Potential Treatment of Retinal Diseases with Iron Chelators

{kind=link}
Abstract
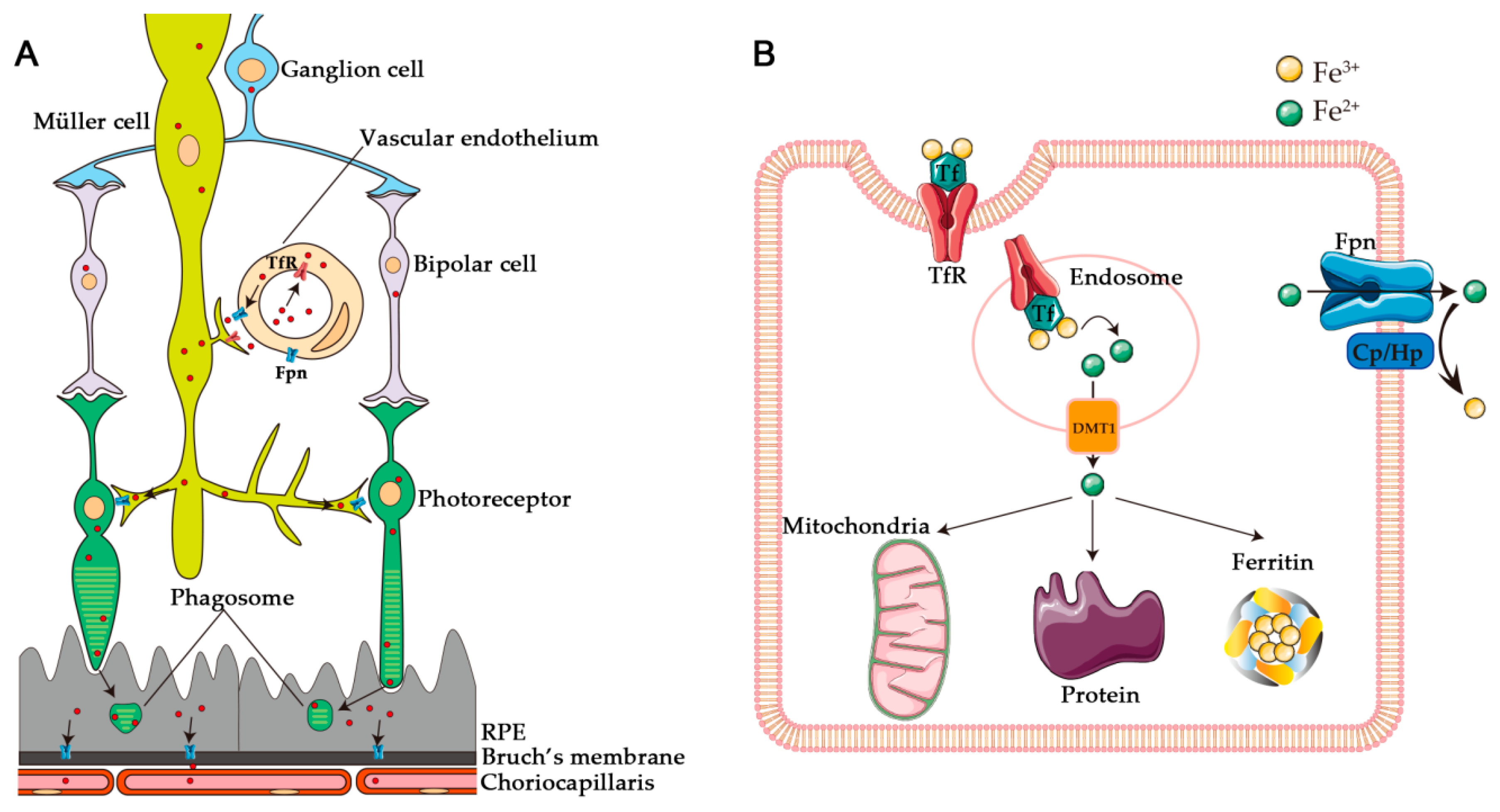
1. Introduction
2. Retinal Degeneration Resulting from Iron Dysregulation
2.1. Hereditary Iron Overload
2.1.1. Aceruloplasminemia
2.1.2. Hereditary Hemochromatosis
2.1.3. Friedreich’s Ataxia (FRDA)
2.1.4. Pantothenate Kinase Associated Neurodegeneration (PKAN)
2.2. Age-Related Macular Degeneration (AMD)
2.3. Iron Overload from Supplementation
2.4. Siderosis
2.5. Subretinal Hemorrhage
3. The Potential for Retinal Protection by Iron Chelators
3.1. Deferoxamine
3.2. Deferasirox
3.3. Deferiprone
3.4. Salicylaldehyde Isonicotinoyl Hydrazine
4. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Shichi, H. Microsomal electron transfer system of bovine retinal pigment epithelium. Exp. Eye Res. 1969, 8, 60–68. [Google Scholar] [CrossRef]
- Moiseyev, G.; Chen, Y.; Takahashi, Y.; Wu, B.X.; Ma, J.X. Rpe65 is the isomerohydrolase in the retinoid visual cycle. Proc. Natl. Acad. Sci. USA 2005, 102, 12413–12418. [Google Scholar] [CrossRef] [PubMed]
- Moiseyev, G.; Takahashi, Y.; Chen, Y.; Gentleman, S.; Redmond, T.M.; Crouch, R.K.; Ma, J.X. Rpe65 is an iron(II)-dependent isomerohydrolase in the retinoid visual cycle. J. Biol. Chem. 2006, 281, 2835–2840. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J. 1984, 219, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Harris, P.L.; Sayre, L.M.; Perry, G. Iron accumulation in alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. USA 1997, 94, 9866–9868. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shachar, D.; Riederer, P.; Youdim, M.B. Iron-melanin interaction and lipid peroxidation: Implications for parkinson’s disease. J. Neurochem. 1991, 57, 1609–1614. [Google Scholar] [CrossRef] [PubMed]
- Grolez, G.; Moreau, C.; Sablonniere, B.; Garcon, G.; Devedjian, J.C.; Meguig, S.; Gele, P.; Delmaire, C.; Bordet, R.; Defebvre, L.; et al. Ceruloplasmin activity and iron chelation treatment of patients with parkinson’s disease. BMC Neurol. 2015, 15, 74. [Google Scholar] [CrossRef] [PubMed]
- Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C.L.; Targum, S.; Bush, A.I.; Murdoch, R.; et al. Safety, efficacy, and biomarker findings of pbt2 in targeting abeta as a modifying therapy for alzheimer’s disease: A phase iia, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008, 7, 779–786. [Google Scholar] [CrossRef]
- Velasco-Sanchez, D.; Aracil, A.; Montero, R.; Mas, A.; Jimenez, L.; O’Callaghan, M.; Tondo, M.; Capdevila, A.; Blanch, J.; Artuch, R.; et al. Combined therapy with idebenone and deferiprone in patients with friedreich’s ataxia. Cerebellum 2011, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Harris, Z.L.; Takahashi, Y.; Miyajima, H.; Serizawa, M.; MacGillivray, R.T.; Gitlin, J.D. Aceruloplasminemia: Molecular characterization of this disorder of iron metabolism. Proc. Natl. Acad. Sci. USA 1995, 92, 2539–2543. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Pin, S.; Gathinji, M.; Fuchs, R.; Harris, Z.L. Aceruloplasminemia: An inherited neurodegenerative disease with impairment of iron homeostasis. Ann. N. Y. Acad. Sci. 2004, 1012, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Takahashi, S.; Kawanami, T.; Kato, T.; Sasaki, H. Retinal degeneration in hereditary ceruloplasmin deficiency. Ophthalmologica 1998, 212, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Dunaief, J.L.; Richa, C.; Franks, E.P.; Schultze, R.L.; Aleman, T.S.; Schenck, J.F.; Zimmerman, E.A.; Brooks, D.G. Macular degeneration in a patient with aceruloplasminemia, a disease associated with retinal iron overload. Ophthalmology 2005, 112, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Wolkow, N.; Song, Y.; Wu, T.D.; Qian, J.; Guerquin-Kern, J.L.; Dunaief, J.L. Aceruloplasminemia: Retinal histopathologic manifestations and iron-mediated melanosome degradation. Arch. Ophthalmol. 2011, 129, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Hahn, P.; Qian, Y.; Dentchev, T.; Chen, L.; Beard, J.; Harris, Z.L.; Dunaief, J.L. Disruption of ceruloplasmin and hephaestin in mice causes retinal iron overload and retinal degeneration with features of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 13850–13855. [Google Scholar] [CrossRef] [PubMed]
- Feder, J.N.; Gnirke, A.; Thomas, W.; Tsuchihashi, Z.; Ruddy, D.A.; Basava, A.; Dormishian, F.; Domingo, R., Jr.; Ellis, M.C.; Fullan, A.; et al. A novel mhc class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 1996, 13, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Feder, J.N.; Penny, D.M.; Irrinki, A.; Lee, V.K.; Lebron, J.A.; Watson, N.; Tsuchihashi, Z.; Sigal, E.; Bjorkman, P.J.; Schatzman, R.C. The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc. Natl. Acad. Sci. USA 1998, 95, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Non-hfe hemochromatosis. Semin. Liver Dis. 2005, 25, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Ganz, T. Regulation of iron metabolism by hepcidin. Annu. Rev. Nutr. 2006, 26, 323–342. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Hereditary hemochromatosis. Biochim. Biophys. Acta 2006, 1763, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.M.; Foos, R.Y. Ocular pathologic changes in primary hemochromatosis. Arch. Ophthalmol. 1972, 87, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.M.; Gnana-Prakasam, J.P.; Roon, P.; Smith, R.G.; Smith, S.B.; Ganapathy, V. Expression and polarized localization of the hemochromatosis gene product hfe in retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4238–4244. [Google Scholar] [CrossRef] [PubMed]
- Gnana-Prakasam, J.P.; Zhang, M.; Martin, P.M.; Atherton, S.S.; Smith, S.B.; Ganapathy, V. Expression of the iron-regulatory protein haemojuvelin in retina and its regulation during cytomegalovirus infection. Biochem. J. 2009, 419, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Gnana-Prakasam, J.P.; Tawfik, A.; Romej, M.; Ananth, S.; Martin, P.M.; Smith, S.B.; Ganapathy, V. Iron-mediated retinal degeneration in haemojuvelin-knockout mice. Biochem. J. 2012, 441, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Gnana-Prakasam, J.P.; Thangaraju, M.; Liu, K.; Ha, Y.; Martin, P.M.; Smith, S.B.; Ganapathy, V. Absence of iron-regulatory protein Hfe results in hyperproliferation of retinal pigment epithelium: Role of cystine/glutamate exchanger. Biochem. J. 2009, 424, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Hadziahmetovic, M.; Song, Y.; Ponnuru, P.; Iacovelli, J.; Hunter, A.; Haddad, N.; Beard, J.; Connor, J.R.; Vaulont, S.; Dunaief, J.L. Age-dependent retinal iron accumulation and degeneration in hepcidin knockout mice. Investig. Ophthalmol. Vis. Sci. 2011, 52, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Abrahao, A.; Pedroso, J.L.; Braga-Neto, P.; Bor-Seng-Shu, E.; de Carvalho Aguiar, P.; Barsottini, O.G. Milestones in friedreich ataxia: More than a century and still learning. Neurogenetics 2015, 16, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Porter, N.; Downes, S.M.; Fratter, C.; Anslow, P.; Nemeth, A.H. Catastrophic visual loss in a patient with friedreich ataxia. Arch. Ophthalmol. 2007, 125, 273–274. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Dickson, A.C. Iron in the hallervorden-spatz syndrome. Pediatr. Neurol. 2001, 25, 148–155. [Google Scholar] [CrossRef]
- Zhou, B.; Westaway, S.K.; Levinson, B.; Johnson, M.A.; Gitschier, J.; Hayflick, S.J. A novel pantothenate kinase gene (pank2) is defective in hallervorden-spatz syndrome. Nat. Genet. 2001, 28, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Newell, F.W.; Johnson, R.O., 2nd; Huttenlocher, P.R. Pigmentary degeneration of the retina in the hallervorden-spatz syndrome. Am. J. Ophthalmol. 1979, 88, 467–471. [Google Scholar] [CrossRef]
- Luckenbach, M.W.; Green, W.R.; Miller, N.R.; Moser, H.W.; Clark, A.W.; Tennekoon, G. Ocular clinicopathologic correlation of hallervorden-spatz syndrome with acanthocytosis and pigmentary retinopathy. Am. J. Ophthalmol. 1983, 95, 369–382. [Google Scholar] [CrossRef]
- Kuo, Y.M.; Duncan, J.L.; Westaway, S.K.; Yang, H.; Nune, G.; Xu, E.Y.; Hayflick, S.J.; Gitschier, J. Deficiency of pantothenate kinase 2 (pank2) in mice leads to retinal degeneration and azoospermia. Hum. Mol. Genet. 2005, 14, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.S.; Mitchell, P.; Seddon, J.M.; Holz, F.G.; Wong, T.Y. Age-related macular degeneration. Lancet 2012, 379, 1728–1738. [Google Scholar] [CrossRef]
- Coleman, H.R.; Chan, C.C.; Ferris, F.L., 3rd; Chew, E.Y. Age-related macular degeneration. Lancet 2008, 372, 1835–1845. [Google Scholar] [CrossRef]
- Zarbin, M.A. Current concepts in the pathogenesis of age-related macular degeneration. Arch. Ophthalmol. 2004, 122, 598–614. [Google Scholar] [CrossRef] [PubMed]
- Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: Areds report No. 8. Arch. Ophthalmol. 2001, 119, 1417–1436. [Google Scholar] [CrossRef]
- Chew, E.Y.; Clemons, T.E.; Agron, E.; Sperduto, R.D.; Sangiovanni, J.P.; Kurinij, N.; Davis, M.D.; Age-Related Eye Disease Study Research Group. Long-term effects of vitamins C and E, beta-carotene, and zinc on age-related macular degeneration: Areds report No. 35. Ophthalmology 2013, 120, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Hahn, P.; Milam, A.H.; Dunaief, J.L. Maculas affected by age-related macular degeneration contain increased chelatable iron in the retinal pigment epithelium and bruch’s membrane. Arch. Ophthalmol. 2003, 121, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Biesemeier, A.; Yoeruek, E.; Eibl, O.; Schraermeyer, U. Iron accumulation in bruch’s membrane and melanosomes of donor eyes with age-related macular degeneration. Exp. Eye Res. 2015, 137, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Dentchev, T.; Hahn, P.; Dunaief, J.L. Strong labeling for iron and the iron-handling proteins ferritin and ferroportin in the photoreceptor layer in age-related macular degeneration. Arch. Ophthalmol. 2005, 123, 1745–1746. [Google Scholar] [CrossRef] [PubMed]
- Junemann, A.G.; Stopa, P.; Michalke, B.; Chaudhri, A.; Reulbach, U.; Huchzermeyer, C.; Schlotzer-Schrehardt, U.; Kruse, F.E.; Zrenner, E.; Rejdak, R. Levels of aqueous humor trace elements in patients with non-exsudative age-related macular degeneration: A case-control study. PLoS ONE 2013, 8, e56734. [Google Scholar] [CrossRef] [PubMed]
- Hahn, P.; Ying, G.S.; Beard, J.; Dunaief, J.L. Iron levels in human retina: Sex difference and increase with age. Neuroreport 2006, 17, 1803–1806. [Google Scholar] [CrossRef] [PubMed]
- Ugarte, M.; Grime, G.W.; Lord, G.; Geraki, K.; Collingwood, J.F.; Finnegan, M.E.; Farnfield, H.; Merchant, M.; Bailey, M.J.; Ward, N.I.; et al. Concentration of various trace elements in the rat retina and their distribution in different structures. Metallomics 2012, 4, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Chowers, I.; Wong, R.; Dentchev, T.; Farkas, R.H.; Iacovelli, J.; Gunatilaka, T.L.; Medeiros, N.E.; Presley, J.B.; Campochiaro, P.A.; Curcio, C.A.; et al. The iron carrier transferrin is upregulated in retinas from patients with age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2135–2140. [Google Scholar] [CrossRef] [PubMed]
- Fraenkel, P.G. Anemia of inflammation: A review. Med. Clin. N. Am. 2017, 101, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Fan, M.; Du, F.; Gong, Q.; Bi, Z.G.; Zhu, Z.J.; Zhu, L.L.; Ke, Y. Hypoxic preconditioning increases iron transport rate in astrocytes. Biochim. Biophys. Acta 2012, 1822, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, A.M.; Pereira, C.F.; Porto, G.; Arosa, F.A. Red blood cells upregulate cytoprotective proteins and the labile iron pool in dividing human t cells despite a reduction in oxidative stress. Free Radic. Biol. Med. 2003, 35, 1404–1416. [Google Scholar] [CrossRef] [PubMed]
- Bhisitkul, R.B.; Winn, B.J.; Lee, O.T.; Wong, J.; Pereira Dde, S.; Porco, T.C.; He, X.; Hahn, P.; Dunaief, J.L. Neuroprotective effect of intravitreal triamcinolone acetonide against photoreceptor apoptosis in a rabbit model of subretinal hemorrhage. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4071–4077. [Google Scholar] [CrossRef] [PubMed]
- McLean, E.; Cogswell, M.; Egli, I.; Wojdyla, D.; de Benoist, B. Worldwide prevalence of anaemia, who vitamin and mineral nutrition information system, 1993-2005. Public Health Nutr. 2009, 12, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Stevens, G.A.; Finucane, M.M.; De-Regil, L.M.; Paciorek, C.J.; Flaxman, S.R.; Branca, F.; Pena-Rosas, J.P.; Bhutta, Z.A.; Ezzati, M.; Nutrition Impact Model Study Group. Global, regional, and national trends in haemoglobin concentration and prevalence of total and severe anaemia in children and pregnant and non-pregnant women for 1995-2011: A systematic analysis of population-representative data. Lancet Glob. Health 2013, 1, e16–e25. [Google Scholar] [CrossRef]
- Kassebaum, N.J.; Jasrasaria, R.; Naghavi, M.; Wulf, S.K.; Johns, N.; Lozano, R.; Regan, M.; Weatherall, D.; Chou, D.P.; Eisele, T.P.; et al. A systematic analysis of global anemia burden from 1990 to 2010. Blood 2014, 123, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.B.; Hurrell, R.F. Nutritional iron deficiency. Lancet 2007, 370, 511–520. [Google Scholar] [CrossRef]
- Kumar, P.; Nag, T.C.; Jha, K.A.; Dey, S.K.; Kathpalia, P.; Maurya, M.; Gupta, C.L.; Bhatia, J.; Roy, T.S.; Wadhwa, S. Experimental oral iron administration: Histological investigations and expressions of iron handling proteins in rat retina with aging. Toxicology 2017, 392, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Bhoiwala, D.L.; Song, Y.; Cwanger, A.; Clark, E.; Zhao, L.L.; Wang, C.; Li, Y.; Song, D.; Dunaief, J.L. Cd1 mouse retina is shielded from iron overload caused by a high iron diet. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5344–5352. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.J.; Pollock, J.R.; Bingham, S.A. Haem, not protein or inorganic iron, is responsible for endogenous intestinal n-nitrosation arising from red meat. Cancer Res. 2003, 63, 2358–2360. [Google Scholar] [PubMed]
- Chong, E.W.; Simpson, J.A.; Robman, L.D.; Hodge, A.M.; Aung, K.Z.; English, D.R.; Giles, G.G.; Guymer, R.H. Red meat and chicken consumption and its association with age-related macular degeneration. Am. J. Epidemiol. 2009, 169, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Onken, J.E.; Bregman, D.B.; Harrington, R.A.; Morris, D.; Acs, P.; Akright, B.; Barish, C.; Bhaskar, B.S.; Smith-Nguyen, G.N.; Butcher, A.; et al. A multicenter, randomized, active-controlled study to investigate the efficacy and safety of intravenous ferric carboxymaltose in patients with iron deficiency anemia. Transfusion 2014, 54, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Vadhan-Raj, S.; Strauss, W.; Ford, D.; Bernard, K.; Boccia, R.; Li, J.; Allen, L.F. Efficacy and safety of iv ferumoxytol for adults with iron deficiency anemia previously unresponsive to or unable to tolerate oral iron. Am. J. Hematol. 2014, 89, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, Y.; Song, D.; Song, Y.; Theurl, M.; Wang, C.; Cwanger, A.; Su, G.; Dunaief, J.L. A high serum iron level causes mouse retinal iron accumulation despite an intact blood-retinal barrier. Am. J. Pathol. 2014, 184, 2862–2867. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Kanu, L.N.; Li, Y.; Kelly, K.L.; Bhuyan, R.K.; Aleman, T.; Morgan, J.I.; Dunaief, J.L. Amd-like retinopathy associated with intravenous iron. Exp. Eye Res. 2016, 151, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Bhoiwala, D.L.; Dunaief, J.L. Retinal abnormalities in beta-thalassemia major. Surv. Ophthalmol. 2016, 61, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Cibis, P.A.; Yamashita, T.; Rodriguez, F. Clinical aspects of ocular siderosis and hemosiderosis. AMA Arch. Ophthalmol. 1959, 62, 180–187. [Google Scholar] [PubMed]
- Talamo, J.H.; Topping, T.M.; Maumenee, A.E.; Green, W.R. Ultrastructural studies of cornea, iris and lens in a case of siderosis bulbi. Ophthalmology 1985, 92, 1675–1680. [Google Scholar] [CrossRef]
- Sneed, S.R. Ocular siderosis. Arch. Ophthalmol. 1988, 106, 997. [Google Scholar] [CrossRef] [PubMed]
- Knave, B. Long-term changes in retinal function induced by short, high intensity flashes. Experientia 1969, 25, 379–380. [Google Scholar] [CrossRef] [PubMed]
- Masciulli, L.; Anderson, D.R.; Charles, S. Experimental ocular siderosis in the squirrel monkey. Am. J. Ophthalmol. 1972, 74, 638–661. [Google Scholar] [CrossRef]
- Declercq, S.S.; Meredith, P.C.; Rosenthal, A.R. Experimental siderosis in the rabbit: Correlation between electroretinography and histopathology. Arch. Ophthalmol. 1977, 95, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Gillies, A.; Lahav, M. Absorption of retinal and subretinal hemorrhages. Ann. Ophthalmol. 1983, 15, 1068–1074. [Google Scholar] [PubMed]
- Glatt, H.; Machemer, R. Experimental subretinal hemorrhage in rabbits. Am. J. Ophthalmol. 1982, 94, 762–773. [Google Scholar] [CrossRef]
- Youssef, T.A.; Trese, M.T.; Hartzer, M.; Mahgoub, M.; Raza, H.; Azrak, M.; Allredge, C. Deferoxamine reduces retinal toxicity from subretinal blood. Investig. Ophthalmol. Vis. Sci. 2002, 43, U845. [Google Scholar]
- Ito, T.; Nakano, M.; Yamamoto, Y.; Hiramitsu, T.; Mizuno, Y. Hemoglobin-induced lipid peroxidation in the retina: A possible mechanism for macular degeneration. Arch. Biochem. Biophys. 1995, 316, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Hunt, R.C.; Handy, I.; Smith, A. Heme-mediated reactive oxygen species toxicity to retinal pigment epithelial cells is reduced by hemopexin. J. Cell. Physiol. 1996, 168, 81–86. [Google Scholar] [CrossRef]
- Zheng, H.; Youdim, M.B.; Weiner, L.M.; Fridkin, M. Novel potential neuroprotective agents with both iron chelating and amino acid-based derivatives targeting central nervous system neurons. Biochem. Pharmacol. 2005, 70, 1642–1652. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R. Novel chelators for central nervous system disorders that involve alterations in the metabolism of iron and other metal ions. Ann. N. Y. Acad. Sci. 2004, 1012, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Brittenham, G.M. Iron-chelating therapy for transfusional iron overload. N. Engl. J. Med. 2011, 364, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.C.; Marcus, R.E.; Hungerford, J.L.; Miller, M.H.; Arden, G.B.; Huehns, E.R. Ocular toxicity of high-dose intravenous desferrioxamine. Lancet 1983, 2, 181–184. [Google Scholar] [CrossRef]
- Lakhanpal, V.; Schocket, S.S.; Jiji, R. Deferoxamine (desferal)-induced toxic retinal pigmentary degeneration and presumed optic neuropathy. Ophthalmology 1984, 91, 443–451. [Google Scholar] [CrossRef]
- Bene, C.; Manzler, A.; Bene, D.; Kranias, G. Irreversible ocular toxicity from single “challenge” dose of deferoxamine. Clin. Nephrol. 1989, 31, 45–48. [Google Scholar] [PubMed]
- Obolensky, A.; Berenshtein, E.; Lederman, M.; Bulvik, B.; Alper-Pinus, R.; Yaul, R.; Deleon, E.; Chowers, I.; Chevion, M.; Banin, E. Zinc-desferrioxamine attenuates retinal degeneration in the rd10 mouse model of retinitis pigmentosa. Free Radic. Biol. Med. 2011, 51, 1482–1491. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.L.; Lam, S.; Tso, M.O. Desferrioxamine ameliorates retinal photic injury in albino rats. Curr. Eye Res. 1991, 10, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Vanorden, H.E.; Hagemann, T.M. Deferasirox--an oral agent for chronic iron overload. Ann. Pharmacother. 2006, 40, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Galanello, R.; Piga, A.; Alberti, D.; Rouan, M.C.; Bigler, H.; Sechaud, R. Safety, tolerability, and pharmacokinetics of icl670, a new orally active iron-chelating agent in patients with transfusion-dependent iron overload due to beta-thalassemia. J. Clin. Pharmacol. 2003, 43, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Porter, J.; El-Beshlawy, A.; Li, C.K.; Seymour, J.F.; Elalfy, M.; Gattermann, N.; Giraudier, S.; Lee, J.W.; Chan, L.L.; et al. Tailoring iron chelation by iron intake and serum ferritin: The prospective epic study of deferasirox in 1744 patients with transfusion-dependent anemias. Haematologica 2010, 95, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Cohen, A.; Piga, A.; Bejaoui, M.; Perrotta, S.; Agaoglu, L.; Aydinok, Y.; Kattamis, A.; Kilinc, Y.; Porter, J.; et al. A phase 3 study of deferasirox (icl670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood 2006, 107, 3455–3462. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Bejaoui, M.; Agaoglu, L.; Canatan, D.; Capra, M.; Cohen, A.; Drelichman, G.; Economou, M.; Fattoum, S.; Kattamis, A.; et al. Iron chelation with deferasirox in adult and pediatric patients with thalassemia major: Efficacy and safety during 5 years’ follow-up. Blood 2011, 118, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Arora, E.; Singh, H. Hypersensitivity reaction with deferasirox. J. Pharmacol. Pharmacother. 2015, 6, 105–106. [Google Scholar] [CrossRef] [PubMed]
- Gartaganis, S.; Ismiridis, K.; Papageorgiou, O.; Beratis, N.G.; Papanastasiou, D. Ocular abnormalities in patients with beta thalassemia. Am. J. Ophthalmol. 1989, 108, 699–703. [Google Scholar] [CrossRef]
- Sorcinelli, R.; Sitzia, A.; Figus, A.; Lai, M.E. Ocular findings in beta-thalassemia. Metab. Pediatr. Syst. Ophthalmol. 1990, 13, 23–25. [Google Scholar]
- Bloomfield, S.E.; Markenson, A.L.; Miller, D.R.; Peterson, C.M. Lens opacities in thalassemia. J. Pediatr. Ophthalmol. Strabismus 1978, 15, 154–156. [Google Scholar] [PubMed]
- De Virgiliis, S.; Congia, M.; Turco, M.P.; Frau, F.; Dessi, C.; Argiolu, F.; Sorcinelli, R.; Sitzia, A.; Cao, A. Depletion of trace elements and acute ocular toxicity induced by desferrioxamine in patients with thalassaemia. Arch. Dis. Child. 1988, 63, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Galanello, R. Deferiprone in the treatment of transfusion-dependent thalassemia: A review and perspective. Ther. Clin. Risk Manag. 2007, 3, 795–805. [Google Scholar] [PubMed]
- Hadziahmetovic, M.; Song, Y.; Wolkow, N.; Iacovelli, J.; Grieco, S.; Lee, J.; Lyubarsky, A.; Pratico, D.; Connelly, J.; Spino, M.; et al. The oral iron chelator deferiprone protects against iron overload-induced retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Zhao, L.; Li, Y.; Hadziahmetovic, M.; Song, Y.; Connelly, J.; Spino, M.; Dunaief, J.L. The oral iron chelator deferiprone protects against systemic iron overload-induced retinal degeneration in hepcidin knockout mice. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4525–4532. [Google Scholar] [CrossRef] [PubMed]
- Hadziahmetovic, M.; Pajic, M.; Grieco, S.; Song, Y.; Song, D.; Li, Y.; Cwanger, A.; Iacovelli, J.; Chu, S.; Ying, G.S.; et al. The oral iron chelator deferiprone protects against retinal degeneration induced through diverse mechanisms. Transl. Vis. Sci. Technol. 2012, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Song, Y.; Hadziahmetovic, M.; Zhong, Y.; Dunaief, J.L. Systemic administration of the iron chelator deferiprone protects against light-induced photoreceptor degeneration in the mouse retina. Free Radic. Biol. Med. 2012, 53, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Jansova, H.; Bures, J.; Machacek, M.; Haskova, P.; Jirkovska, A.; Roh, J.; Wang, Q.; Franz, K.J.; Kovarikova, P.; Simunek, T. Characterization of cytoprotective and toxic properties of iron chelator sih, prochelator bsih and their degradation products. Toxicology 2016, 350–352, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Haskova, P.; Kovarikova, P.; Koubkova, L.; Vavrova, A.; Mackova, E.; Simunek, T. Iron chelation with salicylaldehyde isonicotinoyl hydrazone protects against catecholamine autoxidation and cardiotoxicity. Free Radic. Biol. Med. 2011, 50, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Sterba, M.; Popelova, O.; Simunek, T.; Mazurova, Y.; Potacova, A.; Adamcova, M.; Guncova, I.; Kaiserova, H.; Palicka, V.; Ponka, P.; et al. Iron chelation-afforded cardioprotection against chronic anthracycline cardiotoxicity: A study of salicylaldehyde isonicotinoyl hydrazone (sih). Toxicology 2007, 235, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Lukinova, N.; Iacovelli, J.; Dentchev, T.; Wolkow, N.; Hunter, A.; Amado, D.; Ying, G.S.; Sparrow, J.R.; Dunaief, J.L. Iron chelation protects the retinal pigment epithelial cell line arpe-19 against cell death triggered by diverse stimuli. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1440–1447. [Google Scholar] [CrossRef] [PubMed]
- Caro, A.A.; Commissariat, A.; Dunn, C.; Kim, H.; Garcia, S.L.; Smith, A.; Strang, H.; Stuppy, J.; Desrochers, L.P.; Goodwin, T.E. Prooxidant and antioxidant properties of salicylaldehyde isonicotinoyl hydrazone iron chelators in hepg2 cells. Biochim. Biophys. Acta 2015, 1850, 2256–2264. [Google Scholar] [CrossRef] [PubMed]
- Buss, J.L.; Ponka, P. Hydrolysis of pyridoxal isonicotinoyl hydrazone and its analogs. Biochim. Biophys. Acta 2003, 1619, 177–186. [Google Scholar] [CrossRef]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shu, W.; Dunaief, J.L. Potential Treatment of Retinal Diseases with Iron Chelators. Pharmaceuticals 2018, 11, 112. https://doi.org/10.3390/ph11040112
Shu W, Dunaief JL. Potential Treatment of Retinal Diseases with Iron Chelators. Pharmaceuticals. 2018; 11(4):112. https://doi.org/10.3390/ph11040112
Chicago/Turabian StyleShu, Wanting, and Joshua L. Dunaief. 2018. "Potential Treatment of Retinal Diseases with Iron Chelators" Pharmaceuticals 11, no. 4: 112. https://doi.org/10.3390/ph11040112
APA StyleShu, W., & Dunaief, J. L. (2018). Potential Treatment of Retinal Diseases with Iron Chelators. Pharmaceuticals, 11(4), 112. https://doi.org/10.3390/ph11040112