Cyclooxygenase-1 (COX-1) and COX-1 Inhibitors in Cancer: A Review of Oncology and Medicinal Chemistry Literature

{kind=link}
Abstract
1. Introduction
2. COX-1 Involvement in Neoplastic Diseases
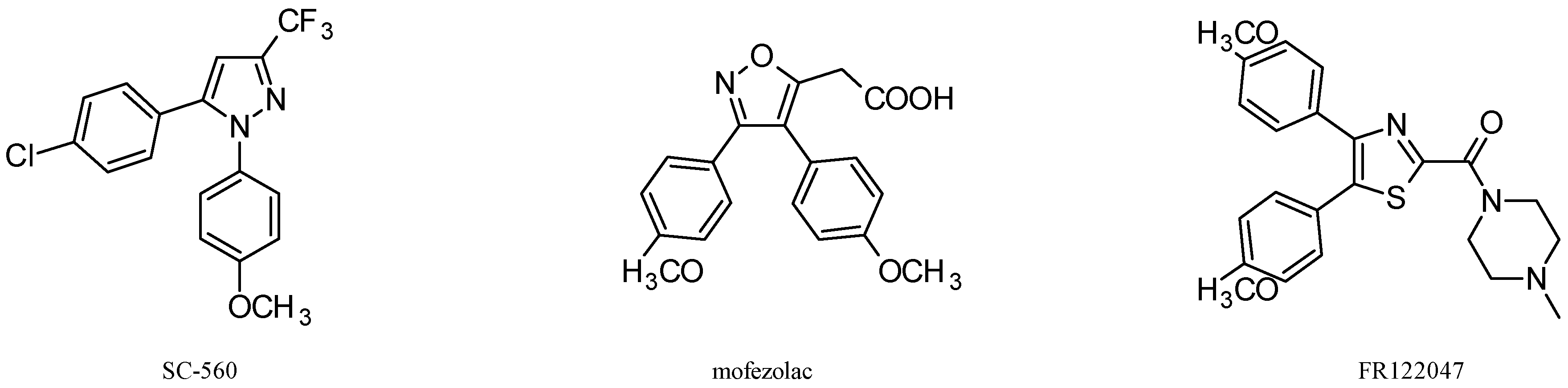
3. Antitumor Activity of COX-1 Selective Inhibitors
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Zitvogel, L.; Palucka, A.K. Neutralizing tumor-promoting chronic inflammation: A magic bullet? Science 2013, 339, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Hirata, T.; Narumiya, S. Prostanoid receptors. Chem. Rev. 2011, 111, 6209–6230. [Google Scholar] [CrossRef] [PubMed]
- Botha, J.H.; Bobinson, K.M.; Ramchurren, N.; Reddi, K.; Norman, R.J. Human esophageal carcinoma cell lines: Prostaglandin production, biological properties, and behavior in nude mice. J. Natl. Cancer Inst. 1986, 76, 1053–1056. [Google Scholar] [PubMed]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Rosenberg, D.W. Multifaceted roles of PGE2 in inflammation and cancer. Semin. Immunopathol. 2013, 35, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Sha, W.; Brüne, B.; Weigert, A. The multi-faceted roles of prostaglandin E2 in cancer-infiltrating mononuclear phagocyte biology. Immunobiology 2012, 217, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Oshima, H.; Oshima, M. The inflammatory network in the gastrointestinal tumor microenvironment: Lessons from mouse models. J. Gastroenterol. 2012, 47, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Vong, L.; Ferraz, J.G.; Panaccione, R.; Beck, P.L.; Wallace, J.L. A pro-resolution mediator, prostaglandin D(2), is specifically up-regulated in individuals in long-term remission from ulcerative colitis. Proc. Natl. Acad. Sci. USA 2010, 107, 12023–12027. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, K.; Boardman, L.A.; Zhao, Y.; Wang, L.; Sheng, Y.; Oi, N.; Limburg, P.J.; Bode, A.M.; Dong, Z. Circulating prostaglandin biosynthesis in colorectal cancer and potential clinical significance. EBioMedicine 2015, 2, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Dovizio, M.; Tacconelli, S.; Ricciotti, E.; Bruno, A.; Maier, T.J.; Anzellotti, P.; Di Francesco, L.; Sala, P.; Signoroni, S.; Bertario, L.; et al. Effects of celecoxib on prostanoid biosyntesis and circulating angiogenesis proteins in familial adenomatous polyposis. J. Pharmacol. Exp. Ther. 2012, 341, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Tao, B.; Liu, G.; Chen, G.; Zhu, Q.; Yu, Y.; Yu, Y.; Xiong, H. Thromboxane A2 receptor inhibition suppresses multiple myeloma cell proliferation by inducing p38/c-Jun N-terminal Kinase (JNK) Mitogen-activated Protein Kinase (MAPK)-mediated G2/M progression delay and cell apoptosis. J. Biol. Chem. 2016, 291, 4779–4792. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tai, H.H. Activation of thromboxane A(2) receptors induces orphan nuclear receptor Nurr1 expression and stimulates cell proliferation in human lung cancer cells. Carcinogenesis 2009, 30, 1606–1613. [Google Scholar] [CrossRef] [PubMed]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef] [PubMed]
- Zidar, N.; Odar, K.; Glavac, D.; Jerse, M.; Zupanc, T.; Stajer, D. Cyclooxygenase in normal human tissues—Is COX-1 really a constitutive isoform, and COX-2 an inducible isoform? J. Cell. Mol. Med. 2009, 13, 3753–3763. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Mbonye, U.R.; DeLong, C.J.; Wada, M.; Smith, W.L. Regulation of intracellular cyclooxygenase levels by gene transcription and protein degradation. Prog. Lipid Res. 2007, 46, 108–125. [Google Scholar] [CrossRef] [PubMed]
- Lutz, C.S.; Cornett, A.L. Regulation of genes in the arachidonic acid metabolic pathway by RNA processing and RNA-mediated mechanisms. Wiley Interdiscip. Rev. RNA 2013, 4, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhou, S.; Zhang, L.; Ye, W.; Wen, Q.; Wang, J. Role of cancer-related inflammation in esophageal cancer. Crit. Rev. Eukaryot. Gene Expr. 2013, 23, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Fan, X.M. Role of cyclooxygenase-2 in gastric cancer development and progression. World J. Gastroenterol. 2013, 19, 7361–7368. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, M.C.; O’Byrne, K.J.; Reynolds, J.V.; O’Sullivan, J.; Pidgeon, G.P. COX-derived prostanoid pathways in gastrointestinal cancer development and progression: Novel targets for prevention and intervention. Biochim. Biophys. Acta 2012, 1825, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Knab, L.M.; Grippo, P.J.; Bentrem, D.J. Involvement of eicosanoids in the pathogenesis of pancreatic cancer: The roles of cyclooxygenase-2 and 5-lipoxygenase. World J. Gastroenterol. 2014, 20, 10729–10739. [Google Scholar] [CrossRef] [PubMed]
- Glover, J.A.; Hughes, C.M.; Cantwell, M.M.; Murray, L.J. A systematic review to establish the frequency of cyclooxygenase-2 expression in normal breast epithelium, ductal carcinoma in situ, microinvasive carcinoma of the breast and invasive breast cancer. Br. J. Cancer 2011, 105, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Parida, S.; Mandal, M. Inflammation induced by human papillomavirus in cervical cancer and its implication in prevention. Eur. J. Cancer Prev. 2014, 23, 432–448. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, K.; Szczylik, C.; Lian, F.; Czarnecka, A.M. The role of prostaglandin E2 in renal cell cancer development: Future implications for prognosis and therapy. Future Oncol. 2014, 10, 2177–2187. [Google Scholar] [CrossRef] [PubMed]
- Shao, N.; Feng, N.; Wang, Y.; Mi, Y.; Li, T.; Hua, L. Systematic review and meta-analysis of COX-2 expression and polymorphisms in prostate cancer. Mol. Biol. Rep. 2012, 39, 10997–11004. [Google Scholar] [CrossRef] [PubMed]
- Gakis, G. The role of inflammation in bladder cancer. Adv. Exp. Med. Biol. 2014, 816, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Elmets, C.A.; Ledet, J.J.; Athar, M. Cyclooxygenases: Mediators of UV-induced skin cancer and potential targets for prevention. J. Investig. Dermatol. 2014, 134, 2497–2502. [Google Scholar] [CrossRef] [PubMed]
- Rundhaug, J.E.; Simper, M.S.; Surh, I.; Fischer, S.M. The role of the EP receptors for prostaglandin E2 in skin and skin cancer. Cancer Metastasis Rev. 2011, 30, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Mendes, R.A.; Carvalho, J.F.; van der Waal, I. An overview on the expression of cyclooxygenase-2 in tumors of the head and neck. Oral Oncol. 2009, 45, e124–e128. [Google Scholar] [CrossRef] [PubMed]
- Ramon, S.; Woeller, C.F.; Phipps, R.P. The influence of Cox-2 and bioactive lipids on hematological cancers. Curr. Angiogenes. 2013, 2, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Nuvoli, B.; Galati, R. Cyclooxygenase-2, epidermal growth factor receptor, and aromatase signaling in inflammation and mesothelioma. Mol. Cancer Ther. 2013, 12, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Greenhough, A.; Smartt, H.J.; Moore, A.E.; Roberts, H.R.; Williams, A.C.; Paraskeva, C.; Kaidi, A. The COX-2/PGE2 pathway: Key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009, 30, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.E. Cyclooxygenase-2 (cox-2) and the inflammogenesis of cancer. Subcell. Biochem. 2007, 42, 93–126. [Google Scholar] [CrossRef] [PubMed]
- Dixon, D.A.; Blanco, F.F.; Bruno, A.; Patrignani, P. Mechanistic aspects of COX-2 expression in colorectal neoplasia. Recent Results Cancer Res. 2013, 191, 7–37. [Google Scholar] [CrossRef] [PubMed]
- Cebola, I.; Peinado, M.A. Epigenetic deregulation of the COX pathway in cancer. Prog. Lipid Res. 2012, 51, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.M.; Hawk, E.T.; Lubet, R.A. Coxibs and other nonsteroidal anti-inflammatory drugs in animal models of cancer chemoprevention. Cancer Prev. Res. 2011, 4, 1728–1735. [Google Scholar] [CrossRef] [PubMed]
- Gurpinar, E.; Grizzle, W.E.; Piazza, G.A. NSAIDs inhibit tumorigenesis, but how? Clin. Cancer Res. 2014, 20, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Rouzer, C.A.; Marnett, L.J. Cyclooxygenases: Structural and functional insights. J. Lipid Res. 2009, 50, S29–S34. [Google Scholar] [CrossRef] [PubMed]
- Chulada, P.C.; Thompson, M.B.; Mahler, J.F.; Doyle, C.M.; Gaul, B.W.; Lee, C.; Tiano, H.F.; Morham, S.G.; Smithies, O.; Langenbach, R. Genetic disruption of Ptgs-1, as well as Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Res. 2000, 60, 4705–4708. [Google Scholar] [PubMed]
- Tiano, H.F.; Loftin, C.D.; Akunda, J.; Lee, C.A.; Spalding, J.; Sessoms, A.; Dunson, D.B.; Rogan, E.G.; Morham, S.G.; Smart, R.C.; et al. Deficiency of either cyclooxygenase (COX)-1 or COX-2 alters epidermal differentiation and reduces mouse skin tumorigenesis. Cancer Res. 2002, 62, 3395–3401. [Google Scholar] [PubMed]
- Okamoto, T.; Hara, A.; Hino, O. Down-regulation of cyclooxygenase-2 expression but up-regulation of cyclooxygenase-1 in renal carcinomas of the Eker (TSC2 gene mutant) rat model. Cancer Sci. 2003, 94, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.H.; Zhang, Q.; Wang, Y.D.; Chen, J.; Jiang, Z.M.; Shi, M.; Guo, X.; Qin, J.; Cui, G.H.; Cai, Z.M.; et al. Overexpression of cyclooxygenase-1 correlates with poor prognosis in renal cell carcinoma. Asian Pac. J. Cancer Prev. 2013, 14, 3729–3734. [Google Scholar] [CrossRef] [PubMed]
- Osman, W.M.; Youssef, N.S. Combined use of COX-1 and VEGF immunohistochemistry refines the histopathologic prognosis of renal cell carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 8165–8177. [Google Scholar] [PubMed]
- Tang, J.Y.; Aszterbaum, M.; Athar, M.; Barsanti, F.; Cappola, C.; Estevez, N.; Hebert, J.; Hwang, J.; Khaimskiy, Y.; Kim, A.; et al. Basal cell carcinoma chemoprevention with nonsteroidal anti-inflammatory drugs in genetically predisposed PTCH1+/− humans and mice. Cancer Prev. Res. 2010, 3, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Müller-Decker, K. Cyclooxygenase-dependent signaling is causally linked to non-melanoma skin carcinogenesis: Pharmacological, genetic, and clinical evidence. Cancer Metastasis Rev. 2011, 30, 343–361. [Google Scholar] [CrossRef] [PubMed]
- Erovic, B.M.; Woegerbauer, M.; Pammer, J.; Selzer, E.; Grasl, M.C.; Thurnher, D. Strong evidence for up-regulation of cyclooxygenase-1 in head and neck cancer. Eur. J. Clin. Investig. 2008, 38, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A.; Lipari, L.; Leone, A.; Tortorici, S.; Burruano, F.; Provenzano, S.; Gerbino, A.; Buscemi, M. Expression of cyclooxygenase-1 and cyclooxygenase-2 in normal and pathological human oral mucosa. Folia Histochem. Cytobiol. 2010, 48, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Pannone, G.; Sanguedolce, F.; De Maria, S.; Farina, E.; Lo Muzio, L.; Serpico, R.; Emanuelli, M.; Rubini, C.; De Rosa, G.; Staibano, S.; et al. Cyclooxygenase isozymes in oral squamous cell carcinoma: A real-time RT-PCR study with clinic pathological correlations. Int. J. Immunopathol. Pharmacol. 2007, 20, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Erovic, B.M.; Al Habeeb, A.; Harris, L.; Goldstein, D.P.; Kim, D.; Ghazarian, D.; Irish, J.C. Identification of novel target proteins in sebaceous gland carcinoma. Head Neck 2013, 35, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Kim, H.S.; Hah, J.H.; Kim, K.H.; Heo, D.S.; Sung, M.W. Differential effects between cyclooxygenase-2 inhibitors and siRNA on vascular endothelial growth factor production in head and neck squamous cell carcinoma cell lines. Head Neck 2010, 32, 1534–1543. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Kim, H.S.; Choi, M.S.; Kim, J.E.; Jeong, W.J.; Heo, D.S.; Sung, M.W. The influence of cyclooxygenase-1 expression on the efficacy of cyclooxygenase-2 inhibition in head and neck squamous cell carcinoma cell lines. Anticancer Drugs 2011, 22, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, J.; Alexandre, L.; Baruah, A.; Buttar, N.; Chandra, R.; Clark, A.B.; Hart, A.R.; Hawk, E.; Kandioler, D.; Kappel, S.; et al. Strategy for prevention of cancers of the esophagus. Ann. N. Y. Acad. Sci. 2014, 1325, 108–126. [Google Scholar] [CrossRef] [PubMed]
- Tsibouris, P.; Vlachou, E.; Isaacs, P.E. Role of chemoprophylaxis with either NSAIDs or statins in patients with Barrett’s esophagus. World J. Gastrointest. Pharmacol. Ther. 2014, 5, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.D.; Armstrong, G.R.; Bigley, G.; Green, H.; Attwood, S.E. Cyclooxygenase-2 expression in the Barrett’s metaplasia–dysplasia–adenocarcinoma sequence. Am. J. Gastroenterol. 2001, 96, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Taddei, A.; Fabbroni, V.; Pini, A.; Lucarini, L.; Ringressi, M.N.; Fantappiè, O.; Bani, D.; Messerini, L.; Masini, E.; Bechi, P. Cyclooxygenase-2 and inflammation mediators have a crucial role in reflux-related esophageal histological changes and Barrett’s esophagus. Dig. Dis. Sci. 2014, 59, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Von Rahden, B.H.; Stein, H.J.; Pühringer, F.; Koch, I.; Langer, R.; Piontek, G.; Siewert, J.R.; Höfler, H.; Sarbia, M. Coexpression of cyclooxygenases (COX-1, COX-2) and vascular endothelial growth factors (VEGF-A, VEGF-C) in esophageal adenocarcinoma. Cancer Res. 2005, 65, 5038–5044. [Google Scholar] [CrossRef] [PubMed]
- Piazuelo, E.; Santander, S.; Cebrián, C.; Jiménez, P.; Pastor, C.; García-González, M.A.; Esteva, F.; Esquivias, P.; Ortego, J.; Lanas, A. Characterization of the prostaglandin E2 pathway in a rat model of esophageal adenocarcinoma. Curr. Cancer Drug Targets 2012, 12, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Esquivias, P.; Morandeira, A.; Escartín, A.; Cebrián, C.; Santander, S.; Esteva, F.; García-González, M.A.; Ortego, J.; Lanas, A.; Piazuelo, E. Indomethacin but not a selective cyclooxygenase-2 inhibitor inhibits esophageal adenocarcinogenesis in rats. World J. Gastroenterol. 2012, 18, 4866–4874. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, X.Q.; Ding, X.W.; Yang, R.K.; Huang, S.L.; Kastelein, F.; Bruno, M.; Yu, X.J.; Zhou, D.; Zou, X.P. Cyclooxygenase inhibitors use is associated with reduced risk of esophageal adenocarcinoma in patients with Barrett’s esophagus: A meta-analysis. Br. J. Cancer 2014, 110, 2378–2388. [Google Scholar] [CrossRef] [PubMed]
- Janakiram, N.B.; Rao, C.V. The role of inflammation in colon cancer. Adv. Exp. Med. Biol. 2014, 816, 25–52. [Google Scholar] [CrossRef] [PubMed]
- Arber, N.; Eagle, C.J.; Spicak, J.; Rácz, I.; Dite, P.; Hajer, J.; Zavoral, M.; Lechuga, M.J.; Gerletti, P.; Tang, J.; et al. PreSAP Trial Investigators. Celecoxib for the prevention of colorectal adenomatous polyps. N. Engl. J. Med. 2006, 355, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Kraus, S.; Naumov, I.; Arber, N. COX-2 active agents in the chemoprevention of colorectal cancer. Recent Results Cancer Res. 2013, 191, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Church, R.D.; Yu, J.; Fleshman, J.W.; Shannon, W.D.; Govindan, R.; McLeod, H.L. RNA profiling of cyclooxygenases 1 and 2 in colorectal cancer. Br. J. Cancer 2004, 91, 1015–1018. [Google Scholar] [CrossRef] [PubMed]
- Von Rahden, B.H.; Brücher, B.L.; Langner, C.; Siewert, J.R.; Stein, H.J.; Sarbia, M. Expression of cyclo-oxygenase 1 and 2, prostaglandin E synthase and transforming growth factor beta1, and their relationship with vascular endothelial growth factors A and C, in primary adenocarcinoma of the small intestine. Br. J. Surg. 2006, 93, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Riehl, T.E.; George, R.J.; Sturmoski, M.A.; May, R.; Dieckgraefe, B.; Anant, S.; Houchen, C.W. Azoxymethane protects intestinal stem cells and reduces crypt epithelial mitosis through a COX-1 dependent mechanism. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G1062–G1070. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Sonoshita, M.; Oshima, H.; Sugihara, K.; Chulada, P.C.; Langenbach, R.; Oshima, M.; Taketo, M.M. Cooperation of cyclooxygenase 1 and cyclooxygenase 2 in intestinal polyposis. Cancer Res. 2003, 63, 4872–4877. [Google Scholar] [PubMed]
- Smartt, H.J.; Greenhough, A.; Ordóñez-Morán, P.; Talero, E.; Cherry, C.A.; Wallam, C.A.; Parry, L.; Al Kharusi, M.; Roberts, H.R.; Mariadason, J.M.; et al. β-catenin represses expression of the tumour suppressor 15-prostaglandin dehydrogenase in the normal intestinal epithelium and colorectal tumour cells. Gut 2012, 61, 1306–13014. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, F.; Chen, H.; Cheng, K.W.; Zykova, T.; Oi, N.; Lubet, R.A.; Bode, A.M.; Wang, M.; Dong, Z. 6-C-(E-phenylethenyl)-naringenin suppresses colorectal cancer growth by inhibiting cyclooxygenase-1. Cancer Res. 2014, 74, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Vona-Davis, L.; Rose, D.P. The obesity-inflammation-eicosanoid axis in breast cancer. J. Mammary Gland Biol. Neoplasia 2013, 18, 291–307. [Google Scholar] [CrossRef] [PubMed]
- Hoellen, F.; Kelling, K.; Dittmer, C.; Diedrich, K.; Friedrich, M.; Thill, M. Impact of cyclooxygenase-2 in breast cancer. Anticancer Res. 2011, 31, 4359–4367. [Google Scholar] [PubMed]
- Hwang, D.; Scollard, D.; Byrne, J.; Levine, E. Expression of cyclooxygenase-1 and cyclooxygenase-2 in human breast cancer. J. Natl. Cancer Inst. 1998, 90, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Fahlén, M.; Zhang, H.; Löfgren, L.; Masironi, B.; von Schoultz, E.; von Schoultz, B.; Sahlin, L. Expression of cyclooxygenase-1 and cyclooxygenase-2, syndecan-1 and connective tissue growth factor in benign and malignant breast tissue from premenopausal women. Gynecol. Endocrinol. 2017, 33, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Haakensen, V.D.; Bjøro, T.; Lüders, T.; Riis, M.; Bukholm, I.K.; Kristensen, V.N.; Troester, M.A.; Homen, M.M.; Ursin, G.; Børresen-Dale, A.L.; et al. Serum estradiol levels associated with specific gene expression patterns in normal breast tissue and in breast carcinomas. BMC Cancer 2011, 11, 332. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.S.; Kim, J.H.; Choi, H.Y.; Lee, E.R.; Cho, S.G. Induction of cell growth arrest and apoptotic cell death in human breast cancer MCF-7 cells by the COX-1 inhibitor FR122047. Oncol. Rep. 2010, 24, 351–356. [Google Scholar] [CrossRef] [PubMed]
- McFadden, D.W.; Riggs, D.R.; Jackson, B.J.; Cunningham, C. Additive effects of Cox-1 and Cox-2 inhibition on breast cancer in vitro. Int. J. Oncol. 2006, 29, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Kundu, N.; Fulton, A.M. Selective cyclooxygenase (COX)-1 or COX-2 inhibitors control metastatic disease in a murine model of breast cancer. Cancer Res. 2002, 62, 2343–2346. [Google Scholar] [PubMed]
- Boccardo, E.; Lepique, A.P.; Villa, L.L. The role of inflammation in HPV carcinogenesis. Carcinogenesis 2010, 31, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Subbaramaiah, K.; Dannenberg, A.J. Cyclooxygenase-2 transcription is regulated by human papillomavirus 16 E6 and E7 oncoproteins: Evidence of a corepressor/coactivator exchange. Cancer Res. 2007, 67, 3976–3985. [Google Scholar] [CrossRef] [PubMed]
- Young, J.L.; Jazaeri, A.A.; Darus, C.J.; Modesitt, S.C. Cyclooxygenase-2 in cervical neoplasia: A review. Gynecol. Oncol. 2008, 109, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Grabosch, S.M.; Shariff, O.M.; Wulff, J.L.; Helm, C.W. Non-steroidal anti-inflammatory agents to induce regression and prevent the progression of cervical intraepithelial neoplasia. Cochrane Database Syst. Rev. 2014, CD004121. [Google Scholar] [CrossRef] [PubMed]
- Sales, K.J.; Katz, A.A.; Howard, B.; Soeters, R.P.; Millar, R.P.; Jabbour, H.N. Cyclooxygenase 1 is up-regulated in cervical carcinomas: Autocrine/paracrine regulation of cyclooxygenase-2, prostaglandin E receptors and angiogenic factors by Cyclooxygenase-1. Cancer Res. 2002, 62, 424–432. [Google Scholar] [PubMed]
- Sutherland, J.R.; Sales, K.J.; Jabbour, H.N.; Katz, A.A. Seminal plasma enhances cervical adenocarcinoma cell proliferation and tumour growth in vivo. PLoS ONE 2012, 7, e33848. [Google Scholar] [CrossRef] [PubMed]
- Sales, K.J.; Sutherland, J.R.; Jabbour, H.N.; Katz, A.A. Seminal plasma induces angiogenic chemokine expression in cervical cancer cells and regulates vascular function. Biochim. Biophys. Acta 2012, 1823, 1789–1795. [Google Scholar] [CrossRef] [PubMed]
- Radilova, H.; Libra, A.; Holasova, S.; Safarova, M.; Viskova, A.; Kunc, F.; Buncek, M. COX-1 is coupled with mPGES-1 and ABCC4 in human cervix cancer cells. Mol. Cell. Biochem. 2009, 330, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.W.; Kim, S.W.; Kim, S.; Kim, J.H.; Cho, N.H.; Kim, J.W.; Kim, Y.T. Prevalence and clinical relevance of cyclooxygenase-1 and -2 expression in stage IIB cervical adenocarcinoma. Eur. J. Obstet. Gynecol. Reprod. Biol. 2010, 148, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Athavale, R.; Clooney, K.; O’Hagan, J.; Shawki, H.; Clark, A.H.; Green, J.A. COX-1 and COX-2 expression in stage I and II invasive cervical carcinoma: Relationship to disease relapse and long-term survival. Int. J. Gynecol. Cancer 2006, 16, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.T.; Song, Y.C.; Kim, S.H.; Wu, H.G.; Kim, I.H.; Park, I.A.; Kim, J.W.; Park, N.H.; Kang, S.B.; Lee, H.P.; et al. Influences of cyclooxygenase-1 and -2 expression on the radiosensitivities of human cervical cancer cell lines. Cancer Lett. 2007, 256, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, T.; Kim, M.K.; Suh, D.H.; Chung, H.H.; Song, YS. Cyclooxygenase-1 and -2: Molecular targets for cervical neoplasia. J. Cancer Prev. 2013, 18, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Ohno, Y.; Suzuki, N.; Inagawa, H.; Kohchi, C.; Soma, G.; Inoue, M. Multiple roles of cyclooxygenase-2 in endometrial cancer. Anticancer Res. 2005, 25, 3679–3687. [Google Scholar] [PubMed]
- Nasir, A.; Boulware, D.; Kaiser, H.E.; Lancaster, J.M.; Coppola, D.; Smith, P.V.; Hakam, A.; Siegel, S.E.; Bodey, B. Cyclooxygenase-2 (COX-2) expression in human endometrial carcinoma and precursor lesions and its possible use in cancer chemoprevention and therapy. In Vivo 2007, 21, 35–43. [Google Scholar] [PubMed]
- Hasegawa, K.; Torii, Y.; Ishii, R.; Oe, S.; Kato, R.; Udagawa, Y. Effects of a selective COX-2 inhibitor in patients with uterine endometrial cancers. Arch. Gynecol. Obstet. 2011, 284, 1515–1521. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, T.; Koizumi, T.; Sudo, T.; Yamaguchi, S.; Kojima, A.; Kumagai, S.; Nishimura, R. Correlative expression of cyclooxygenase-1 (Cox-1) and human epidermal growth factor receptor type-2 (Her-2) in endometrial cancer. Kobe J. Med. Sci. 2007, 53, 177–187. [Google Scholar] [PubMed]
- Déry, M.C.; Chaudhry, P.; Leblanc, V.; Parent, S.; Fortier, A.M.; Asselin, E. Oxytocin increases invasive properties of endometrial cancer cells through phosphatidylinositol 3-kinase/AKT-dependent up-regulation of cyclooxygenase-1, -2, and X-linked inhibitor of apoptosis protein. Biol. Reprod. 2011, 85, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, S.; Coward, J.I.; Bast, R.C., Jr.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Liu, J. Inflammation, a hidden path to breaking the spell of ovarian cancer. Cell Cycle 2009, 8, 3107–3111. [Google Scholar] [CrossRef] [PubMed]
- Macciò, A.; Madeddu, C. Inflammation and ovarian cancer. Cytokine 2012, 58, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Ng, H.T. Clinical evaluations of a new ovarian cancer marker, COX-1. Int. J. Gynaecol. Obstet. 1995, 49, S27–S32. [Google Scholar] [CrossRef]
- Gupta, R.A.; Tejada, L.V.; Tong, B.J.; Das, S.K.; Morrow, J.D.; Dey, S.K.; DuBois, R.N. Cyclooxygenase-1 is overexpressed and promotes angiogenic growth factor production in ovarian cancer. Cancer Res. 2003, 63, 906–911. [Google Scholar] [PubMed]
- Daikoku, T.; Wang, D.; Tranguch, S.; Morrow, J.D.; Orsulic, S.; DuBois, R.N.; Dey, S.K. Cyclooxygenase-1 is a potential target for prevention and treatment of ovarian epithelial cancer. Cancer Res. 2005, 65, 3735–3744. [Google Scholar] [CrossRef] [PubMed]
- Daikoku, T.; Tranguch, S.; Trofimova, I.N.; Dinulescu, D.M.; Jacks, T.; Nikitin, A.Y.; Connolly, D.C.; Dey, S.K. Cyclooxygenase-1 is overexpressed in multiple genetically engineered mouse models of epithelial ovarian cancer. Cancer Res. 2006, 66, 2527–2531. [Google Scholar] [CrossRef] [PubMed]
- Hales, D.B.; Zhuge, Y.; Lagman, J.A.; Ansenberger, K.; Mahon, C.; Barua, A.; Luborsky, J.L.; Bahr, J.M. Cyclooxygenases expression and distribution in the normal ovary and their role in ovarian cancer in the domestic hen (Gallus domesticus). Endocrine 2008, 33, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Urick, M.E.; Johnson, P.A. Cyclooxygenase 1 and 2 mRNA and protein expression in the Gallus domesticus model of ovarian cancer. Gynecol. Oncol. 2006, 103, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Eilati, E.; Pan, L.; Bahr, J.M.; Hales, D.B. Age dependent increase in prostaglandin pathway coincides with onset of ovarian cancer in laying hens. Prostaglandins Leukot. Essent. Fat. Acids 2012, 87, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Kino, Y.; Kojima, F.; Kiguchi, K.; Igarashi, R.; Ishizuka, B.; Kawai, S. Prostaglandin E2 production in ovarian cancer cell lines is regulated by cyclooxygenase-1, not cyclooxygenase-2. Prostaglandins Leukot. Essent. Fat. Acids 2005, 73, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Lau, M.T.; Wong, A.S.; Leung, P.C. Gonadotropins induce tumor cell migration and invasion by increasing cyclooxygenases expression and prostaglandin E(2) production in human ovarian cancer cells. Endocrinology 2010, 151, 2985–2993. [Google Scholar] [CrossRef] [PubMed]
- Ali-Fehmi, R.; Semaan, A.; Sethi, S.; Arabi, H.; Bandyopadhyay, S.; Hussein, Y.R.; Diamond, M.P.; Saed, G.; Morris, R.T.; Munkarah, A.R. Molecular typing of epithelial ovarian carcinomas using inflammatory markers. Cancer 2011, 117, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Matsumura, N.; Mandai, M.; Li, K.; Yagi, H.; Baba, T.; Suzuki, A.; Hamanishi, J.; Fukuhara, K.; Konishi, I. Classification using hierarchical clustering of tumor-infiltrating immune cells identifies poor prognostic ovarian cancers with high levels of COX expression. Mod. Pathol. 2009, 22, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Abiko, K.; Matsumura, N.; Baba, T.; Yoshioka, Y.; Kosaka, K.; Konishi, I. The comprehensive assessment of local immune status of ovarian cancer by the clustering of multiple immune factors. Clin. Immunol. 2011, 141, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Myung, S.K.; Song, Y.S. Prognostic role of cyclooxygenase-2 in epithelial ovarian cancer: A meta-analysis of observational studies. Gynecol. Oncol. 2013, 129, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Yim, G.W.; Nam, E.J.; Kim, Y.T. Synergistic effect of COX-2 inhibitor on paclitaxel-induced apoptosis in the human ovarian cancer cell line OVCAR-3. Cancer Res. Treat. 2014, 46, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.J.; Tian, J.Y.; Jin, Y.M.; Wang, L.; Yang, R.Q.; Cui, M.H. Effects of cyclooxygenase-2 gene silencing on the biological behavior of SKOV3 ovarian cancer cells. Mol. Med. Rep. 2015, 11, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Fadare, O.; Beeghly-Fadiel, A.; Son, D.S.; Liu, Q.; Zhao, S.; Saskowski, J.; Uddin, M.J.; Daniel, C.; Crews, B.; et al. Aberrant over-expression of COX-1 intersects multiple pro-tumorigenic pathways in high-grade serous ovarian cancer. Oncotarget 2015, 6, 21353–21368. [Google Scholar] [CrossRef] [PubMed]
- Daikoku, T.; Tranguch, S.; Chakrabarty, A.; Wang, D.; Khabele, D.; Orsulic, S.; Morrow, J.D.; Dubois, R.N.; Dey, S.K. Extracellular signal-regulated kinase is a target of cyclooxygenase-1-peroxisome proliferator-activated receptor-delta signaling in epithelial ovarian cancer. Cancer Res. 2007, 67, 5285–5292. [Google Scholar] [CrossRef] [PubMed]
- Sonnemann, J.; Hüls, I.; Sigler, M.; Palani, C.D.; Hong, L.T.T.; Völker, U.; Kroemer, H.K.; Beck, J.F. Histone deacetylase inhibitors and aspirin interact synergistically to induce cell death in ovarian cancer cells. Oncol. Rep. 2008, 20, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.; Kabir, S.M.; Dong, Y.; Lee, E.; Rice, V.M.; Khabele, D.; Son, D.S. Aspirin blocks EGF-stimulated cell viability in a COX-1 dependent manner in ovarian cancer cells. J. Cancer 2013, 4, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Son, D.S.; Wilson, A.J.; Parl, A.K.; Khabele, D. The effects of the histone deacetylase inhibitor romidepsin (FK228) are enhanced by aspirin (ASA) in COX-1 positive ovarian cancer cells through augmentation of p21. Cancer Biol. Ther. 2010, 9, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Truffinet, V.; Donnard, M.; Vincent, C.; Faucher, J.L.; Bordessoule, D.; Turlure, P.; Trimoreau, F.; Denizot, Y. Cyclooxygenase-1, but not -2, in blast cells of patients with acute leukemia. Int. J. Cancer 2007, 121, 924–927. [Google Scholar] [CrossRef] [PubMed]
- Fiancette, R.; Vincent-Fabert, C.; Guerin, E.; Trimoreau, F.; Denizot, Y. Lipid mediators and human leukemic blasts. J. Oncol. 2011, 2011, 389021. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, D.; Brown, C.; Iland, H. Retinoic acid and arsenic trioxide in the treatment of acute promyelocytic leukemia: Current perspectives. Onco Targets Ther. 2017, 10, 1585–1601. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.B.; Li, G.; Chen, S.J.; Chen, Z. From dissection of disease pathogenesis to elucidation of mechanisms of targeted therapies: Leukemia research in the genomic era. Acta Pharmacol. Sin. 2007, 28, 1434–1449. [Google Scholar] [CrossRef] [PubMed]
- Rocca, B.; Morosetti, R.; Habib, A.; Maggiano, N.; Zassadowski, F.; Ciabattoni, G.; Chomienne, C.; Papp, B.; Ranelletti, F.O. Cyclooxygenase-1, but not -2, is upregulated in NB4 leukemic cells and human primary promyelocytic blasts during differentiation. Leukemia 2004, 18, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Habib, A.; Hamade, E.; Mahfouz, R.; Nasrallah, M.S.; de Thé, H.; Bazarbachi, A. Arsenic trioxide inhibits ATRA-induced prostaglandin E2 and cyclooxygenase-1 in NB4 cells, a model of acute promyelocytic leukemia. Leukemia 2008, 22, 1125–1130. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.Y.; Ho, N.P.; Lee, T.K. Cancer stem cells and their microenvironment: Biology and therapeutic implications. Stem Cells Int. 2017, 2017, 3714190. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Tyutyunnykova, A.; Pantel, K.; Dubrovska, A. Cancer stem cells: The root of tumor recurrence and metastases. Semin. Cancer Biol. 2017, 44, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.Y.; Hurst, E.A.; Argyle, D.J. Cyclooxygenase-2: A role in cancer stem cell survival and repopulation of cancer cells during therapy. Stem Cells Int. 2016, 2016, 2048731. [Google Scholar] [CrossRef] [PubMed]
- Goessling, W.; North, T.E.; Loewer, S.; Lord, A.M.; Lee, S.; Stoick-Cooper, C.L.; Weidinger, G.; Puder, M.; Daley, G.Q.; Moon, R.T.; et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell 2009, 136, 1136–1147. [Google Scholar] [CrossRef] [PubMed]
- Kanojia, D.; Zhou, W.; Zhang, J.; Jie, C.; Lo, P.K.; Wang, Q.; Chen, H. Proteomic profiling of cancer stem cells derived from primary tumors of HER2/Neu transgenic mice. Proteomics 2012, 12, 3407–3415. [Google Scholar] [CrossRef] [PubMed]
- Lazennec, G.; Lam, P.Y. Recent discoveries concerning the tumor—Mesenchymal stem cell interactions. Biochim. Biophys. Acta 2016, 1866, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Roodhart, J.M.; Daenen, L.G.; Stigter, E.C.; Prins, H.J.; Gerrits, J.; Houthuijzen, J.M.; Gerritsen, M.G.; Schipper, H.S.; Backer, M.J.; van Amersfoort, M.; et al. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell 2011, 20, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Loftin, C.D.; Tiano, H.F.; Langenbanch, R. Phenotypes of the COX-deficient mice indicate physiological and pathophysiological roles for COX-1 and COX-2. Prostaglandins Other Lipid Mediat. 2002, 68–69, 177–185. [Google Scholar] [CrossRef]
- Islam, A.B.; Dave, M.; Amin, S.; Jensen, R.V.; Amin, A.R. Genomic, lipidomic and metabolomic analysis of cyclooxygenase-null cells: Eicosanoid storm, cross talk, and compensation by COX-1. Genom. Proteom. Bioinform. 2016, 14, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Tortorella, M.D.; Zhang, Y.; Talley, J. Desirable Properties for 3rd Generation Cyclooxygenase-2 Inhibitors. Mini Rev. Med. Chem. 2016, 16, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Vitale, P.; Panella, A.; Scilimati, A.; Perrone, M.G. COX-1 inhibitors: Beyond structure toward therapy. Med. Res. Rev. 2016, 36, 641–671. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Zhang, Y.; Koboldt, C.M.; Muhammad, J.; Zweifel, B.S.; Shaffer, A.; Talley, J.J.; Masferrer, J.L.; Seibert, K.; Isakson, P.C. Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc. Natl. Acad. Sci. USA 1998, 95, 13313–13318. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xu, R.J.; Lin, Z.Y.; Zhuo, G.C.; Zhang, H.H. Effects of a cyclooxygenase-1-selective inhibitor in a mouse model of ovarian cancer, administered alone or in combination with ibuprofen, a nonselective cyclooxygenase inhibitor. Med. Oncol. 2009, 26, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ji, Z.L.; Zhuo, G.C.; Xu, R.J.; Wang, J.; Jiang, H.R. Effects of a selective cyclooxygenase-1 inhibitor in SKOV-3 ovarian carcinoma xenograft-bearing mice. Med. Oncol. 2010, 27, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, J.; Jiang, H.R.; Xu, X.L.; Zhang, J.; Liu, M.L.; Zhai, L.Y. Combined effects of cyclooxygenase-1 and cyclooxygenase-2 selective inhibitors on ovarian carcinoma in vivo. Int. J. Mol. Sci. 2011, 12, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, M.L.; Cai, J.H.; Tang, Y.X.; Zhai, L.Y.; Zhang, J. Effect of the combination of a cyclooxygenase-1 selective inhibitor and taxol on proliferation, apoptosis and angiogenesis of ovarian cancer in vivo. Oncol. Lett. 2012, 4, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cai, J.H.; Zhang, J.; Tang, Y.X.; Wan, L. Effects of cyclooxygenase inhibitors in combination with taxol on expression of cyclin D1 and Ki-67 in a xenograft model of ovarian carcinoma. Int. J. Mol. Sci. 2012, 13, 9741–9753. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Tang, Y.X.; Wan, L.; Cai, J.H.; Zhang, J. Effects of combining Taxol and cyclooxygenase inhibitors on the angiogenesis and apoptosis in human ovarian cancer xenografts. Oncol. Lett. 2013, 5, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wan, L.; Zhai, L.Y.; Wang, J. Effects of SC-560 in combination with cisplatin or taxol on angiogenesis in human ovarian cancer xenografts. Int. J. Mol. Sci. 2014, 5, 19265–19280. [Google Scholar] [CrossRef] [PubMed]
- Brenneis, C.; Maier, T.J.; Schmidt, R.; Hofacker, A.; Zulauf, L.; Jakobsson, P.J.; Scholich, K.; Geisslinger, G. Inhibition of prostaglandin E2 synthesis by SC-560 is independent of cyclooxygenase 1 inhibition. FASEB J. 2006, 20, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Saed, G.M. Immunohistochemical staining of cyclooxygenases with monoclonal antibodies. Methods Mol. Biol. 2008, 477, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.P.; Hahn, H.S.; Hwang, S.J.; Choi, J.Y.; Park, J.S.; Lee, I.H.; Kim, T.J. Selective cyclooxygenase inhibitors increase paclitaxel sensitivity in taxane-resistant ovarian cancer by suppressing P-glycoprotein expression. J. Gynecol. Oncol. 2013, 24, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ju, B.; Tian, J.; Liu, F.; Yu, H.; Xiao, H.; Liu, X.; Liu, W.; Yao, Z.; Hao, Q. Ovarian cancer stem cell-specific gene expression profiling and targeted drug prescreening. Oncol. Rep. 2014, 31, 1235–1248. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Havaleshko, D.M.; Cho, H.; Weinstein, J.N.; Kaldjian, E.P.; Karpovich, J.; Grimshaw, A.; Theodorescu, D. A strategy for predicting the chemosensitivity of human cancer and its application to drug discovery. Proc. Natl. Acad. Sci. USA 2007, 104, 13086–13091. [Google Scholar] [CrossRef] [PubMed]
- Grösch, S.; Tegeder, I.; Niederberger, E.; Bräutigam, L.; Geisslinger, G. COX-2 independent induction of cell cycle arrest and apoptosis in colon cancer cells by the selective COX-2 inhibitor celecoxib. FASEB J. 2001, 15, 2742–2744. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.K.; Sung, J.J.; Wu, Y.C.; Li, H.T.; Yu, L.; Li, Z.J.; Cho, C.H. Inhibition of cyclooxygenase-1 lowers proliferation and induces macroautophagy in colon cancer cells. Biochem. Biophys. Res. Commun. 2009, 382, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Sheng, H.; Shao, J.; Kirkland, S.C.; Isakson, P.; Coffey, R.J.; Morrow, J.; Beauchamp, R.D.; DuBois, R.N. Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase-2. J. Clin. Investig. 1997, 99, 2254–2259. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Choi, M.K.; Han, I.O.; Lim, S.J. Role of p21CIP1 as a determinant of SC560 response in human HCT116 colon carcinoma cells. Exp. Mol. Med. 2006, 38, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Sakoguchi-Okada, N.; Takahashi-Yanaga, F.; Fukada, K.; Shiraishi, F.; Taba, Y.; Miwa, Y.; Morimoto, S.; Iida, M.; Sasaguri, T. Celecoxib inhibits the expression of survivin via the suppression of promoter activity in human colon cancer cells. Biochem. Pharmacol. 2007, 73, 1318–1329. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Moore, S.M.; Yamaguchi, K.; Eling, T.E.; Baek, S.J. Selective nonsteroidal anti-inflammatory drugs induce thymosin beta-4 and alter actin cytoskeletal organization in human colorectal cancer cells. J. Pharmacol. Exp. Ther. 2004, 311, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y. NSAID-activated gene 1 and its implications for mucosal integrity and intervention beyond NSAIDs. Pharmacol. Res. 2017, 121, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.S.; Chen, P.M.; Hsiao, H.L.; Wang, H.S.; Liang, W.Y.; Su, Y. Overexpression of the thymosin beta-4 gene is associated with increased invasion of SW480 colon carcinoma cells and the distant metastasis of human colorectal carcinoma. Oncogene 2004, 23, 6666–6671. [Google Scholar] [CrossRef] [PubMed]
- Piao, Z.; Hong, C.S.; Jung, M.R.; Choi, C.; Park, Y.K. Thymosin β4 induces invasion and migration of human colorectal cancer cells through the ILK/AKT/β-catenin signaling pathway. Biochem. Biophys. Res. Commun. 2014, 452, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Gemoll, T.; Strohkamp, S.; Schillo, K.; Thorns, C.; Habermann, J.K. MALDI-imaging reveals thymosin beta-4 as an independent prognostic marker for colorectal cancer. Oncotarget 2015, 6, 43869–43880. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Choi, M.K.; Youk, H.J.; Kim, C.H.; Han, I.O.; Yoo, B.C.; Lee, M.K.; Lim, S.J. 5-(4-chlorophenyl)-1-(4-methoxyphenyl)-3-trifluoromethylpyrazole acts in a reactive oxygen species-dependent manner to suppress human lung cancer growth. J. Cancer Res. Clin. Oncol. 2006, 132, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Tsuboi, K.; Hoshikawa, H.; Goto, R.; Mori, N.; Katsukawa, M.; Hiraki, E.; Yamamoto, S.; Abe, M.; Ueda, N. Cyclooxygenase isozymes are expressed in human myeloma cells but not involved in anti-proliferative effect of cyclooxygenase inhibitors. Mol. Carcinog. 2006, 45, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Lampiasi, N.; Foderà, D.; D’Alessandro, N.; Cusimano, A.; Azzolina, A.; Tripodo, C.; Florena, A.M.; Minervini, M.I.; Notarbartolo, M.; Montalto, G.; et al. The selective cyclooxygenase-1 inhibitor SC-560 suppresses cell proliferation and induces apoptosis in human hepatocellular carcinoma cells. Int. J. Mol. Med. 2006, 17, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Ochi, H.; Yasunaga, Y.; Matsuyuki, H.; Imayoshi, T.; Kusuhara, H.; Okumoto, T. Analgesic effect of mofezolac, a non-steroidal anti-inflammatory drug, against phenylquinone-induced acute pain in mice. Prostaglandins Other Lipid Mediat. 1998, 56, 245–254. [Google Scholar] [CrossRef]
- Kusuhara, H.; Matsuyuki, H.; Matsuura, M.; Imayoshi, T.; Okumoto, T.; Matsui, H. Induction of apoptotic DNA fragmentation by nonsteroidal anti-inflammatory drugs in cultured rat gastric mucosal cells. Eur. J. Pharmacol. 1998, 360, 273–280. [Google Scholar] [CrossRef]
- Kusuhara, H.; Komatsu, H.; Sumichika, H.; Sugahara, K. Reactive oxygen species are involved in the apoptosis induced by nonsteroidal anti-inflammatory drugs in cultured gastric cells. Eur. J. Pharmacol. 1999, 383, 331–337. [Google Scholar] [CrossRef]
- Kitamura, T.; Kawamori, T.; Uchiya, N.; Itoh, M.; Noda, T.; Matsuura, M.; Sugimura, T.; Wakabayashi, K. Inhibitory effects of mofezolac, a cyclooxygenase-1 selective inhibitor, on intestinal carcinogenesis. Carcinogenesis 2002, 23, 1463–1466. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Itoh, M.; Noda, T.; Matsuura, M.; Wakabayashi, K. Combined effects of cyclooxygenase-1 and cyclooxygenase-2 selective inhibitors on intestinal tumorigenesis in adenomatous polyposis coli gene knockout mice. Int. J. Cancer 2004, 109, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Niho, N.; Kitamura, T.; Takahashi, M.; Mutoh, M.; Sato, H.; Matsuura, M.; Sugimura, T.; Wakabayashi, K. Suppression of azoxymethane-induced colon cancer development in rats by a cyclooxygenase-1 selective inhibitor, mofezolac. Cancer Sci. 2006, 97, 1011–1014. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Shiraishi, R.; Fujise, T.; Kuroki, T.; Kakimoto, T.; Sakata, Y.; Takashima, T.; Iwakiri, R.; Fujimoto, K.; Shi, R.; et al. Chemopreventive effect of mofezolac on beef tallow diet/azoxymethane-induced colon carcinogenesis in rats. Hepatogastroenterology 2011, 58, 81–88. [Google Scholar] [PubMed]
- Tanaka, A.; Sakai, H.; Motoyama, Y.; Ishikawa, T.; Takasugi, H. Antiplatelet agents based on cyclooxygenase inhibition without ulcerogenesis. Evaluation and synthesis of 4,5-bis(4-methoxyphenyl)-2-substituted-thiazoles. J. Med. Chem. 1994, 37, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Ochi, T.; Motoyama, Y.; Goto, T. The analgesic effect profile of FR122047, a selective cyclooxygenase-1 inhibitor, in chemical nociceptive models. Eur. J. Pharmacol. 2000, 391, 49–54. [Google Scholar] [CrossRef]
- Ochi, T.; Goto, T. Differential effect of FR122047, a selective cyclo-oxygenase-1 inhibitor, in rat chronic models of arthritis. Br. J. Pharmacol. 2002, 135, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Ochi, T.; Ohkubo, Y.; Mutoh, S. Role of cyclooxygenase-2, but not cyclooxygenase-1, on type II collagen-induced arthritis in DBA/1J mice. Biochem. Pharmacol. 2003, 66, 1055–1060. [Google Scholar] [CrossRef]
- Jeong, H.S.; Choi, H.Y.; Lee, E.R.; Kim, J.H.; Jeon, K.; Lee, H.J.; Cho, S.G. Involvement of caspase-9 in autophagy-mediated cell survival pathway. Biochim. Biophys. Acta 2011, 1813, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Boccarelli, A.; Pannunzio, A.; Coluccia, M. The Challenge of Establishing Reliable Screening Tests for Selecting Anticancer Metal Compounds. In Bioinorganic Medicinal Chemistry; Alessio, E., Ed.; Wiley-VCH Verlag GmbH & Co., KGaA: Weinheim, Germany, 2011; pp. 175–196. ISBN 978-3-527-32631-0. [Google Scholar]
- Lin, J.H.; Cocchetto, D.M.; Duggan, D.E. Protein binding as a primary determinant of the clinical pharmacokinetic properties of non-steroidal anti-inflammatory drugs. Clin. Pharmacokinet. 1987, 12, 402–432. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Zhang, L. Cyclooxygenase inhibitors not inhibit resting lung cancer A549 cell proliferation. Prostaglandins Leukot. Essent. Fat. Acids. 2006, 74, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Cancer Cell Line Encyclopedia (CCLE). Available online: http://www.broadinstitute.org/ccle (accessed on 4 September 2018).
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Bray, M.A.; Singh, S.; Han, H.; Davis, C.T.; Borgeson, B.; Hartland, C.; Kost-Alimova, M.; Gustafsdottir, S.M.; Gibson, C.C.; Carpenter, A.E. Cell Painting a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat. Protoc. 2016, 11, 1757–1774. [Google Scholar] [CrossRef] [PubMed]
- Grösch, S.; Maier, T.J.; Schiffmann, S.; Geisslinger, G. Cyclooxygenase-2 (COX-2)-independent anticarcinogenic effects of selective COX-2 inhibitors. J. Natl. Cancer Inst. 2006, 98, 736–747. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem BioAssay Database, AID=624141. Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/624141 (accessed on 4 September 2018).

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pannunzio, A.; Coluccia, M. Cyclooxygenase-1 (COX-1) and COX-1 Inhibitors in Cancer: A Review of Oncology and Medicinal Chemistry Literature. Pharmaceuticals 2018, 11, 101. https://doi.org/10.3390/ph11040101
Pannunzio A, Coluccia M. Cyclooxygenase-1 (COX-1) and COX-1 Inhibitors in Cancer: A Review of Oncology and Medicinal Chemistry Literature. Pharmaceuticals. 2018; 11(4):101. https://doi.org/10.3390/ph11040101
Chicago/Turabian StylePannunzio, Alessandra, and Mauro Coluccia. 2018. "Cyclooxygenase-1 (COX-1) and COX-1 Inhibitors in Cancer: A Review of Oncology and Medicinal Chemistry Literature" Pharmaceuticals 11, no. 4: 101. https://doi.org/10.3390/ph11040101
APA StylePannunzio, A., & Coluccia, M. (2018). Cyclooxygenase-1 (COX-1) and COX-1 Inhibitors in Cancer: A Review of Oncology and Medicinal Chemistry Literature. Pharmaceuticals, 11(4), 101. https://doi.org/10.3390/ph11040101