Protective Role of Histidine Supplementation Against Oxidative Stress Damage in the Management of Anemia of Chronic Kidney Disease



Abstract
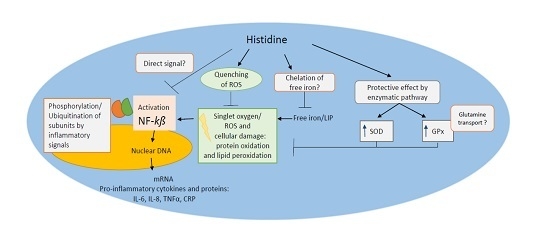
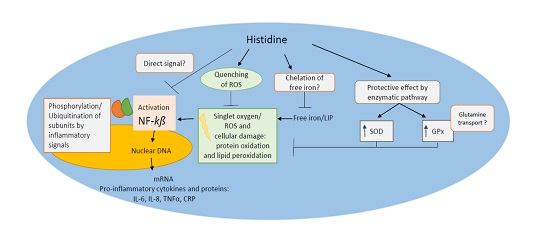{kind=link}
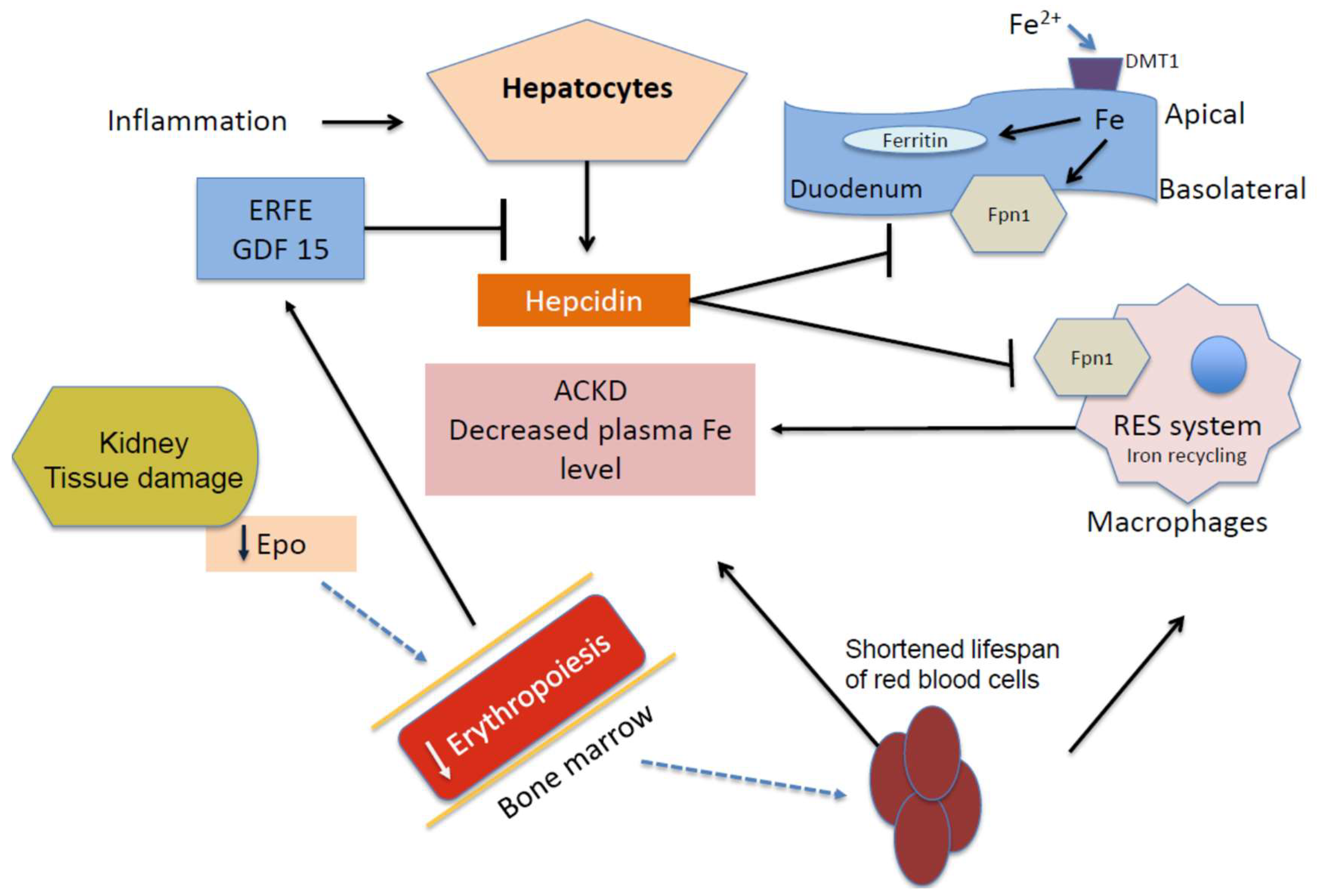{kind=link}
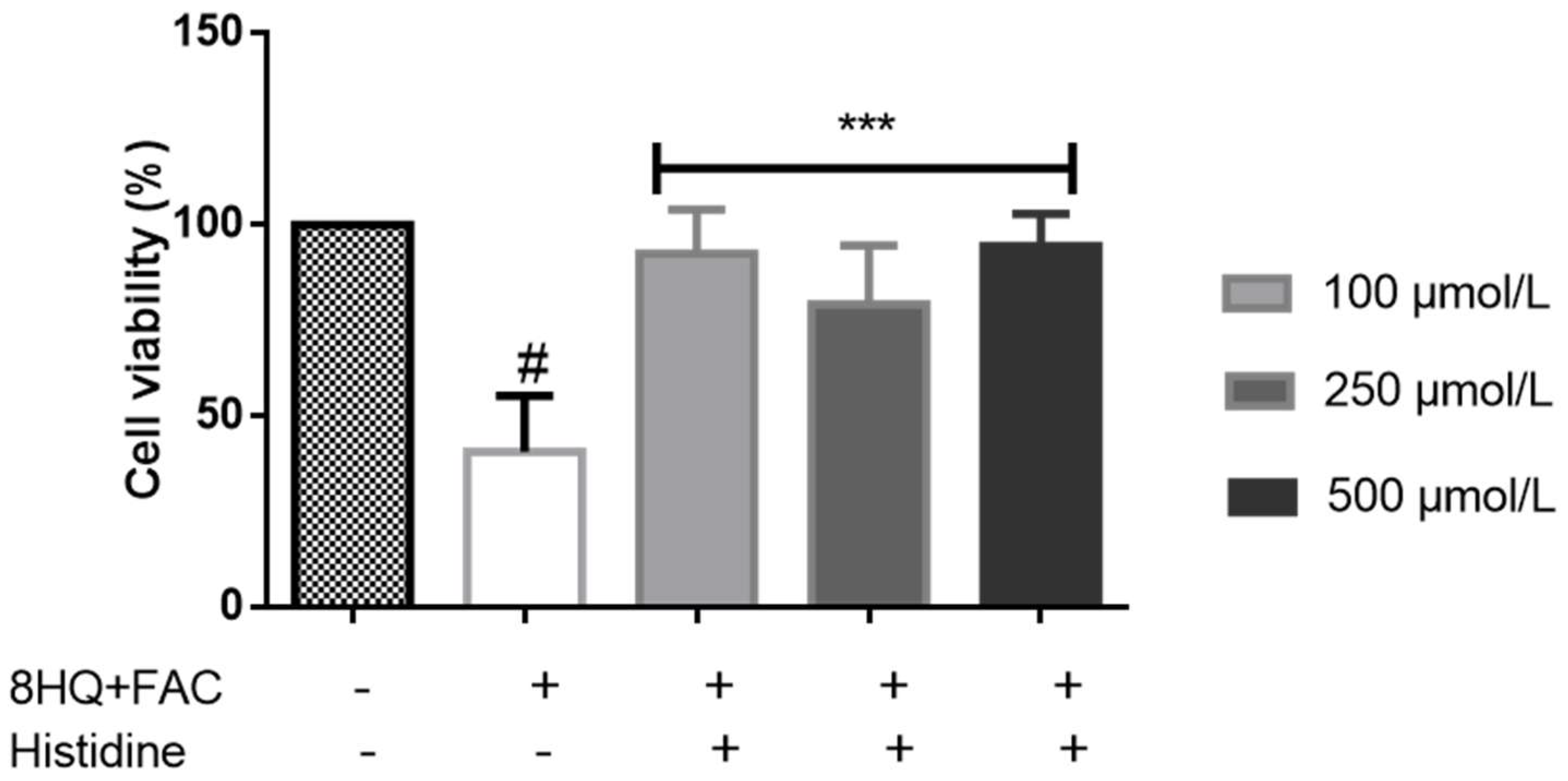{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Anemia of Chronic Kidney Disease (ACKD)
3. Treatment of ACKD
4. Iron, Oxidative Stress, and Anemia
5. Cytoprotective Functions of Histidine against Iron Toxicity during the Treatment of ACKD
6. Antioxidant Function of Histidine against Oxidative Stress
7. Metal Chelation Capacity of Histidine
8. Anti-Inflammatory Potential of Histidine
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stauffer, M.E.; Fan, T. Prevalence of anemia in chronic kidney disease in the united states. PLoS ONE 2014, 9, e84943. [Google Scholar] [CrossRef] [PubMed]
- Macdougall, I.C.; Bircher, A.J.; Eckardt, K.-U.; Obrador, G.T.; Pollock, C.A.; Stenvinkel, P.; Swinkels, D.W.; Wanner, C.; Weiss, G.; Chertow, G.M. Iron management in chronic kidney disease: Conclusions from a “kidney disease: Improving global outcomes” (KDIGO) controversies conference. Kidney Int. 2016, 89, 28–39. [Google Scholar] [CrossRef] [PubMed]
- National Health System. Diabetes with Kidney Disease: Key Facts; Diabetes Kindey Care, Ed.; National Health System England: London, UK, 2011. [Google Scholar]
- McClellan, W.; Aronoff, S.L.; Bolton, W.K.; Hood, S.; Lorber, D.L.; Tang, K.L.; Tse, T.F.; Wasserman, B.; Leiserowitz, M. The prevalence of anemia in patients with chronic kidney disease. Curr. Med. Res. Opin. 2004, 20, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.S.; Kazmi, W.H.; Abichandani, R.; Tighiouart, H.; Pereira, B.J.G.; Kausz, A.T. Health care utilization among patients with chronic kidney disease. Kidney Int. 2002, 62, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Jing, Z.; Wei-jie, Y.; Nan, Z.; Yi, Z.; Ling, W. Hemoglobin targets for chronic kidney disease patients with anemia: A systematic review and meta-analysis. PLoS ONE 2012, 7, e43655. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.; Singer, J.; Thompson, C.R.; Ross, H.; Lewis, M. Prevalent left ventricular hypertrophy in the predialysis population: Identifying opportunities for intervention. Am. J. Kidney Dis. 1996, 27, 347–354. [Google Scholar] [CrossRef]
- Locatelli, F.; Pisoni, R.L.; Combe, C.; Bommer, J.; Andreucci, V.E.; Piera, L.; Greenwood, R.; Feldman, H.I.; Port, F.K.; Held, P.J. Anaemia in haemodialysis patients of five european countries: Association with morbidity and mortality in the dialysis outcomes and practice patterns study (DOPPS). Nephrol. Dial. Transplant. 2004, 19, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.A.; Chin, H.; Blalock, S.; Joy, M.S. Predialysis chronic kidney disease: Evaluation of quality of life in clinic patients receiving comprehensive anemia care. Res. Soc. Adm. Pharm. 2009, 5, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N.; Takasawa, K. Impact of inflammation on ferritin, hepcidin and the management of iron deficiency anemia in chronic kidney disease. Nutrients 2018, 10, 1173. [Google Scholar] [CrossRef] [PubMed]
- Macdougall, I.C. Anaemia of chronic kidney disease. Medicine 2007, 35, 457–460. [Google Scholar] [CrossRef]
- Babitt, J.L.; Lin, H.Y. Mechanisms of anemia in CKD. J. Am. Soc. Nephrol. 2012, 23, 1631–1634. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Bárány, P.; Covic, A.; De Francisco, A.; Del Vecchio, L.; Goldsmith, D.; Hörl, W.; London, G.; Vanholder, R.; Van Biesen, W.; et al. Kidney disease: Improving global outcomes guidelines on anaemia management in chronic kidney disease: A european renal best practice position statement. Nephrol. Dial. Transplant. 2013, 28, 1346–1359. [Google Scholar] [CrossRef] [PubMed]
- Ofsthun, N.; Labrecque, J.; Lacson, E.; Keen, M.; Lazarus, J.M. The effects of higher hemoglobin levels on mortality and hospitalization in hemodialysis patients. Kidney Int. 2003, 63, 1908–1914. [Google Scholar] [CrossRef] [PubMed]
- Phrommintikul, A.; Haas, S.J.; Elsik, M.; Krum, H. Mortality and target haemoglobin concentrations in anaemic patients with chronic kidney disease treated with erythropoietin: A meta-analysis. Lancet 2007, 369, 381–388. [Google Scholar] [CrossRef]
- Abramson, J.L.; Jurkovitz, C.T.; Vaccarino, V.; Weintraub, W.S.; Mcclellan, W. Chronic kidney disease, anemia, and incident stroke in a middle-aged, community-based population: The aric study. Kidney Int. 2003, 64, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Malik, J. Heart disease in chronic kidney disease – Review of the mechanisms and the role of dialysis access. J. Vasc. Access 2018, 19, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Dull, R.B.; Davis, E. Heme iron polypeptide for the management of anaemia of chronic kidney disease. J. Clin. Pharm. Ther. 2015, 40, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, H.; Jiang, X.; Shi, W.; Shen, Z.; Li, M. Hepcidin is directly regulated by insulin and plays an important role in iron overload in streptozotocin-induced diabetic rats. Diabetes 2014, 63, 1506–1518. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Goodnough, L.T. Anemia of chronic disease. N. Engl. J. Med. 2005, 352, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- De Francisco, A.L.M.; Stenvinkel, P.; Vaulont, S. Inflammation and its impact on anaemia in chronic kidney disease: From haemoglobin variability to hyporesponsiveness. NDT Plus 2009, 2, i18–i26. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, D.M. Inflammation in chronic kidney disease: Role in the progression of renal and cardiovascular disease. Pediatr. Nephrol. 2009, 24, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.A.; White, C.T. Hepcidin in anemia of chronic kidney disease: Review for the pediatric nephrologist. Pediatr. Nephrol. 2012, 27, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, D.A.; Roy, C.N.; Fleming, M.D.; Loda, M.F.; Wolfsdorf, J.I.; Andrews, N.C. Inappropriate expression of hepcidin is associated with iron refractory anemia: Implications for the anemia of chronic disease. Blood 2002, 100, 3776–3781. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; McVey Ward, D.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Young, B.; Zaritsky, J. Hepcidin for clinicians. Clin. J. Am. Soc. Nephrol. 2009, 4, 1384–1387. [Google Scholar] [CrossRef] [PubMed]
- Goodnough, L.T.; Nemeth, E.; Ganz, T. Detection, evaluation, and management of iron-restricted erythropoiesis. Blood 2010, 116, 4754–4761. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Fishbane, S.; Block, G.A.; Macdougall, I.C. Targeting hypoxia-inducible factors for the treatment of anemia in chronic kidney disease patients. Am. J. Nephrol. 2017, 45, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Gummer, J.; Trengove, R.; Pascoe, E.M.; Badve, S.V.; Cass, A.; Clarke, P.; McDonald, S.P.; Morrish, A.T.; Pedagogos, E.; Perkovic, V.; et al. Association between serum hepcidin-25 and primary resistance to erythropoiesis-stimulating agents in chronic kidney disease: A secondary analysis of the hero trial. Nephrology 2017, 22, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, C.; Da Silva, J.L.; Bruneval, P.; Fournier, J.G.; Wendling, F.; Casadevall, N.; Camilleri, J.P.; Bariety, J.; Varet, B.; Tambourin, P. Peritubular cells are the site of erythropoietin synthesis in the murine hypoxic kidney. J. Clin. Investig. 1988, 81, 620–623. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Osmond, M.K.; Pugh, C.W.; Heryet, A.; Nicholls, L.G.; Tan, C.C.; Doe, B.G.; Ferguson, D.J.P.; Johnson, M.H.; Ratcliffe, P.J. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int. 1993, 44, 1149–1162. [Google Scholar] [CrossRef] [PubMed]
- Koury, M.J.; Haase, V.H. Anaemia in kidney disease: Harnessing hypoxia responses for therapy. Nat. Rev. Nephrol. 2015, 11, 394–410. [Google Scholar] [CrossRef] [PubMed]
- Kopple, J.D. The national kidney foundation k/doqi clinical practice guidelines for dietary protein intake for chronic dialysis patients. Am. J. Kidney Dis. 2001, 38, S68–S73. [Google Scholar] [CrossRef] [PubMed]
- Apsangikar, P.; Chaudhry, S.; Naik, M.; Deoghare, S.; Joseph, J. Comparative efficacy and safety of biosimilar darbepoetin alfa in adults with anemia of chronic kidney disease. Indian J. Transplant. 2018, 12, 30–34. [Google Scholar] [CrossRef]
- Locatelli, F.; Covic, A.; Eckardt, K.-U.; Wiecek, A.; Vanholder, R. Anaemia management in patients with chronic kidney disease: A position statement by the anaemiaworking group of european renal best practice (ERBP). Nephrol Dial Transplant. 2009, 24, 348–354. [Google Scholar] [CrossRef] [PubMed]
- National Clinical Guideline Centre. Anaemia Management in Chronic Kidney Disease: Partial Update 2015; NICE Guideline, No. 8. Royal College of Physicians (UK); National Clinical Guideline Centre: London, UK, 2015. [Google Scholar]
- Singh, A.K.; Szczech, L.; Tang, K.L.; Barnhart, H.; Sapp, S.; Wolfson, M.; Reddan, D. Correction of anemia with epoetin alfa in chronic kidney disease. N. Engl. J. Med. 2006, 355, 2085–2098. [Google Scholar] [CrossRef] [PubMed]
- Szczech, L.A.; Barnhart, H.X.; Inrig, J.K.; Reddan, D.N.; Sapp, S.; Califf, R.M.; Patel, U.D.; Singh, A.K. Secondary analysis of the choir trial epoetin-alpha dose and achieved hemoglobin outcomes. Kidney Int. 2008, 74, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K. The controversy surrounding hemoglobin and erythropoiesis-stimulating agents: What should we do now? Am. J. Kidney Dis. 2008, 52, S5–S13. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Zhou, X.J. Potential mechanisms of adverse outcomes in trials of anemia correction with erythropoietin in chronic kidney disease. Nephrol. Dial. Transplant. 2009, 24, 1082–1088. [Google Scholar] [CrossRef] [PubMed]
- Krapf, R.; Hulter, H.N. Arterial hypertension induced by erythropoietin and erythropoiesis-stimulating agents (ESA). Clin. J. Am. Soc. Nephrol. 2009, 4, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.A.; Burdmann, E.A.; Chen, C.-Y.; Cooper, M.E.; de Zeeuw, D.; Eckardt, K.-U.; Feyzi, J.M.; Ivanovich, P.; Kewalramani, R.; Levey, A.S.; et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N. Engl. J. Med. 2009, 361, 2019–2032. [Google Scholar] [CrossRef] [PubMed]
- Coyne, D.W.; Kapoian, T.; Suki, W.; Singh, A.K.; Moran, J.E.; Dahl, N.V.; Rizkala, A.R. Ferric gluconate is highly efficacious in anemic hemodialysis patients with high serum ferritin and low transferrin saturation: Results of the dialysis patients’ response to iv iron with elevated ferritin (drive) study. J. Am. Soc. Nephrol. 2007, 18, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Kidney Disease Improving Global Outcomes (KDIGO). Chapter 2: Use of iron to treat anemia in CKD. Kidney Int. Suppl. 2012, 2, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, S.; Belo, L.; Reis, F.; Santos-Silva, A. Iron therapy in chronic kidney disease: Recent changes, benefits and risks. Blood Rev. 2016, 30, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, L.; Longhi, S.; Locatelli, F. Safety concerns about intravenous iron therapy in patients with chronic kidney disease. Clin. Kidney J. 2016, 9, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Bailie, G.R.; Larkina, M.; Goodkin, D.A.; Li, Y.; Pisoni, R.L.; Bieber, B.; Mason, N.; Tong, L.; Locatelli, F.; Marshall, M.R.; et al. Data from the dialysis outcomes and practice patterns study validate an association between high intravenous iron doses and mortality. Kidney Int 2015, 87, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Slotki, I.; Cabantchik, Z.I. The labile side of iron supplementation in CKD. J. Am. Soc. Nephrol. 2015, 26, 2612–2619. [Google Scholar] [CrossRef] [PubMed]
- Macdougall, I.C.; Geisser, P. Use of intravenous iron supplementation in chronic kidney disease. An update. Iran. J. Kidney Dis. 2013, 7, 9–22. [Google Scholar] [PubMed]
- Shah, S.V.; Rajapurkar, M.M.; Baliga, R. The role of catalytic iron in acute kidney injury. Clin. J. Am. Soc. Nephrol. 2011, 6, 2329–2331. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.; Sinclair, A.; Collins, H.; Rice, L.; Jelkmann, W. Progress in detecting cell-surface protein receptors: The erythropoietin receptor example. Ann. Hematol. 2014, 93, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.R.; Zhang, X.; Nixon, A.M.; Webb, B.; Perno, J.R. Comparative evaluation of nephrotoxicity and management by macrophages of intravenous pharmaceutical iron formulations. PLoS ONE 2015, 10, e0125272. [Google Scholar] [CrossRef] [PubMed]
- Geisser, P.; Burckhardt, S. The pharmacokinetics and pharmacodynamics of iron preparations. Pharmaceutics 2011, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Kalra, P.A.; Bhandari, S. Efficacy and safety of iron isomaltoside (monofer(®)) in the management of patients with iron deficiency anemia. Int. J. Nephrol. Renov. Dis. 2016, 9, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Pratt, R.D.; Crumbliss, A.L. Ferrous iron content of intravenous iron formulations. BioMetals 2016, 29, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Mazzaferro, S.; Yee, J. Iron therapy challenges for the treatment of nondialysis ckd patients. Clin. J. Am. Soc. Nephrol. 2016, 11, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Wish, J.B.; Aronoff, G.R.; Bacon, B.R.; Brugnara, C.; Eckardt, K.U.; Ganz, T.; Macdougall, I.C.; Núñez, J.; Perahia, A.J.; Wood, J.C. Positive iron balance in chronic kidney disease: How much is too much and how to tell? Am. J. Nephrol. 2018, 47, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Macdougall, I.C. Intravenous iron therapy in patients with chronic kidney disease: Recent evidence and future directions. Clin. Kidney J. 2017, 10, i16–i24. [Google Scholar] [CrossRef] [PubMed]
- Kruszewski, M. Labile iron pool: The main determinant of cellular response to oxidative stress. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2003, 531, 81–92. [Google Scholar] [CrossRef]
- Meneghini, R. Iron homeostasis, oxidative stress, and DNA damage. Free Radic. Biol. Med. 1997, 23, 783–792. [Google Scholar] [CrossRef]
- Small, D.M.; Coombes, J.S.; Bennett, N.; Johnson, D.W.; Gobe, G.C. Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology 2012, 17, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.-S.; Wei, Y.-H.; Yu, Y.L.; Kho, B. Enhanced oxidative stress in haemodialysis patients receiving intravenous iron therapy. Nephrol. Dial. Transplant. 1999, 14, 2680–2687. [Google Scholar] [CrossRef] [PubMed]
- Drueke, T.; Witko-Sarsat, V.; Massy, Z.; Descamps-Latscha, B.; Guerin, A.P.; Marchais, S.J.; Gausson, V.; London, G.M. Iron therapy, advanced oxidation protein products, and carotid artery intima-media thickness in end-stage renal disease. Circulation 2002, 106, 2212–2217. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.N.; Shahni, R.; Iqbal, M.M. Increased peripheral blood mitochondrial DNA in type 2 diabetic patients with nephropathy. Diabetes Res. Clin. Pract. 2009, 86, e22–e24. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.N.; Czajka, A. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion 2013, 13, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Granata, S.; Dalla Gassa, A.; Tomei, P.; Lupo, A.; Zaza, G. Mitochondria: A new therapeutic target in chronic kidney disease. Nutr. Metab. 2015, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.-L.; Hung, S.-C.; Lin, Y.-P.; Tang, C.-F.; Lee, T.-S.; Lin, C.-P.; Tarng, D.-C. Intravenous ferric chloride hexahydrate supplementation induced endothelial dysfunction and increased cardiovascular risk among hemodialysis patients. PLoS ONE 2012, 7, e50295. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.-L.; Hung, S.-C.; Lee, T.-S.; Tarng, D.-C. Iron sucrose accelerates early atherogenesis by increasing superoxide production and upregulating adhesion molecules in ckd. J. Am. Soc. Nephrol. 2014, 25, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Neven, E.; De Schutter, T.M.; Behets, G.J.; Gupta, A.; D’Haese, P.C. Iron and vascular calcification. Is there a link? Nephrol. Dial. Transplant. 2011, 26, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Kuragano, T.; Nanami, M.; Nagasawa, Y.; Hasuike, Y. Misdistribution of iron and oxidative stress in chronic kidney disease. Free Radic. Biol. Med. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Kawada, S.; Nagasawa, Y.; Kawabe, M.; Ohyama, H.; Kida, A.; Kato-Kogoe, N.; Nanami, M.; Hasuike, Y.; Kuragano, T.; Kishimoto, H.; et al. Iron-induced calcification in human aortic vascular smooth muscle cells through interleukin-24 (il-24), with/without tnf-alpha. Sci. Rep. 2018, 8, 658. [Google Scholar] [CrossRef] [PubMed]
- Eschbach, J.W.; Adamson, J.W. Iron overload in renal failure patients: Changes since the introduction of erythropoietin therapy. Kidney Int. 1999, 55, s-35–S-43. [Google Scholar] [CrossRef]
- Malik, I.A.; Wilting, J.; Ramadori, G.; Naz, N. Reabsorption of iron into acutely damaged rat liver: A role for ferritins. World J. Gastroenterol. 2017, 23, 7347–7358. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Hevi, S.; Chuck, S.L. Regulated secretion of glycosylated human ferritin from hepatocytes. Blood 2004, 103, 2369–2376. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.N.; Eubanks, S.K.; Schaffer, K.J.; Zhou, C.Y.J.; Linder, M.C. Secretion of ferritin by rat hepatoma cells and its regulation by inflammatory cytokines and iron. Blood 1997, 90, 4979–4986. [Google Scholar] [PubMed]
- Giachelli, C.M. Vascular calcification: In vitro evidence for the role of inorganic phosphate. J. Am. Soc. Nephrol. 2003, 14, S300–S304. [Google Scholar] [CrossRef] [PubMed]
- Zarjou, A.; Jeney, V.; Arosio, P.; Poli, M.; Antal-Szalmás, P.; Agarwal, A.; Balla, G.; Balla, J. Ferritin prevents calcification and osteoblastic differentiation of vascular smooth muscle cells. J. Am. Soc. Nephrol. 2009, 20, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Zarjou, A.; Jeney, V.; Arosio, P.; Poli, M.; Zavaczki, E.; Balla, G.; Balla, J. Ferritin ferroxidase activity: A potent inhibitor of osteogenesis. J. Bone Miner. Res. 2010, 25, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Levi, S. Ferritin, iron homeostasis, and oxidative damage. Free Radic. Biol. Med. 2002, 33, 457–463. [Google Scholar] [CrossRef]
- Broyles, R.H.; Belegu, V.; DeWitt, C.R.; Shah, S.N.; Stewart, C.A.; Pye, Q.N.; Floyd, R.A. Specific repression of β-globin promoter activity by nuclear ferritin. Proc. Natl. Acad. Sci. USA 2001, 98, 9145–9150. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Suliman, M.E.; Qureshi, A.R.; Garcia-Lopez, E.; Bárány, P.; Heimbürger, O.; Stenvinkel, P.; Lindholm, B. Consequences of low plasma histidine in chronic kidney disease patients: Associations with inflammation, oxidative stress, and mortality. Am. J. Clin. Nutr. 2008, 87, 1860–1866. [Google Scholar] [CrossRef] [PubMed]
- Kopple, J.D.; Swendseid, M.E. Evidence that histidine is an essential amino-acid in normal and chronically uremic man. J. Clin. Investig. 1975, 55, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Jontofsohn, R.; Trivisas, G.; Katz, N.; Kiuthe, I. Amino acid content of erythrocytes in uremia. Am. J. Clin. Nutr. 1978, 31, 1956–1960. [Google Scholar] [CrossRef] [PubMed]
- Bergström, J.; Furst, P.; Josephson, B.; Norée, L.-O. Improvement of nitrogen balance in a uremic patient by the addition of histidine to essential amino acid solutions given intravenously. Life Sci. 1970, 9, 787–794. [Google Scholar] [CrossRef]
- Blumenkrantz, M.J.; Shapiro, D.J.; Swendseid, M.E.; Kopple, J.D. Histidine supplementation for treatment of anaemia of uraemia. Br. Med. J. 1975, 2, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Jontofsohn, R.; Heinze, V.; Katz, N.; Stuber, U.; Wilke, H.; Kluthe, R. Histidine and iron supplementation in dialysis and pre-dialysis patient. Proc. Eur. Dial. Transpl. Assoc. 1974, 11, 391–397. [Google Scholar]
- Kaplan, P.; Matejovicova, M.; Herijgers, P.; Flameng, W. Effect of free radical scavengers on myocardial function and Na+, K+-atpase activity in stunned rabbit myocardium. Scand. Cardiovasc. J. 2005, 39, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. How to characterize a biological antioxidant. Free Radic. Res. Commun. 1990, 9, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Wade, A.M.; Tucker, H.N. Antioxidant characteristics of L-histidine. J. Nutr. Biochem. 1998, 9, 308–315. [Google Scholar] [CrossRef]
- Pisarenko, O.I. Mechanisms of myocardial protection by amino acids: Facts and hypotheses. Clin. Exp. Pharmacol. Physiol. 1996, 23, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Kim, N.A.; Lim, D.G.; Kim, K.H.; Hada, S.; Jeong, S.H. Evaluation of etanercept degradation under oxidative stress and potential protective effects of various amino acids. Int. J. Pharm. 2015, 492, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Son, D.O.; Satsu, H.; Shimizu, M. Histidine inhibits oxidative stress- and tnf-a-induced interleukin-8 secretion in intestinal epithelial cells. FEBS lett. 2005, 579, 4671–4677. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.C.; Hultin, H.O. A unique role of histidine in fe-catalyzed lipid oxidation by fish sarcoplasmic reticulum. In Oxygen Radicals in Biology and Medicine; Simic, M.G., Taylor, K.A., Ward, J.F., Sonntag, C., Eds.; Springer: Boston, MA, USA, 1988; pp. 307–312. [Google Scholar]
- Erickson, M.C.; Hultin, H.O. Influence of histidine on lipid peroxidation in sarcoplasmic reticulum. Arch. Biochem. Biophys. 1992, 292, 427–432. [Google Scholar] [CrossRef]
- Filho, J.C.D.; Bergström, J.; Stehle, P.; Fürst, P. Simultaneous measurements of free amino acid patternsof plasma, muscle and erythrocytes in healthy human subjects. Clin. Nutr. 1997, 16, 299–305. [Google Scholar] [CrossRef]
- Hobart, L.J.; Seibel, I.; Yeargans, G.S.; Seidler, N.W. Anti-crosslinking properties of carnosine: Significance of histidine. Life Sci. 2004, 75, 1379–1389. [Google Scholar] [CrossRef] [PubMed]
- Rama Rao, K.V.; Reddy, P.V.B.; Tong, X.; Norenberg, M.D. Brain edema in acute liver failure: Inhibition by L-histidine. Am. J. Pathol. 2010, 176, 1400–1408. [Google Scholar] [CrossRef] [PubMed]
- Baldbige, K.C. Effects of thiamine and biboflavin deficiency on histidine metabolism. J. Nutr. 1958, 66, 29–34. [Google Scholar]
- Ruszkiewicz, J.; Albrecht, J. Changes of the thioredoxin system, glutathione peroxidase activity and total antioxidant capacity in rat brain cortex during acute liver failure: Modulation by L-histidine. Neurochem. Res. 2015, 40, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-H.; Liu, T.-C.; Yin, M.-C. Beneficial effects of histidine and carnosine on ethanol-induced chronic liver injury. Food Chem. Toxicol. 2008, 46, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lu, W.; Fang, Y.; Liu, J. Evolution of oxidation dynamics of histidine: Nonreactivity in the gas phase, peroxides in hydrated clusters, and ph dependence in solution. Phys. Chem. Chem. Phys. 2014, 16, 22179–22191. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-C.; Shiau, C.-Y.; Chen, H.-M.; Chiou, T.-K. Antioxidant activities of carnosine, anserine, some freeamino acids and their combination. J. Food Drug Anal. 2003, 11, 148–153. [Google Scholar]
- Milewski, K.; Hilgier, W.; Albrecht, J.; Zielinska, M. The dimethylarginine (adma)/nitric oxide pathway in the brain and periphery of rats with thioacetamide-induced acute liver failure: Modulation by histidine. Neurochem. Int. 2015, 88, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Babizhayev, M.A.; Seguin, M.-C.; Gueynej, J.; Evstigneeva, R.P.; Ageyeva, J.E.A.; Zheltukhina, G.A. L-carnosine (beta-alanyl-L-histidine) and carcinine (beta-alanylhistamine) act as natural antioxidants with hydroxyl-radical-scavenging and lipid-peroxidase activities. Biochem. J. 1994, 304, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Miyawaki, H.; Bobst, E.V.; Hester, J.D.; Ashraf, M.; Bobst, A.M. Improved functional recovery of ischemic rat hearts due to singlet oxygen scavengers histidine and carnosine. J. Mol. Cell Cardiol. 1999, 31, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Vera-Aviles, M.; Latunde-Dada, G.O. Protective effect of histidine against iron-induced toxicity in hek-293 cells. Proc. Nutr. Soc. 2017, 76, E188. [Google Scholar] [CrossRef]
- Tyfield, L.A.; Holton, J.B. The effect of high concentrations of histidine on the level of other amino acids in plasma and brain of the mature rat. J. Neurochem. 1976, 26, 101–105. [Google Scholar] [PubMed]
- Kim, N.H.; Kang, J.H. Protective effects of histidine dipeptides on the modification of neurofilament-l by the cytochrome c/hydrogen peroxide system. J. Biochem. Mol. Biol. 2007, 40, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Tabor, H. Metabolic studies on histidine, histamine, and related imidazoles. Pharmacol. Rev. 1954, 6, 299–343. [Google Scholar] [PubMed]
- Sundberg, R.J.; Martin, R.B. Interactions of histidine and other imidazole derivatives with transition metal ions in chemical and biological systems. Chem. Rev. 1973, 74, 471–516. [Google Scholar] [CrossRef]
- Meyer, J.L.; Bauman, J.; John, E. Copper (II) -histidine complexes. J. Am. Chem. Soc. 1970, 92, 4210–4216. [Google Scholar] [CrossRef] [PubMed]
- Leberman, R.; Rabin, B.R. Metal complexes of histidine. Trans. Faraday Soc. 1959, 55, 1660–1670. [Google Scholar] [CrossRef]
- Williams, D.R. Thermodynamic considerations in co-ordination. Part VII. Solubility of the histidine-H+ system and stability constants, free energies, enthalpies, and entropies of protonation of histidine and tryptophan and of formation of their manganese(II), iron(II), cobalt(II), nickel(II), copper(II), and zinc(II) complexes. J. Chem. Soc. A 1970, 1550–1555. [Google Scholar]
- Zs.-Nagy, I.; Floyd, R.A. Hydroxyl free radical reactions with amino acids and proteins studied by electron spin resonance spectroscopy and spin-trapping. Biochim. Biophys. Acta 1984, 790, 238–250. [Google Scholar] [CrossRef]
- Nair, N.G.; Perry, G.; Smithc, M.A.; Reddya, V.P. Nmr studies of zinc, copper, and iron binding to histidine, the principal metal ion complexing site of amyloid-β peptide. J. Alzheimer’s Dis. 2010, 20, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Tachon, P. DNA single strand breakage by H2O2 and ferric or cupric ions: Its modulation by histidine. Free Radic. Res. Commun. 1990, 9, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Torres-Fuentes, C.; Alaiz, M.; Vioque, J. Iron-chelating activity of chickpea protein hydrolysate peptides. Food Chem. 2012, 134, 1585–1588. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, B.; Davidsson, L.; Cederblad, A.; Lonnerdal, B. Oral iron, dietary ligands and zinc absorption. J. Nutr. 1985, 115, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Van Campen, D.; Gross, E. Effect of histidine and certain other amino acids on the absorption of iron-59 by rats. J. Nutr. 1969, 99, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Fairbanks, V.F.; Beutler, E. Iron metabolism. In Hematology; Beutler, E., Ed.; McGraw-Hill: New York, NY, USA, 1995; pp. 369–380. [Google Scholar]
- Layrisse, M.; Martinez-Torres, C.; Leets, I.; Taylor, P.; Ramirez, J. Effect of histidine, cysteine, glutathione or beef on iron absorption in humans. J. Nutr. 1984, 114, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Van Campen, D. Effect of histidine and ascorbic acid on the absorption and retention of 59Fe by iron-depleted rats. J. Nutr. 1972, 102, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, V.; Pullakhandam, R.; Nair, K.M. Dietary ligands as determinants of iron–zinc interactions at the absorptive enterocyte. J. Food Sci. 2010, 75, H260–H264. [Google Scholar] [CrossRef] [PubMed]
- Glahn, R.P.; Van Campen, D.R. Iron uptake is enhanced in caco-2 cell monolayers by cysteine and reduced cysteinyl glycine. J. Nutr. 1997, 127, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Swain, J.H.; Tabatabai, L.B.; Reddy, M.B. Histidine content of low-molecular-weight beef proteins influences nonheme iron bioavailability in Caco-2 cells. Nutr Interact. Toxic. 2002, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Storcksdieck, S.; Bonsmann, G.; Hurrell, R.F. Iron-binding properties, amino acid composition, and structure of muscle tissue peptides from in vitro digestion of different meat sources. J. Food Sci. 2007, 72, S019–S029. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Hsu, C.C.; Lin, M.H.; Liu, K.S.; Yin, M.C. Histidine and carnosine delay diabetic deterioration in mice and protect human low density lipoprotein against oxidation and glycation. Eur. J. Pharmacol. 2005, 513, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Liakopoulos, V.; Roumeliotis, S.; Gorny, X.; Dounousi, E.; Mertens, P.R. Oxidative stress in hemodialysis patients: A review of the literature. Oxid. Med. Cell. Longev. 2017, 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Baylis, C. Arginine, arginine analogs and nitric oxide production in chronic kidney disease. Nat. Clin. Pract. Nephrol. 2006, 2, 209. [Google Scholar] [CrossRef] [PubMed]
- Coombes, J.S.; Fassett, R.G. Antioxidant therapy in hemodialysis patients: A systematic review. Kidney Int. 2012, 81, 233–246. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vera-Aviles, M.; Vantana, E.; Kardinasari, E.; Koh, N.L.; Latunde-Dada, G.O. Protective Role of Histidine Supplementation Against Oxidative Stress Damage in the Management of Anemia of Chronic Kidney Disease. Pharmaceuticals 2018, 11, 111. https://doi.org/10.3390/ph11040111
Vera-Aviles M, Vantana E, Kardinasari E, Koh NL, Latunde-Dada GO. Protective Role of Histidine Supplementation Against Oxidative Stress Damage in the Management of Anemia of Chronic Kidney Disease. Pharmaceuticals. 2018; 11(4):111. https://doi.org/10.3390/ph11040111
Chicago/Turabian StyleVera-Aviles, Mayra, Eleni Vantana, Emmy Kardinasari, Ngat L. Koh, and Gladys O. Latunde-Dada. 2018. "Protective Role of Histidine Supplementation Against Oxidative Stress Damage in the Management of Anemia of Chronic Kidney Disease" Pharmaceuticals 11, no. 4: 111. https://doi.org/10.3390/ph11040111
APA StyleVera-Aviles, M., Vantana, E., Kardinasari, E., Koh, N. L., & Latunde-Dada, G. O. (2018). Protective Role of Histidine Supplementation Against Oxidative Stress Damage in the Management of Anemia of Chronic Kidney Disease. Pharmaceuticals, 11(4), 111. https://doi.org/10.3390/ph11040111