Tuning the Anti(myco)bacterial Activity of 3-Hydroxy-4-pyridinone Chelators through Fluorophores




Abstract
1. Introduction
1.1. Chelators and Iron
1.2. Iron and Infection
1.2.1. Iron Chelation—A Therapeutic Tool to Tackle Microbial Infection?
1.2.2. Mycobacterial Infections
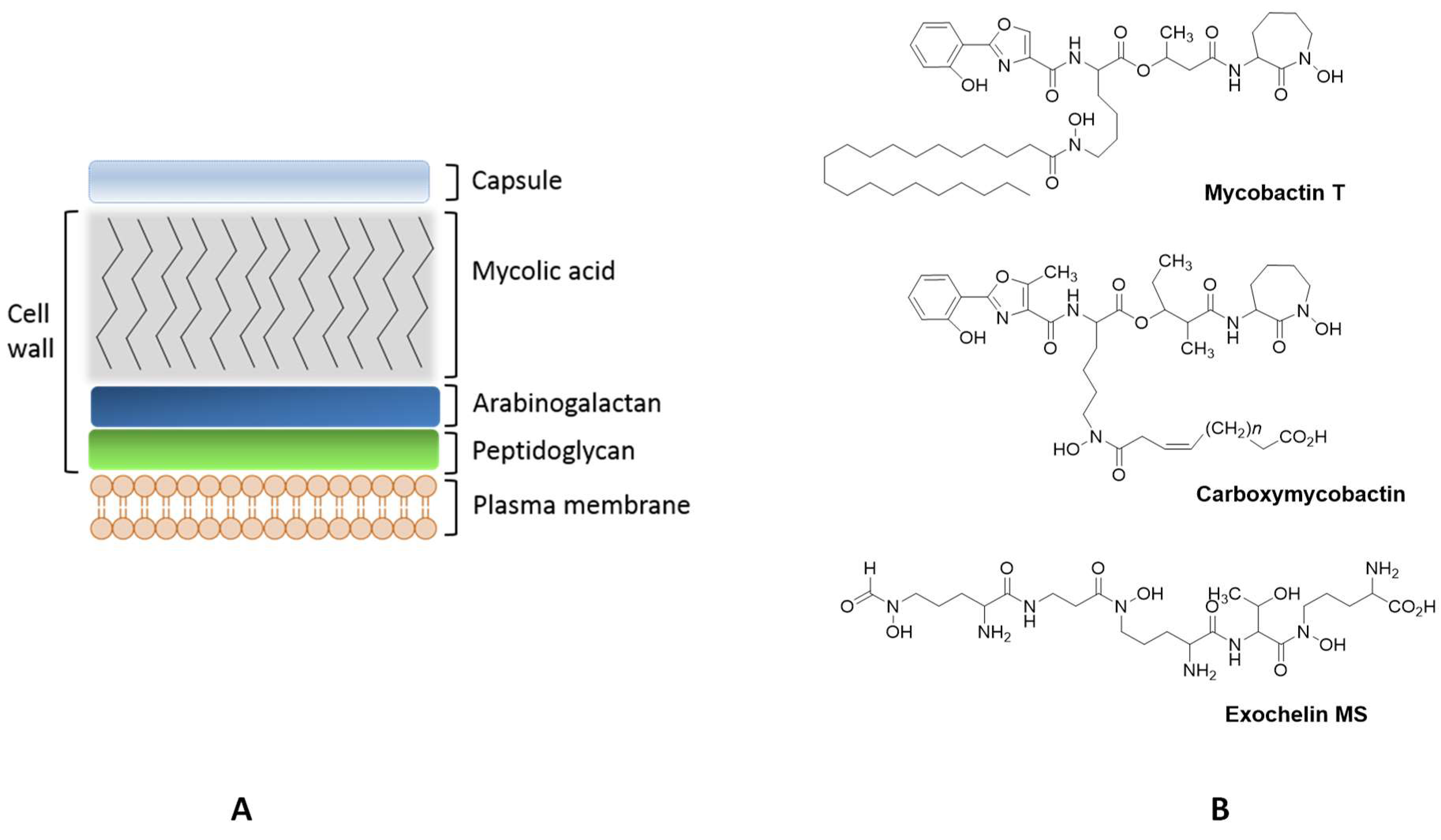
Mycobacteria
Mycobacterial Siderophores
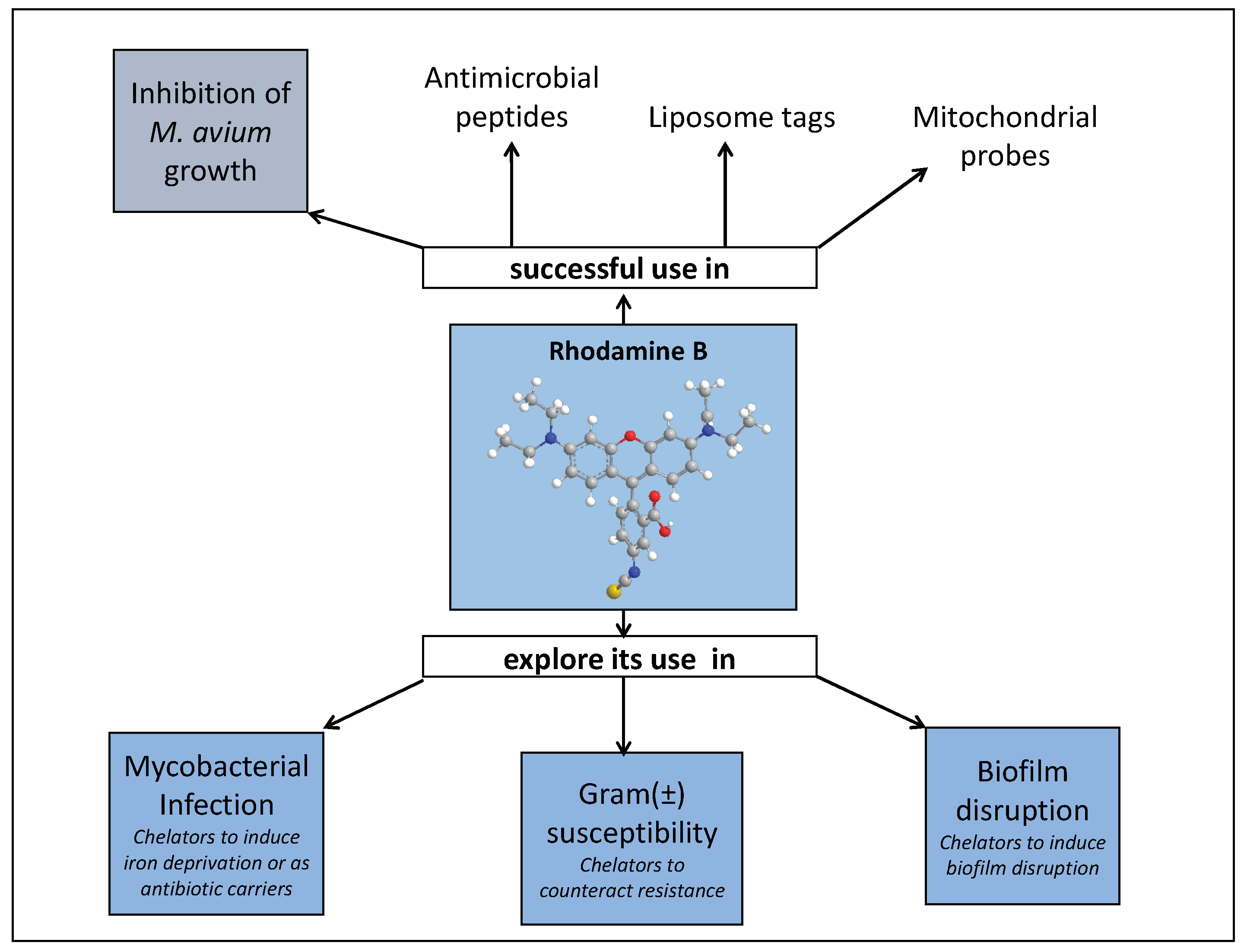
Iron Chelators to Control Mycobacterial Infection
2. Design of 3-Hydroxy-4-pyridinone Chelators to Address Mycobacterial Infections
2.1. Overview
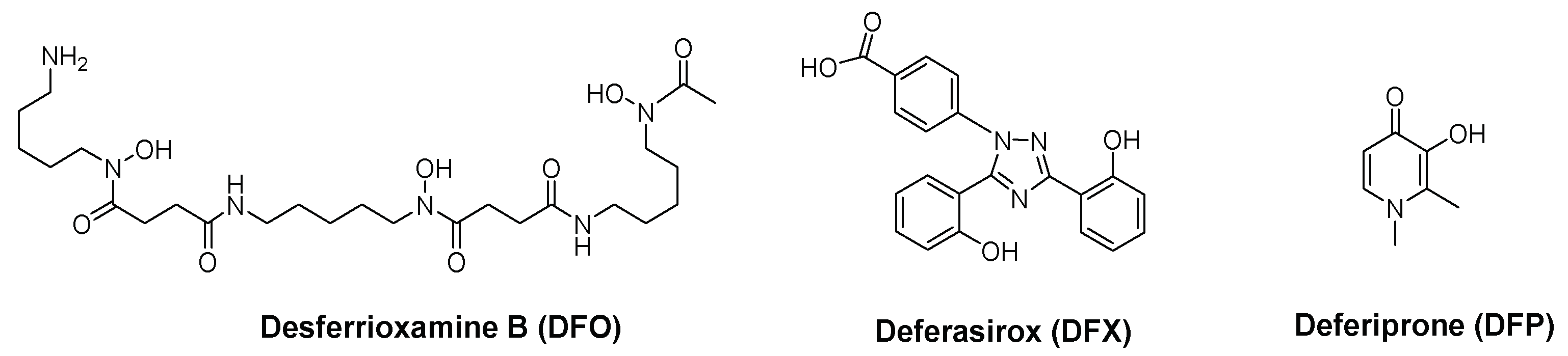
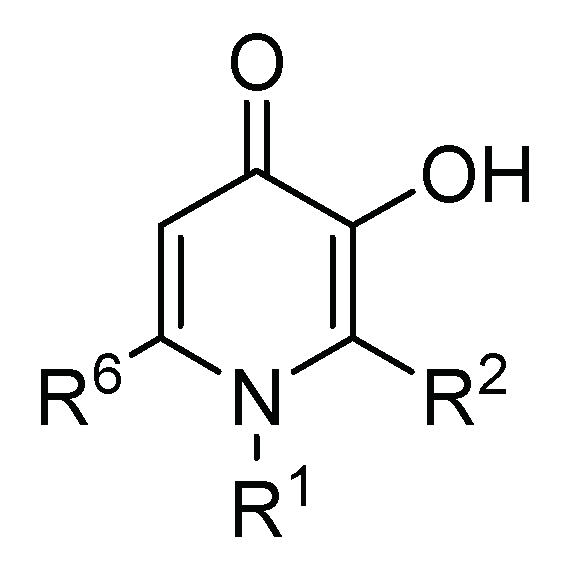
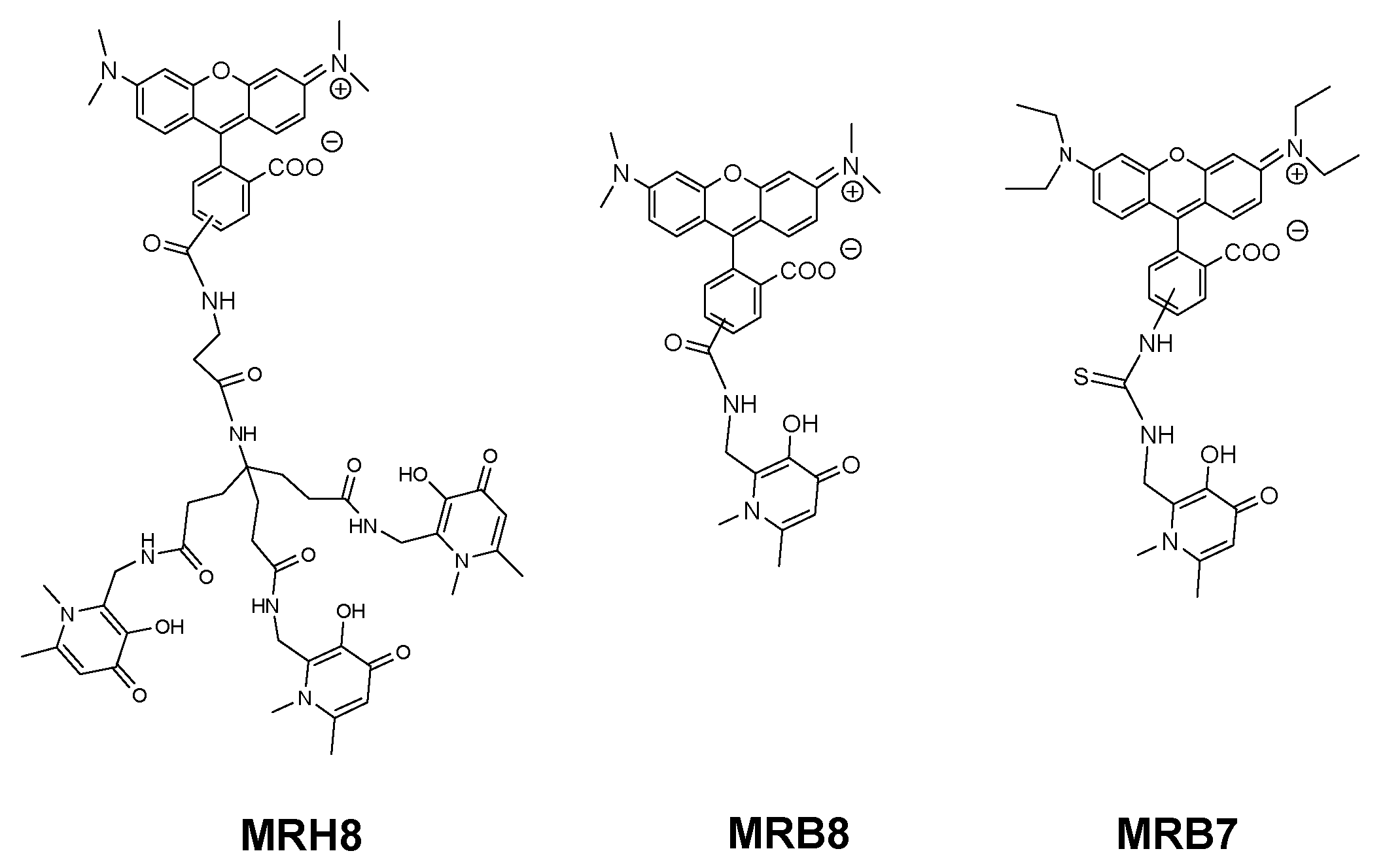
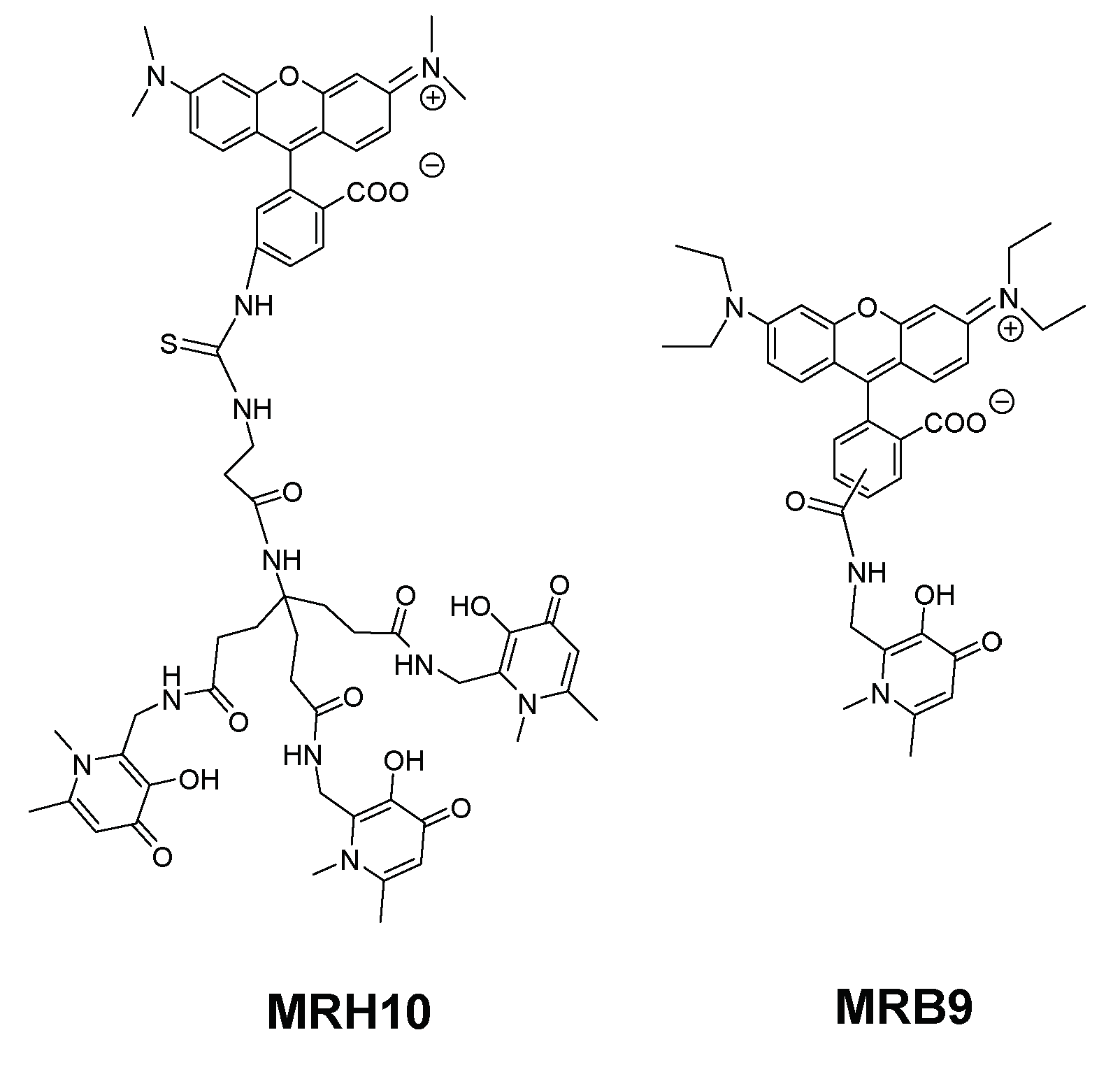
2.2. 3-Hydroxy-4-pyridinone (3,4-HPO) Chelators
2.3. Anti(myco)bacterial Effect in Intramacrophagic Growth of M. avium
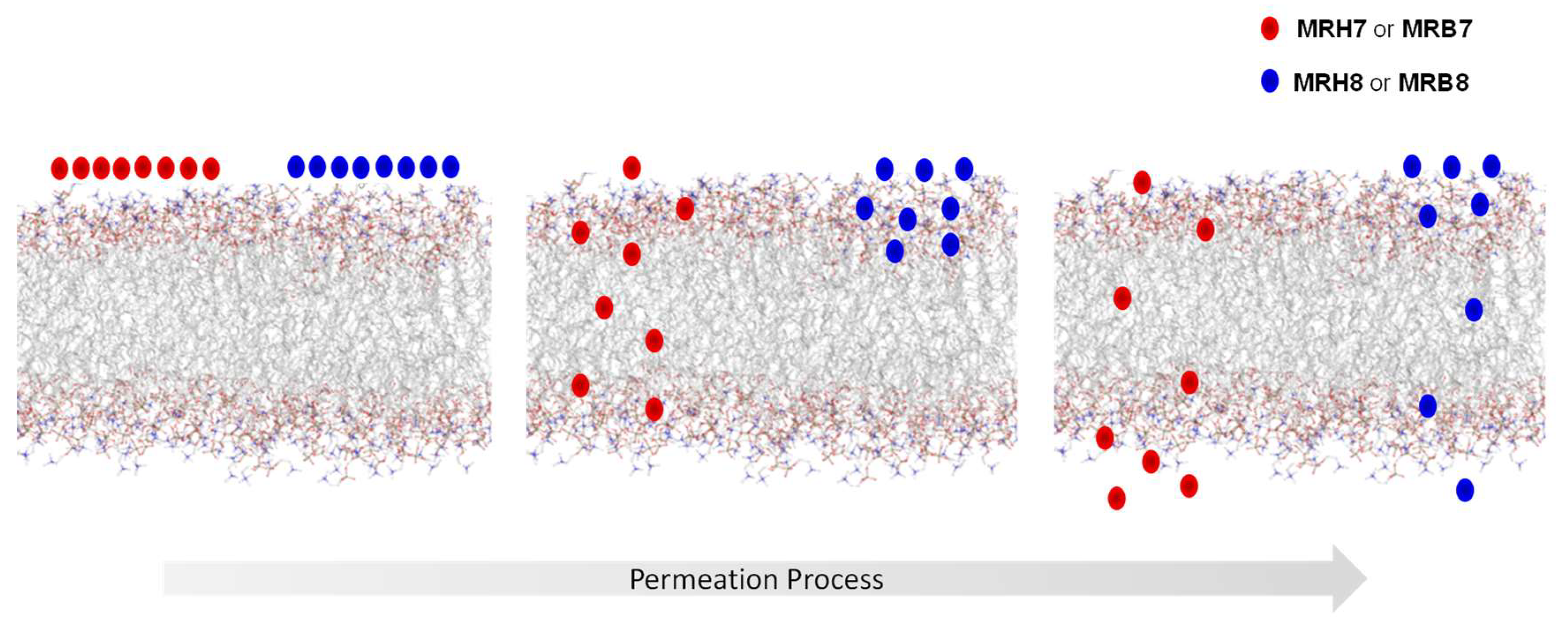
2.4. Chelator Membrane Interactions
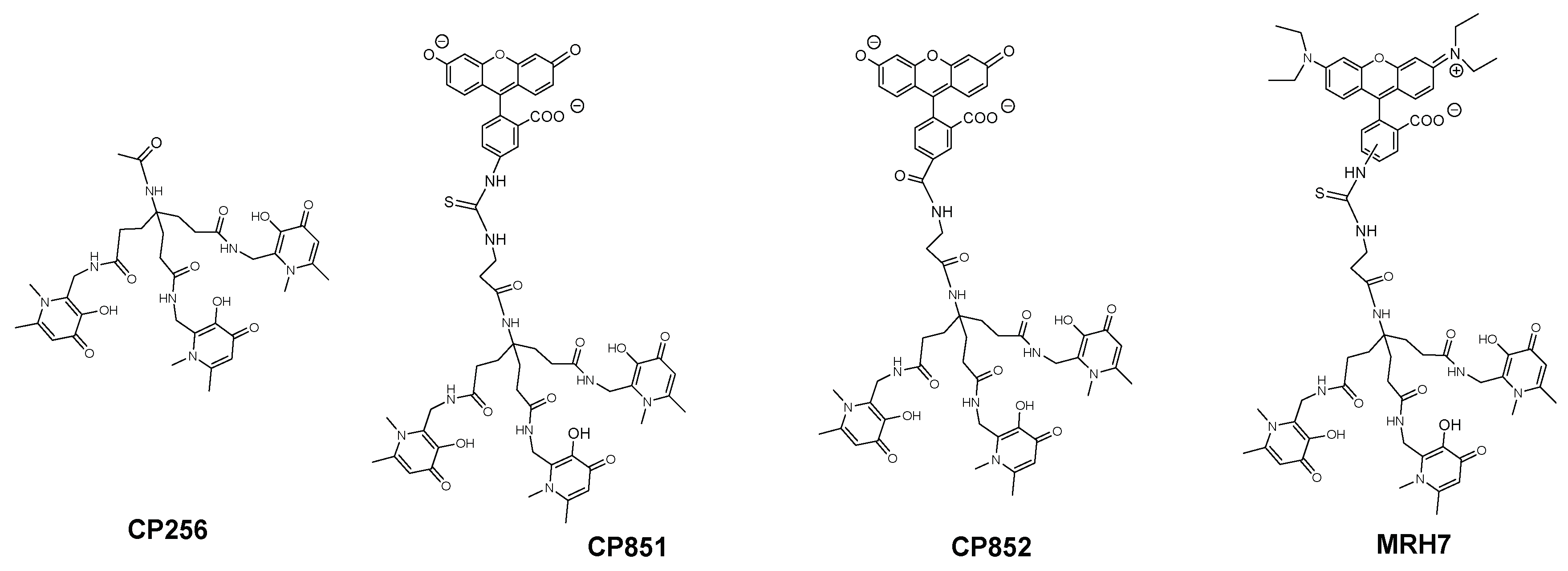
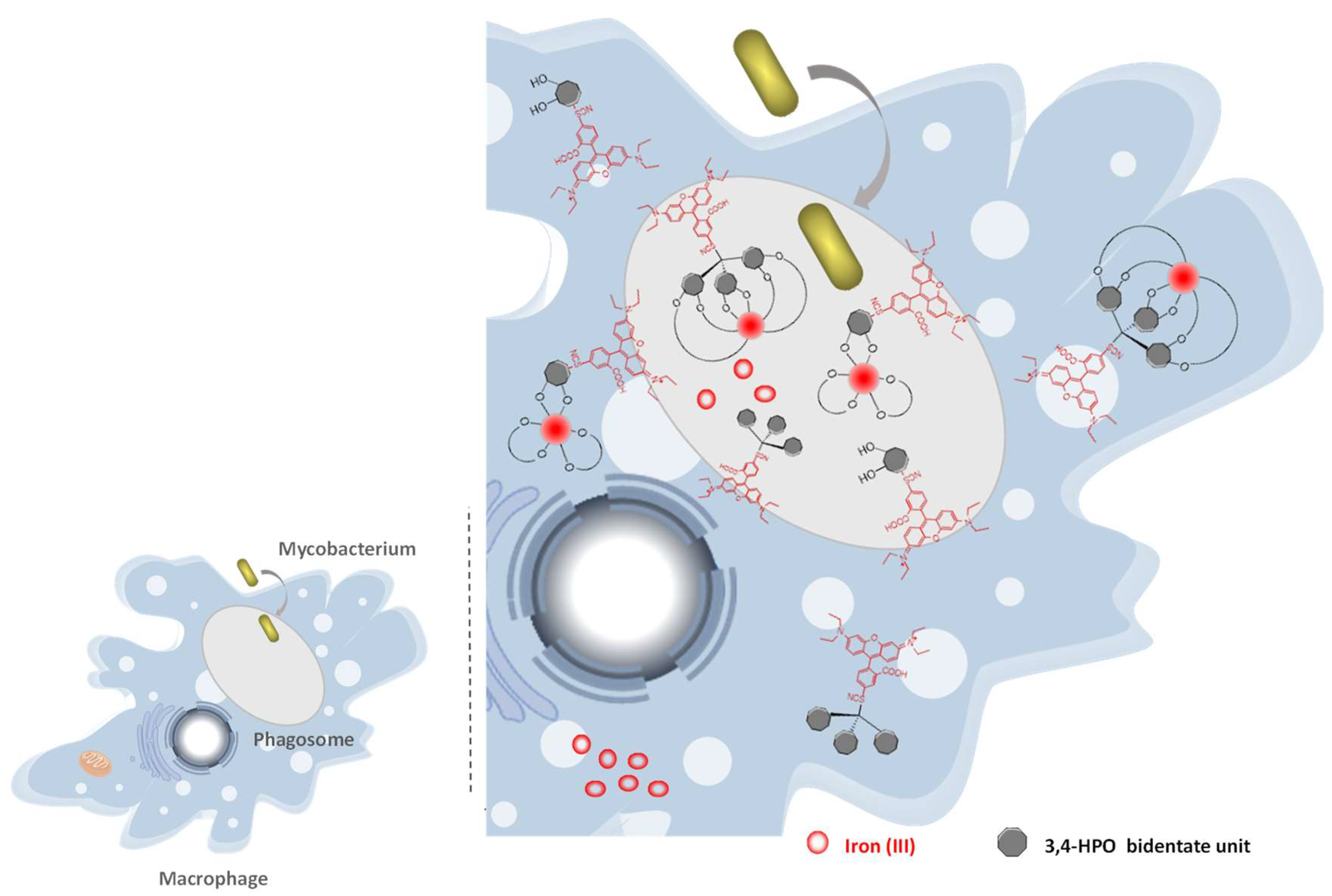
2.5. Intracellular Distribution and Co-Localization Studies of Rhodamine Labelled Chelators in Macrophages
2.6. Suggested Mechanism in M. avium Infection
2.7. First Studies in a Different Infection Scenario
3. Concluding Remarks and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Bertini, P.I.; Bertini, I.; Gray, H.B.; Gray, P.H.; Stiefel, E.I.; Stiefel, P.E.; Valentine, J.S. Biological Inorganic Chemistry: Structure and Reactivity (Tutorial II—Fundamentals of Coordination Chemistry), 1st ed.; University Science Books: Sausalito, CA, USA, 2007; ISBN 1-891389-43-2. [Google Scholar]
- Crichton, R. Iron Metabolism: From Molecular Mechanisms to Clinical Consequences, 3rd ed.; John Wiley & Sons Ltd.: Chichester, West Sussex, UK, 2009; ISBN 978-0-470-01028-0. [Google Scholar]
- Latunde-Dada, G.O. Iron metabolism: Microbes, mouse, and man. Bioessays 2009, 31, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Archibald, F. Lactobacillus plantarum, an organism not requiring iron. FEMS Microbiol. Lett. 1983, 19, 29–32. [Google Scholar] [CrossRef]
- Posey, J.E.; Gherardini, F.C. Lack of a role for iron in the Lyme disease pathogen. Science 2000, 288, 1651–1653. [Google Scholar] [CrossRef] [PubMed]
- Hider, R.C.; Kong, X. Chemistry and biology of siderophores. Nat. Prod. Rep. 2010, 27, 637–657. [Google Scholar] [CrossRef] [PubMed]
- Boukhalfa, H.; Crumbliss, A.L. Chemical aspects of siderophore mediated iron transport. Biometals 2002, 15, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Raymond, K.; Müller, G.; Matzanke, B. Complexation of iron by siderophores a review of their solution and structural chemistry and biological function. Top. Curr. Chem. 1984, 123, 49–102. [Google Scholar]
- Dhungana, S.; Harrington, J.M.; Gebhardt, P.; Mollmann, U.; Crumbliss, A.L. Iron chelation equilibria, redox, and siderophore activity of a saccharide platform ferrichrome analogue. Inorg. Chem. 2007, 46, 8362–8371. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, F.; Rocha, S.; Fernandes, R. Iron metabolism: From health to disease. J. Clin. Lab. Anal. 2014, 28, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Crichton, R.R.; Pierre, J.L. Old iron, young copper: From Mars to Venus. Biometals 2001, 14, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Lippard, S.J.; Berg, J.M. Principles of Bioinorganic Chemistry, 1st ed.; University Science Books: Mill Valley, CA, USA, 1994; ISBN 10 0935702725. [Google Scholar]
- Valko, M.; Morris, H.; Cronin, M.T. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Longo, F.; Nielsen, P.; Engelhardt, R.; Hider, R.C.; Piga, A. Monitoring long-term efficacy of iron chelation therapy by deferiprone and desferrioxamine in patients with beta-thalassaemia major: Application of SQUID biomagnetic liver susceptometry. Br. J. Haematol. 2003, 121, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Cronje, L.; Bornman, L. Iron overload and tuberculosis: A case for iron chelation therapy. Int. J. Tuberc. Lung Dis. 2005, 9, 2–9. [Google Scholar] [PubMed]
- Budimir, A. Metal ions, Alzheimer’s disease and chelation therapy. Acta Pharm. 2011, 61, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Guariglia, R.; Martorelli, M.C.; Villani, O.; Pietrantuono, G.; Mansueto, G.; D’Auria, F.; Grieco, V.; Bianchino, G.; Lerose, R.; Bochicchio, G.B.; et al. Positive effects on hematopoiesis in patients with myelodysplastic syndrome receiving deferasirox as oral iron chelation therapy: A brief review. Leuk. Res. 2011, 35, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Rombout-Sestrienkova, E.; van Kraaij, M.G.J.; Koek, G.H. How we manage patients with hereditary haemochromatosis. Br. J. Haematol. 2016, 175, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Ceci, A.; Baiardi, P.; Felisi, M.; Cappellini, M.D.; Carnelli, V.; De Sanctis, V.; Galanello, R.; Maggio, A.; Masera, G.; Piga, A.; et al. The safety and effectiveness of deferiprone in a large-scale, 3-year study in Italian patients. Br. J. Haematol. 2002, 118, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, P.C.; Richardson, D.R.; Kalinowski, D.S.; Bernhardt, P.V. Synthetic and natural products as iron chelators. Curr. Top. Med. Chem. 2011, 11, 591–607. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.; Gyparaki, M.; Burke, L.; Huehns, E.; Sarpong, P.; Saez, V.; Hider, R. Iron mobilization from hepatocyte monolayer cultures by chelators: The importance of membrane permeability and the iron-binding constant. Blood 1988, 72, 1497–1503. [Google Scholar] [PubMed]
- Weinberg, E.D. Iron Out-of-Balance: A Risk Factor for Acute and Chronic Diseases. Hemoglobin 2008, 32, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, G.; Pantopoulos, K. Disorders associated with systemic or local iron overload: From pathophysiology to clinical practice. Metallomics 2011, 3, 971–986. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Hood, M.I.; Skaar, E.P. Nutritional immunity: Transition metals at the pathogen-host interface. Nat. Rev. Microbiol. 2012, 10, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.W.; Skaar, E.P. Metal limitation and toxicity at the interface between host and pathogen. FEMS Microbiol. Rev. 2014, 38, 1235–1249. [Google Scholar] [CrossRef] [PubMed]
- Skaar, E.P. The Battle for Iron between Bacterial Pathogens and Their Vertebrate Hosts. PLoS Pathog. 2010, 6, e1000949. [Google Scholar] [CrossRef] [PubMed]
- Schaible, U.E.; Kaufmann, S.H.E. Iron and microbial infection. Nat. Rev. Microbiol. 2004, 2, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.A.; Fisher, M.A.; Khakoo, R.A. Association of hemochromatosis with infectious diseases: Expanding spectrum. Int. J. Infect. Dis. 2007, 11, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Höpfner, M.; Nitsche, R.N.; Rohr, A.; Harms, D.; Schubert, S.; Fölsch, U.R. Yersinia enterocolitica Infection with Multiple Liver Abscesses Uncovering a Primary Hemochromatosis. Scand. J. Gastroenterol. 2001, 36, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Barton, J.C.; Acton, R.T. Hemochromatosis and Vibrio vulnificus Wound Infections. J. Clin. Gastroenterol. 2009, 43, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Gangaidzo, I.T.; Moyo, V.M.; Mvundura, E.; Aggrey, G.; Murphree, N.L.; Khumalo, H.; Saungweme, T.; Kasvosve, I.; Gomo, Z.A.; Rouault, T.; et al. Association of pulmonary tuberculosis with increased dietary iron. J. Infect. Dis. 2001, 184, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Pereira, S.; Rodrigues, P.N.; Appelberg, R.; Gomes, M.S. Increased susceptibility to Mycobacterium avium in hemochromatosis protein HFE-deficient mice. Infect. Immun. 2008, 76, 4713–4719. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.S.; Boelaert, J.R.; Appelberg, R. Role of iron in experimental Mycobacterium avium infection. J. Clin. Virol. 2001, 20, 117–122. [Google Scholar] [CrossRef]
- De Monye, C.; Karcher, D.S.; Boelaert, J.R.; Gordeuk, V.R. Bone marrow macrophage iron grade and survival of HIV-seropositive patients. AIDS 1999, 13, 375–380. [Google Scholar] [CrossRef] [PubMed]
- McDermid, J.M.; Hennig, B.J.; van der Sande, M.; Hill, A.V.; Whittle, H.C.; Jaye, A.; Prentice, A.M. Host iron redistribution as a risk factor for incident tuberculosis in HIV infection: An 11-year retrospective cohort study. BMC Infect. Dis. 2013, 13, 48. [Google Scholar] [CrossRef] [PubMed]
- Foley, T.L.; Simeonov, A. Targeting iron assimilation to develop new antibacterials. Expert Opin. Drug Discov. 2012, 7, 831–847. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Spellberg, B.; Gebremariam, T.; Lee, H.; Xiong, Y.Q.; French, S.W.; Bayer, A.; Ibrahim, A.S. Combination therapy with iron chelation and vancomycin in treating murine staphylococcemia. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.F.; Qiu, D.H.; Kong, X.L.; Hider, R.C.; Zhou, T. Synthesis and in-vitro antimicrobial evaluation of a high-affinity iron chelator in combination with chloramphenicol. J. Pharm. Pharmacol. 2013, 65, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Neres, J.; Labello, N.P.; Somu, R.V.; Boshoff, H.I.; Wilson, D.J.; Vannada, J.; Chen, L.; Barry, C.E., III; Bennett, E.M.; Aldrich, C.C. Inhibition of siderophore biosynthesis in Mycobacterium tuberculosis with nucleoside bisubstrate analogues: Structure-activity relationships of the nucleobase domain of 5′-O-[N-(salicyl)sulfamoyl]adenosine. J. Med. Chem. 2008, 51, 5349–5370. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.S.; Dom, G.; Pedrosa, J.; Boelaert, J.R.; Appelberg, R. Effects of iron deprivation on Mycobacterium avium growth. Tuber. Lung Dis. 1999, 79, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Lounis, N.; Truffot-Pernot, C.; Grosset, J.; Gordeuk, V.R.; Boelaert, J.R. Iron and Mycobacterium tuberculosis infection. J. Clin. Virol. 2001, 20, 123–126. [Google Scholar] [CrossRef]
- Douvas, G.S.; May, M.H.; Crowle, A.J. Transferrin, iron, and serum lipids enhance or inhibit Mycobacterium avium replication in human macrophages. J. Infect. Dis. 1993, 167, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Ma, Y.; Kong, X.; Hider, R.C. Design of iron chelators with therapeutic application. Dalton Trans. 2012, 41, 6371–6389. [Google Scholar] [CrossRef] [PubMed]
- Bilitewski, U.; Blodgett, J.A.V.; Duhme-Klair, A.K.; Dallavalle, S.; Laschat, S.; Routledge, A.; Schobert, R. Chemical and Biological Aspects of Nutritional Immunity—Perspectives for New Anti-Infectives that Target Iron Uptake Systems. Angew. Chem. Int. Ed. 2017, 56, 14360–14382. [Google Scholar] [CrossRef] [PubMed]
- Sheth, S. Iron chelation: An update. Curr. Opin. Hematol. 2014, 21, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Juárez-Hernández, R.E.; Miller, M.J. Exploiting bacterial iron acquisition: Siderophore conjugates. Future Med. Chem. 2012, 4, 297–313. [Google Scholar] [CrossRef] [PubMed]
- Albert, A.; Rubbo, S.D.; Goldacre, R.J.; Balfour, B.G. The Influence of Chemical Constitution on Antibacterial Activity. Part III: A Study of 8-Hydroxyquinoline (Oxine) and Related Compounds. Br. J. Exp. Pathol. 1947, 28, 69–87. [Google Scholar] [PubMed]
- Chew, B.P.; Tjoelker, L.W.; Tanaka, T.S. In vitro growth inhibition of mastitis causing bacteria by phenolics and metal chelators. J. Dairy Sci. 1985, 68, 3037–3046. [Google Scholar] [CrossRef]
- Bergan, T.; Klaveness, J.; Aasen, A.J. Chelating Agents. Chemotherapy 2001, 47, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.W.; O’May, C.; Kirov, S.M.; Roddam, L.; Lamont, I.L.; Sanderson, K. Iron chelation directed against biofilms as an adjunct to conventional antibiotics. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, L857–L858. [Google Scholar] [CrossRef] [PubMed]
- Musk, D.J., Jr.; Hergenrother, P.J. Chelated iron sources are inhibitors of Pseudomonas aeruginosa biofilms and distribute efficiently in an in vitro model of drug delivery to the human lung. J. Appl. Microbiol. 2008, 105, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Sobke, A.; Klinger, M.; Hermann, B.; Sachse, S.; Nietzsche, S.; Makarewicz, O.; Keller, P.M.; Pfister, W.; Straube, E. The urinary antibiotic 5-nitro-8-hydroxyquinoline (Nitroxoline) reduces the formation and induces the dispersal of Pseudomonas aeruginosa biofilms by chelation of iron and zinc. Antimicrob. Agents Chemother. 2012, 56, 6021–6025. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, L.; Molin, S. Synergistic activities of an efflux pump inhibitor and iron chelators against Pseudomonas aeruginosa growth and biofilm formation. Antimicrob. Agents Chemother. 2010, 54, 3960–3963. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-H.; Shu, J.-C.; Huang, H.-Y.; Cheng, Y.-C. Involvement of Iron in Biofilm Formation by Staphylococcus aureus. PLoS ONE 2012, 7, e34388. [Google Scholar] [CrossRef]
- Van Asbeck, B.S.; Marcelis, J.H.; van Kats, J.H.; Jaarsma, E.Y.; Verhoef, J. Synergy between the iron chelator deferoxamine and the antimicrobial agents gentamicin, chloramphenicol, cefalothin, cefotiam and cefsulodin. Eur. J. Clin. Microbiol. 1983, 2, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Neupane, G.P.; Kim, D.M. In vitro time-kill activities of ciprofloxacin alone and in combination with the iron chelator deferasirox against Vibrio vulnificus. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.C.; Chan, S.; Ho, P.L.; Ha, S.Y. Effects of chelators (deferoxamine, deferiprone and deferasirox) on the growth of Klebsiella pneumoniae and Aeromonas hydrophila isolated from transfusion-dependent thalassemia patients. Hemoglobin 2009, 33, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Neupane, G.P.; Kim, D.M. Comparison of the effects of deferasirox, deferiprone, and deferoxamine on the growth and virulence of Vibrio vulnificus. Transfusion 2009, 49, 1762–1769. [Google Scholar] [CrossRef] [PubMed]
- Collins, H.L.; Kaufmann, S.H.; Schaible, U.E. Iron chelation via deferoxamine exacerbates experimental salmonellosis via inhibition of the nicotinamide adenine dinucleotide phosphate oxidase-dependent respiratory burst. J. Immunol. 2002, 168, 3458–3463. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.G.; Corey, B.W.; Si, Y.; Craft, D.W.; Zurawski, D.V. Antibacterial activities of iron chelators against common nosocomial pathogens. Antimicrob. Agents Chemother. 2012, 56, 5419–5421. [Google Scholar] [CrossRef] [PubMed]
- Visca, P.; Bonchi, C.; Minandri, F.; Frangipani, E.; Imperi, F. The dual personality of iron chelators: Growth inhibitors or promoters? Antimicrob. Agents Chemother. 2013, 57, 2432–2433. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.M.; Shin, S.H. Effect of iron-chelator deferiprone on the in vitro growth of staphylococci. J. Korean Med. Sci. 2009, 24, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-J.; Liu, M.-S.; Osamah, A.R.; Kong, X.-L.; Alsam, S.; Battah, S.; Xie, Y.-Y.; Hider, R.C.; Zhou, T. Hexadentate 3-hydroxypyridin-4-ones with high iron(III) affinity: Design, synthesis and inhibition on methicillin resistant Staphylococcus aureus and Pseudomonas strains. Eur. J. Med. Chem. 2015, 94, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.X.; Zhu, C.F.; Zhou, Y.J.; Kong, X.L.; Hider, R.C.; Zhou, T. Design, Synthesis, and Antimicrobial Evaluation of Hexadentate Hydroxypyridinones with High Iron(III) Affinity. Chem. Biol. Drug Des. 2014, 84, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.-H.; Huang, Z.-L.; Zhou, T.; Shen, C.; Hider, R.C. In vitro inhibition of bacterial growth by iron chelators. FEMS Microbiol. Lett. 2011, 314, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.-Y.; Liu, M.-S.; Hu, P.-P.; Kong, X.-L.; Qiu, D.-H.; Xu, J.-L.; Hider, R.; Zhou, T. Synthesis, physico-chemical properties, and antimicrobial evaluation of a new series of iron(III) hexadentate chelators. Med. Chem. Res. 2013, 22, 2351–2359. [Google Scholar] [CrossRef]
- Xu, B.; Kong, X.L.; Zhou, T.; Qiu, D.H.; Chen, Y.L.; Liu, M.S.; Yang, R.H.; Hider, R.C. Synthesis, iron(III)-binding affinity and in vitro evaluation of 3-hydroxypyridin-4-one hexadentate ligands as potential antimicrobial agents. Bioorg. Med. Chem. Lett. 2011, 21, 6376–6380. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.J.; Zhang, M.X.; Hider, R.C.; Zhou, T. In vitro antimicrobial activity of hydroxypyridinone hexadentate-based dendrimeric chelators alone and in combination with norfloxacin. FEMS Microbiol. Lett. 2014, 355, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Tortoli, E. Clinical manifestations of nontuberculous mycobacteria infections. Clin. Microbiol. Infect. 2009, 15, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Danelishvili, L.; Wagner, D.; Petrofsky, M.; Bermudez, L.E. Identification of virulence determinants of Mycobacterium avium that impact on the ability to resist host killing mechanisms. J. Med. Microbiol. 2010, 59, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Quesniaux, V.; Fremond, C.; Jacobs, M.; Parida, S.; Nicolle, D.; Yeremeev, V.; Bihl, F.; Erard, F.; Botha, T.; Drennan, M.; et al. Toll-like receptor pathways in the immune responses to mycobacteria. Microbes Infect. 2004, 6, 946–959. [Google Scholar] [CrossRef] [PubMed]
- Guenin-Mace, L.; Simeone, R.; Demangel, C. Lipids of pathogenic Mycobacteria: Contributions to virulence and host immune suppression. Transbound. Emerg. Dis. 2009, 56, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Brennan, P.J. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis 2003, 83, 91–97. [Google Scholar] [CrossRef]
- Ratledge, C.; Dover, L.G. Iron Metabolism in Pathogenic Bacteria. Annu. Rev. Microbiol. 2000, 54, 881–941. [Google Scholar] [CrossRef] [PubMed]
- De Voss, J.J.; Rutter, K.; Schroeder, B.G.; Su, H.; Zhu, Y.; Barry, C.E. The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. USA 2000, 97, 1252–1257. [Google Scholar] [CrossRef] [PubMed]
- Roosenberg, J.M., II; Lin, Y.M.; Lu, Y.; Miller, M.J. Studies and syntheses of siderophores, microbial iron chelators, and analogs as potential drug delivery agents. Curr. Med. Chem. 2000, 7, 159–197. [Google Scholar] [CrossRef] [PubMed]
- Neyrolles, O.; Wolschendorf, F.; Mitra, A.; Niederweis, M. Mycobacteria, metals, and the macrophage. Immunol. Rev. 2015, 264, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.; Maser, J.; Moric, I.; Vogt, S.; Kern, W.V.; Bermudez, L.E. Elemental analysis of the Mycobacterium avium phagosome in Balb/c mouse macrophages. Biochem. Biophys. Res. Commun. 2006, 344, 1346–1351. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.; Maser, J.; Lai, B.; Cai, Z.; Barry, C.E., 3rd; Honer Zu Bentrup, K.; Russell, D.G.; Bermudez, L.E. Elemental analysis of Mycobacterium avium-, Mycobacterium tuberculosis-, and Mycobacterium smegmatis-containing phagosomes indicates pathogen-induced microenvironments within the host cell’s endosomal system. J. Immunol. 2005, 174, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Schaible, U.E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 2015, 264, 182–203. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Farhana, A.; Ehtesham, N.Z.; Hasnain, S.E. Iron acquisition, assimilation and regulation in mycobacteria. Infect. Genet. Evol. 2011, 11, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Sritharan, M. Iron Homeostasis in Mycobacterium tuberculosis: Mechanistic Insights into Siderophore-Mediated Iron Uptake. J. Bacteriol. 2016, 198, 2399–2409. [Google Scholar] [CrossRef] [PubMed]
- Eiglmeier, K.; Parkhill, J.; Honore, N.; Garnier, T.; Tekaia, F.; Telenti, A.; Klatser, P.; James, K.D.; Thomson, N.R.; Wheeler, P.R.; et al. The decaying genome of Mycobacterium leprae. Lepr Rev. 2001, 72, 387–398. [Google Scholar] [PubMed]
- Dhungana, S.; Ratledge, C.; Crumbliss, A.L. Iron chelation properties of an extracellular siderophore exochelin MS. Inorg. Chem. 2004, 43, 6274–6283. [Google Scholar] [CrossRef] [PubMed]
- Ratledge, C. Iron, mycobacteria and tuberculosis. Tuberculosis 2004, 84, 110–130. [Google Scholar] [CrossRef] [PubMed]
- De Voss, J.J.; Rutter, K.; Schroeder, B.G.; Barry, C.E., III. Iron Acquisition and Metabolism by Mycobacteria. J. Bacteriol. 1999, 181, 4443–4451. [Google Scholar] [PubMed]
- Nunes, A.; Podinovskaia, M.; Leite, A.; Gameiro, P.; Zhou, T.; Ma, Y.; Kong, X.; Schaible, U.E.; Hider, R.C.; Rangel, M. Fluorescent 3-hydroxy-4-pyridinone hexadentate iron chelators: Intracellular distribution and the relevance to antimycobacterial properties. J. Biol. Inorg. Chem. 2010, 15, 861–877. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Fadeev, E.A.; Groves, J.T. Mycobactin-mediated iron acquisition within macrophages. Nat. Chem. Biol. 2005, 1, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Dhungana, S.; Miller, M.J.; Dong, L.; Ratledge, C.; Crumbliss, A.L. Iron Chelation Properties of an Extracellular Siderophore Exochelin MN. J. Am. Chem. Soc. 2003, 125, 7654–7663. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.K.; Gobin, J.; Horwitz, M.A.; Gibson, B.W. Characterization of exochelins of Mycobacterium avium: Evidence for saturated and unsaturated and for acid and ester forms. J. Bacteriol. 1996, 178, 6394–6398. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.G. Mycobacterium tuberculosis: Here today, and here tomorrow. Nat. Rev. Mol. Cell Biol. 2001, 2, 569–586. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, S.S.; Nunes, A.; Gomes, A.R.; de Castro, B.; Hider, R.C.; Rangel, M.; Appelberg, R.; Gomes, M.S. Identification of a new hexadentate iron chelator capable of restricting the intramacrophagic growth of Mycobacterium avium. Microbes Infect. 2010, 12, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Moniz, T.; Nunes, A.; Silva, A.M.G.; Queirós, C.; Ivanova, G.; Gomes, M.S.; Rangel, M. Rhodamine labeling of 3-hydroxy-4-pyridinone iron chelators is an important contribution to target Mycobacterium avium infection. J. Inorg. Biochem. 2013, 121, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Bosne-David, S.; Bricard, L.; Ramiandrasoa, F.; DeRoussent, A.; Kunesch, G.; Andremont, A. Evaluation of growth promotion and inhibition from mycobactins and nonmycobacterial siderophores (Desferrioxamine and FR160) in Mycobacterium aurum. Antimicrob. Agents Chemother. 1997, 41, 1837–1839. [Google Scholar] [CrossRef] [PubMed]
- Cronjé, L.; Edmondson, N.; Eisenach, K.D.; Bornman, L. Iron and iron chelating agents modulate Mycobacterium tuberculosis growth and monocyte-macrophage viability and effector functions. FEMS Immunol. Med. Microbiol. 2005, 45, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; Arai, M.; Niikawa, H.; Kobayashi, M. Inhibitory effect of cyclic trihydroxamate siderophore, desferrioxamine E, on the biofilm formation of Mycobacterium species. Biol. Pharm. Bull. 2011, 34, 917–920. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.R.; Perumal, S.; Senthilkumar, P.; Yogeeswari, P.; Sriram, D. A highly atom economic, chemo-, regio- and stereoselective synthesis, and discovery of spiro-pyrido-pyrrolizines and pyrrolidines as antimycobacterial agents. Tetrahedron 2008, 64, 2962–2971. [Google Scholar] [CrossRef]
- Kumar, R.R.; Perumal, S.; Senthilkumar, P.; Yogeeswari, P.; Sriram, D. An atom efficient, solvent-free, green synthesis and antimycobacterial evaluation of 2-amino-6-methyl-4-aryl-8-[(E)-arylmethylidene]-5,6,7,8-tetrahydro-4H-pyrano[3,2-c]pyridine-3-carbonitriles. Bioorg. Med. Chem. Lett. 2007, 17, 6459–6462. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.R.; Perumal, S.; Senthilkumar, P.; Yogeeswari, P.; Sriram, D. Discovery of antimycobacterial spiro-piperidin-4-ones: An atom economic, stereoselective synthesis, and biological intervention. J. Med. Chem. 2008, 51, 5731–5735. [Google Scholar] [CrossRef] [PubMed]
- Bairwa, R.; Tawari, N.R.; Alate, A.; Anam, S.; Degani, M.S.; Ray, M.; Rajan, R.M. Novel 2-hydrazino-pyrimidin-4(3H)-one derivatives with pseudofunctional- similarity to siderophores as potential antimycobacterial agents. Med. Chem. 2013, 9, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Douvas, G.S.; May, M.H.; Kolnagou, A.; Kontoghiorghes, G.J. Effects on Mycobacterium avium replication in normal human macrophages by deferiprone (L1) and other iron chelators. Possible implications on toxicity. Arzneimittelforschung 2002, 52, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Raja, V.P.A.; Perumal, S.; Yogeeswari, P.; Sriram, D. Synthesis and antimycobacterial activity of highly functionalized tetrahydro-4(1H)-pyridinones. Bioorg. Med. Chem. Lett. 2011, 21, 3881–3884. [Google Scholar] [CrossRef] [PubMed]
- Yacoby, I.; Benhar, I. Targeted Anti Bacterial Therapy. Infect. Disord. Drug Targets 2007, 7, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Cooper, M.A. Antibiotics in the clinical pipeline in 2011. J. Antibiot. 2011, 64, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Nigam, A.; Gupta, D.; Sharma, A. Treatment of infectious disease: Beyond antibiotics. Microbiol. Res. 2014, 169, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, R.B.R.; Suárez, R.; Cerdà, V.; Rangel, M.; Rangel, A.O.S.S. Exploiting the use of 3,4-HPO ligands as nontoxic reagents for the determination of iron in natural waters with a sequential injection approach. Talanta 2013, 108, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Moniz, T.; Silva, D.; Silva, T.; Gomes, M.S.; Rangel, M. Antimycobacterial activity of rhodamine 3,4-HPO iron chelators against Mycobacterium avium: Analysis of the contribution of functional groups and of chelator’s combination with ethambutol. MedChemComm 2015, 6, 2194–2203. [Google Scholar] [CrossRef]
- Suarez, R.; Mesquita, R.B.; Rangel, M.; Cerda, V.; Rangel, A.O. Iron speciation by microsequential injection solid phase spectrometry using 3-hydroxy-1(H)-2-methyl-4-pyridinone as chromogenic reagent. Talanta 2015, 133, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.S.; Carvalho, S.M.P.; Leite, A.; Moniz, T.; Roriz, M.; Rangel, A.O.S.S.; Rangel, M.; Vasconcelos, M.W. Effect of tris(3-hydroxy-4-pyridinonate) iron(III) complexes on iron uptake and storage in soybean (Glycine max L.). Plant Physiol. Biochem. 2016, 106, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Cilibrizzi, A.; Abbate, V.; Chen, Y.-L.; Ma, Y.; Zhou, T.; Hider, R.C. Hydroxypyridinone Journey into Metal Chelation. Chem. Rev. 2018, 118, 7657–7701. [Google Scholar] [CrossRef] [PubMed]
- Chaves, S.; Piemontese, L.; Hiremathad, A.; Santos, M.A. Hydroxypyridinone Derivatives: A Fascinating Class of Chelators with Therapeutic Applications—An Update. Curr. Med. Chem. 2018, 25, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Botzenhardt, S.; Li, N.Y.; Chan, E.W.; Sing, C.W.; Wong, I.C.K.; Neubert, A. Safety profiles of iron chelators in young patients with haemoglobinopathies. Eur. J. Haematol. 2017, 98, 198–217. [Google Scholar] [CrossRef] [PubMed]
- Bollig, C.; Schell, L.K.; Rucker, G.; Allert, R.; Motschall, E.; Niemeyer, C.M.; Bassler, D.; Meerpohl, J.J. Deferasirox for managing iron overload in people with thalassaemia. Cochrane Database Syst. Rev. 2017, 8, CD007476. [Google Scholar] [CrossRef] [PubMed]
- Azman, N.F.; Abdullah, W.Z.; Mohamad, N.; Bahar, R.; Johan, M.F.; Diana, R.; Sarifah, B.H.; Yusoff, S.; Nasir, A.; Othman, A.; et al. Practice of iron chelation therapy for transfusion-dependent thalassemia in Southeast Asia. Asian Biomed. 2016, 10, 537–547. [Google Scholar] [CrossRef]
- Borgna-Pignatti, C.; Marsella, M. Iron Chelation in Thalassemia Major. Clin. Ther. 2015, 37, 2866–2877. [Google Scholar] [CrossRef] [PubMed]
- Bucki, R.; Pastore, J.J.; Randhawa, P.; Vegners, R.; Weiner, D.J.; Janmey, P.A. Antibacterial Activities of Rhodamine B-Conjugated Gelsolin-Derived Peptides Compared to Those of the Antimicrobial Peptides Cathelicidin LL37, Magainin II, and Melittin. Antimicrob. Agents Chemother. 2004, 48, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, I.; Mishra, A.K. Fluorophore tagged bio-molecules and their applications: A brief review. Appl. Spectrosc. Rev. 2018, 53, 552–601. [Google Scholar] [CrossRef]
- Birch, D.; Christensen, M.V.; Staerk, D.; Franzyk, H.; Nielsen, H.M. Fluorophore labeling of a cell-penetrating peptide induces differential effects on its cellular distribution and affects cell viability. Biochim. Biophys. Acta Biomembr. 2017, 1859, 2483–2494. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.-C.; Cai, L.; Zhang, S.; Zhang, X. Cell-Penetrating Peptide Spirolactam Derivative as a Reversible Fluorescent pH Probe for Live Cell Imaging. Anal. Chem. 2017, 89, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Hedegaard, S.F.; Derbas, M.S.; Lind, T.K.; Kasimova, M.R.; Christensen, M.V.; Michaelsen, M.H.; Campbell, R.A.; Jorgensen, L.; Franzyk, H.; Cárdenas, M.; et al. Fluorophore labeling of a cell-penetrating peptide significantly alters the mode and degree of biomembrane interaction. Sci. Rep. 2018, 8, 6327. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.T.; Sayers, E.J. Cell entry of cell penetrating peptides: Tales of tails wagging dogs. J. Control. Release 2012, 161, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Hirose, H.; Takeuchi, T.; Osakada, H.; Pujals, S.; Katayama, S.; Nakase, I.; Kobayashi, S.; Haraguchi, T.; Futaki, S. Transient focal membrane deformation induced by arginine-rich peptides leads to their direct penetration into cells. Mol. Ther. 2012, 20, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Bucki, R.; Janmey, P.A. Interaction of the Gelsolin-Derived Antibacterial PBP 10 Peptide with Lipid Bilayers and Cell Membranes. Antimicrob. Agents Chemother. 2006, 50, 2932–2940. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.A.; Marques, S.M.; Chaves, S. Hydroxypyridinones as “privileged” chelating structures for the design of medicinal drugs. Coord. Chem. Rev. 2012, 256, 240–259. [Google Scholar] [CrossRef]
- Leite, A.; Silva, A.M.G.; Nunes, A.; Andrade, M.; Sousa, C.; Cunha-Silva, L.; Gameiro, P.; De Castro, B.; Rangel, M. Novel tetradentate chelators derived from 3-hydroxy-4-pyridinone units: Synthesis, characterization and aqueous solution properties. Tetrahedron 2011, 67, 4009–4016. [Google Scholar] [CrossRef]
- Kim, H.W.; Lee, C.H.; Kim, M.G.; Lee, H.S. Antibacterial activities of phenethyl isothiocyanate and its derivatives against human oral pathogens. J. Korean Soc. Appl. Biol. Chem. 2009, 52, 555–559. [Google Scholar] [CrossRef]
- Jang, M.; Hong, E.; Kim, G.H. Evaluation of antibacterial activity of 3-butenyl, 4-pentenyl, 2-phenylethyl, and benzyl isothiocyanate in Brassica vegetables. J. Food Sci. 2010, 75, M412–M416. [Google Scholar] [CrossRef] [PubMed]
- Sofrata, A.; Santangelo, E.M.; Azeem, M.; Borg-Karlson, A.K.; Gustafsson, A.; Pütsep, K. Benzyl isothiocyanate, a major component from the roots of Salvadora persica is highly active against Gram-Negative bacteria. PLoS ONE 2011, 6, e23045. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.S.; Gebremariam, T.; French, S.W.; Edwards, J.E., Jr.; Spellberg, B. The iron chelator deferasirox enhances liposomal amphotericin B efficacy in treating murine invasive pulmonary aspergillosis. J. Antimicrob. Chemother. 2010, 65, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, S.S.; Pinto, E.G.; Steverding, D.; Pleban, K.; Tempone, A.G.; Hider, R.C.; Wagner, G.K. Conjugation to 4-aminoquinoline improves the anti-trypanosomal activity of Deferiprone-type iron chelators. Bioorg. Med. Chem. 2013, 21, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.E.; Mikusova, K.; Brennan, P.J.; Besra, G.S. Synthesis of the mycobacterial arabinose donor β-D-arabinofuranosyl-1-monophosphoryldecaprenol, development of a basic arabinosyl-transferase assay, and identification of ethambutol as an arabinosyl transferase inhibitor. J. Am. Chem. Soc. 1995, 117, 11829–11832. [Google Scholar] [CrossRef]
- Lety, M.A.; Nair, S.; Berche, P.; Escuyer, V. A single point mutation in the embB gene is responsible for resistance to ethambutol in Mycobacterium smegmatis. Antimicrob. Agents Chemother. 1997, 41, 2629–2633. [Google Scholar] [CrossRef] [PubMed]
- Goude, R.; Amin, A.G.; Chatterjee, D.; Parish, T. The arabinosyltransferase EmbC is inhibited by ethambutol in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2009, 53, 4138–4146. [Google Scholar] [CrossRef] [PubMed]
- Moniz, T.; Leite, A.; Silva, T.; Gameiro, P.; Gomes, M.S.; de Castro, B.; Rangel, M. The influence of functional groups on the permeation and distribution of antimycobacterial rhodamine chelators. J. Inorg. Biochem. 2017, 175, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, J.T.S.; Moniz, T.; Brás, N.F.; Ivanova, G.; Fernandes, P.A.; Ramos, M.J.; Rangel, M. Relevant Interactions of Antimicrobial Iron Chelators and Membrane Models Revealed by Nuclear Magnetic Resonance and Molecular Dynamics Simulations. J. Phys. Chem. B 2014, 118, 14590–14601. [Google Scholar] [CrossRef] [PubMed]
- Moniz, T.; de Castro, B.; Rangel, M.; Ivanova, G. NMR study of the interaction of fluorescent 3-hydroxy-4-pyridinone chelators with DMPC liposomes. Phys. Chem. Chem. Phys. 2016, 18, 5027–5033. [Google Scholar] [CrossRef] [PubMed]
- Lucio, M.; Lima, J.L.; Reis, S. Drug-membrane interactions: Significance for medicinal chemistry. Curr. Med. Chem. 2010, 17, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Santos, N.C.; Prieto, M.; Castanho, M.A.R.B. Quantifying molecular partition into model systems of biomembranes: An emphasis on optical spectroscopic methods. Biochim. Biophys. Acta 2003, 1612, 123–135. [Google Scholar] [CrossRef]
- Epand, R.M.; Epand, R.F. Bacterial membrane lipids in the action of antimicrobial agents. J. Pept. Sci. 2011, 17, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Moniz, T.; Feio, M.; Silva, D.; de Castro, B.; Rangel, M. Study of the effect of thiourea and N-ethyl groups on antibacterial activity of rhodamine-labeled 3,4-HPO iron chelators against Gram (plus/-) bacteria. Med. Chem. Res. 2018, 27, 1472–1477. [Google Scholar] [CrossRef]
- Novais, Â.; Moniz, T.; Rebelo, A.R.; Silva, A.M.G.; Rangel, M.; Peixe, L. New fluorescent rosamine chelator showing promising antibacterial activity against Gram-positive bacteria. Bioorg. Chem. 2018, 79, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Moniz, T. Design of Novel 3-Hydroxy-4-Pyridinone Iron Chelators to Fight Mycobacterium Infection. Ph.D. Thesis, Department of Chemistry and Biochemistry, Faculty of Sciences, University of Porto, Porto, Portugal, 2016. [Google Scholar]
- Meerovich, I.; Koshkaryev, A.; Thekkedath, R.; Torchilin, V.P. Screening and Optimization of Ligand Conjugates for Lysosomal Targeting. Bioconjug. Chem. 2011, 22, 2271–2282. [Google Scholar] [CrossRef] [PubMed]
- Rauen, U.; Springer, A.; Weisheit, D.; Petrat, F.; Korth, H.G.; de Groot, H.; Sustmann, R. Assessment of chelatable mitochondrial iron by using mitochondrion-selective fluorescent iron indicators with different iron-binding affinities. ChemBioChem 2007, 8, 341–352. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chelator | Structural Features | Antibacterial Activity | Membrane Interaction | Ref. | |||
|---|---|---|---|---|---|---|---|
| Type of Fluorophore | Linker | Charge (at pH = 7.4) | M. avium | Gram (+/−) | |||
| MRB7 |  | Thiourea | Neutral | ++++ | 0 | ++ | a |
| MRH7 | Thiourea | Neutral | ++++ | +++ | +++ | ||
| MRB8 |  | Amide | Neutral | + | 0 | + | |
| MRH8 | Amide | Neutral | + | + | + | ||
| MRB9 |  | Amide | Neutral | ++ | 0 | nd | b |
| MRH10 |  | Thiourea | Neutral | +++ | ++ | nd | |
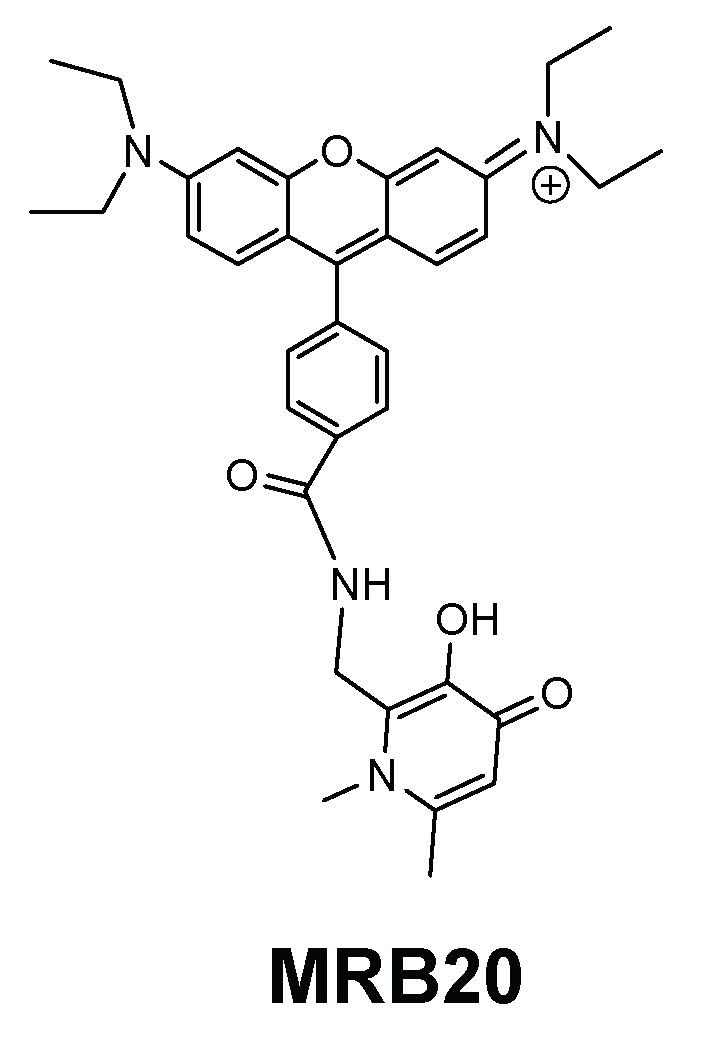
| MRB20 |  | Amide | Positive | 0 | ++++ | nd | c |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rangel, M.; Moniz, T.; Silva, A.M.N.; Leite, A. Tuning the Anti(myco)bacterial Activity of 3-Hydroxy-4-pyridinone Chelators through Fluorophores. Pharmaceuticals 2018, 11, 110. https://doi.org/10.3390/ph11040110
Rangel M, Moniz T, Silva AMN, Leite A. Tuning the Anti(myco)bacterial Activity of 3-Hydroxy-4-pyridinone Chelators through Fluorophores. Pharmaceuticals. 2018; 11(4):110. https://doi.org/10.3390/ph11040110
Chicago/Turabian StyleRangel, Maria, Tânia Moniz, André M. N. Silva, and Andreia Leite. 2018. "Tuning the Anti(myco)bacterial Activity of 3-Hydroxy-4-pyridinone Chelators through Fluorophores" Pharmaceuticals 11, no. 4: 110. https://doi.org/10.3390/ph11040110
APA StyleRangel, M., Moniz, T., Silva, A. M. N., & Leite, A. (2018). Tuning the Anti(myco)bacterial Activity of 3-Hydroxy-4-pyridinone Chelators through Fluorophores. Pharmaceuticals, 11(4), 110. https://doi.org/10.3390/ph11040110