Influence of Iron on Bone Homeostasis

{kind=link}
Abstract
1. Introduction
2. Bone Homeostasis
2.1. Osteoclasts
2.2. Osteoblasts
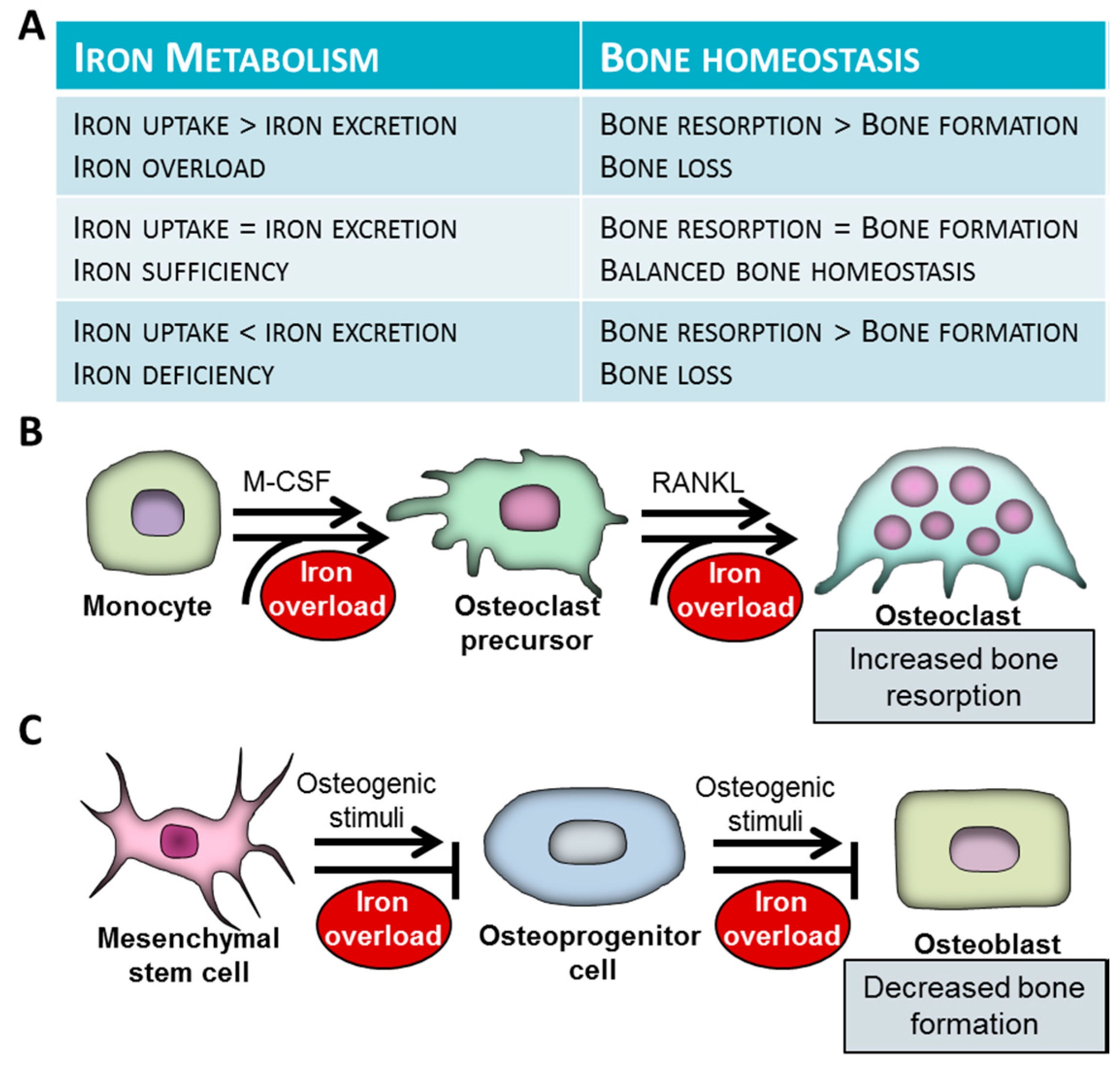
3. Bone Homeostasis in Iron Overload
3.1. Iron Overload
3.2. Bone Phenotype in Association with Iron Overload
4. Bone Homeostasis in Iron Deficiency
5. Cellular Mechanisms Underlying Bone Loss in Iron-Overload
5.1. Iron Overload and Bone Resorption
5.2. Iron Overload and Bone Formation
6. Effect of Iron Deficiency on Osteoclast and Osteoblast Differentiation and Function
6.1. Iron Deficiency and Bone Resorption
6.2. Iron Deficiency and Bone Formation
7. Targeting Iron as a Therapeutic Approach to Treat Bone Loss in Association with Iron Ovreload
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Florencio-Silva, R.; da Silva Sasso, G.R.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. Biomed. Res. Int. 2015, 421746. [Google Scholar] [CrossRef] [PubMed]
- Hadjidakis, D.J.; Androulakis, I.I. Bone remodeling. Ann. N. Y. Acad. Sci. 2006, 1092, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Soltanoff, C.S.; Chen, W.; Yang, S.; Li, Y.P. Signaling Networks that Control the Lineage Commitment and Differentiation of Bone Cells. Crit. Rev. Eukaryot. Gene Expr. 2009, 19, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Teitelbaum, S.L.; Ross, F.P. Genetic regulation of osteoclast development and function. Nat. Rev. Genet. 2003, 4, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Takeshita, S. The role of osteoclast differentiation and function in skeletal homeostasis. J. Biochem. 2016, 159, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Amarasekara, D.S.; Yun, H.; Kim, S.; Lee, N.; Kim, H.; Rho, J. Regulation of Osteoclast Differentiation by Cytokine Networks. Immune Netw. 2018, 18, e8. [Google Scholar] [CrossRef] [PubMed]
- Kodama, H.; Yamasaki, A.; Nose, M.; Niida, S.; Ohgame, Y.; Abe, M.; Kumegawa, M.; Suda, T. Congenital osteoclast deficiency in osteopetrotic (op/op) mice is cured by injections of macrophage colony-stimulating factor. J. Exp. Med. 1991, 173, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegerin osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA 1998, 95, 3597–3602. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Udagawa, N.; Suda, T. A new member of tumor necrosis factor ligand family, ODF/OPGL/TRANCE/RANKL, regulates osteoclast differentiation and function. Biochem. Biophys. Res. Commun. 1999, 256, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.Y.; Yoshida, H.; Sarosi, I.; Tan, H.L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H. Osteoimmunology: Shared mechanisms and crosstalk between the immune and bone systems. Nat. Rev. Immunol. 2007, 7, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, N. Regulation of NFATc1 in Osteoclast Differentiation. J. Bone Metab. 2014, 21, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Xing, L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res. Ther. 2007, 9, S1. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, J.; Röderer, G.; Günther, K.P.; Brenner, R.E. BMP-2, BMP-4, and PDGF-bb stimulate chemotactic migration of primary human mesenchymal progenitor cells. J. Cell Biochem. 2002, 87, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Pederson, L.; Ruan, M.; Westendorf, J.J.; Khosla, S.; Oursler, M.J. Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proc. Natl. Acad. Sci. USA 2008, 105, 20764–20769. [Google Scholar] [CrossRef] [PubMed]
- Otto, F.; Thornell, A.P.; Crompton, T.; Denzel, A.; Gilmour, K.C.; Rosewell, I.R.; Stamp, G.W.H.; Beddington, R.S.P.; Mundlos, S.; Olsen, B.R.; et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997, 89, 765–771. [Google Scholar] [CrossRef]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.H.; Inada, M.; et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef]
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef]
- Hayrapetyan, A.; Jansen, J.A.; van den Beucken, J.J. Signaling pathways involved in osteogenesis and their application for bone regenerative medicine. Tissue Eng. Part B Rev. 2015, 21, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, R.T.; Xiao, G.; Jiang, D.; Gopalakrishnan, R.; Yang, S.; Reith, E. Multiple signaling pathways converge on the Cbfa1/Runx2 transcription factor to regulate osteoblast differentiation. Connect. Tissue Res. 2003, 44, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Atashi, F.; Modarressi, A.; Pepper, M.S. The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: A review. Stem Cells Dev. 2015, 24, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, V.; Wagener, F.; Immenschuh, S. The macrophage heme-heme oxygenase-1 system and its role in inflammation. Biochem. Pharmacol. 2018, 153, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, C.M.; Looker, A.C. Laboratory methodologies for indicators of iron status: Strengths, limitations, and analytical challenges. Am. J. Clin. Nutr. 2017, 106, 1606S–1614S. [Google Scholar] [CrossRef] [PubMed]
- McLaren, G.D.; Gordeuk, V.R. Hereditary hemochromatosis: Insights from the Hemochromatosis and Iron Overload Screening (HEIRS) Study. Hematol. Am. Soc. Hematol. 2009, 2009, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Feder, J.N.; Gnirke, A.; Thomas, W.; Tsuchihashi, Z.; Ruddy, D.A.; Basava, A.; Dormishian, F.; Domingo, R., Jr.; Ellis, M.C.; Fullan, A.; et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 1996, 13, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Roetto, A.; Calì, A.; De Gobbi, M.; Garozzo, G.; Carella, M.; Majorano, N.; Totaro, A.; Gasparini, P. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat. Genet. 2000, 25, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, G.; Samuels, M.E.; Ludwig, E.H.; MacDonald, M.L.E.; Franchini, P.L.; Dubé, M.P.; Andres, L.; MacFarlane, J.; Sakellaropoulos, N.; Politou, M.; et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat. Genet. 2004, 36, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Roetto, A.; Papanikolaou, G.; Politou, M.; Alberti, F.; Girelli, D.; Christakis, J.; Loukopoulos, D.; Camaschella, C. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat. Genet. 2003, 33, 21–22. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. The ferroportin disease. Blood Cells Mol. Dis. 2004, 32, 131–138. [Google Scholar] [CrossRef] [PubMed]
- ElAlfy, M.S.; Elsherif, N.H.K.; Ebeid, F.S.E.; Ismail, E.A.R.; Ahmed, K.A.; Darwish, Y.W.; Ibrahim, A.S.; Elghamry, I.R.F.; Shokrey, N.A.; Alajeil, D.N. Renal iron deposition by magnetic resonance imaging in pediatric beta-thalassemia major patients: Relation to renal biomarkers, total body iron and chelation therapy. Eur. J. Radiol. 2018, 103, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Ebert, E.C.; Nagar, M.; Hagspiel, K.D. Gastrointestinal and hepatic complications of sickle cell disease. Clin. Gastroenterol. Hepatol. 2010, 8, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Allali, S.; de Montalembert, M.; Brousse, V.; Chalumeau, M.; Karim, Z. Management of iron overload in hemoglobinopathies. Transfus. Clin. Biol. 2017, 24, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Milic, S.; Mikolasevic, I.; Orlic, L.; Devcic, E.; Starcevic-Cizmarevic, N.; Stimac, D.; Kapovic, M.; Ristic, S. The Role of Iron and Iron Overload in Chronic Liver Disease. Med. Sci. Monit. 2016, 22, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- Kato, I.; Dnistrian, A.; Schwartz, M.; Toniolo, P.; Koenig, K.; Shore, R.; Zeleniuch-Jacquotte, A.; Akhmedkhanov, A.; Riboli, E. Risk of iron overload among middle-aged women. Int. J. Vitam. Nutr. Res. 2000, 70, 119–125. [Google Scholar] [CrossRef]
- Cade, J.E.; Moreton, J.A.; O’Hara, B.; Greenwood, D.C.; Moor, J.; Burley, V.J.; Kukalizch, K.; Bishop, D.T.; Worwood, M. Diet and genetic factors associated with iron status in middle-aged women. Am. J. Clin. Nutr. 2005, 82, 813–820. [Google Scholar] [CrossRef]
- Liu, J.M.; Hankinson, S.E.; Stampfer, M.J.; Rifai, N.; Willett, W.C.; Ma, J. Body iron stores and their determinants in healthy postmenopausal US women. Am. J. Clin. Nutr. 2003, 78, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Milman, N.; Byg, K.E.; Ovesen, L.; Kirchhoff, M.; Jürgensen, K.S.L. Iron status in Danish women, 1984–1994: A cohort comparison of changes in iron stores and the prevalence of iron deficiency and iron overload. Eur. J. Haematol. 2003, 71, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Jeney, V. Clinical Impact and Cellular Mechanisms of Iron Overload-Associated Bone Loss. Front. Pharmacol. 2017, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Diamond, T.; Stiel, D.; Posen, S. Osteoporosis in hemochromatosis: Iron excess, gonadal deficiency, or other factors? Ann. Intern. Med. 1989, 110, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Sinigaglia, L.; Fargion, S.; Fracanzani, A.L.; Binelli, L.; Battafarano, N.; Varenna, M.; Piperno, A.; Fiorelli, G. Bone and joint involvement in genetic hemochromatosis: Role of cirrhosis and iron overload. J. Rheumatol. 1997, 24, 1809–1813. [Google Scholar] [PubMed]
- Guggenbuhl, P.; Deugnier, Y.; Boisdet, J.F.; Rolland, Y.; Perdriger, A.; Pawlotsky, Y.; Chalès, G. Bone mineral density in men with genetic hemochromatosis and HFE gene mutation. Osteoporos. Int. 2005, 16, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Angelopoulos, N.G.; Goula, A.K.; Papanikolaou, G.; Tolis, G. Osteoporosis in HFE2 juvenile hemochromatosis. A case report and review of the literature. Osteoporos. Int. 2006, 17, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Varenna, M.; Fracanzani, A.L.; Rossi, V.; Fargion, S.; Sinigaglia, L. Association between iron overload and osteoporosis in patients with hereditary hemochromatosis. Osteoporos. Int. 2009, 20, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Richette, P.; Ottaviani, S.; Vicaut, E.; Bardin, T. Musculoskeletal complications of hereditary hemochromatosis: A case-control study. J. Rheumatol. 2010, 37, 2145–2150. [Google Scholar] [CrossRef] [PubMed]
- Eyres, K.S.; McCloskey, E.V.; Fern, E.D.; Rogers, S.; Beneton, M.; Aaron, J.E.; Kanis, J.A. Osteoporotic fractures: An unusual presentation of haemochromatosis. Bone 1992, 13, 431–433. [Google Scholar] [CrossRef]
- Duquenne, M.; Rohmer, V.; Legrand, E.; Chappard, D.; Barbot, N.W.; Basle, M.F.; Audran, M.; Bigorgne, J.C. Spontaneous multiple vertebral fractures revealed primary haemochromatosis. Osteoporos. Int. 1996, 6, 338–340. [Google Scholar] [CrossRef] [PubMed]
- Weatherall, D.J. The thalassaemias. Br. Med. J. 1997, 314, 1675–1678. [Google Scholar] [CrossRef]
- Bayanzay, K.; Alzoebie, L. Reducing the iron burden and improving survival in transfusion-dependent thalassemia patients: Current perspectives. J. Blood Med. 2016, 7, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Dede, A.D.; Trovas, G.; Chronopoulos, E.; Triantafyllopoulos, I.K.; Dontas, I.; Papaioannou, N.; Tournis, S. Thalassemia-associated osteoporosis: A systematic review on treatment and brief overview of the disease. Osteoporos. Int. 2016, 27, 3409–3425. [Google Scholar] [CrossRef] [PubMed]
- Gaudio, A.; Morabito, N.; Catalano, A.; Rapisarda, R.; Xourafa, A.; Lasco, A. Pathogenesis of Thalassemia Major-Associated Osteoporosis: Review of the Literature and Our Experience. J. Clin. Res. Pediatr. Endocrinol. 2018. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, V.; Soliman, A.T.; Elsefdy, H.; Soliman, N.; Bedair, E.; Fiscina, B.; Kattamis, C. Bone disease in beta thalassemia patients: Past, present and future perspectives. Metabolism 2018, 80, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Exarchou, E.; Politou, C.; Vretou, E.; Pasparakis, D.; Madessis, G.; Caramerou, A. Fractures and epiphyseal deformities in beta-thalassemia. Clin. Orthop. Relat. Res. 1984, 189, 229–233. [Google Scholar] [CrossRef]
- Finsterbush, A.; Farber, I.; Mogle, P.; Goldfarb, A. Fracture patterns in thalassemia. Clin. Orthop. Relat. Res. 1985, 192, 132–136. [Google Scholar] [CrossRef]
- Dines, D.M.; Canale, V.C.; Arnold, W.D. Fractures in thalassemia. J. Bone Joint Surg. Am. 1976, 58, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Fung, E.B.; Harmatz, P.R.; Milet, M.; Coates, T.D.; Thompson, A.A.; Ranalli, M.; Mignaca, R.; Scher, C.; Giardina, P.; Robertson, S.; et al. Fracture prevalence and relationship to endocrinopathy in iron overloaded patients with sickle cell disease and thalassemia. Bone 2008, 43, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, L.; De Sanctis, V. Multicentre study on prevalence of fractures in transfusion-dependent thalassaemic patients. J. Pediatr. Endocrinol. Metab. 1998, 11, 773–778. [Google Scholar] [PubMed]
- Vogiatzi, M.G.; Macklin, E.A.; Fung, E.B.; Vichinsky, E.; Olivieri, N.; Kwiatkowski, J.; Cohen, A.; Neufeld, E.; Giardina, P.J. Prevalence of fractures among the Thalassemia syndromes in North. America. Bone 2006, 38, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Baldini, M.; Ulivieri, F.M.; Forti, S.; Serafino, S.; Seghezzi, S.; Marcon, A.; Giarda, F.; Messina, C.; Cassinerio, E.; Aubry-Rozier, B.; et al. Spine bone texture assessed by trabecular bone score (TBS) to evaluate bone health in thalassemia major. Calcif. Tissue Int. 2014, 95, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.G.; Segal, J.B.; Ashar, B.H.; Leung, S.; Ahmed, S.; Siddique, S.; Rice, T.; Lanzkron, S. High prevalence and correlates of low bone mineral density in young adults with sickle cell disease. Am. J. Hematol. 2006, 81, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Sadat-Ali, M.; Al-Elq, A.H.; Sultan, O.; Al-Turki, H.; Bukhari, R.; Al-Mulhim, E. Low bone mass due to sickle cell anemia: Is it becoming a real issue? West Afr. J. Med. 2008, 27, 218–223. [Google Scholar] [PubMed]
- Sadat-Ali, M.; Al Elq, A.H. Sickle cell anaemia: Is it a cause for secondary osteoporosis? West Afr. J. Med. 2007, 26, 134–137. [Google Scholar] [PubMed]
- Sarrai, M.; Duroseau, H.; D’Augustine, J.; Moktan, S.; Bellevue, R.; et al. Bone mass density in adults with sickle cell disease. Br. J. Haematol. 2007, 136, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Roberts, I. Bone involvement in sickle cell disease. Br. J. Haematol. 2005, 129, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Osunkwo, I. An update on the recent literature on sickle cell bone disease. Curr. Opin. Endocrinol. Diabetes Obes. 2013, 20, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Baldanzi, G.; Traina, F.; Marques Neto, J.F.; Santos, A.O.; Ramos, C.D.; Saad, S.T.; et al. Low bone mass density is associated with hemolysis in Brazilian patients with sickle cell disease. Clinics 2011, 66, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Koren, A.; Fink, D.; Admoni, O.; Tennenbaum-Rakover, Y.; Levin, C.; et al. Non-transferrin-bound labile plasma iron and iron overload in sickle-cell disease: A comparative study between sickle-cell disease and beta-thalassemic patients. Eur. J. Haematol. 2010, 84, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Koduri, P.R. Iron in sickle cell disease: A review why less is better. Am. J. Hematol. 2003, 73, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Natta, C.; Creque, L.; Navarro, C. Compartmentalization of iron in sickle cell anemia—An autopsy study. Am. J. Clin. Pathol. 1985, 83, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Sadat-Ali, M.; Sultan, O.; Al-Turki, H.; Alelq, A. Does high serum iron level induce low bone mass in sickle cell anemia? Biometals 2011, 24, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Milman, N.; Kirchhoff, M. Iron stores in 1359, 30- to 60-year-old Danish women: Evaluation by serum ferritin and hemoglobin. Ann. Hematol. 1992, 64, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Zacharski, L.R.; Ornstein, D.L.; Woloshin, S.; Schwartz, L.M. Association of age, sex, and race with body iron stores in adults: Analysis of NHANES III data. Am. Heart J. 2000, 140, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Jian, J.; Pelle, E.; Huang, X. Iron and menopause: Does increased iron affect the health of postmenopausal women? Antioxid. Redox Signal. 2009, 11, 2939–2943. [Google Scholar] [CrossRef] [PubMed]
- Black, D.M.; Rosen, C.J. Postmenopausal Osteoporosis. N. Engl. J. Med. 2016, 374, 2096–2097. [Google Scholar] [CrossRef] [PubMed]
- Cummings, S.R.; Black, D.M.; Rubin, S.M. Lifetime risks of hip, Colles’, or vertebral fracture and coronary heart disease among white postmenopausal women. Arch. Intern. Med. 1989, 149, 2445–2448. [Google Scholar] [CrossRef] [PubMed]
- Cummings, S.R.; Melton, L.J. Epidemiology and outcomes of osteoporotic fractures. Lancet 2002, 359, 1761–1767. [Google Scholar] [CrossRef]
- Kim, B.J.; Lee, S.H.; Koh, J.M.; Kim, G.S. The association between higher serum ferritin level and lower bone mineral density is prominent in women ≥45 years of age (KNHANES 2008–2010). Osteoporos. Int. 2013, 24, 2627–2637. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Ahn, S.H.; Bae, S.J.; Kim, E.H.; Lee, S.H.; Kim, H.K.; Choe, J.W.; Koh, J.M.; Kim, G.S. Iron overload accelerates bone loss in healthy postmenopausal women and middle-aged men: A 3-year retrospective longitudinal study. J. Bone Miner. Res. 2012, 27, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Babaei, M.; Bijani, A.; Heidari, P.; Hosseini, S.R.; Heidari, B. Serum ferritin levels and bone mineral density in the elderly. Caspian J. Intern. Med. 2018, 9, 232–238. [Google Scholar] [PubMed]
- Tsay, J.; Yang, Z.; Ross, F.P.; Cunningham-Rundles, S.; Lin, H.; Coleman, R.; Mayer-Kuckuk, P.; Doty, S.B.; Grady, R.W.; Giardina, P.J.; et al. Bone loss caused by iron overload in a murine model: Importance of oxidative stress. Blood 2010, 116, 2582–2589. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xu, J.; Qiu, J.; Xing, C.; Li, X.; Leng, B.; Su, Y.; Lin, J.; Lin, J.; Mei, X.; et al. Novel and rapid osteoporosis model established in zebrafish using high iron stress. Biochem. Biophys. Res. Commun. 2018, 496, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.S.; Yang, Q.; Jian, J.L.; Zhao, G.Y.; Liu, L.L.; Wang, X.; Zhang, W.; Huang, X.; Xu, Y.J. Hepcidin1 knockout mice display defects in bone microarchitecture and changes of bone formation markers. Calcif. Tissue Int. 2014, 94, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Guo, W.; Yin, C.; Zhang, S.; Qu, G.; Hou, Y.; Rong, H.; Ji, H.; Liu, S. Hepcidin deficiency undermines bone load-bearing capacity through inducing iron overload. Gene 2014, 543, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Guggenbuhl, P.; Fergelot, P.; Doyard, M.; Libouban, H.; Roth, M.P.; Gallois, Y.; Chales, G.; Loreal, O.; Chappard, D. Bone status in a mouse model of genetic hemochromatosis. Osteoporos. Int. 2011, 22, 2313–2319. [Google Scholar] [CrossRef] [PubMed]
- Doyard, M.; Chappard, D.; Leroyer, P.; Roth, M.P.; Loreal, O.; Guggenbuhl, P. Decreased Bone Formation Explains Osteoporosis in a Genetic Mouse Model of Hemochromatosiss. PLoS ONE 2016, 11, e0148292. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yan, Y.; Wang, X.; Zhu, G.; Xu, Y.J. Hepcidin inhibition on the effect of osteogenesis in zebrafish. Biochem. Biophys. Res. Commun. 2016, 476, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Keel, S.B.; Doty, R.T.; Yang, Z.; Quigley, J.G.; Chen, J.; Knoblaugh, S.; Kingsley, P.D.; De Domenico, I.; Vaughn, M.B.; Kaplan, J.; et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 2008, 319, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Marro, S.; Mercurio, S.; Giorgi, C.; Petrillo, S.; Vinchi, F.; Fiorito, V.; Fagoonee, S.; Camporeale, A.; Turco, E.; et al. The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J. Clin. Investig. 2012, 122, 4569–4579. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, M.G.; Tsay, J.; Verdelis, K.; Rivella, S.; Grady, R.W.; Doty, S.; Giardina, P.J.; Boskey, A.L. Changes in bone microarchitecture and biomechanical properties in the th3 thalassemia mouse are associated with decreased bone turnover and occur during the period of bone accrual. Calcif. Tissue Int. 2010, 86, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Thongchote, K.; Svasti, S.; Teerapornpuntakit, J.; Krishnamra, N.; Charoenphandhu, N. Running exercise alleviates trabecular bone loss and osteopenia in hemizygous beta-globin knockout thalassemic mice. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1406–E1417. [Google Scholar] [CrossRef] [PubMed]
- Thongchote, K.; Svasti, S.; Teerapornpuntakit, J.; Suntornsaratoon, P.; Krishnamra, N.; Charoenphandhu, N. Bone microstructural defects and osteopenia in hemizygous βIVSII-654 knockin thalassemic mice: Sex-dependent changes in bone density and osteoclast function. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E936–E948. [Google Scholar] [CrossRef] [PubMed]
- Green, M.; Akinsami, I.; Lin, A.; Banton, S.; Ghosh, S.; Chen, B.; Platt, M.; Osunkwo, I.; Ofori-Acquah, S.; Guldberg, R.; et al. Microarchitectural and mechanical characterization of the sickle bone. J. Mech. Behav. Biomed. Mater. 2015, 48, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Andemariam, B.; Taxel, P.; Adams, D.J.; Zempsky, W.T.; Dorcelus, V.; Hurley, M.M. Loss of Bone in Sickle Cell Trait and Sickle Cell Disease Female Mice Is Associated with Reduced IGF-1 in Bone and Serum. Endocrinology 2016, 157, 3036–3046. [Google Scholar] [CrossRef] [PubMed]
- Guanabens, N.; Pares, A. Osteoporosis in chronic liver disease. Liver Int. 2018, 38, 776–785. [Google Scholar] [CrossRef] [PubMed]
- Van Wagner, L.B.; Rinella, M.E. Extrahepatic Manifestations of Nonalcoholic Fatty Liver Disease. Curr. Hepatol. Rep. 2016, 15, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Gaspa, S.; Martinez-Ferrer, A.; Guanabens, N.; Dubreuil, M.; Peris, P.; Enjuanes, A.; Martinez de Osaba, M.J.; Alvarez, L.; Monegal, A.; Combalia, A.; et al. Effects of bilirubin and sera from jaundiced patients on osteoblasts: Contribution to the development of osteoporosis in liver diseases. Hepatology 2011, 54, 2104–2113. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Beibei, F.; Guangsi, S.; Yu, J.; Wen, Z.; Xi, H.; Youjia, X. Iron overload increases osteoclastogenesis and aggravates the effects of ovariectomy on bone mass. J. Endocrinol. 2015, 226, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Isomura, H.; Fujie, K.; Shibata, K.; Inoue, N.; Iizuka, T.; Takebe, G.; Takahashi, K.; Nishihira, J.; Izumi, H.; Sakamoto, W. Bone metabolism and oxidative stress in postmenopausal rats with iron overload. Toxicology 2004, 197, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Grosbois, B.; Decaux, O.; Cador, B.; Cazalets, C.; Jego, P. Human iron deficiency. Bull. Acad. Natl. Med. 2005, 189, 1649–1663. [Google Scholar] [PubMed]
- Toxqui, L.; Vaquero, M.P. Chronic Iron Deficiency as an Emerging Risk Factor for Osteoporosis: A Hypothesis. Nutrients 2015, 7, 2324–2344. [Google Scholar] [CrossRef] [PubMed]
- Zofkova, I.; Davis, M.; Blahos, J. Trace Elements Have Beneficial, as Well as Detrimental Effects on Bone Homeostasis. Physiol. Res. 2017, 66, 391–402. [Google Scholar] [PubMed]
- Gorres, K.L.; Raines, R.T. Prolyl 4-hydroxylase. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 106–124. [Google Scholar] [PubMed]
- Tuderman, L.; Myllyla, R.; Kivirikko, K.I. Mechanism of the prolyl hydroxylase reaction. 1. Role of co-substrates. Eur. J. Biochem. 1977, 80, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Goltzman, D. Functions of vitamin D in bone. Histochem. Cell Biol. 2018, 149, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Prosser, D.E.; Kaufmann, M. Thematic Review Series: Fat-Soluble Vitamins: Vitamin D Cytochrome P450-mediated metabolism of vitamin D. J. Lipid Res. 2014, 55, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, D.M.; Stoecker, B.; Plattner, A.; Jennings, D.; Haub, M. Iron deficiency negatively affects vertebrae and femurs of rats independently of energy intake and body weight. J. Nutr. 2004, 134, 3061–3067. [Google Scholar] [CrossRef] [PubMed]
- Katsumata, S.I.; Katsumata-Tsuboi, R.; Uehara, M.; Suzuki, K. Severe Iron Deficiency Decreases Both Bone Formation and Bone Resorption in Rats. J. Nutr. 2009, 139, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Castro, J.; Lopez-Frias, M.R.; Campos, M.S.; Lopez-Frias, M.; Alferez, M.J.M.; Nestares, T.; Ojeda, M.L.; Lopez-Aliaga, I. Severe nutritional iron-deficiency anaemia has a negative effect on some bone turnover biomarkers in rats. Eur. J. Nutr. 2012, 51, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, D.M. Copper, iron, and selenium dietary deficiencies negatively impact skeletal integrity: A review. Exp. Biol. Med. 2016, 241, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Toxqui, L.; Perez-Granados, A.M.; Blanco-Rojo, R.; Wright, I.; de la Piedra, C.; Vaquero, M.P. Low iron status as a factor of increased bone resorption and effects of an iron and vitamin D-fortified skimmed milk on bone remodelling in young Spanish women. Eur. J. Nutr. 2014, 53, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Wright, I.; Blanco-Rojo, R.; Fernandez, M.C.; Toxqui, L.; Moreno, G.; Perez-Granados, A.M.; de la Piedra, C.; Remacha, A.F.; Vaquero, M.P. Bone remodelling is reduced by recovery from iron-deficiency anaemia in premenopausal women. J. Physiol. Biochem. 2013, 69, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, L.C.; Kuhne, C.A.; Viereck, V. The OPG/RANKL/RANK system in metabolic bone diseases. J. Musculoskelet. Neuronal Interact. 2004, 4, 268–275. [Google Scholar] [PubMed]
- Jia, P.; Xu, Y.J.; Zhang, Z.L.; Li, K.; Li, B.; Zhang, W.; Yang, H. Ferric ion could facilitate osteoclast differentiation and bone resorption through the production of reactive oxygen species. J. Orthop. Res. 2012, 30, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.A.; Fumoto, T.; Iwai, K.; Takeshita, S.; Ito, M.; Shimohata, N.; Aburatani, H.; Taketani, S.; Lelliott, C.J.; Vidal-Puig, A.; et al. Coordination of PGC-1β and iron uptake in mitochondrial biogenesis and osteoclast activation. Nat. Med. 2009, 15, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Roodman, G.D. Osteoclasts pump iron. Cell Metab. 2009, 9, 405–406. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Wang, H.; Xia, J.; Yang, Y.; Jin, Z.; Xu, H.; Shi, J.; De Domenico, I.; Tricot, G.; Zhan, F. Decreased ferroportin promotes myeloma cell growth and osteoclast differentiation. Cancer Res. 2015, 75, 2211–2221. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ye, S.Q.; Fujiwara, T.; Manolagas, S.C.; Zhao, H.B. Steap4 Plays a Critical Role in Osteoclastogenesis in Vitro by Regulating Cellular Iron/Reactive Oxygen Species (ROS) Levels and cAMP Response Element-binding Protein (CREB) Activation. J. Biol. Chem. 2013, 288, 30064–30074. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.J.; Lorenz, S.; Dolder, S.; Hofstetter, W. Extracellular Iron is a Modulator of the Differentiation of Osteoclast Lineage Cells. Calcif. Tissue Int. 2016, 98, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fang, B.; Fujiwara, T.; Krager, K.; Gorantla, A.; Li, C.; Feng, J.Q.; Jennings, M.L.; Zhou, J.; Aykin-Burns, N.; et al. Deletion of ferroportin in murine myeloid cells increases iron accumulation and stimulates osteoclastogenesis in vitro and in vivo. J. Biol. Chem. 2018, 293, 9248–9264. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, A.; Oda, Y.; Yachie, A.; Koizumi, S.; Nakanishi, I. Heme oxygenase-1 deficiency: The first autopsy case. Hum. Pathol. 2002, 33, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Kovtunovych, G.; Eckhaus, M.A.; Ghosh, M.C.; Ollivierre-Wilson, H.; Rouault, T.A. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: Effects on macrophage viability and tissue iron distribution. Blood 2010, 116, 6054–6062. [Google Scholar] [CrossRef] [PubMed]
- Zwerina, J.; Tzima, S.; Hayer, S.; Redlich, K.; Hoffmann, O.; Hanslik-Schnabel, B.; Smolen, J.S.; Kollias, G.; Schett, G. Heme oxygenase 1 (HO-1) regulates osteoclastogenesis and bone resorption. FASEB J. 2005, 19, 2011–2013. [Google Scholar] [CrossRef] [PubMed]
- Florczyk-Soluch, U.; Jozefczuk, E.; Stepniewski, J.; Bukowska-Strakova, K.; Mendel, M.; Viscardi, M.; Nowak, W.N.; Jozkowicz, A.; Dulak, J. Various roles of heme oxygenase-1 in response of bone marrow macrophages to RANKL and in the early stage of osteoclastogenesis. Sci. Rep. 2018, 8, 10797. [Google Scholar] [CrossRef] [PubMed]
- Lemma, S.; Sboarina, M.; Porporato, P.E.; Zini, N.; Sonveaux, P.; Di Pompo, G.; Baldini, N.; Avnet, S. Energy metabolism in osteoclast formation and activity. Int. J. Biochem. Cell Biol. 2016, 79, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Arnett, T.R.; Orriss, I.R. Metabolic properties of the osteoclast. Bone 2018, 115, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, B.; Sun, J.; Jiang, Y.; Zhang, H.; Zhang, P.; Fei, B.; Xu, Y. Iron-induced oxidative stress stimulates osteoclast differentiation via NF-κB signaling pathway in mouse model. Metabolism 2018, 83, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Ek-Rylander, B.; Flores, M.; Wendel, M.; Heinegard, D.; Andersson, G. Dephosphorylation of osteopontin and bone sialoprotein by osteoclastic tartrate-resistant acid phosphatase. Modulation of osteoclast adhesion in vitro. J. Biol. Chem. 1994, 269, 14853–14856. [Google Scholar] [PubMed]
- Zaidi, M.; Moonga, B.; Moss, D.W.; MacIntyre, I. Inhibition of osteoclastic acid phosphatase abolishes bone resorption. Biochem. Biophys. Res. Commun. 1989, 159, 68–71. [Google Scholar] [CrossRef]
- Hayman, A.R.; Cox, T.M. Tartrate-resistant acid phosphatase knockout mice. J. Bone Miner. Res. 2003, 18, 1905–1907. [Google Scholar] [CrossRef] [PubMed]
- Hayman, A.R.; Warburton, M.J.; Pringle, J.A.; Coles, B.; Chambers, T.J. Purification and characterization of a tartrate-resistant acid phosphatase from human osteoclastomas. Biochem. J. 1989, 261, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Hayman, A.R.; Cox, T.M. Purple acid phosphatase of the human macrophage and osteoclast. Characterization, molecular properties, and crystallization of the recombinant di-iron-oxo protein secreted by baculovirus-infected insect cells. J. Biol. Chem. 1994, 269, 1294–1300. [Google Scholar] [PubMed]
- Alcantara, O.; Reddy, S.V.; Roodman, G.D.; Boldt, D.H. Transcriptional regulation of the tartrate-resistant acid phosphatase (TRAP) gene by iron. Biochem. J. 1994, 298, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Balogh, E.; Tolnai, E.; Nagy, B., Jr.; Nagy, B.; Balla, G.; Balla, J.; Jeney, V. Iron overload inhibits osteogenic commitment and differentiation of mesenchymal stem cells via the induction of ferritin. Biochim. Biophys. Acta 2016, 1862, 1640–1649. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.K.; Liu, Y.P.; Ho, J.H.; Hsu, S.C.; Lee, O.K. Amine-surface-modified superparamagnetic iron oxide nanoparticles interfere with differentiation of human mesenchymal stem cells. J. Orthop. Res. 2012, 30, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Hsiao, J.K.; Liu, H.M.; Lai, I.Y.; Yao, M.; Hsu, S.C.; Ko, B.S.; Chen, Y.C.; Yang, C.S.; Huang, D.M. The inhibitory effect of superparamagnetic iron oxide nanoparticle (Ferucarbotran) on osteogenic differentiation and its signaling mechanism in human mesenchymal stem cells. Toxicol. Appl. Pharmacol. 2010, 245, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Messer, J.G.; Kilbarger, A.K.; Erikson, K.M.; Kipp, D.E. Iron overload alters iron-regulatory genes and proteins, down-regulates osteoblastic phenotype, and is associated with apoptosis in fetal rat calvaria cultures. Bone 2009, 45, 972–979. [Google Scholar] [CrossRef] [PubMed]
- Diamond, T.; Pojer, R.; Stiel, D.; Alfrey, A.; Posen, S. Does Iron Affect Osteoblast Function—Studies In vitro and in Patients with Chronic Liver-Disease. Calcif. Tissue Int. 1991, 48, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Zarjou, A.; Jeney, V.; Arosio, P.; Poli, M.; Zavaczki, E.; Balla, G.; Balla, J. Ferritin ferroxidase activity: A potent inhibitor of osteogenesis. J. Bone Miner. Res. 2010, 25, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Zarjou, A.; Jeney, V.; Arosio, P.; Poli, M.; Antal-Szalmas, P.; Agarwal, A.; Balla, G.; Balla, J. Ferritin prevents calcification and osteoblastic differentiation of vascular smooth muscle cells. J. Am. Soc. Nephrol. 2009, 20, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Doyard, M.; Fatih, N.; Monnier, A.; Island, M.L.; Aubry, M.; Leroyer, P.; Bouvet, R.; Chales, G.; Mosser, J.; Loreal, O.; et al. Iron excess limits HHIPL-2 gene expression and decreases osteoblastic activity in human MG-63 cells. Osteoporos. Int. 2012, 23, 2435–2445. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Jian, J.; Abramson, S.B.; Huang, X. Inhibitory effects of iron on bone morphogenetic protein 2-induced osteoblastogenesis. J. Bone Miner. Res. 2011, 26, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Becs, G.; Zarjou, A.; Agarwal, A.; Kovacs, K.E.; Becs, A.; Nyitrai, M.; Balogh, E.; Banyai, E.; Eaton, J.W.; Arosio, P.; et al. Pharmacological induction of ferritin prevents osteoblastic transformation of smooth muscle cells. J. Cell. Mol. Med. 2016, 20, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Cornish, J.; Callon, K.E.; Naot, D.; Palmano, K.P.; Banovic, T.; Bava, U.; Watson, M.; Lin, J.M.; Tong, P.C.; Chen, Q.; et al. Lactoferrin is a potent regulator of bone cell activity and increases bone formation in vivo. Endocrinology 2004, 145, 4366–4374. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.M.; Xue, Y.; Lin, Q.M. Bovine lactoferrin improves bone mass and microstructure in ovariectomized rats via OPG/RANKL/RANK pathway. Acta Pharmacol. Sin. 2012, 33, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Yan, Y.; Jia, P.; Yang, K.; Guo, C.; Chen, H.; Qi, J.; Qian, N.; Xu, X.; Wang, F.; et al. Desferrioxamine reduces ultrahigh-molecular-weight polyethylene-induced osteolysis by restraining inflammatory osteoclastogenesis via heme oxygenase-1. Cell Death Dis. 2016, 7, e2435. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.P.; Pan, J.X.; Xiong, L.; Xia, W.F.; Cui, S.; Xiong, W.C. Iron Chelation Inhibits Osteoclastic Differentiation in Vitro and in Tg2576 Mouse Model of Alzheimer’s Disease. PLoS ONE 2015, 10, e0139395. [Google Scholar] [CrossRef] [PubMed]
- Drager, J.; Sheikh, Z.; Zhang, Y.L.; Harvey, E.J.; Barralet, J.E. Local delivery of iron chelators reduces in vivo remodeling of a calcium phosphate bone graft substitute. Acta Biomater. 2016, 42, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 1999, 15, 551–578. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Knowles, H.J.; Cleton-Jansen, A.M.; Korsching, E.; Athanasou, N.A. Hypoxia-inducible factor regulates osteoclast-mediated bone resorption: Role of angiopoietin-like 4. FASEB J. 2010, 24, 4648–4659. [Google Scholar] [CrossRef] [PubMed]
- Hulley, P.A.; Bishop, T.; Vernet, A.; Schneider, J.E.; Edwards, J.R.; Athanasou, N.A.; Knowles, H.J. Hypoxia-inducible factor 1-alpha does not regulate osteoclastogenesis but enhances bone resorption activity via prolyl-4-hydroxylase 2. J. Pathol. 2017, 242, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Arnett, T.R.; Gibbons, D.C.; Utting, J.C.; Orriss, I.R.; Hoebertz, A.; Rosendaal, M.; Meghji, S. Hypoxia is a major stimulator of osteoclast formation and bone resorption. J. Cell. Physiol. 2003, 196, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Parelman, M.; Stoecker, B.; Baker, A.; Medeiros, D. Iron restriction negatively affects bone in female rats and mineralization of hFOB osteoblast cells. Exp. Biol. Med. 2006, 231, 378–386. [Google Scholar] [CrossRef]
- Messer, J.G.; Cooney, P.T.; Kipp, D.E. Iron chelator deferoxamine alters iron-regulatory genes and proteins and suppresses osteoblast phenotype in fetal rat calvaria cells. Bone 2010, 46, 1408–1415. [Google Scholar] [CrossRef] [PubMed]
- Baschant, U.; Rauner, M.; Bulycheva, E.; Weidner, H.; Roetto, A.; Platzbecker, U.; Hofbauer, L.C. Wnt5a is a key target for the pro-osteogenic effects of iron chelation on osteoblast progenitors. Haematologica 2016. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.H.; Zhang, X.L.; Tang, T.T.; Dai, K.R. Promotion of osteogenesis through beta-catenin signaling by desferrioxamine. Biochem. Biophys. Res. Commun. 2008, 370, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.Y.; Zhao, L.P.; He, Y.F.; Li, G.F.; Gao, C.; Li, K.; Xu, Y.J. A comparison of the biological activities of human osteoblast hFOB1.19 between iron excess and iron deficiency. Biol. Trace Elem. Res. 2012, 150, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Bo, L.; Liu, Z.; Zhong, Y.; Huang, J.; Chen, B.; Wang, H.; Xu, Y. Iron deficiency anemia’s effect on bone formation in zebrafish mutant. Biochem. Biophys. Res. Commun. 2016, 475, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Maggio, A.; Filosa, A.; Vitrano, A.; Aloj, G.; Kattamis, A.; Ceci, A.; Fucharoen, S.; Cianciulli, P.; Grady, R.W.; Prossomariti, L.; et al. Iron chelation therapy in thalassemia major: A systematic review with meta-analyses of 1520 patients included on randomized clinical trials. Blood Cells Mol. Dis. 2011, 47, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Fabio, G.; Minonzio, F.; Delbini, P.; Bianchi, A.; Cappellini, M.D. Reversal of cardiac complications by deferiprone and deferoxamine combination therapy in a patient affected by a severe type of juvenile hemochromatosis (JH). Blood 2007, 109, 362–364. [Google Scholar] [CrossRef] [PubMed]
- Kalpatthi, R.; Peters, B.; Kane, I.; Holloman, D.; Rackoff, E.; Disco, D.; Jackson, S.; Laver, J.H.; Abboud, M.R. Safety and efficacy of high dose intravenous desferrioxamine for reduction of iron overload in sickle cell disease. Pediatr. Blood Cancer 2010, 55, 1338–1342. [Google Scholar] [CrossRef] [PubMed]
- Christoforidis, A.; Kazantzidou, E.; Tsatra, I.; Tsantali, H.; Koliakos, G.; Hatzipantelis, E.; Katzos, G.; Athanassiou-Metaxa, M. Normal lumbar bone mineral density in optimally treated children and young adolescents with beta-thalassaemia major. Hormones 2007, 6, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Casale, M.; Citarella, S.; Filosa, A.; De Michele, E.; Palmieri, F.; Ragozzino, A.; Amendola, G.; Pugliese, U.; Tartaglione, I.; Della Rocca, F.; et al. Endocrine function and bone disease during long-term chelation therapy with deferasirox in patients with beta-thalassemia major. Am. J. Hematol. 2014, 89, 1102–1106. [Google Scholar] [CrossRef] [PubMed]
- Poggi, M.; Sorrentino, F.; Pugliese, P.; Smacchia, M.P.; Daniele, C.; Equitani, F.; Terlizzi, F.; Guitarrini, M.R.; Monti, S.; Maffei, L.; et al. Longitudinal changes of endocrine and bone disease in adults with beta-thalassemia major receiving different iron chelators over 5 years. Ann. Hematol. 2016, 95, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, M.G.; Macklin, E.A.; Fung, E.B.; Cheung, A.M.; Vichinsky, E.; Olivieri, N.; Kirby, M.; Kwiatkowski, J.L.; Cunningham, M.; Holm, I.A.; et al. Bone disease in thalassemia: A frequent and still unresolved problem. J. Bone Miner. Res. 2009, 24, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Li, G.F.; Pan, Y.Z.; Sirois, P.; Li, K.; Xu, Y.J. Iron homeostasis in osteoporosis and its clinical implications. Osteoporos. Int. 2012, 23, 2403–2408. [Google Scholar] [CrossRef] [PubMed]
- Capulli, M.; Paone, R.; Rucci, N. Osteoblast and osteocyte: Games without frontiers. Arch. Biochem. Biophys. 2014, 561, 3–12. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balogh, E.; Paragh, G.; Jeney, V. Influence of Iron on Bone Homeostasis. Pharmaceuticals 2018, 11, 107. https://doi.org/10.3390/ph11040107
Balogh E, Paragh G, Jeney V. Influence of Iron on Bone Homeostasis. Pharmaceuticals. 2018; 11(4):107. https://doi.org/10.3390/ph11040107
Chicago/Turabian StyleBalogh, Enikő, György Paragh, and Viktória Jeney. 2018. "Influence of Iron on Bone Homeostasis" Pharmaceuticals 11, no. 4: 107. https://doi.org/10.3390/ph11040107
APA StyleBalogh, E., Paragh, G., & Jeney, V. (2018). Influence of Iron on Bone Homeostasis. Pharmaceuticals, 11(4), 107. https://doi.org/10.3390/ph11040107