Oxidative Stress and Cardiovascular Complications in Chronic Kidney Disease, the Impact of Anaemia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Oxidative Stress in Chronic Kidney Disease
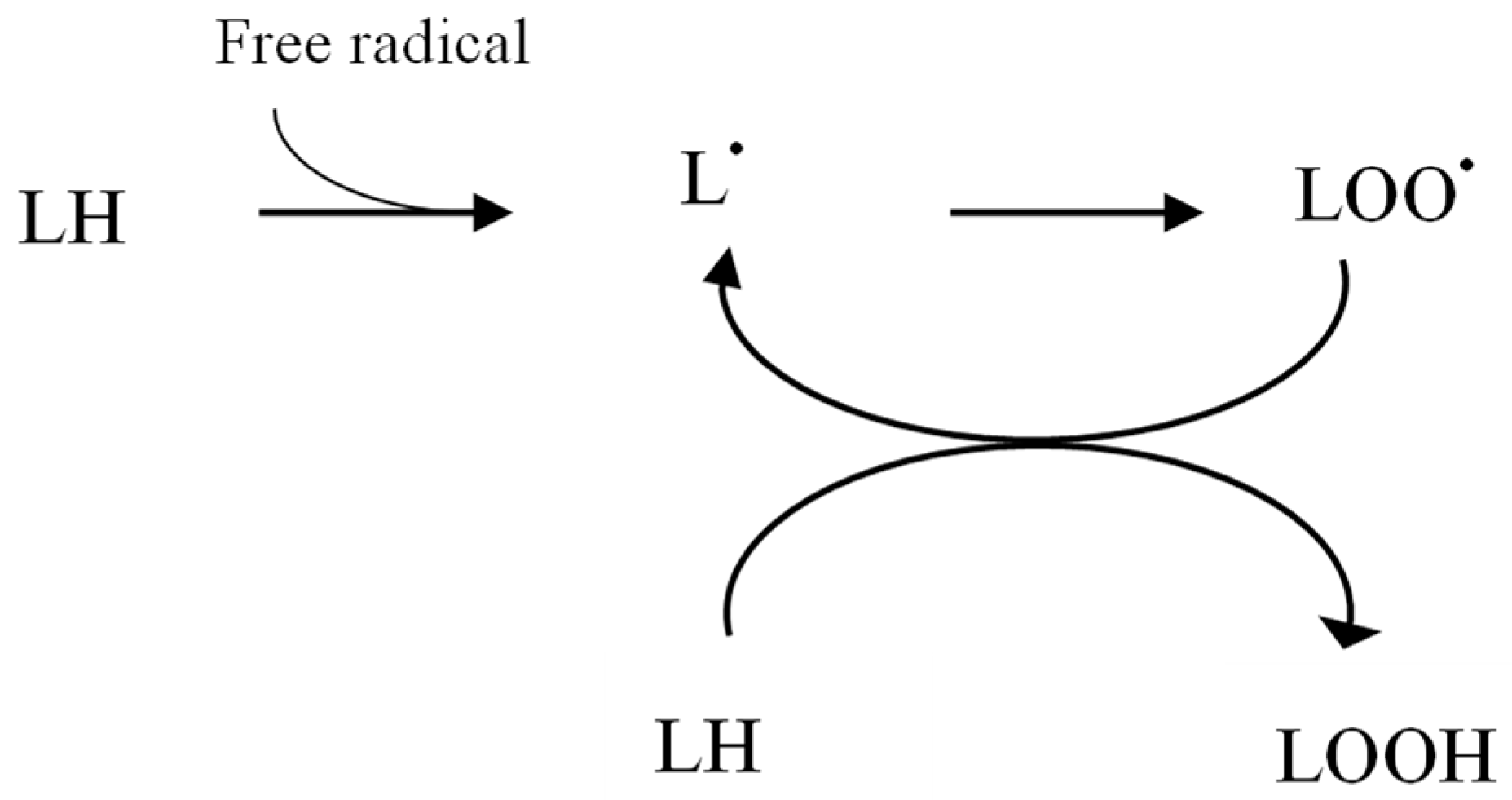
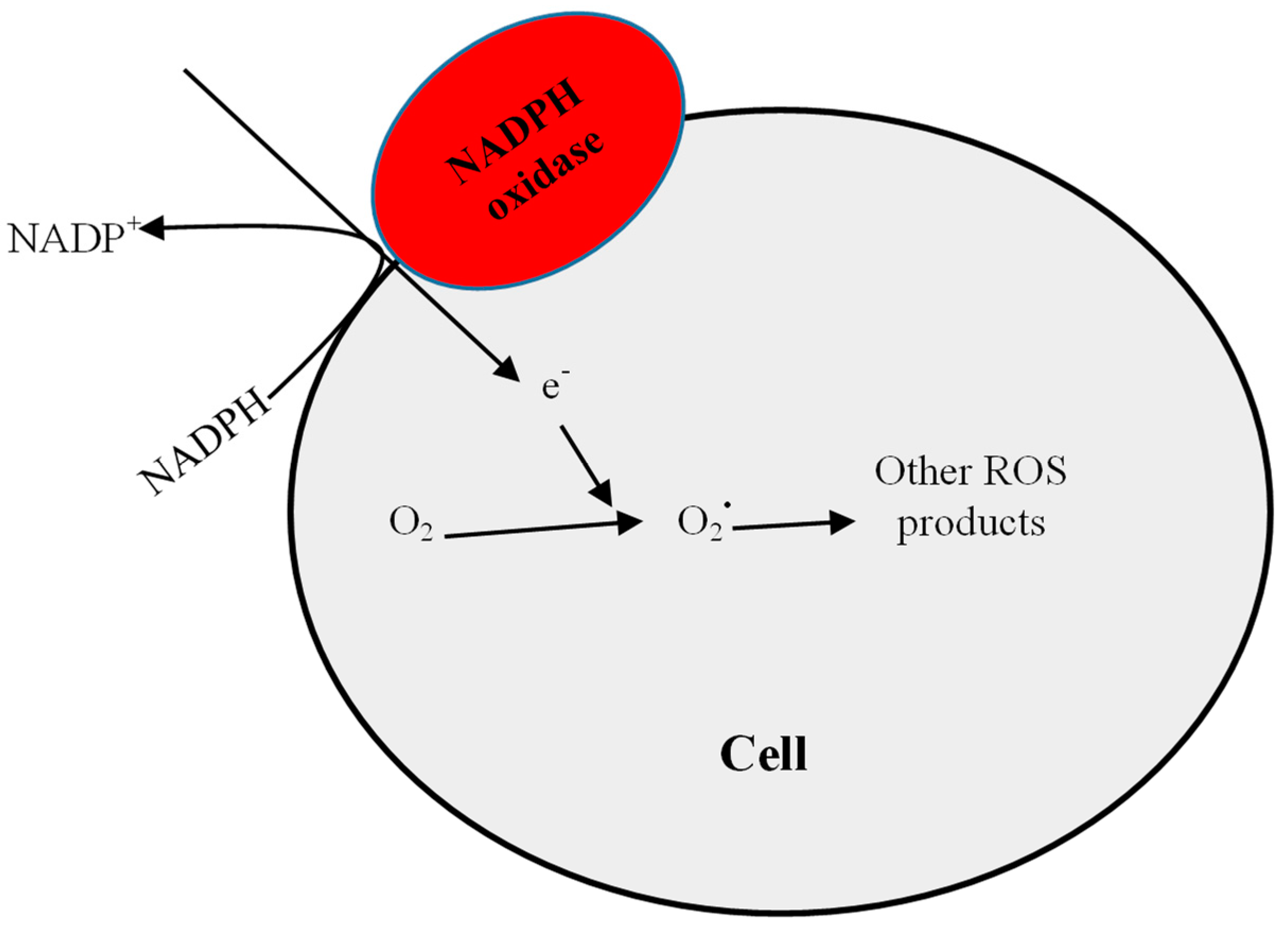
2.1. Increased Pro-Oxidant Activity
2.2. Uraemic Toxins
2.3. Inflammation
2.4. Impaired Antioxidant System
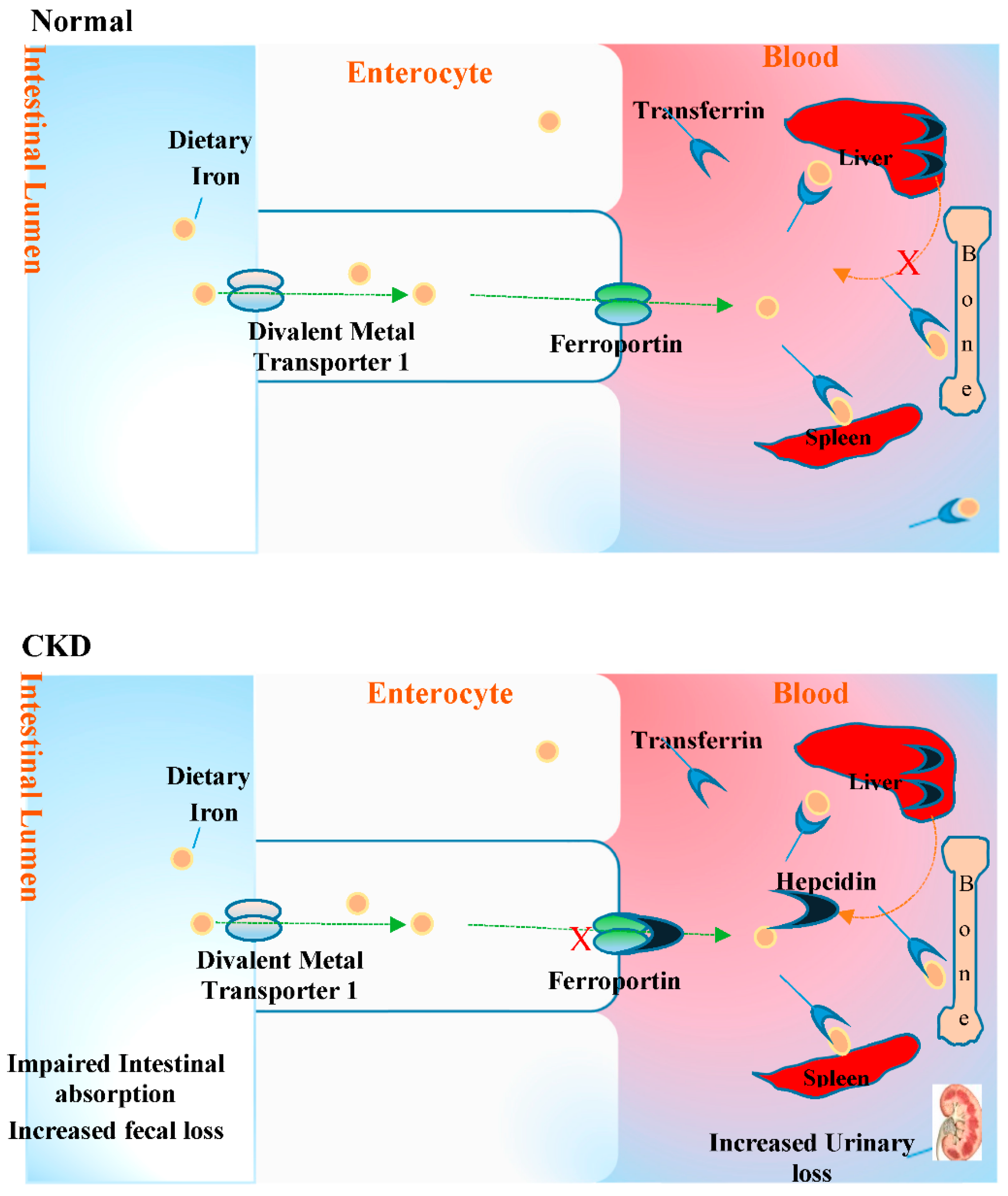
2.5. Anaemia and Oxidative Stress in CKD
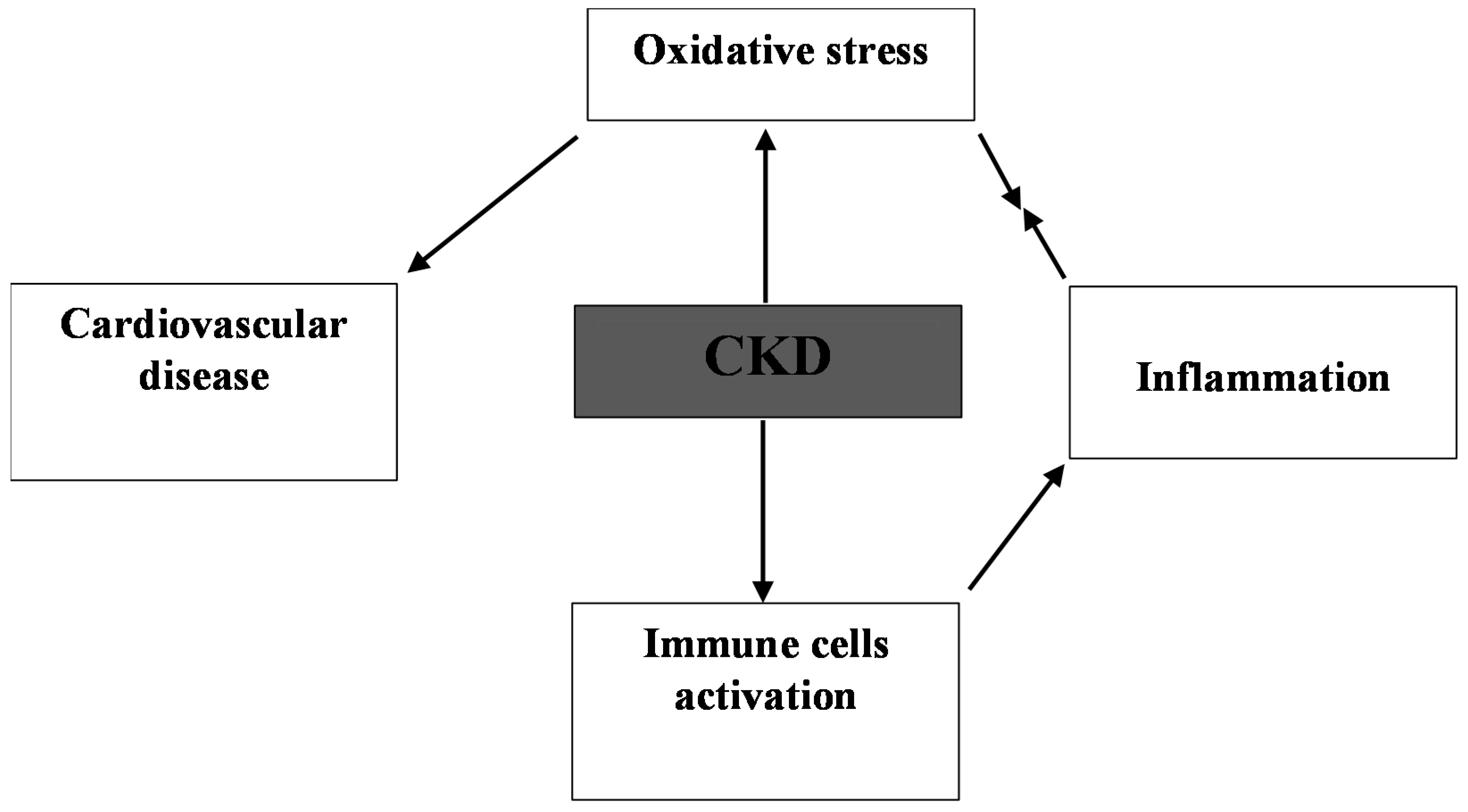
3. Consequence of Oxidative Stress in CKD
4. Cardio-Renal Oxidative Stress
5. Future Questions
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cachofeiro, V.; Goicochea, M.; Garca de Vinuesa, S.; Oubina, P.; Lahera, V.; Luno, J. Oxidative stress and inflammation, a link between chronic kidney disease and cardiovascular disease. Kidney Int. 2008, 74, S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Sinha, N.; Dabla, P.K. Oxidative stress and antioxidants in hypertension—A current review. Curr. Hypertens. Rev. 2015, 11, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Blair, A.I. DNA adducts with lipid peroxidation products. J. Biol. Chem. 2008, 283, 15545–15549. [Google Scholar] [CrossRef] [PubMed]
- Kabel, A.M. Free Radicals and Antioxidants: Role of Enzymes and Nutrition. World J. Nutr. Health 2014, 2, 35–38. [Google Scholar]
- Panth, N.; Paudel, K.R.; Parajuli, K. Reactive Oxygen Species: A Key Hallmark of Cardiovascular Disease. Adv. Med. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Kao, M.P.C.; Ang, D.S.C.; Pall, A.; Struthers, A.D. Oxidative stress in renal dysfunction: Mechanisms, clinical sequelae and therapeutic options. J. Hum. Hypertens. 2010, 24, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dennis, J.M.; Witting, P.K. Protective Role for Antioxidants in Acute Kidney Disease. Nutrients 2017, 97, 718. [Google Scholar] [CrossRef] [PubMed]
- Sung, C.C.; Hsu, Y.C.; Chen, C.C.; Lin, Y.F.; Wu, C.C. Oxidative stress and nucleic acid oxidation in patients with chronic kidney disease. Oxid. Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D. Roles of oxidative stress and antioxidant therapy in chronic kidney disease and hypertension. Curr. Opin. Nephrol. Hypertens. 2004, 13, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Oberg, B.P.; McMenamin, E.; Lucas, F.L.; McMonagle, E.; Morrowm, J.; Ikizler, T.A.; Himmelfarb, J. Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int. 2004, 65, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Perianayagam, M.C.; Liangos, O.; Kolyadam, A.Y.; Wald, R.; MacKinnon, R.W.; Li, L.; Rao, M.; Balakrishnan, V.S.; Bonventre, J.V.; Pereira, B.J.; et al. NADPH oxidase p22phox and catalase gene variants are associated with biomarkers of oxidative stress and adverse outcomes in acute renal failure. J. Am. Soc. Nephrol. 2007, 18, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Castilla, P.; Dávalos, A.; Teruel, J.L.; Cerrato, F.; Fernández-Lucas, M.; Merino, J.L.; Sánchez-Martín, C.C.; Ortuño, J.; Lasunción, M.A. Comparative effects of dietary supplementation with red grape juice and vitamin E on production of superoxide by circulating neutrophil NADPH oxidase in hemodialysis patients. Am. J. Clin. Nutr. 2008, 87, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Onozato, M.L.; Tojo, A.; Goto, A.; Fujita, T.; Wilcox, C.S. Oxidative stress and nitric oxide synthase in rat diabetic nephropathy: Effects of ACEI and ARB. Kidney Int. 2002, 61, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Thallas-Bonke, V.; Thorpe, S.R.; Coughlan, M.T.; Fukami, K.; Yap, F.Y.; Sourris, K.; Penfold, S.; Bach, L.A.; Cooper, M.E.; Forbesm, J.M. Inhibition of NADPH oxidase prevents AGE-mediated damage in diabetic nephropathy through a protein kinase C-α-dependent pathway. Diabetes 2007, 57, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Bryk, D.; Olejarz, W.; Zapolska-Downar, D. The role of oxidative stress and NADPH oxidase in the pathogenesis of atherosclerosis. Postepy Hig. Med. Dosw. 2017, 28, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Schulz, A.M.; Terne, C.; Jankowski, V.; Cohen, G.; Schaefer, M.; Boehringer, F.; Tepel, M.; Kunkel, D.; Zidek, W.; Jankowski, J. Modulation of NADPH oxidase activity by known uraemic retention solutes. Eur. J. Clin. Investig. 2014, 44, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Shimoishi, K.; Anraku, M.; Kitamura, K.; Tasaki, Y.; Taguchi, K.; Hashimoto, M.; Fukunaga, E.; Maruyama, T.; Otagiri, M. An oral adsorbent, AST-120 protects against the progression of oxidative stress by reducing the accumulation of indoxyl sulfate in the systemic circulation in renal failure. Pharm. Res. 2007, 24, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- Sirker, A.; Zhang, M.; Shah, A.M. NADPH oxidases in cardiovascular disease: Insights from in vivo models and clinical studies. Basic Res. Cardiol. 2011, 106, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Sachse, A.; Wolf, G. Angiotensin II-Induced Reactive Oxygen Species and the Kidney. J. Am. Soc. Nephrol. 2007, 18, 2439–2446. [Google Scholar] [CrossRef] [PubMed]
- Flyvbjerg, A.; Denner, L.; Schrijvers, B.F.; Tilton, R.G.; Mogensen, T.H.; Paludan, S.R.; Rasch, R. Long-term renal effects of a neutralizing RAGE antibody in obese type 2 diabetic mice. Diabetes 2004, 53, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Yoon, Y.J.; Choi, H.J.; Park, S.H.; Kim, C.D.; Kim, I.S.; Kwon, T.H.; Do, J.Y.; Kim, S.H.; Ryu, D.H.; et al. Dialysis modality-dependent changes in serum metabolites: Accumulation of inosine and hypoxanthine in patients undergoing hemodialysis. Nephrol. Dial. Transplant. 2011, 26, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Xu, B.; Yan, B.; Qiao, X.; Wang, L. Effects of uric acid-lowering therapy in patients with chronic kidney disease: A meta-analysis. PLoS ONE 2017, 12, e0187550. [Google Scholar] [CrossRef] [PubMed]
- Oshima, N.; Onimaru, H.; Matsubara, H.; Uchida, T.; Watanabe, A.; Takechi, H.; Nishida, Y.; Kumagai, H. Uric acid, indoxyl sulfate, and methylguanidine activate bulbospinal neurons in the rvlm via their specific transporters and by producing oxidative stress. Neuroscience 2015, 304, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Evenepoel, P.; Meijers, B.K.; Bammens, B.R.; Verbeke, K. Uremic toxins originating from colonic microbial metabolism. Kidney Int. 2009, 76, S12–S19. [Google Scholar] [CrossRef] [PubMed]
- Tumur, Z.; Shimizu, H.; Enomoto, A.; Miyazaki, H.; Niwa, T. Indoxyl sulfate upregulates expression of ICAM-1 and MCP-1 by oxidative stress-induced NF-kappaB activation. Am. J. Nephrol. 2010, 31, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Miyamoto, Y.; Honda, D.; Tanaka, H.; Wu, Q.; Endo, M.; Noguchi, T.; Kadowaki, D.; Ishima, Y.; Kotani, S.; et al. P-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress through the activation of NADPH oxidase. Kidney Int. 2013, 83, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Campbell, K.L.; Johnson, D.W.; Stanton, T.; Vesey, D.A.; Coombes, J.S.; Weston, K.S.; Hawley, C.M.; McWhinney, B.C.; Ungerer, J.P.J.; et al. Protein-bound Uremic Toxins, Inflammation and Oxidative Stress: A Cross-sectional Study in Stage 3–4 Chronic Kidney Disease. Arch. Med. Res. 2014, 45, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.I.; Evenepoel, P. The gut-kidney axis: Indoxyl sulfate, p-cresyl sulfate and CKD progression. Nephrol. Dial. Transplant. 2011, 26, 759–761. [Google Scholar] [CrossRef] [PubMed]
- Motojima, M.; Hosokawa, A.; Yamato, H.; Muraki, T.; Yoshioka, T. Uremic toxins of organic anions up-regulate PAI-1 expression by induction of NF-kappaB and free radical in proximal tubular cells. Kidney Int. 2003, 63, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Kim, Y.J.; Kang, D.H. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin. J. Am. Soc. Nephrol. 2011, 6, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Kazama, J.J.; Omori, K.; Matsuo, K.; Takahashi, Y.; Kawamura, K.; Matsuto, T.; Watanabe, H.; Maruyama, T.; Narita, I. Continuous Reduction of Protein-Bound Uraemic Toxins with Improved Oxidative Stress by Using the Oral Charcoal Adsorbent AST-120 in Haemodialysis Patients. Sci. Rep. 2015, 5, 14381. [Google Scholar] [CrossRef] [PubMed]
- Taki, K.; Niwa, T. Indoxyl sulfate-lowering capacity of oral sorbents affects prognosis of kidney function and oxidative stress in chronic kidney disease. J. Ren. Nutr. 2007, 17, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Tumur, Z.; Niwa, T. An oral sorbent AST-120 increases renal NO synthesis in uremic rats. J. Ren. Nutr. 2008, 18, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Tanaka, A.; Oyama, J.; Yamasaki, A.; Shimomura, M.; Hiwatashi, A.; Ueda, Y.; Amaha, M.; Nomura, M.; Matsumura, D.; et al. Long-term effects of AST-120 on the progression and prognosis of pre-dialysis chronic kidney disease: A 5-year retrospective study. Heart Vessels 2016, 31, 1625–1632. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.A.; Lu, L.F.; Yu, T.H.; Hung, W.C.; Chung, F.M.; Tsai, I.T.; Yang, C.Y.; Hsu, C.C.; Lu, Y.C.; Wang, C.P.; et al. Increased levels of total P-cresylsulphate and indoxyl sulphate are associated with coronary artery disease in patients with diabetic nephropathy. Rev. Diabet. Stud. 2010, 7, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.Y.; Hsu, H.H.; Wu, M.S. p-Cresol sulfate and indoxyl sulfate induce similar cellular inflammatory gene expressions in cultured proximal renal tubular cells. Nephrol. Dial. Transplant. 2012, 28, 70–78. [Google Scholar] [CrossRef] [PubMed]
- DeFIlippi, C.R.; Herzog, C.A. Interpreting Cardiac Biomarkers in the Setting of Chronic Kidney Disease. Clin. Chem. 2017, 63, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Ori, Y.; Bergman, M.; Bessler, H.; Zingerman, B.; Levy-Drummer, R.S.; Gafter, U.; Salman, H. Cytokine secretion and markers of inflammation in relation to acidosis among chronic hemodialysis patients. Blood Purif. 2013, 35, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Taetzsch, T.; Levesque, S.; McGraw, C.; Bookins, S.; Luqa, R.; Bonini, G.M.; Mason, P.R.; Oh, U.; Block, L.M. Redox regulation of NF-κB p50 and M1 polarization in microglia. Glia 2015, 63, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Giam, B.; Kaye, D.M.; Rajapakse, N.W. Role of Renal Oxidative Stress in the Pathogenesis of the Cardiorenal Syndrome. Heart Lung Circ. 2016, 25, 874–880. [Google Scholar] [CrossRef] [PubMed]
- Oberg, B.P.; McMenamin, E.; Lucas, F.L.; McMonagle, E.; Morrow, J.; Ikizler, T.A.; Himmelfarb, J. Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int. 2004, 65, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.D.; Ryan, M.J. Immune and Inflammatory Role in Renal Disease. Compr. Physiol. 2013, 3, 957–976. [Google Scholar] [PubMed]
- Forbes, J.M.; Coughlan, M.T.; Cooper, M.E. Oxidative Stress as a Major Culprit in Kidney Disease in Diabetes. Diabetes 2008, 5, 1446–1454. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Brennan, M.L.; Hazen, S.L. Serum myeloperoxidase and mortality in maintenance hemodialysis patients. Am. J. Kidney Dis. 2006, 48, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Zargari, M.; Sedighi, O. Influence of hemodialysis on lipid peroxidation, enzymatic and non-enzymatic antioxidant capacity in chronic renal failure patients. Nephro-Urol. Mon. 2015, 7, e28526. [Google Scholar] [CrossRef] [PubMed]
- Tbahriti, H.F.; Kaddous, A.; Bouchenak, M.; Mekki, K. Effect of Different Stages of Chronic Kidney Disease and Renal Replacement Therapies on Oxidant-Antioxidant Balance in Uremic Patients. Biochem. Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rico, M.; Puchades, M.J.; Garcia, R.; Saez, G.; Tormos, M.C.; Miguel, A. Effect of oxidative stress in patients with chronic renal failure. Nefrologia 2006, 26, 218–225. [Google Scholar] [PubMed]
- Niwa, T.; Tsukushi, S. 3-Deoxyglucosone and AGEs in uraemic complications: Inactivation of glutathione peroxidase by 3-deoxyglucosone. Kidney Int. 2001, 59, S37–S41. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Yu, Y.; Xing, R.; Guo, X.; Liu, D.; Wei, J.; Song, H. Unglycosylated recombinant human glutathione peroxidase 3 mutant from Escherichia coli is active as a monomer. Sci. Rep. 2014, 4, 6698. [Google Scholar] [CrossRef] [PubMed]
- Labunskyy, M.V.; Hatfield, L.D.; Gladyshev, N.V. Selenoproteins: Molecular Pathways and Physiological Roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef] [PubMed]
- Avissar, N.; Ornt, D.B.; Yagil, Y.; Horowitz, S.; Watkins, R.H.; Kerl, E.A.; Takahashi, K.; Palmer, I.S.; Cohen, H.J. Human kidney proximal tubules are the main source of plasma glutathione peroxidase. Am. J. Physiol. 1994, 266, C367–C375. [Google Scholar] [CrossRef] [PubMed]
- Kuchta, A.; Pacanis, A.; Kortas-Stempak, B.; Ćwiklińska, A.; Ziętkiewicz, M.; Renke, M.; Rutkowski, B. Estimation of Oxidative Stress Markers in Chronic Kidney Disease. Kidney Blood Press Res. 2011, 34, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Papavasiliou, E.C.; Gouva, C.; Siamopoulos, K.C.; Tselepis, A.D. Erythrocyte PAF-acetylhydrolase activity in various stages of chronic kidney disease: Effect of long-term therapy with erythropoietin. Kidney Int. 2005, 68, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Michea, L.; Villagrán, A.; Urzúa, A.; Kuntsmann, S.; Venegas, P.; Carrasco, L.; González, M.; Marusic, E. Mineralocorticoid receptor antagonism attenuates cardiac hypertrophy and prevents oxidative stress in uremic rats. Hypertension 2008, 52, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Oveisi, F.; Ding, Y. Role of increased oxygen free radical activity in the pathogenesis of uremic hypertension. Kidney Int. 1998, 53, 1748–1754. [Google Scholar] [CrossRef] [PubMed]
- Kalk, P.; Godes, M.; Relle, K.; Rothkegel, C.; Hucke, A.; Stasch, J.P.; Hocher, B. NO-independent activation of soluble guanylate cyclase prevents disease progression in rats with 5/6 nephrectomy. Br. J. Pharmacol. 2006, 148, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Goodnough, L.T.; Nemeth, E.; Ganz, T. Detection, evaluation, and management of iron-restricted erythropoiesis. Blood 2010, 116, 4754–4761. [Google Scholar] [CrossRef] [PubMed]
- Sathyan, S.; George, S.; Vijayan, P. Prevalence of anemia and cardiovascular diseases in chronic kidney disease patients: A single tertiary care centre study. Int. J. Adv. Med. 2017, 4, 247–251. [Google Scholar] [CrossRef]
- Mehdi, U.; Toto, R.D. Anemia, Diabetes, and Chronic Kidney Disease. Diabetes Care 2009, 32, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Babitt, J.L.; Lin, H.Y. Mechanisms of anemia in CKD. J. Am. Soc. Nephrol. 2012, 23, 1631–1634. [Google Scholar] [CrossRef] [PubMed]
- Franchini, M.; Montagnana, M.; Lippi, G. Hepcidin and iron metabolism: From laboratory to clinical implications. Clin. Chim. Acta 2010, 411, 1565–1569. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. Biochim. Biophys. Acta 2012, 1823, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A.; Dierssen, U.; Valli, L.; Garuti, C.; Rump, A.; Corradini, E.; Ernst, M.; Klein, C.; Trautwein, C. STAT3 is required for IL-6-gp130-dependent activation of hepcidin in vivo. Gastroenterology 2007, 132, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Falzacappa, M.V.; Vujic, S.M.; Kessler, R.; Stolte, J.; Hentze, M.W.; Muckenthaler, M.U. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood 2007, 109, 353–358. [Google Scholar] [CrossRef] [PubMed]
- David, V.; Martin, A.; Isakova, T.; Spaulding, C.; Qi, L.; Ramirez, V.; Zumbrennen-Bullough, K.B.; Sun, C.C.; Lin, H.Y.; Babitt, J.L.; et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016, 89, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Kempe, D.S.; Lang, P.A.; Duranton, C.; Akel, A.; Lang, K.S.; Huber, S.M.; Wieder, T.; Lang, F. Enhanced programmed cell death of iron-deficient erythrocytes. Faseb. J. 2006, 20, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Malorni, W.; Straface, E.; Pagano, G.; Monti, D.; Zatterale, A.; Del Principe, D.; Deeva, I.B.; Franceschi, C.; Masella, R.; Korkina, L.G. Cytoskeleton alterations of erythrocytes from patients with Fanconi’s anemia. FEBS Lett. 2000, 468, 125–128. [Google Scholar] [CrossRef]
- Ghosh, S.; Bandyopadhyay, S.; Bhattacharya, D.K.; Mandal, C. Altered erythrocyte membrane characteristics during anemia in childhood acute lymphoblastic leukemia. Ann. Hematol. 2005, 84, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Ghoshal, A.; Bhattacharya, K.; Saha, B.; Walden, P.; Mandal, C. Sialoglycosylation of RBC in Visceral Leishmaniasis Leads to Enhanced Oxidative Stress, Calpain-Induced Fragmentation of Spectrin and Hemolysis. PLoS ONE 2012, 7, e42361. [Google Scholar] [CrossRef] [PubMed]
- Snyder, L.M.; Fortier, N.L.; Trainor, J.; Jacobs, J.; Leb, L.; Lubin, B.; Chiu, D.; Shohet, S.; Mohandas, N. Effect of hydrogen peroxide exposure on normal human erythrocyte deformability, morphology, surface characteristics, and spectrin-hemoglobin cross-linking. J. Clin. Investig. 1985, 76, 1971–1977. [Google Scholar] [CrossRef] [PubMed]
- Vives-Corrons, J.L.; Miguel-Garcia, A.; Pujades, M.A.; Miguel-Sosa, A.; Cambiazzo, S.; Linares, M.; Dibarrart, M.T.; Calvo, M.A. Increased susceptibility of microcytic red blood cells to in vitro oxidative stress. Eur. J. Haematol. 1995, 55, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Nagababu, E.; Gulyani, S.; Earley, C.J.; Cutler, R.G.; Mattson, M.P.; Rifkind, J.M. Iron-deficiency anaemia enhances red blood cell oxidative stress. Free Radic. Res. 2008, 42, 824–829. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.C.; Van der Linden, P.; Biro, G.; Hu, L.Q. Physiologic aspects of anemia. Crit. Care Clin. 2004, 20, 187–212. [Google Scholar] [CrossRef] [PubMed]
- Rifkind, J.M.; Nagababu, E. Hemoglobin Redox Reactions and Red Blood Cell Aging. Antioxid. Redox Signal. 2013, 18, 2274–2283. [Google Scholar] [CrossRef] [PubMed]
- Lazarte, S.S.; Mónaco, M.E.; Jimenez, C.L.; Achem, M.E.L.; Terán, M.M.; Issé, B.A. Erythrocyte Catalase Activity in More Frequent Microcytic Hypochromic Anemia: Beta-Thalassemia Trait and Iron Deficiency Anemia. Adv. Hematol. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Yetgin, S.; Hincal, F.; Basaran, N.; Ciliv, G. Serum selenium status in children with iron deficiency anemia. Acta Haematol. 1992, 88, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Prats, M.; Font, R.; García, C.; Muñoz-Cortés, M.; Cabré, C.; Jariod, M.; Romeu, M.; Giralt, M.; Martinez-Vea, A. Oxidative stress markers in predicting response to treatment with ferric carboxymaltose in nondialysis chronic kidney disease patients. Clin. Nephrol. 2014, 81, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, H.T. The role of intravenous iron in the treatment of anemia in cancer patients. Ther. Adv. Hematol. 2012, 3, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Kumerova, A.; Lece, A.; Skesters, A.; Silova, A.; Petuhovs, V. Anaemia and antioxidant defence of the red blood cells. Mater. Med. Pol. 1998, 30, 12–15. [Google Scholar] [PubMed]
- Kidney International KDIGO Clinical. Practice Guidelines for the Diagnosis, Evaluation, Prevention and Treatment of Chronic Kidney Disease-Mineral and Bone Disorders. J. Int. Soc. Nephrol. 2009, 7, S22–S31. [Google Scholar]
- Bhandari, S. Beyond efficacy and safety-the need for convenient and cost-effective iron therapy in health care. NDT Plus 2011, 4, i14–i19. [Google Scholar] [CrossRef] [PubMed]
- Hayat, A. Safety Issues with Intravenous Iron Products in the Management of Anemia in Chronic Kidney Disease. Clin. Med. Res. 2008, 6, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Ganguli, A.; Kohli, H.S.; Khullar, M.; Lal Gupta, K.; Jha, V.; Sakhuja, V. Lipid peroxidation products formation with various intravenous iron preparations in chronic kidney disease. Ren. Fail. 2009, 31, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.; Butcher, A. Evaluation of Serum Oxidative Stress Indices Following Intravenous Iron Delivery in Women with Iron Deficiency Anemia. Blood 2014, 124, 2683. [Google Scholar]
- Bailie, G.R.; Schuler, C.; Leggett, R.E.; Li, H.; Li, H.D.; Patadia, H.; Levin, R. Oxidative effect of several intravenous iron complexes in the rat. Biometals 2013, 26, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Swarnalatha, G.; Ram, R.; Neela, P.; Naidu, M.U.; Dakshina Murty, K.V. Oxidative stress in hemodialysis patients receiving intravenous iron therapy and the role of N-acetylcysteine in preventing oxidative stress. Saudi J. Kidney Dis. Transpl. 2010, 21, 852–858. [Google Scholar] [PubMed]
- Schaller, G.; Scheiber-Mojdehkar, B.; Wolzt, M.; Puttinger, H.; Mittermayer, F.; Hörl, W.H.; Fodinger, M.; Sunder-Plassmann, G.; Vychytil, A. Intravenous iron increases labile serum iron but does not impair forearm blood flow reactivity in dialysis patients. Kidney Int. 2005, 68, 2814–2822. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Mechanisms of iron hepatotoxicity. Hepatology 2016, 65, 226–227. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.M.; Bertoncini, C.R.A.; Boreky, J.; Souza-Pinto, N.C.; Vercesi, A.E. Mitochondrial DNA damage associated with lipid peroxidation of the mitochondrial membrane induced by Fe2+-citrate. Ann. Braz. Acad. Sci. 2006, 78, 505–514. [Google Scholar] [CrossRef]
- Bhandari, S.; Pereira, D.I.A.; Chappell, H.F.; Drakesmith, H. Intravenous irons: From basic science to clinical practice. Pharmaceuticals 2018, 11, 82. [Google Scholar] [CrossRef] [PubMed]
- Brewster, U.C.; Perazella, M.A. Intravenous iron and the risk of infection in end-stage renal disease patients. Semin. Dial. 2004, 17, 57–60. [Google Scholar] [PubMed]
- Diamond, J.R. The role of reactive oxygen species in animal models of glomerular disease. Am. J. Kidney Dis. 1992, 19, 292–300. [Google Scholar] [CrossRef]
- Takimoto, E.; Kass, D.A. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007, 49, 241. [Google Scholar] [CrossRef] [PubMed]
- Siragy, H.M.; Carey, R.M. Role of the Intrarenal Renin-Angiotensin-Aldosterone System in Chronic Kidney Disease. Am. J. Nephrol. 2010, 31, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Toma, I.; Kang, J.J.; Sipos, A.; Vargas, S.; Bansal, E.; Hanner, F.; Meer, E.; Peti-Peterdi, J. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J. Clin. Investig. 2008, 118, 2526–2534. [Google Scholar] [CrossRef] [PubMed]
- Kobori, H.; Alper, A.B.; Shenava, R.; Katsurada, A.; Saito, T.; Ohashi, N.; Urushihara, M.; Miyata, K.; Satou, R.; Hamm, L.L.; et al. Urinary angiotensinogen as a novel biomarker of the intrarenal renin-angiotensin system status in hypertensive patients. Hypertension 2009, 53, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Urushihara, M.; Kotani, Y.; Kagami, S.; Kobori, H. Increased urinary angiotensinogen is precedent to increased urinary albumin in patients with type 1 diabetes. Am. J. Med. Sci. 2009, 338, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Ruster, C.; Wolf, G. Renin-angiotensin-aldosterone system and progression of renal disease. J. Am. Soc. Nephrol. 2009, 17, 2985–2991. [Google Scholar] [CrossRef] [PubMed]
- Manrique, C.; Lastra, G.; Gardner, M.; Sowers, J.R. The Renin Angiotensin Aldosterone System in Hypertension: Roles of Insulin Resistance and Oxidative Stress. Med. Clin. N. Am. 2009, 93, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Morrone, D.; Marzilli, M. Role of RAAS inhibition in preventing left ventricular remodeling in patients post myocardial infarction. Heart Metab. 2010, 47, 9–13. [Google Scholar]
- Montezano, A.C.; Callera, G.E.; Yogi, A.; He, Y.; Tostes, R.C.; He, G.; Schiffrin, E.L.; Touyz, R.M. Aldosterone and Angiotensin II Synergistically Stimulate Migration in Vascular Smooth Muscle Cells Through c-Src-Regulated Redox-Sensitive RhoA Pathways. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Cat, A.N.D.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH Oxidase, and Redox Signaling in the Vasculature. Antioxid. Redox Signal. 2013, 19, 1110–1120. [Google Scholar]
- Mollnau, H.; Wendt, M.; Szocs, K.; Lassegue, B.; Schulz, E.; Oelze, M.; Li, H.; Bodenschatz, M.; August, M.; Kleschyov, A.L.; et al. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ. Res. 2002, 90, E58–E65. [Google Scholar] [CrossRef] [PubMed]
- Herbert, K.E.; Mistry, Y.; Hastings, R.; Poolman, T.; Niklason, L.; Williams, B. Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circ. Res. 2008, 102, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Lovett, D.; Lehrer, R.I.; Couser, W.G.; Klebanoff, S.J. Role of oxidants and protease in glomerular injury. Kidney Int. 1994, 45, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Magagna, A.; Salvetti, A. Vitamin C improves endothelium-dependent vasodilation by restoring nitric oxide activity in essential hypertension. Circulation 1998, 97, 2222–2229. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, E.; Bellino, D.; Cassottana, P.; Rolla, D.; Cannella, G. Left ventricular hypertrophy in nondiabetic predialysis CKD. Am. J. Kidney Dis. 2005, 46, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Cave, A.C.; Grieve, D.J.; Johar, S.; Zhang, M.; Shah, A.M. NADPH oxidase-derived reactive oxygen species in cardiac pathophysiology. Philos. Trans. R. Soc. 2005, 360, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Li, J.M.; Gall, N.P.; Grieve, D.J.; Chen, M.; Shah, A.M. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension 2002, 40, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Kuster, G.M.; Pimentel, D.R.; Adachi, T.; Ido, Y.; Brenner, D.A.; Cohen, R.A.; Liao, R.; Siwik, D.A.; Colucci, W.S. Alpha-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes is mediated via thioredoxin-1-sensitive oxidative modification of thiols on Ras. Circulation 2005, 111, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Chen, T.; Szeto, H.; Nieves-Cintrón, M.; Kutyavin, V.; Santana, L.F.; Rabinovitch, P.S. Mitochondrial targeted antioxidant peptide ameliorates hypertensive cardiomyopathy. J. Am. Coll. Cardiol. 2011, 58, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Alani, H.; Tamimi, A.; Tamimi, N. Cardiovascular co-morbidity in chronic kidney disease: Current knowledge and future research needs. World J. Nephrol. 2014, 3, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.N.; Greaves, K. The Cardiorenal Syndrome: A Review. Int. J. Nephrol. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Kon, V.; Yang, H.; Fazio, S. Residual Cardiovascular Risk in Chronic Kidney Disease: Role of High-density Lipoprotein. Arch. Med. Res. 2015, 46, 379–391. [Google Scholar] [CrossRef] [PubMed]
- McAlister, F.A.; Ezekowitz, J.; Tonelli, M.; Armstrong, P.W. Renal insufficiency and heart failure: Prognostic and therapeutic implications from a prospective cohort study. Circulation 2004, 109, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Haapio, M.; House, A.A.; Anavekar, N.; Bellomo, R. Cardiorenal syndrome. J. Am. Coll. Cardiol. 2008, 52, 1527–1539. [Google Scholar] [CrossRef] [PubMed]
- Ronco, F.; Ronco, C. Cardiorenal syndrome, current understanding. Recenti Prog. Med. 2009, 100, 202–213. [Google Scholar] [PubMed]
- Rosner, M.H.; Ronco, C.; Okusa, M.D. The role of inflammation in the cardio-renal syndrome: A focus on cytokines and inflammatory mediators. Semin. Nephrol. 2012, 32, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Small, D.M.; Gobe, G.C. Oxidative Stress and Antioxidant Therapy in Chronic Kidney and Cardiovascular Disease. InTech 2013, 233–264. [Google Scholar] [CrossRef]
- Josephson, R.A.; Silverman, H.S.; Lakatta, E.G.; Stern, M.D.; Zweier, J.L. Study of the mechanisms of hydrogen peroxide and hydroxyl free radical-induced cellular injury and calcium overload in cardiac myocytes. J. Biol. Chem. 1991, 266, 2354–2361. [Google Scholar] [PubMed]
- Boaz, M.; Smetana, S.; Weinstein, T.; Matas, Z.; Gafter, U.; Iaina, A.; Knecht, A.; Weissgarten, Y.; Brunner, D.; Fainaru, M.; et al. Secondary prevention with antioxidants of cardiovascular disease in endstage renal disease (SPACE): Randomised placebo-controlled trial. Lancet 2000, 356, 1213–1218. [Google Scholar] [CrossRef]
- Tojo, A.; Onozato, M.L.; Kobayashi, N. Angiotensin II and oxidative stress in Dahl Salt-sensitive rat with heart failure. Hypertension 2002, 40, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Koya, D.; Sugimoto, T.; Isono, M.; Araki, S.; Kashiwagi, A.; Haneda, M. Translocation of Glomerular p47phox and p67phox by Protein Kinase C-β Activation Is Required for Oxidative Stress in Diabetic Nephropathy. Diabetes 2003, 52, 2603–2614. [Google Scholar] [CrossRef] [PubMed]
- De Blasio, M.J.; Ramalingam, A.; Cao, A.H.; Prakoso, D.; Ye, J.M.; Pickering, R.; Watson, A.M.D.; de Haan, J.B.; Kaye, D.M.; Ritchie, R.H. The superoxide dismutase mimetic tempol blunts diabetes-induced upregulation of NADPH oxidase and endoplasmic reticulum stress in a rat model of diabetic nephropathy. Eur. J. Pharmacol. 2017, 15, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Tepel, M.; van der Giet, M.; Statz, M.; Jankowski, J.; Zidek, W. The antioxidant acetylcysteine reduces cardiovascular events in patients with end-stage renal failure: A randomized, controlled trial. Circulation 2003, 107, 992–995. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nuhu, F.; Bhandari, S. Oxidative Stress and Cardiovascular Complications in Chronic Kidney Disease, the Impact of Anaemia. Pharmaceuticals 2018, 11, 103. https://doi.org/10.3390/ph11040103
Nuhu F, Bhandari S. Oxidative Stress and Cardiovascular Complications in Chronic Kidney Disease, the Impact of Anaemia. Pharmaceuticals. 2018; 11(4):103. https://doi.org/10.3390/ph11040103
Chicago/Turabian StyleNuhu, Faisal, and Sunil Bhandari. 2018. "Oxidative Stress and Cardiovascular Complications in Chronic Kidney Disease, the Impact of Anaemia" Pharmaceuticals 11, no. 4: 103. https://doi.org/10.3390/ph11040103
APA StyleNuhu, F., & Bhandari, S. (2018). Oxidative Stress and Cardiovascular Complications in Chronic Kidney Disease, the Impact of Anaemia. Pharmaceuticals, 11(4), 103. https://doi.org/10.3390/ph11040103