Links Between Iron and Lipids: Implications in Some Major Human Diseases


{kind=link}
Abstract
1. Introduction
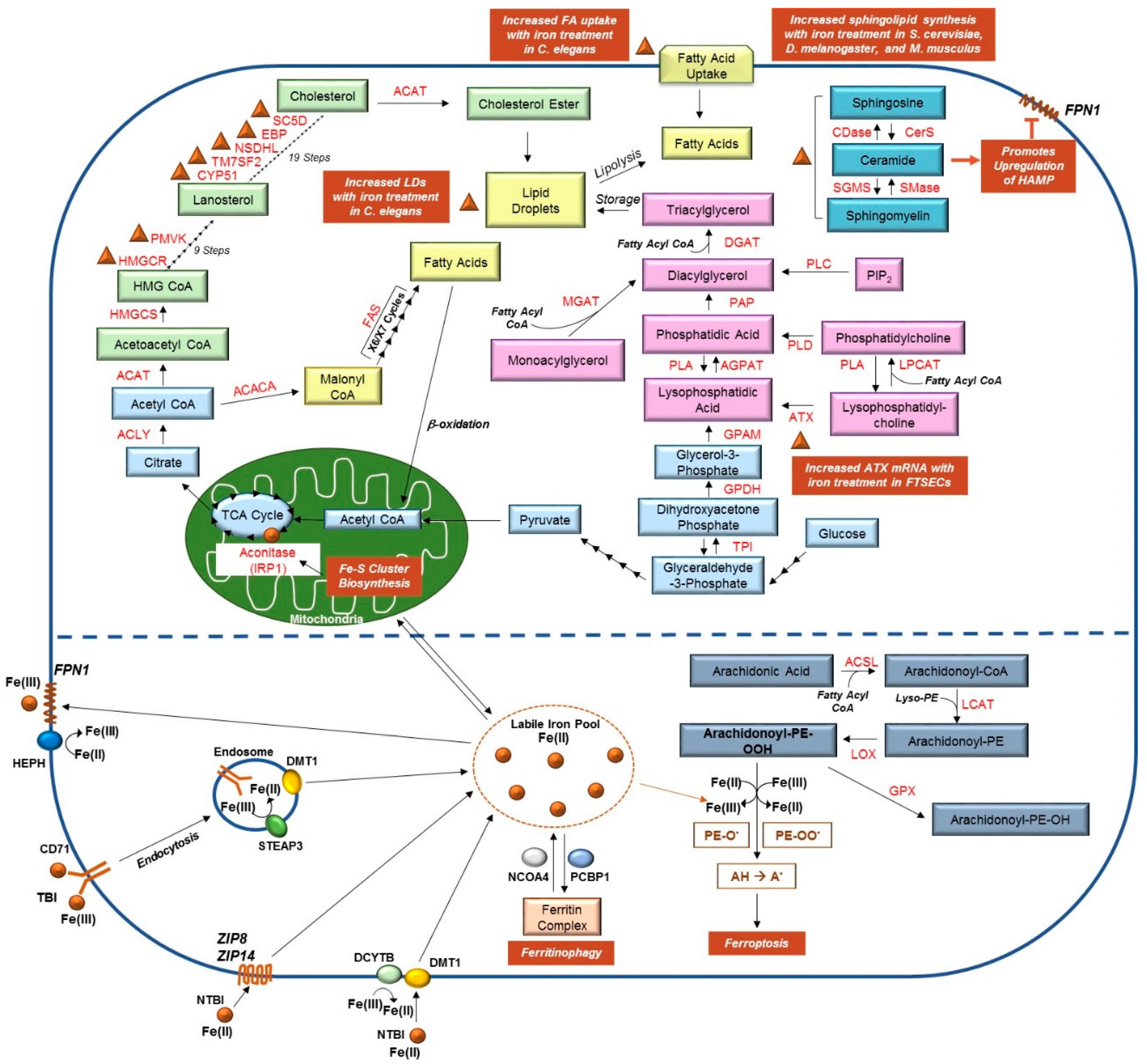
2. Interactions between Iron and Lipids in Model Systems
2.1. Iron and Cholesterol
2.2. Iron and Sphingolipids
2.3. Iron-Sulfur Cluster and Lipids
2.4. Iron, Lipid Droplets, and Leptin
2.5. Iron, LPP1, and Other Enzymes Involved in Lysophospholipid Metabolism
2.6. Iron and Fatty Acid Metabolism
2.7. Ferroptosis and Lipids
3. Iron and Lipids: Neurodegenerative Diseases
3.1. Brain Iron Localization
3.2. Iron-Mediated Lipid Peroxidation and Ferroptosis
3.3. Iron and the Sphingolipid Pathway
4. Treatments and Concluding Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abbaspour, N.; Hurrell, R.; Kelishadi, R. Review on iron and its importance for human health. J. Res. Med. Sci. 2014, 19, 164–174. [Google Scholar] [PubMed]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Eid, R.; Arab, N.T.; Greenwood, M.T. Iron mediated toxicity and programmed cell death: A review and a re-examination of existing paradigms. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 399–430. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.C. Disorders of iron metabolism. N. Engl. J. Med. 1999, 341, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Sadrzadeh, S.M.; Graf, E.; Panter, S.S.; Hallaway, P.E.; Eaton, J.W. Hemoglobin. A biologic fenton reagent. J. Biol. Chem. 1984, 259, 14354–14356. [Google Scholar] [PubMed]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef]
- Wallace, D.F. The Regulation of Iron Absorption and Homeostasis. Clin. Biochem. Rev. 2016, 37, 51–62. [Google Scholar] [PubMed]
- Manz, D.H.; Blanchette, N.L.; Paul, B.T.; Torti, F.M.; Torti, S.V. Iron and cancer: Recent insights. Ann. N. Y. Acad. Sci. 2016, 1368, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Rockfield, S.; Raffel, J.; Mehta, R.; Rehman, N.; Nanjundan, M. Iron overload and altered iron metabolism in ovarian cancer. Biol. Chem. 2017, 398, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, T.B.; Cousins, R.J. The Multiple Faces of the Metal Transporter ZIP14 (SLC39A14). J. Nutr. 2018, 148, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. PNAS 2006, 103, 13612–13617. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Jenkitkasemwong, S.; Duarte, S.; Sparkman, B.K.; Shawki, A.; Mackenzie, B.; Knutson, M.D. ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J. Biol. Chem. 2012, 287, 34032–34043. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.; Bae, D.H.; Merlot, A.M.; Sahni, S.; Richardson, D.R. Duodenal cytochrome b (DCYTB) in iron metabolism: An update on function and regulation. Nutrients 2015, 7, 2274–2296. [Google Scholar] [CrossRef] [PubMed]
- Sendamarai, A.K.; Ohgami, R.S.; Fleming, M.D.; Lawrence, C.M. Structure of the membrane proximal oxidoreductase domain of human Steap3, the dominant ferrireductase of the erythroid transferrin cycle. PNAS 2008, 105, 7410–7415. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Bencze, K.Z.; Stemmler, T.L.; Philpott, C.C. A cytosolic iron chaperone that delivers iron to ferritin. Science 2008, 320, 1207–1210. [Google Scholar] [CrossRef] [PubMed]
- Dowdle, W.E.; Nyfeler, B.; Nagel, J.; Elling, R.A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014, 16, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Petrak, J.; Vyoral, D. Hephaestin—A ferroxidase of cellular iron export. Int. J. Biochem. Cell Biol. 2005, 37, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Ropert, M.; Le Lan, C.; Loreal, O. Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochim. Biophys. Acta 2012, 1820, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Craven, C.M.; Alexander, J.; Eldridge, M.; Kushner, J.P.; Bernstein, S.; Kaplan, J. Tissue distribution and clearance kinetics of non-transferrin-bound iron in the hypotransferrinemic mouse: A rodent model for hemochromatosis. PNAS 1987, 84, 3457–3461. [Google Scholar] [CrossRef] [PubMed]
- Iancu, T.C.; Shiloh, H.; Raja, K.B.; Simpson, R.J.; Peters, T.J.; Perl, D.P.; Hsu, A.; Good, P.F. The hypotransferrinaemic mouse: Ultrastructural and laser microprobe analysis observations. J. Pathol. 1995, 177, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.; Wang, C.Y.; Zhang, L.; Zhang, W.; Hojyo, S.; Fukada, T.; Knutson, M.D. ZIP14 and DMT1 in the liver, pancreas, and heart are differentially regulated by iron deficiency and overload: Implications for tissue iron uptake in iron-related disorders. Haematologica 2013, 98, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.K.; Karmakar, S.; Asthana, A.; Ashok, A.; Desai, V.; Baksi, S.; Singh, N. Transport of Non-Transferrin Bound Iron to the Brain: Implications for Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyaya, R. Oxidative damage to DNA constituents by iron-mediated Fenton reactions--the thymidine family. J. Biomol. Struct. Dyn. 2014, 32, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyaya, R.; Goswami, B. Oxidative damage to DNA constituents by iron-mediated Fenton reactions: The deoxyadenosine family. J. Biomol. Struct. Dyn. 2012, 30, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.S.; Moreau, C. Striking while the iron is hot: Iron metabolism and Ferroptosis in neurodegeneration. Free Radic Biol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Yamada, Y.; Kanayama, S.; Furukawa, N.; Noguchi, T.; Haruta, S.; Yoshida, S.; Sakata, M.; Sado, T.; Oi, H. The role of iron in the pathogenesis of endometriosis. Gynecol. Endocrinol. 2009, 25, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Mandai, M.; Toyokuni, S.; Hamanishi, J.; Higuchi, T.; Takakura, K.; Fujii, S. Contents of endometriotic cysts, especially the high concentration of free iron, are a possible cause of carcinogenesis in the cysts through the iron-induced persistent oxidative stress. Clin. Cancer Res. 2008, 14, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.; Papaleo, E.; Corti, L.; Santambrogio, P.; Levi, S.; Vigano, P.; Candiani, M.; Panina-Bordignon, P. Iron availability is increased in individual human ovarian follicles in close proximity to an endometrioma compared with distal ones. Hum Reprod. 2014, 29, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Vercellini, P.; Crosignani, P.; Somigliana, E.; Vigano, P.; Buggio, L.; Bolis, G.; Fedele, L. The ‘incessant menstruation’ hypothesis: A mechanistic ovarian cancer model with implications for prevention. Hum. Reprod. 2011, 26, 2262–2273. [Google Scholar] [CrossRef] [PubMed]
- Ashmore, J.H.; Rogers, C.J.; Kelleher, S.L.; Lesko, S.M.; Hartman, T.J. Dietary Iron and Colorectal Cancer Risk: A Review of Human Population Studies. Crit. Rev. Food Sci. Nutr. 2016, 56, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.C.; Farese, R.V., Jr. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef] [PubMed]
- Petan, T.; Jarc, E.; Jusovic, M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef] [PubMed]
- Kuri-Harcuch, W.; Velez-delValle, C.; Vazquez-Sandoval, A.; Hernandez-Mosqueira, C.; Fernandez-Sanchez, V. A cellular perspective of adipogenesis transcriptional regulation. J. Cell. Phys. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.; Merlot, A.M.; Huang, M.L.; Bae, D.H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta 2015, 1853, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Duncan, R.E.; Jaworski, K.; Sarkadi-Nagy, E.; Sul, H.S. Triacylglycerol metabolism in adipose tissue. Futur. Lipidol. 2007, 2, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer. 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Luo, Q.; Halim, A.; Song, G. Targeting lipid metabolism of cancer cells: A promising therapeutic strategy for cancer. Cancer Lett. 2017, 401, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Nakanaga, K.; Hama, K.; Aoki, J. Autotaxin—An LPA producing enzyme with diverse functions. J. Biochem. 2010, 148, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.; Debose-Boyd, R.A. Control of cholesterol synthesis through regulated ER-associated degradation of HMG CoA reductase. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Stoyanovsky, D.A.; Tyurina, Y.Y.; Shrivastava, I.; Bahar, I.; Tyurin, V.A.; Protchenko, O.; Jadhav, S.; Bolevich, S.B.; Kozlov, A.V.; Vladimirov, Y.A.; et al. Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Radic Biol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ingolfsson, H.I.; Melo, M.N.; van Eerden, F.J.; Arnarez, C.; Lopez, C.A.; Wassenaar, T.A.; Periole, X.; de Vries, A.H.; Tieleman, D.P.; Marrink, S.J. Lipid organization of the plasma membrane. J. Am. Chem. Soc. 2014, 136, 14554–14559. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.M.; Chua, A.C.; Carter, K.W.; Delima, R.D.; Johnstone, D.; Herbison, C.E.; Firth, M.J.; O’Leary, R.; Milward, E.A.; Olynyk, J.K.; et al. Hepatic iron loading in mice increases cholesterol biosynthesis. Hepatology 2010, 52, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Finkelstein, D.I.; Adlard, P.A. Interactions of metals and Apolipoprotein E in Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 121. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Perreau, V.M.; Dent, K.A.; Bush, A.I.; Finkelstein, D.I.; Adlard, P.A. Iron Regulates Apolipoprotein E Expression and Secretion in Neurons and Astrocytes. J. Alzheimers Dis. 2016, 51, 471–487. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Faux, N.G.; Bush, A.I. Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat. Commun. 2015, 6, 6760. [Google Scholar] [CrossRef] [PubMed]
- Mahoney-Sanchez, L.; Belaidi, A.A.; Bush, A.I.; Ayton, S. The Complex Role of Apolipoprotein E in Alzheimer’s Disease: An Overview and Update. J. Mol. Neurosci. 2016, 60, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Wood, H. Alzheimer disease: Iron—The missing link between ApoE and Alzheimer disease? Nat. Rev. Neurol. 2015, 11, 369. [Google Scholar] [CrossRef] [PubMed]
- Britton, L.J.; Bridle, K.; Jaskowski, L.A.; He, J.; Ng, C.; Ruelcke, J.E.; Mohamed, A.; Reiling, J.; Santrampurwala, N.; Hill, M.M.; et al. Iron Inhibits the Secretion of Apolipoprotein E in Cultured Human Adipocytes. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 215–217.e8. [Google Scholar] [CrossRef] [PubMed]
- Kraft, M.L. Sphingolipid Organization in the Plasma Membrane and the Mechanisms That Influence It. Front. Cell. Dev. Biol. 2016, 4, 154. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Huang, X.; Kropat, J.; Henras, A.; Merchant, S.S.; Dickson, R.C.; Chanfreau, G.F. Sphingolipid signaling mediates iron toxicity. Cell Metab. 2012, 16, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Ho, T.S.; Lin, G.; Tan, K.L.; Rasband, M.N.; Bellen, H.J. Loss of Frataxin activates the iron/sphingolipid/PDK1/Mef2 pathway in mammals. eLife 2016, 5, e20732. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Lin, G.; Haelterman, N.A.; Ho, T.S.; Li, T.; Li, Z.; Duraine, L.; Graham, B.H.; Jaiswal, M.; Yamamoto, S.; et al. Loss of Frataxin induces iron toxicity, sphingolipid synthesis, and Pdk1/Mef2 activation, leading to neurodegeneration. eLife 2016, 5, e16043. [Google Scholar] [CrossRef] [PubMed]
- Vaubel, R.A.; Isaya, G. Iron-sulfur cluster synthesis, iron homeostasis and oxidative stress in Friedreich ataxia. Mol. Cell. Neurosci. 2013, 55, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, T.; Kotamraju, S.; Kalivendi, S.V.; Dhanasekaran, A.; Joseph, J.; Kalyanaraman, B. Ceramide-induced intracellular oxidant formation, iron signaling, and apoptosis in endothelial cells: Protective role of endogenous nitric oxide. J. Biol. Chem. 2004, 279, 28614–28624. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Natarajan, S.K.; Mott, J.L.; Kharbanda, K.K.; Harrison-Findik, D.D. Ceramide Induces Human Hepcidin Gene Transcription through JAK/STAT3 Pathway. PLoS ONE 2016, 11, e0147474. [Google Scholar] [CrossRef] [PubMed]
- Shakor, A.B.; Taniguchi, M.; Kitatani, K.; Hashimoto, M.; Asano, S.; Hayashi, A.; Nomura, K.; Bielawski, J.; Bielawska, A.; Watanabe, K.; et al. Sphingomyelin synthase 1-generated sphingomyelin plays an important role in transferrin trafficking and cell proliferation. J. Biol. Chem. 2011, 286, 36053–36062. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A. Mammalian iron-sulphur proteins: Novel insights into biogenesis and function. Nat. Rev. Mol. Cell Biol. 2015, 16, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.R.; Matyushov, D.V. Electron-transfer chain in respiratory complex I. Sci. Rep. 2017, 7, 5495. [Google Scholar] [CrossRef] [PubMed]
- Crooks, D.R.; Maio, N.; Lane, A.N.; Jarnik, M.; Higashi, R.M.; Haller, R.G.; Yang, Y.; Fan, T.W.; Linehan, W.M.; Rouault, T.A. Acute loss of iron-sulfur clusters results in metabolic reprogramming and generation of lipid droplets in mammalian cells. J. Biol. Chem. 2018, 293, 8297–8311. [Google Scholar] [CrossRef] [PubMed]
- Berteau, O. A missed Fe-S cluster handoff causes a metabolic shakeup. J. Biol. Chem. 2018, 293, 8312–8313. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta 2014, 1837, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.A.; Fox, J.L.; Gohil, V.M.; Winge, D.R.; Greenberg, M.L. Loss of cardiolipin leads to perturbation of mitochondrial and cellular iron homeostasis. J. Biol. Chem. 2013, 288, 1696–1705. [Google Scholar] [CrossRef] [PubMed]
- WHO. Obesity and Overweight Fact Sheet. 2018. Available online: http://www.who.int/en/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 21 October 2018).
- Lee, M.J.; Wu, Y.; Fried, S.K. Adipose tissue remodeling in pathophysiology of obesity. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Obin, M.S. Obesity and the role of adipose tissue in inflammation and metabolism. Am. J. Clin. Nutr. 2006, 83, 461S–465S. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Parton, R.G. Not just fat: The structure and function of the lipid droplet. Cold Spring Harb Perspect. Biol. 2011, 3, a004838. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jiang, X.; Wu, J.; Zhang, L.; Huang, J.; Zhang, Y.; Zou, X.; Liang, B. Iron Overload Coordinately Promotes Ferritin Expression and Fat Accumulation in Caenorhabditis elegans. Genetics 2016, 203, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, S.; Catarino, C.; Santos-Silva, A. The role of adipocytes in the modulation of iron metabolism in obesity. Obes. Rev. 2013, 14, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.; Matak, P.; McKie, A.T.; Sharp, P. Leptin increases the expression of the iron regulatory hormone hepcidin in HuH7 human hepatoma cells. J. Nutr. 2007, 137, 2366–2370. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Kuragano, T.; Kimura, T.; Nanami, M.; Hasuike, Y.; Nakanishi, T. Interplay of adipocyte and hepatocyte: Leptin upregulates hepcidin. Biochem. Biophys. Res. Commun. 2018, 495, 1548–1554. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Chen, M.; Liu, G.; Xu, E.; Chen, H. Ablation of hephaestin and ceruloplasmin results in iron accumulation in adipocytes and type 2 diabetes. FEBS Lett. 2018, 592, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Citelli, M.; Fonte-Faria, T.; Nascimento-Silva, V.; Renovato-Martins, M.; Silva, R.; Luna, A.S.; Silva, S.V.; Barja-Fidalgo, C. Obesity promotes alterations in iron recycling. Nutrients 2015, 7, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, S.; Sun, P.; Cao, F.; Li, H.; Sun, J.; Peng, M.; Liu, W.; Shi, P. Iron depletion participates in the suppression of cell proliferation induced by lipin1 overexpression. Metallomics 2018. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Murph, M.; Panupinthu, N.; Mills, G.B. ATX-LPA receptor axis in inflammation and cancer. Cell Cycle 2009, 8, 3695–3701. [Google Scholar] [CrossRef] [PubMed]
- Valdes-Rives, S.A.; Gonzalez-Arenas, A. Autotaxin-Lysophosphatidic Acid: From Inflammation to Cancer Development. Mediators Inflamm. 2017, 2017, 9173090. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.T.; Chang, R.; Wang, H.; Xiao, F.J.; Ge, R.L.; Wang, L.S. ENPP2 protects cardiomyocytes from erastin-induced ferroptosis. Biochem. Biophys. Res. Commun. 2018, 499, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Basuli, D.; Tesfay, L.; Deng, Z.; Paul, B.; Yamamoto, Y.; Ning, G.; Xian, W.; McKeon, F.; Lynch, M.; Crum, C.P.; et al. Iron addiction: A novel therapeutic target in ovarian cancer. Oncogene 2017, 36, 4089–4099. [Google Scholar] [CrossRef] [PubMed]
- Konstorum, A.; Lynch, M.L.; Torti, S.V.; Torti, F.M.; Laubenbacher, R.C. A Systems Biology Approach to Understanding the Pathophysiology of High-Grade Serous Ovarian Cancer: Focus on Iron and Fatty Acid Metabolism. OMICS 2018, 22, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Agmon, E.; Stockwell, B.R. Lipid homeostasis and regulated cell death. Curr. Opin. Chem. Biol. 2017, 39, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Mao, C.; Yang, R.; Yan, B.; Shi, Y.; Liu, X.; Lai, W.; Liu, Y.; Wang, X.; Xiao, D.; et al. EGLN1/c-Myc Induced Lymphoid-Specific Helicase Inhibits Ferroptosis through Lipid Metabolic Gene Expression Changes. Theranostics 2017, 7, 3293–3305. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 2016, 478, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Jodeiri Farshbaf, M.; Ghaedi, K. Does any drug to treat cancer target mTOR and iron hemostasis in neurodegenerative disorders? Biometals. 2017, 30, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Fretham, S.J.; Carlson, E.S.; Georgieff, M.K. The role of iron in learning and memory. Adv. Nutr. 2011, 2, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Piñero, D.J.; Connor, J.R. Iron in the Brain: An Important Contributor in Normal and Diseased States. Neuroscientist 2000, 6, 435–453. [Google Scholar] [CrossRef]
- Bettencourt, C.; Forabosco, P.; Wiethoff, S.; Heidari, M.; Johnstone, D.M.; Botia, J.A.; Collingwood, J.F.; Hardy, J.; Milward, E.A.; Ryten, M.; et al. Gene co-expression networks shed light into diseases of brain iron accumulation. Neurobiol. Dis. 2016, 87, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Obulesu, M.; Venu, R.; Somashekhar, R. Lipid peroxidation in Alzheimer’s disease: Emphasis on metal-mediated neurotoxicity. Acta Neurol. Scand. 2011, 124, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.R.; Ayton, S.; Bush, A.I. Iron and Alzheimer’s Disease: An Update on Emerging Mechanisms. J. Alzheimers Dis. 2018, 64, S379–S395. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.R.; Tuo, Q.Z.; Lei, P. Ferroptosis, a Recent Defined Form of Critical Cell Death in Neurological Disorders. J. Mol. Neurosci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Czapski, G.A.; Czubowicz, K.; Strosznajder, J.B.; Strosznajder, R.P. The Lipoxygenases: Their Regulation and Implication in Alzheimer’s Disease. Neurochem. Res. 2016, 41, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhao, W.; Yu, J.; Li, S.; Lin, L.; Chen, X. Induction of ferroptosis and mitochondrial dysfunction by oxidative stress in PC12 cells. Sci. Rep. 2018, 8, 574. [Google Scholar] [CrossRef] [PubMed]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef] [PubMed]
- Alessenko, A.V.; Shupik, M.A.; Bugrova, A.E.; Dudnik, L.B.; Shingarova, L.N.; Mikoyan, A.; Vanin, A.F. The relation between sphingomyelinase activity, lipid peroxide oxidation and NO-releasing in mice liver and brain. FEBS Lett. 2005, 579, 5571–5576. [Google Scholar] [CrossRef] [PubMed]
- Levenson, C.W.; Cutler, R.G.; Ladenheim, B.; Cadet, J.L.; Hare, J.; Mattson, M.P. Role of dietary iron restriction in a mouse model of Parkinson’s disease. Exp. Neurol. 2004, 190, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Wang, T.; Zheng, W.; Shan, Z.Y.; Teng, W.P.; Wang, Z.Y. Intranasal deferoxamine reverses iron-induced memory deficits and inhibits amyloidogenic APP processing in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Dusek, P.; Schneider, S.A.; Aaseth, J. Iron chelation in the treatment of neurodegenerative diseases. J. Trace. Elem. Med. Biol. 2016, 38, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R.; Kalinowski, D.S.; Lau, S.; Jansson, P.J.; Lovejoy, D.B. Cancer cell iron metabolism and the development of potent iron chelators as anti-tumour agents. Biochim. Biophys. Acta 2009, 1790, 702–717. [Google Scholar] [CrossRef] [PubMed]
- Aigner, E.; Theurl, I.; Theurl, M.; Lederer, D.; Haufe, H.; Dietze, O.; Strasser, M.; Datz, C.; Weiss, G. Pathways underlying iron accumulation in human nonalcoholic fatty liver disease. Am. J. Clin. Nutr. 2008, 87, 1374–1383. [Google Scholar] [CrossRef] [PubMed]
- Houschyar, K.S.; Ludtke, R.; Dobos, G.J.; Kalus, U.; Broecker-Preuss, M.; Rampp, T.; Brinkhaus, B.; Michalsen, A. Effects of phlebotomy-induced reduction of body iron stores on metabolic syndrome: Results from a randomized clinical trial. BMC Med. 2012, 10, 54. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Real, J.M.; Penarroja, G.; Castro, A.; Garcia-Bragado, F.; Hernandez-Aguado, I.; Ricart, W. Blood letting in high-ferritin type 2 diabetes: Effects on insulin sensitivity and beta-cell function. Diabetes 2002, 51, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Zacharski, L.R.; Chow, B.K.; Howes, P.S.; Shamayeva, G.; Baron, J.A.; Dalman, R.L.; Malenka, D.J.; Ozaki, C.K.; Lavori, P.W. Decreased cancer risk after iron reduction in patients with peripheral arterial disease: Results from a randomized trial. J. Natl. Cancer Inst. 2008, 100, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Edgren, G.; Tran, T.N.; Hjalgrim, H.; Rostgaard, K.; Shanwell, A.; Titlestad, K.; Wikman, A.; Norda, R.; Jersild, C.; Wideroff, L.; et al. Improving health profile of blood donors as a consequence of transfusion safety efforts. Transfusion 2007, 47, 2017–2024. [Google Scholar] [CrossRef] [PubMed]
- Edgren, G.; Reilly, M.; Hjalgrim, H.; Tran, T.N.; Rostgaard, K.; Adami, J.; Titlestad, K.; Shanwell, A.; Melbye, M.; Nyren, O. Donation frequency, iron loss, and risk of cancer among blood donors. J. Natl. Cancer Inst. 2008, 100, 572–579. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rockfield, S.; Chhabra, R.; Robertson, M.; Rehman, N.; Bisht, R.; Nanjundan, M. Links Between Iron and Lipids: Implications in Some Major Human Diseases. Pharmaceuticals 2018, 11, 113. https://doi.org/10.3390/ph11040113
Rockfield S, Chhabra R, Robertson M, Rehman N, Bisht R, Nanjundan M. Links Between Iron and Lipids: Implications in Some Major Human Diseases. Pharmaceuticals. 2018; 11(4):113. https://doi.org/10.3390/ph11040113
Chicago/Turabian StyleRockfield, Stephanie, Ravneet Chhabra, Michelle Robertson, Nabila Rehman, Richa Bisht, and Meera Nanjundan. 2018. "Links Between Iron and Lipids: Implications in Some Major Human Diseases" Pharmaceuticals 11, no. 4: 113. https://doi.org/10.3390/ph11040113
APA StyleRockfield, S., Chhabra, R., Robertson, M., Rehman, N., Bisht, R., & Nanjundan, M. (2018). Links Between Iron and Lipids: Implications in Some Major Human Diseases. Pharmaceuticals, 11(4), 113. https://doi.org/10.3390/ph11040113