z.; Du, G. Lipopolysaccharide induced serotonin transporter up-regulation involves pkg-i and p38mapk activation partially through a3 adenosine receptor. Biosci. Trends 2015, 9, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Lin, Y.S.; Cheng, J.T.; Lin, C.F.; Wu, H.T.; Wu, S.R.; Tsai, W.H. Interferon-alpha- induced serotonin uptake in Jurkat T cells via mitogen-activated protein kinase and transcriptional regulation of the serotonin transporter. J. Psychopharmacol. 2008, 22, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Haase, J.; Brown, E. Integrating the monoamine, neurotrophin and cytokine hypotheses of depression—A central role for the serotonin transporter? Pharmacol. Ther. 2015, 147, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.B.; Blakely, R.D.; Hewlett, W.A. The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology 2006, 31, 2121–2131. [Google Scholar] [CrossRef]
- Zhu, C.B.; Lindler, K.M.; Owens, A.W.; Daws, L.C.; Blakely, R.D.; Hewlett, W.A. Interleukin-1 receptor activation by systemic lipopolysaccharide induces behavioral despair linked to MAPK regulation of CNS serotonin transporters. Neuropsychopharmacology 2010, 35, 2510–2520.34. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.B.; Hewlett, W.A.; Feoktistov, I.; Biaggioni, I.; Blakely, R.D. Adenosine receptor, protein kinase, G.; and p38 mitogen-activated protein kinase-dependent up-regulation of serotonin transporters involves both transporter trafficking and activation. Mol. Pharmacol. 2004, 65, 1462–1474. [Google Scholar] [CrossRef]
- Zhu, C.B.; Carneiro, A.M.; Dostmann, W.R.; Hewlett, W.A.; Blakely, R.D. P38 MAPK activation elevates serotonin transport activity via a trafficking-independent, protein phosphatase 2a-dependent process. J. Biol. Chem. 2005, 280, 15649–15658. [Google Scholar] [CrossRef]
- Zhu, C.B.; Steiner, J.A.; Munn, J.L.; Daws, L.C.; Hewlett, W.A.; Blakely, R.D. Rapid stimulation of presynaptic serotonin transport by A(3) adenosine receptors. J. Pharmacol. Exp. Ther. 2007, 322, 332–340. [Google Scholar] [CrossRef]
- Morón, J.A.; Zakharova, I.; Ferrer, J.V.; Merrill, G.A.; Hope, B.; Lafer, E.M.; Lin, Z.C.; Wang, J.B.; Javitch, J.A.; Galli, A.; et al. Mitogen-activated protein kinase regulates dopamine transporter surface expression and dopamine transport capacity. J. Neurosci. 2003, 23, 8480–8488. [Google Scholar] [CrossRef] [PubMed]
- Krishnadas, R.; Nicol, A.; Sassarini, J.; Puri, N.; Burden, A.D.; Leman, J.; Combet, E.; Pimlott, S.; Hadley, D.; McInnes, I.B.; et al. Circulating tumour necrosis factor is highly correlated with brainstem serotonin transporter availability in humans. Brain Behav. Immun. 2016, 51, 29–38. [Google Scholar] [CrossRef]
- Korte-Bouws, G.A.H.; van Heesch, F.; Westphal, K.G.C.; Ankersmit, L.M.J.; van Oosten, E.M.; Güntürkün, O.; Korte, S.M. Bacterial Lipopolysaccharide Increases Serotonin Metabolism in Both Medial Prefrontal Cortex and Nucleus Accumbens in Male Wild Type Rats, but Not in Serotonin Transporter Knockout Rats. Pharmaceuticals 2018, 11, 66. [Google Scholar] [CrossRef]
- Weinberger, J.F.; Raison, C.L.; Rye, D.B.; Montague, A.R.; Woolwine, B.J.; Felger, J.C.; Haroon, E.; Miller, A.H. Inhibition of tumor necrosis factor improves sleep continuity in patients with treatment resistant depression and high inflammation. Brain Behav. Immun. 2015, 47, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Raison, C.L.; Rutherford, R.E.; Woolwine, B.J.; Shuo, C.; Schettler, P.; Drake, D.F.; Haroon, E.; Miller, A.H. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: The role of baseline inflammatory biomarkers. JAMA Psychiatry 2013, 70, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Mangge, H.; Becker, K.; Fuchs, D.; Gostner, J.M. Antioxidants, inflammation and cardiovascular disease. World J. Cardiol. 2014, 6, 462–477. [Google Scholar]
- Murr, C.; Winklhofer-Roob, B.M.; Schroecksnadel, K.; Maritschnegg, M.; Mangge, H.; Böhm, B.O.; Winkelmann, B.R.; März, W.; Fuchs, D. Inverse association between serum concentrations of neopterin and antioxidants in patients with and without angiographic coronary artery disease. Atherosclerosis 2009, 202, 543–549. [Google Scholar] [CrossRef]
- McCarty, M.F. Supplementation with Phycocyanobilin, Citrulline, Taurine, and Supranutritional Doses of Folic Acid and Biotin-Potential for Preventing or Slowing the Progression of Diabetic Complications. Healthcare 2017, 5, 15. [Google Scholar] [CrossRef]
- Miller, A.L. The methylation, neurotransmitter, and antioxidant connections between folate and depression. Altern. Med Rev. 2008, 13, 216–226. [Google Scholar]
- Papakostas, G.I.; Shelton, R.C.; Zajecka, J.M.; Etemad, B.; Rickels, K.; Clain, A.; Baer, L.; Dalton, E.D.; Sacco, G.R.; Schoenfeld, D.; et al. l-methylfolate as adjunctive therapy for SSRI-resistant major depression: Results of two randomized, double-blind, parallel-sequential trials. Am. J. Psychiatry 2012, 169, 1267–1274. [Google Scholar] [CrossRef]
- Popoli, M.; Yan, Z.; McEwen, B.S.; Sanacora, G. The stressed synapse: The impact of stress and glucocorticoids on glutamate transmission. Nat. Rev. Neurosci. 2011, 13, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Monteggia, L.M. A neurotrophic model for stress-related mooddisorders. Biol. Psychiatry 2006, 59, 1116–1127. [Google Scholar] [CrossRef]
- McEwen, B.S.; Bowles, N.P.; Gray, J.D.; Hill, M.N.; Hunter, R.G.; Karatsoreos, I.N.; Nasca, C. Mechanisms of stress in the brain. Nat. Neurosci. 2015, 18, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Nasca, C.; Xenos, D.; Barone, Y.; Caruso, A.; Scaccianoce, S.; Matrisciano, F.; Battaglia, G.; Mathé, A.A.; Pittaluga, A.; Lionetto, L.; et al. L-acetylcarnitine causes rapid antidepressant effects through the epigenetic induction of mGlu2 receptors. Proc. Natl. Acad. Sci. USA 2013, 110, 4804–4809. [Google Scholar] [CrossRef] [PubMed]
- Bigio, B.; Mathé, A.A.; Sousa, V.C.; Zelli, D.; Svenningsson, P.; McEwen, B.S.; Nasca, C. Epigenetics and energetics in ventral hippocampus mediate rapid antidepressant action: Implications for treatment resistance. Proc. Natl. Acad. Sci. USA 2016, 113, 7906–7911. [Google Scholar] [CrossRef] [PubMed]
- Nasca, C.; Bigio, B.; Zelli, D.; de Angelis, P.; Lau, T.; Okamoto, M.; Soya, H.; Ni, J.; Brichta, L.; Greengard, P.; et al. Role of the astroglial glutamate exchanger xCT in ventral hippocampus in resilience to stress. Neuron 2017, 96, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Nasca, C.; Bigio, B.; Lee, F.S.; Young, S.P.; Kautz, M.M.; Albright, A.; Beasley, J.; Millington, D.S.; Mathé, A.A.; Kocsis, J.H.; et al. Acetyl-l-carnitine deficiency in patients with major depressive disorder. Proc. Natl. Acad. Sci. USA 2018, 115, 8627–8632. [Google Scholar] [CrossRef] [PubMed]
- Maalouf, M.; Sullivan, P.G.; Davis, L.; Kim, D.Y.; Rho, J.M. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 2007, 145, 256–264. [Google Scholar] [CrossRef]
- Brietzke, E.; Mansur, R.B.; Subramaniapillai, M.; Balanzá-Martínez, V.; Vinberg, M.; González-Pinto, A.; Rosenblat, J.D.; Ho, R.; McIntyre, R.S. Ketogenic diet as a metabolic therapy for mood disorders: Evidence and developments. Neurosci. Biobehav. Rev. 2018, 94, 11–16. [Google Scholar] [CrossRef]
- Consolaro, A.; Negro, G.; Chiara Gallo, M.; Bracciolini, G.; Ferrari, C.; Schiappapietra, B.; Pistorio, A.; Bovis, F.; Ruperto, N.; Martini, A.; et al. Defining criteria for disease activity states in nonsystemic juvenile idiopathic arthritis based on a three-variable juvenile arthritis disease activity score. Arthritis Care Res. 2014, 66, 1703–1709. [Google Scholar] [CrossRef]
- Swart, J.F.; van Dijkhuizen, E.H.P.; Wulffraat, N.M.; de Roock, S. Clinical Juvenile Arthritis Disease Activity Score proves to be a useful tool in treat-to-target therapy in juvenile idiopathic arthritis. Ann. Rheum. Dis. 2018, 77, 336–342. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DISEASE ACTIVITY | High Disease Activity (HDA) | High Disease Activity (HDA) | Clinically Inactive (IA) | Clinically Inactive (IA) |
|---|---|---|---|---|
| MTX | No | Yes | No | Yes |
| cJADAS | 18.7 ± 2.5 * | 14.3 ± 0.4 * | 0 | 0 |
| CRP | 90.2 ± 38.8 *# | 31.8 ± 10.6 * | 0.3 ± 0.1 | 0.1 ± 0.1 |
| VAS | 4.98 ± 1.26 * | 6,82 ± 0.9 * | 0.0 ± 0.0 | 0.08 ± 0.04 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korte-Bouws, G.A.H.; Albers, E.; Voskamp, M.; Hendriksen, H.; De Leeuw, L.R.; Güntürkün, O.; De Roock, S.; Vastert, S.J.; Korte, S.M. Juvenile Arthritis Patients Suffering from Chronic Inflammation Have Increased Activity of Both IDO and GTP-CH1 Pathways But Decreased BH4 Efficacy: Implications for Well-Being, Including Fatigue, Cognitive Impairment, Anxiety, and Depression. Pharmaceuticals 2019, 12, 9. https://doi.org/10.3390/ph12010009
Korte-Bouws GAH, Albers E, Voskamp M, Hendriksen H, De Leeuw LR, Güntürkün O, De Roock S, Vastert SJ, Korte SM. Juvenile Arthritis Patients Suffering from Chronic Inflammation Have Increased Activity of Both IDO and GTP-CH1 Pathways But Decreased BH4 Efficacy: Implications for Well-Being, Including Fatigue, Cognitive Impairment, Anxiety, and Depression. Pharmaceuticals. 2019; 12(1):9. https://doi.org/10.3390/ph12010009
Chicago/Turabian StyleKorte-Bouws, Gerdien A. H., Eline Albers, Marije Voskamp, Hendrikus Hendriksen, Lidewij R. De Leeuw, Onur Güntürkün, Sytze De Roock, Sebastiaan J. Vastert, and S. Mechiel Korte. 2019. "Juvenile Arthritis Patients Suffering from Chronic Inflammation Have Increased Activity of Both IDO and GTP-CH1 Pathways But Decreased BH4 Efficacy: Implications for Well-Being, Including Fatigue, Cognitive Impairment, Anxiety, and Depression" Pharmaceuticals 12, no. 1: 9. https://doi.org/10.3390/ph12010009
APA StyleKorte-Bouws, G. A. H., Albers, E., Voskamp, M., Hendriksen, H., De Leeuw, L. R., Güntürkün, O., De Roock, S., Vastert, S. J., & Korte, S. M. (2019). Juvenile Arthritis Patients Suffering from Chronic Inflammation Have Increased Activity of Both IDO and GTP-CH1 Pathways But Decreased BH4 Efficacy: Implications for Well-Being, Including Fatigue, Cognitive Impairment, Anxiety, and Depression. Pharmaceuticals, 12(1), 9. https://doi.org/10.3390/ph12010009