Role of Microbiota and Tryptophan Metabolites in the Remote Effect of Intestinal Inflammation on Brain and Depression


{kind=link}
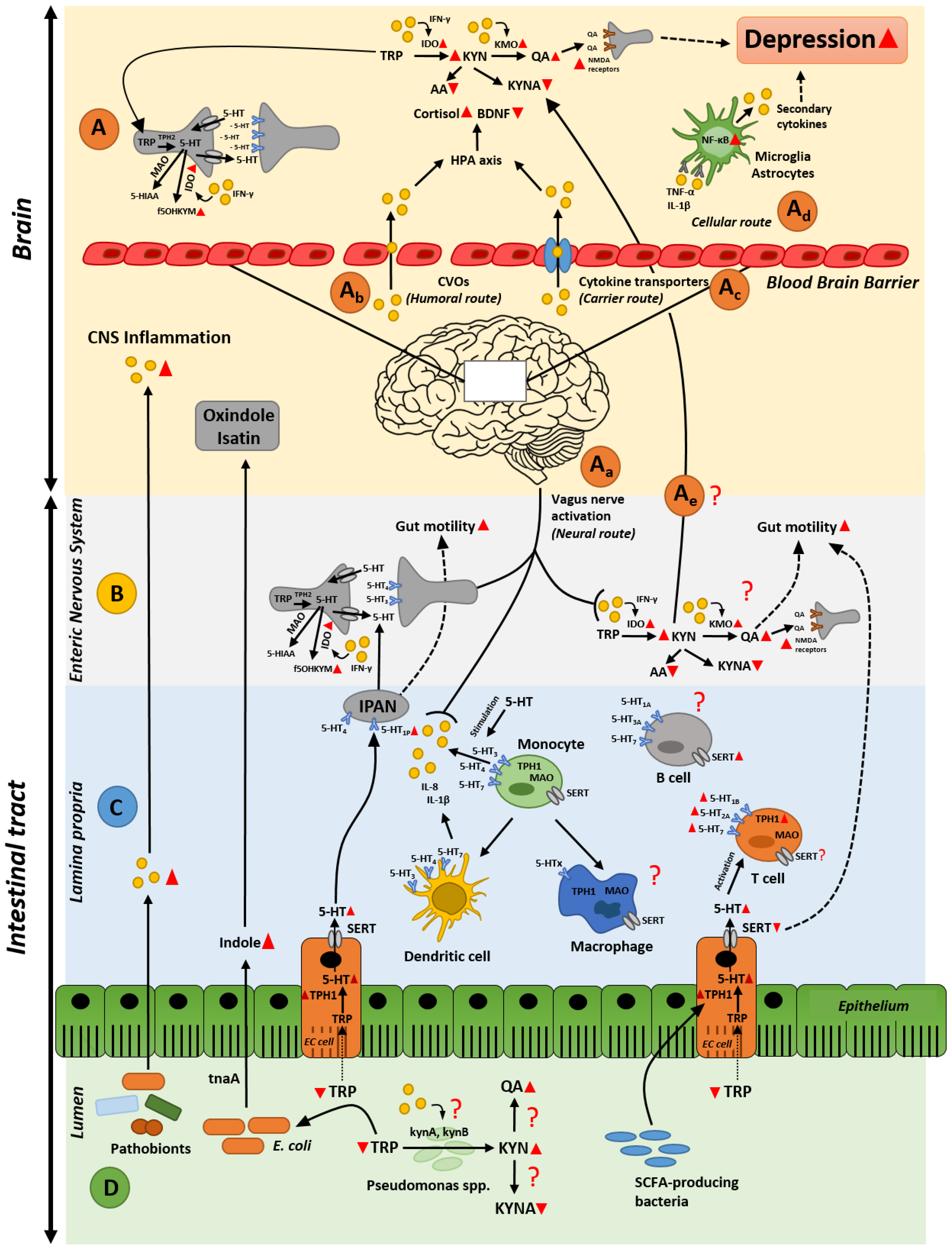
Abstract
1. Introduction
2. Gastrointestinal Inflammation and Depression
2.1. A Gut Perspective on the Role of Tryptophan Metabolites in Depression
2.2. Gut Inflammation-Induced Kynurenine Biosynthesis; Possible Cause of Altered Kynurenine Pathway in the Brain during Depression?
2.3. Intestinal Inflammation and Disrupted Serotonin Signaling System: From Alterated Gut Functionality to Development of Depression
3. Microbiota as an Orchestrator in the Crosstalk between Inflammation and Serotonin Imbalances
3.1. Gut Microbiota and Intestinal Immune (Hyper)-Stimulation
3.2. Gut Microbiota and Serotonin Production
3.3. The Dual Effect of Gut Microbiota and Its Metabolites in Depression
4. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- El Aidy, S.; Stilling, R.; Dinan, T.G.; Cryan, J.F. Microbiome to Brain: Unravelling the Multidirectional Axes of Communication. In Microbial Endocrinology: Interkingdom Signaling in Infectious Disease and Health. Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2016; pp. 301–336. ISBN 978-3-319-20214-3. [Google Scholar]
- Shepherd, E.S.; DeLoache, W.C.; Pruss, K.M.; Whitaker, W.R.; Sonnenburg, J.L. An Exclusive Metabolic Niche Enables Strain Engraftment in the Gut Microbiota. Nature 2018, 557, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.; Pruteanu, M.; Kuhn, M.; Zeller, G.; Telzerow, A.; Anderson, E.E.; Brochado, A.R.; Fernandez, K.C.; Dose, H.; Mori, H.; et al. Extensive Impact of Non-Antibiotic Drugs on Human Gut Bacteria. Nature 2018, 555, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Dodd, D.; Spitzer, M.H.; Van Treuren, W.; Merrill, B.D.; Hryckowian, A.J.; Higginbottom, S.K.; Le, A.; Cowan, T.M.; Nolan, G.P.; Fischbach, M.A.; et al. A Gut Bacterial Pathway Metabolizes Aromatic Amino Acids into Nine Circulating Metabolites. Nature 2017, 551, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Blander, J.M.; Longman, R.S.; Iliev, I.D.; Sonnenberg, G.F.; Artis, D. Regulation of Inflammation by Microbiota Interactions with the Host. Nat. Immunol. 2017, 18, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Berk, M.; Williams, L.J.; Jacka, F.N.; O’Neil, A.; Pasco, J.A.; Moylan, S.; Allen, N.B.; Stuart, A.L.; Hayley, A.C.; Byrne, M.L.; et al. So Depression Is an Inflammatory Disease, but Where Does the Inflammation Come From? BMC Med. 2013, 11, 200. [Google Scholar] [CrossRef] [PubMed]
- Kiecolt-Glaser, J.K.; Derry, H.M.; Fagundes, C.P. Inflammation: Depression Fans the Flames and Feasts on the Heat. Am. J. Psychiatry 2015, 172, 1075–1091. [Google Scholar] [CrossRef] [PubMed]
- Koopman, M.; El Aidy, S. Depressed Gut? The Microbiota-Diet-Inflammation Trialogue in Depression. Curr. Opin. Psychiatry 2017, 30, 369–377. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Depression. Available online: http://www.who.int/en/news-room/fact-sheets/detail/depression (accessed on 20 June 2018).
- Raison, C.L.; Borisov, A.S.; Majer, M.; Drake, D.F.; Pagnoni, G.; Woolwine, B.J.; Vogt, G.J.; Massung, B.; Miller, A.H. Activation of Central Nervous System Inflammatory Pathways by Interferon-Alpha: Relationship to Monoamines and Depression. Biol. Psychiatry 2009, 65, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Khanam, R.; Vohora, D. Augmentation of Effect of Venlafaxine by Folic Acid in Behavioral Paradigms of Depression in Mice: Evidence of Serotonergic and pro-Inflammatory Cytokine Pathways. Pharmacol. Rep. 2016, 68, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Lebeña, A.; Vegas, O.; Gómez-Lázaro, E.; Arregi, A.; Garmendia, L.; Beitia, G.; Azpiroz, A. Melanoma Tumors Alter Proinflammatory Cytokine Production and Monoamine Brain Function, and Induce Depressive-like Behavior in Male Mice. Behav. Brain Res. 2014, 272, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jia, Y.; Li, G.; Wang, B.; Zhou, T.; Zhu, L.; Chen, T.; Chen, Y. The Dopamine Receptor D3 Regulates Lipopolysaccharide-Induced Depressive-Like Behavior in Mice. Int. J. Neuropsychopharmacol. 2018, 21, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Coates, M.D.; Johnson, A.C.; Greenwood-van Meerveld, B.; Mawe, G.M. Effects of Serotonin Transporter Inhibition on Gastrointestinal Motility and Colonic Sensitivity in the Mouse. Neurogastroenterol. Motil. 2006, 18, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Coates, M.D.; Mahoney, C.R.; Linden, D.R.; Sampson, J.E.; Chen, J.; Blaszyk, H.; Crowell, M.D.; Sharkey, K.A.; Gershon, M.D.; Mawe, G.M. Molecular Defects in Mucosal Serotonin Content and Decreased Serotonin Reuptake Transporter in Ulcerative Colitis and Irritable Bowel Syndrome. Gastroenterology 2004, 126, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Kidd, M.; Gustafsson, B.I.; Drozdov, I.; Modlin, I.M. IL1β- and LPS-Induced Serotonin Secretion Is Increased in EC Cells Derived from Crohn’s Disease. Neurogastroenterol. Motil. 2009, 21, 439–450. [Google Scholar] [CrossRef] [PubMed]
- El Aidy, S.; Dinan, T.G.; Cryan, J.F. Immune Modulation of the Brain-Gut-Microbe Axis. Front. Microbiol. 2014, 5, 146. [Google Scholar] [CrossRef] [PubMed]
- Linden, D.R.; Chen, J.-X.; Gershon, M.D.; Sharkey, K.A.; Mawe, G.M. Serotonin Availability Is Increased in Mucosa of Guinea Pigs with TNBS-Induced Colitis. Am. J. Physiol. Liver Physiol. 2003, 285, G207–G216. [Google Scholar] [CrossRef] [PubMed]
- Mawe, G.M.; Hoffman, J.M. Serotonin Signalling in the Gut—functions, Dysfunctions and Therapeutic Targets. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R. Cytokine, Sickness Behavior, and Depression. Immunol. Allergy Clin. N. Am. 2009, 29, 247–264. [Google Scholar] [CrossRef] [PubMed]
- Van Heesch, F. Inflammation-Induced Depression. Studying the Role of Proinflammatory Cytokines in Anhedonia; Utrecht University: Utrecht, The Netherlands, 2014. [Google Scholar]
- Capuron, L.; Miller, A.H. Immune System to Brain Signaling: Neuropsychopharmacological Implications. Pharmacol. Ther. 2011, 130, 226–238. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, C.; Swain, M.G. Immune-to-Brain Communication Pathways in Inflammation-Associated Sickness and Depression. In Inflammation-Associated Depression: Evidence, Mechanisms and Implications. Current Topics in Behavioral Neurosciences; Springer: Cham, Switzerland, 2016; pp. 73–94. [Google Scholar]
- Ransohoff, R.M.; Kivisäkk, P.; Kidd, G. Three or More Routes for Leukocyte Migration into the Central Nervous System. Nat. Rev. Immunol. 2003, 3, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From Inflammation to Sickness and Depression: When the Immune System Subjugates the Brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. The Blood–brain Barrier as a Regulatory Interface in the Gut–brain Axes. Physiol. Behav. 2006, 89, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.J.; Massie, A.; De Keyser, J. Immune Players in the CNS: The Astrocyte. J. Neuroimmune Pharmacol. 2013, 8, 824–839. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.; Han, S.J.; Kaur, G.; Crane, C.; Parsa, A.T. The Role of Microglia in Central Nervous System Immunity and Glioma Immunology. J. Clin. Neurosci. 2010, 17, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, S.; Rivest, S. Effects of Circulating Tumor Necrosis Factor on the Neuronal Activity and Expression of the Genes Encoding the Tumor Necrosis Factor Receptors (P55 and P75) in the Rat Brain: A View from the Blood–brain Barrier. Neuroscience 1999, 93, 1449–1464. [Google Scholar] [CrossRef]
- Rivest, S.; Lacroix, S.; Vallières, L.; Nadeau, S.; Zhang, J.; Laflamme, N. How the Blood Talks to the Brain Parenchyma and the Paraventricular Nucleus of the Hypothalamus during Systemic Inflammatory and Infectious Stimuli. Proc. Soc. Exp. Biol. Med. 2000, 223, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Capuron, L.; Gumnick, J.F.; Musselman, D.L.; Lawson, D.H.; Reemsnyder, A.; Nemeroff, C.B.; Miller, A.H. Neurobehavioral Effects of Interferon-α in Cancer Patients Phenomenology and Paroxetine Responsiveness of Symptom Dimensions. Neuropsychopharmacology 2002, 26, 643–652. [Google Scholar] [CrossRef]
- Howren, M.B.; Lamkin, D.M.; Suls, J. Associations of Depression with C-Reactive Protein, IL-1, and IL-6: A Meta-Analysis. Psychosom. Med. 2009, 71, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Kałużna-Czaplińska, J.; Gątarek, P.; Chirumbolo, S.; Chartrand, M.S.; Bjørklund, G. How Important Is Tryptophan in Human Health? Crit. Rev. Food Sci. Nutr. 2017, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Le Floc’h, N.; Otten, W.; Merlot, E. Tryptophan Metabolism, from Nutrition to Potential Therapeutic Applications. Amino Acids 2011, 41, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Ruddick, J.P.; Evans, A.K.; Nutt, D.J.; Lightman, S.L.; Rook, G.A.W.; Lowry, C.A. Tryptophan Metabolism in the Central Nervous System: Medical Implications. Expert Rev. Mol. Med. 2006, 8, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Kynurenine Pathway Metabolism and the Microbiota-Gut-Brain Axis. Neuropharmacology 2017, 112, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Spiller, R. Serotonin and GI Clinical Disorders. Neuropharmacology 2008, 55, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Gershon, M.D. 5-Hydroxytryptamine (Serotonin) in the Gastrointestinal Tract. Curr. Opin. Endocrinol. Diabetes Obes. 2013, 20, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Nakahama, T.; Le, D.H.; Van Son, L.; Chu, H.H.; Kishimoto, T. Aryl Hydrocarbon Receptor and Kynurenine: Recent Advances in Autoimmune Disease Research. Front. Immunol. 2014, 5, 551. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, Tryptophan Metabolism and the Brain-Gut-Microbiome Axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A.-B. Tryptophan Availability for Kynurenine Pathway Metabolism across the Life Span: Control Mechanisms and Focus on Aging, Exercise, Diet and Nutritional Supplements. Neuropharmacology 2017, 112, 248–263. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.M.; Dawes, M.A.; Mathias, C.W.; Acheson, A.; Hill-Kapturczak, N.; Dougherty, D.M. L-Tryptophan: Basic Metabolic Functions, Behavioral Research and Therapeutic Indications. Int. J. Tryptophan Res. 2009, 2, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Keszthelyi, D.; Troost, F.J.; Jonkers, D.M.; van Donkelaar, E.L.; Dekker, J.; Buurman, W.A.; Masclee, A.A. Does Acute Tryptophan Depletion Affect Peripheral Serotonin Metabolism in the Intestine? Am. J. Clin. Nutr. 2012, 95, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Gál, E.M.; Sherman, A.D. L-Kynurenine: Its Synthesis and Possible Regulatory Function in Brain. Neurochem. Res. 1980, 5, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Catena-Dell’Osso, M.; Rotella, F.; Dell’Osso, A.; Fagiolini, A.; Marazziti, D. Inflammation, Serotonin and Major Depression. Curr. Drug Targets 2013, 14, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.; Pogson, C.I. The Role of Tryptophan 2,3-Dioxygenase in the Hormonal Control of Tryptophan Metabolism in Isolated Rat Liver Cells. Effects of Glucocorticoids and Experimental Diabetes. Biochem. J. 1985, 229, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Yeung, A.W.S.; Terentis, A.C.; King, N.J.C.; Thomas, S.R. Role of Indoleamine 2,3-Dioxygenase in Health and Disease. Clin. Sci. 2015, 129, 601–672. [Google Scholar] [CrossRef] [PubMed]
- Jurgens, B.; Hainz, U.; Fuchs, D.; Felzmann, T.; Heitger, A. Interferon-γ-Triggered Indoleamine 2,3-Dioxygenase Competence in Human Monocyte-Derived Dendritic Cells Induces Regulatory Activity in Allogeneic T Cells. Blood 2009, 114, 3235–3243. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R. Role of the Kynurenine Metabolism Pathway in Inflammation-Induced Depression: Preclinical Approaches. In Inflammation-Associated Depression: Evidence, Mechanisms and Implications. Current Topics in Behavioral Neurosciences; Springer: Cham, Switzerland, 2016; pp. 117–138. ISBN 978-3-319-51152-8. [Google Scholar]
- Pertz, H.; Back, W. Synthesis and Resolution of Chiral Ring-Opened Serotonin Analogs of the 5-Hydroxykynuramine Type. Pharm. Acta Helv. 1988, 63, 128–131. [Google Scholar] [PubMed]
- Jeon, S.W.; Kim, Y.-K. Inflammation-Induced Depression: Its Pathophysiology and Therapeutic Implications. J. Neuroimmunol. 2017, 313, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Keszthelyi, D.; Troost, F.J.; Masclee, A.A.M. Understanding the Role of Tryptophan and Serotonin Metabolism in Gastrointestinal Function. Neurogastroenterol. Motil. 2009, 21, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Fukui, S.; Schwarcz, R.; Rapoport, S.I.; Takada, Y.; Smith, Q.R. Blood-Brain Barrier Transport of Kynurenines: Implications for Brain Synthesis and Metabolism. J. Neurochem. 1991, 56, 2007–2017. [Google Scholar] [CrossRef] [PubMed]
- Savitz, J.; Drevets, W.C.; Wurfel, B.E.; Ford, B.N.; Bellgowan, P.S.F.; Victor, T.A.; Bodurka, J.; Teague, T.K.; Dantzer, R. Reduction of Kynurenic Acid to Quinolinic Acid Ratio in Both the Depressed and Remitted Phases of Major Depressive Disorder. Brain Behav. Immun. 2015, 46, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Bay-Richter, C.; Linderholm, K.R.; Lim, C.K.; Samuelsson, M.; Träskman-Bendz, L.; Guillemin, G.J.; Erhardt, S.; Brundin, L. A Role for Inflammatory Metabolites as Modulators of the Glutamate N-Methyl-d-Aspartate Receptor in Depression and Suicidality. Brain Behav. Immun. 2015, 43, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Bryleva, E.Y.; Brundin, L. Kynurenine Pathway Metabolites and Suicidality. Neuropharmacology 2017, 112, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Wurfel, B.E.; Drevets, W.C.; Bliss, S.A.; McMillin, J.R.; Suzuki, H.; Ford, B.N.; Morris, H.M.; Teague, T.K.; Dantzer, R.; Savitz, J.B. Serum Kynurenic Acid Is Reduced in Affective Psychosis. Transl. Psychiatry 2017, 7, e1115. [Google Scholar] [CrossRef] [PubMed]
- Connor, T.J.; Starr, N.; O’Sullivan, J.B.; Harkin, A. Induction of Indolamine 2,3-Dioxygenase and Kynurenine 3-Monooxygenase in Rat Brain Following a Systemic Inflammatory Challenge: A Role for IFN-γ? Neurosci. Lett. 2008, 441, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Molteni, R.; Macchi, F.; Zecchillo, C.; Dell’Agli, M.; Colombo, E.; Calabrese, F.; Guidotti, G.; Racagni, G.; Riva, M.A. Modulation of the Inflammatory Response in Rats Chronically Treated with the Antidepressant Agomelatine. Eur. Neuropsychopharmacol. 2013, 23, 1645–1655. [Google Scholar] [CrossRef] [PubMed]
- Erhardt, S.; Lim, C.K.; Linderholm, K.R.; Janelidze, S.; Lindqvist, D.; Samuelsson, M.; Lundberg, K.; Postolache, T.T.; Träskman-Bendz, L.; Guillemin, G.J.; et al. Connecting Inflammation with Glutamate Agonism in Suicidality. Neuropsychopharmacology 2013, 38, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Husi, H. NMDA Receptors, Neural Pathways, and Protein Interaction Databases. Int. Rev. Neurobiol. 2004, 61, 49–77. [Google Scholar] [CrossRef] [PubMed]
- Giaroni, C.; Zanetti, E.; Chiaravalli, A.M.; Albarello, L.; Dominioni, L.; Capella, C.; Lecchini, S.; Frigo, G. Evidence for a Glutamatergic Modulation of the Cholinergic Function in the Human Enteric Nervous System via NMDA Receptors. Eur. J. Pharmacol. 2003, 476, 63–69. [Google Scholar] [CrossRef]
- Kirchgessner, A. Glutamate in the Enteric Nervous System. Curr. Opin. Pharmacol. 2001, 1, 591–596. [Google Scholar] [CrossRef]
- Zhou, Q.; Nicholas Verne, G. NMDA Receptors and Colitis: Basic Science and Clinical Implications. Rev. Analg. 2008, 10, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Varga, G.; Érces, D.; Fazekas, B.; Fülöp, M.; Kovács, T.; Kaszaki, J.; Fülöp, F.; Vécsei, L.; Boros, M. N-Methyl-d-Aspartate Receptor Antagonism Decreases Motility and Inflammatory Activation in the Early Phase of Acute Experimental Colitis in the Rat. Neurogastroenterol. Motil. 2010, 22, 217-e68. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, S.V.; Meller, S.T.; Gebhart, G.F. Intracolonic Zymosan Produces Visceral Hyperalgesia in the Rat That Is Mediated by Spinal NMDA and Non-NMDA Receptors. Brain Res. 1996, 736, 7–15. [Google Scholar] [CrossRef]
- Berger, M.; Gray, J.A.; Roth, B.L. The Expanded Biology of Serotonin. Annu. Rev. Med. 2009, 60, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Wade, P.R.; Tamir, H.; Kirchgessner, A.L.; Gershon, M.D. Analysis of the Role of 5-HT in the Enteric Nervous System Using Anti-Idiotopic Antibodies to 5-HT Receptors. Am. J. Physiol. Liver Physiol. 1994, 266, G403–G416. [Google Scholar] [CrossRef] [PubMed]
- Gershon, M.D.; Ross, L.L. Studies on the Relationship of 5-Hydroxytryptamine and the Enterochromaffin Cell to Anaphylactic Shock in Mice. J. Exp. Med. 1962, 115, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.A.; Green, E.M.; Gouaux, E. X-Ray Structures and Mechanism of the Human Serotonin Transporter. Nature 2016, 532, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.L.; Lerner, A.; Rudnick, G.; Lesch, K.-P. Serotonin Transporter: Gene, Genetic Disorders, and Pharmacogenetics. Mol. Interv. 2004, 4, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Hannon, J.; Hoyer, D. Molecular Biology of 5-HT Receptors. Behav. Brain Res. 2008, 195, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Tutton, P.J. The Influence of Serotonin on Crypt Cell Proliferation in the Jejunum of Rat. Virchows Arch. B Cell Pathol. 1974, 16, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Gershon, M.D. Nerves, Reflexes, and the Enteric Nervous System. J. Clin. Gastroenterol. 2005, 39, S184–S193. [Google Scholar] [CrossRef] [PubMed]
- Mazzia, C.; Hicks, G.; Clerc, N. Neuronal Location of 5-Hydroxytryptamine3 Receptor-like Immunoreactivity in the Rat Colon. Neuroscience 2003, 116, 1033–1041. [Google Scholar] [CrossRef]
- Grider, J.R. Desensitization of the Peristaltic Reflex Induced by Mucosal Stimulation with the Selective 5-HT4 Agonist Tegaserod. Am. J. Physiol. Liver Physiol. 2006, 290, G319–G327. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Galligan, J.J. 5-HT1A and 5-HT4 Receptors Mediate Inhibition and Facilitation of Fast Synaptic Transmission in Enteric Neurons. Am. J. Physiol. 1994, 266, G230-8. [Google Scholar] [CrossRef] [PubMed]
- Galligan, J.J.; Pan, H.; Messori, E. Signalling Mechanism Coupled to 5-Hydroxytryptamine4 Receptor-Mediated Facilitation of Fast Synaptic Transmission in the Guinea-Pig Ileum Myenteric Plexus. Neurogastroenterol. Motil. 2003, 15, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Baganz, N.L.; Blakely, R.D. A Dialogue between the Immune System and Brain, Spoken in the Language of Serotonin. ACS Chem. Neurosci. 2013, 4, 48–63. [Google Scholar] [CrossRef] [PubMed]
- Herr, N.; Bode, C.; Duerschmied, D. The Effects of Serotonin in Immune Cells. Front. Cardiovasc. Med. 2017, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Leon-Ponte, M.; Ahern, G.P.; O’Connell, P.J. Serotonin Provides an Accessory Signal to Enhance T-Cell Activation by Signaling through the 5-HT7 Receptor. Blood 2007, 109, 3139–3146. [Google Scholar] [CrossRef] [PubMed]
- Medina-Martel, M.; Urbina, M.; Fazzino, F.; Lima, L. Serotonin Transporter in Lymphocytes of Rats Exposed to Physical Restraint Stress. Neuroimmunomodulation 2013, 20, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Meredith, E.J.; Holder, M.J.; Chamba, A.; Challa, A.; Drake-Lee, A.; Bunce, C.M.; Drayson, M.T.; Pilkington, G.; Blakely, R.D.; Dyer, M.J.S.; et al. The Serotonin Transporter (SLC6A4) Is Present in B-Cell Clones of Diverse Malignant Origin: Probing a Potential Anti-Tumor Target for Psychotropics. FASEB J. 2005, 19, 1187–1189. [Google Scholar] [CrossRef] [PubMed]
- Idzko, M.; Panther, E.; Stratz, C.; Muller, T.; Bayer, H.; Zissel, G.; Durk, T.; Sorichter, S.; Di Virgilio, F.; Geissler, M.; et al. The Serotoninergic Receptors of Human Dendritic Cells: Identification and Coupling to Cytokine Release. J. Immunol. 2004, 172, 6011–6019. [Google Scholar] [CrossRef] [PubMed]
- Dürk, T.; Panther, E.; Müller, T.; Sorichter, S.; Ferrari, D.; Pizzirani, C.; Di Virgilio, F.; Myrtek, D.; Norgauer, J.; Idzko, M. 5-Hydroxytryptamine Modulates Cytokine and Chemokine Production in LPS-Primed Human Monocytes via Stimulation of Different 5-HTR Subtypes. Int. Immunol. 2005, 17, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Fiorica-Howells, E.; Liu, M.-T.; Ponimaskin, E.G.; Li, Z.-S.; Compan, V.; Hen, R.; Gingrich, J.A.; Gershon, M.D. Distribution of 5-HT4 Receptors in Wild-Type Mice and Analysis of Intestinal Motility in 5-HT4 Knockout Mice. Gastroenterology 2003, 124, A342. [Google Scholar] [CrossRef]
- Compan, V. Attenuated Response to Stress and Novelty and Hypersensitivity to Seizures in 5-HT4 Receptor Knock-Out Mice. J. Neurosci. 2004, 24, 412–419. [Google Scholar] [CrossRef] [PubMed]
- El Aidy, S.; Ramsteijn, A.S.; Dini-Andreote, F.; van Eijk, R.; Houwing, D.J.; Salles, J.F.; Olivier, J.D.A. Serotonin Transporter Genotype Modulates the Gut Microbiota Composition in Young Rats, an Effect Augmented by Early Life Stress. Front. Cell. Neurosci. 2017, 11, 222. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Li, Z.; Pan, H.; Murphy, D.L.; Tamir, H.; Koepsell, H.; Gershon, M.D. Maintenance of Serotonin in the Intestinal Mucosa and Ganglia of Mice That Lack the High-Affinity Serotonin Transporter: Abnormal Intestinal Motility and the Expression of Cation Transporters. J. Neurosci. 2001, 21, 6348–6361. [Google Scholar] [CrossRef] [PubMed]
- Gershon, M.D.; Tack, J. The Serotonin Signaling System: From Basic Understanding To Drug Development for Functional GI Disorders. Gastroenterology 2007, 132, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Canli, T.; Lesch, K.-P. Long Story Short: The Serotonin Transporter in Emotion Regulation and Social Cognition. Nat. Neurosci. 2007, 10, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Scheerens, C.; Tack, J.; Rommel, N. Buspirone, a New Drug for the Management of Patients with Ineffective Esophageal Motility? United Eur. Gastroenterol. J. 2015, 3, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Di Sabatino, A.; Giuffrida, P.; Vanoli, A.; Luinetti, O.; Manca, R.; Biancheri, P.; Bergamaschi, G.; Alvisi, C.; Pasini, A.; Salvatore, C.; et al. Increase in Neuroendocrine Cells in the Duodenal Mucosa of Patients with Refractory Celiac Disease. Am. J. Gastroenterol. 2014, 109, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Bruce-Keller, A.J.; Salbaum, J.M.; Berthoud, H.-R. Harnessing Gut Microbes for Mental Health: Getting From Here to There. Biol. Psychiatry 2018, 83, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; O’Connell, R.M.; Mazmanian, S.K. Coordination of Tolerogenic Immune Responses by the Commensal Microbiota. J. Autoimmun. 2010, 34. [Google Scholar] [CrossRef] [PubMed]
- El Aidy, S.; van Baarlen, P.; Derrien, M.; Lindenbergh-Kortleve, D.J.; Hooiveld, G.; Levenez, F.; Doré, J.; Dekker, J.; Samsom, J.N.; Nieuwenhuis, E.E.S.; et al. Temporal and Spatial Interplay of Microbiota and Intestinal Mucosa Drive Establishment of Immune Homeostasis in Conventionalized Mice. Mucosal Immunol. 2012, 5, 567–579. [Google Scholar] [CrossRef] [PubMed]
- El Aidy, S.; Derrien, M.; Aardema, R.; Hooiveld, G.; Richards, S.E.; Dane, A.; Dekker, J.; Vreeken, R.; Levenez, F.; Doré, J.; et al. Transient Inflammatory-like State and Microbial Dysbiosis Are Pivotal in Establishment of Mucosal Homeostasis during Colonisation of Germ-Free Mice. Benef. Microbes 2014, 5, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An Immunomodulatory Molecule of Symbiotic Bacteria Directs Maturation of the Host Immune System. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Gaboriau-Routhiau, V.; Rakotobe, S.; Lécuyer, E.; Mulder, I.; Lan, A.; Bridonneau, C.; Rochet, V.; Pisi, A.; De Paepe, M.; Brandi, G.; et al. The Key Role of Segmented Filamentous Bacteria in the Coordinated Maturation of Gut Helper T Cell Responses. Immunity 2009, 31, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.; Mazmanian, S.K. A Pathobiont of the Microbiota Balances Host Colonization and Intestinal Inflammation. Cell Host Microbe 2010, 7, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Emge, J.R.; Huynh, K.; Miller, E.N.; Kaur, M.; Reardon, C.; Barrett, K.E.; Gareau, M.G. Modulation of the Microbiota-Gut-Brain Axis by Probiotics in a Murine Model of Inflammatory Bowel Disease. Am. J. Physiol. Liver Physiol. 2016, 310, G989–G998. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Tonkonogy, S.L.; Albright, C.A.; Tsang, J.; Balish, E.J.; Braun, J.; Huycke, M.M.; Sartor, R.B. Variable Phenotypes of Enterocolitis in Interleukin 10-Deficient Mice Monoassociated with Two Different Commensal Bacteria. Gastroenterology 2005, 128, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Bloom, S.M.; Bijanki, V.N.; Nava, G.M.; Sun, L.; Malvin, N.P.; Donermeyer, D.L.; Dunne, W.M.; Allen, P.M.; Stappenbeck, T.S. Commensal Bacteroides Species Induce Colitis in Host-Genotype-Specific Fashion in a Mouse Model of Inflammatory Bowel Disease. Cell Host Microbe 2011, 9, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Garrett, W.S.; Gallini, C.A.; Yatsunenko, T.; Michaud, M.; DuBois, A.; Delaney, M.L.; Punit, S.; Karlsson, M.; Bry, L.; Glickman, J.N.; et al. Enterobacteriaceae Act in Concert with the Gut Microbiota to Induce Spontaneous and Maternally Transmitted Colitis. Cell Host Microbe 2010, 8, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Dharmani, P.; Strauss, J.; Ambrose, C.; Allen-Vercoe, E.; Chadee, K. Fusobacterium Nucleatum Infection of Colonic Cells Stimulates MUC2 Mucin and Tumor Necrosis Factor Alpha. Infect. Immun. 2011, 79, 2597–2607. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Menezes, J.S.; Umesaki, Y.; Mazmanian, S.K. Proinflammatory T-Cell Responses to Gut Microbiota Promote Experimental Autoimmune Encephalomyelitis. Proc. Natl. Acad. Sci. USA 2011, 108, 4615–4622. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Reparaz, J.; Mielcarz, D.W.; Ditrio, L.E.; Burroughs, A.R.; Foureau, D.M.; Haque-Begum, S.; Kasper, L.H. Role of Gut Commensal Microflora in the Development of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2009, 183, 6041–6050. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous Bacteria from the Gut Microbiota Regulate Host Serotonin Biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Reigstad, C.S.; Salmonson, C.E.; Rainey, J.F.; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut Microbes Promote Colonic Serotonin Production through an Effect of Short-Chain Fatty Acids on Enterochromaffin Cells. FASEB J. 2015, 29, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics Analysis Reveals Large Effects of Gut Microflora on Mammalian Blood Metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The Microbiome-Gut-Brain Axis during Early Life Regulates the Hippocampal Serotonergic System in a Sex-Dependent Manner. Mol. Psychiatry 2013, 18, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Rooks, M.G.; Veiga, P.; Wardwell-Scott, L.H.; Tickle, T.; Segata, N.; Michaud, M.; Gallini, C.A.; Beal, C.; van Hylckama-Vlieg, J.E.; Ballal, S.A.; et al. Gut Microbiome Composition and Function in Experimental Colitis during Active Disease and Treatment-Induced Remission. ISME J. 2014, 8, 1403–1417. [Google Scholar] [CrossRef] [PubMed]
- Robertson, B.R. Mucispirillum Schaedleri Gen. Nov., Sp. Nov., a Spiral-Shaped Bacterium Colonizing the Mucus Layer of the Gastrointestinal Tract of Laboratory Rodents. Int. J. Syst. Evol. Microbiol. 2005, 55, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.; Schwab, C.; Milinovich, G.; Reichert, J.; Ben Mahfoudh, K.; Decker, T.; Engel, M.; Hai, B.; Hainzl, E.; Heider, S.; et al. Phylotype-Level 16S RRNA Analysis Reveals New Bacterial Indicators of Health State in Acute Murine Colitis. ISME J. 2012, 6, 2091–2106. [Google Scholar] [CrossRef] [PubMed]
- Carbonero, F.; Benefiel, A.C.; Gaskins, H.R. Contributions of the Microbial Hydrogen Economy to Colonic Homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 504–518. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium Nucleatum Potentiates Intestinal Tumorigenesis and Modulates the Tumor-Immune Microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.; Kaplan, G.G.; Beck, P.L.; Rioux, K.; Panaccione, R.; DeVinney, R.; Lynch, T.; Allen-Vercoe, E. Invasive Potential of Gut Mucosa-Derived Fusobacterium Nucleatum Positively Correlates with IBD Status of the Host. Inflamm. Bowel Dis. 2011, 17, 1971–1978. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-P.; Chen, Y.-T.; Tsai, C.-F.; Li, S.-Y.; Luo, J.-C.; Wang, S.-J.; Tang, C.-H.; Liu, C.-J.; Lin, H.-C.; Lee, F.-Y.; et al. Short-Term Use of Serotonin Reuptake Inhibitors and Risk of Upper Gastrointestinal Bleeding. Am. J. Psychiatry 2014, 171, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Macedo, D.; Filho, A.J.M.C.; Soares de Sousa, C.N.; Quevedo, J.; Barichello, T.; Júnior, H.V.N.; Freitas de Lucena, D. Antidepressants, Antimicrobials or Both? Gut Microbiota Dysbiosis in Depression and Possible Implications of the Antimicrobial Effects of Antidepressant Drugs for Antidepressant Effectiveness. J. Affect. Disord. 2017, 208, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Coban, A.Y.; Tanriverdi Cayci, Y.; Keleş Uludağ, S.; Durupinar, B. Investigation of Antibacterial Activity of Sertralin. Mikrobiyol. Bul. 2009, 43, 651–656. [Google Scholar] [PubMed]
- Munoz-Bellido, J.; Munoz-Criado, S.; Garcìa-Rodrìguez, J. Antimicrobial Activity of Psychotropic Drugs: Selective Serotonin Reuptake Inhibitors. Int. J. Antimicrob. Agents 2000, 14, 177–180. [Google Scholar] [CrossRef]
- Kruszewska, H.; Zarȩba, T.; Tyski, S. Examination of Antimicrobial Activity of Selected Non-Antibiotic Medicinal Preparations. Acta Pol. Pharm. Drug Res. 2012, 69, 1368–1371. [Google Scholar]
- Cenit, M.C.; Sanz, Y.; Codoñer-Franch, P. Influence of Gut Microbiota on Neuropsychiatric Disorders. World J. Gastroenterol. 2017, 23, 5486–5498. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.R.; Borre, Y.; O’Brien, C.; Patterson, E.; El Aidy, S.; Deane, J.; Kennedy, P.J.; Beers, S.; Scott, K.; Moloney, G.; et al. Transferring the Blues: Depression-Associated Gut Microbiota Induces Neurobehavioural Changes in the Rat. J. Psychiatr. Res. 2016, 82, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, E.; Tsuji, H.; Asahara, T.; Takahashi, T.; Teraishi, T.; Yoshida, S.; Ota, M.; Koga, N.; Hattori, K.; Kunugi, H. Possible Association of Bifidobacterium and Lactobacillus in the Gut Microbiota of Patients with Major Depressive Disorder. J. Affect. Disord. 2016, 202, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, M.; Lalonde, R.; Violle, N.; Javelot, H.; Desor, D.; Nejdi, A.; Bisson, J.-F.; Rougeot, C.; Pichelin, M.; Cazaubiel, M.; et al. Assessment of Psychotropic-like Properties of a Probiotic Formulation (Lactobacillus Helveticus R0052 and Bifidobacterium Longum R0175) in Rats and Human Subjects. Br. J. Nutr. 2011, 105, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Benton, D.; Williams, C.; Brown, A. Impact of Consuming a Milk Drink Containing a Probiotic on Mood and Cognition. Eur. J. Clin. Nutr. 2007, 61, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Steenbergen, L.; Sellaro, R.; van Hemert, S.; Bosch, J.A.; Colzato, L.S. A Randomized Controlled Trial to Test the Effect of Multispecies Probiotics on Cognitive Reactivity to Sad Mood. Brain Behav. Immun. 2015, 48, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Tillisch, K.; Labus, J.; Kilpatrick, L.; Jiang, Z.; Stains, J.; Ebrat, B.; Guyonnet, D.; Legrain-Raspaud, S.; Trotin, B.; Naliboff, B.; et al. Consumption of Fermented Milk Product With Probiotic Modulates Brain Activity. Gastroenterology 2013, 144, 1394–1401.e4. [Google Scholar] [CrossRef] [PubMed]
- Marin, I.A.; Goertz, J.E.; Ren, T.; Rich, S.S.; Onengut-Gumuscu, S.; Farber, E.; Wu, M.; Overall, C.C.; Kipnis, J.; Gaultier, A. Microbiota Alteration Is Associated with the Development of Stress-Induced Despair Behavior. Sci. Rep. 2017, 7, 43859. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the Gut Microbiota on Intestinal Immunity Mediated by Tryptophan Metabolism. Front. Cell. Infect. Microbiol. 2018, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Genestet, C.; Le Gouellec, A.; Chaker, H.; Polack, B.; Guery, B.; Toussaint, B.; Stasia, M.J. Scavenging of Reactive Oxygen Species by Tryptophan Metabolites Helps Pseudomonas Aeruginosa Escape Neutrophil Killing. Free Radic. Biol. Med. 2014, 73, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Xie, G.; Zhao, A.; Zhao, L.; Yao, C.; Chiu, N.H.L.; Zhou, Z.; Bao, Y.; Jia, W.; Nicholson, J.K.; et al. The Footprints of Gut Microbial–Mammalian Co-Metabolism. J. Proteome Res. 2011, 10, 5512–5522. [Google Scholar] [CrossRef] [PubMed]
- El Aidy, S.; Merrifield, C.A.; Derrien, M.; van Baarlen, P.; Hooiveld, G.; Levenez, F.; Doré, J.; Dekker, J.; Holmes, E.; Claus, S.P.; et al. The Gut Microbiota Elicits a Profound Metabolic Reorientation in the Mouse Jejunal Mucosa during Conventionalisation. Gut 2013, 62, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Jaglin, M.; Rhimi, M.; Philippe, C.; Pons, N.; Bruneau, A.; Goustard, B.; Daugé, V.; Maguin, E.; Naudon, L.; Rabot, S. Indole, a Signaling Molecule Produced by the Gut Microbiota, Negatively Impacts Emotional Behaviors in Rats. Front. Neurosci. 2018, 12, 216. [Google Scholar] [CrossRef] [PubMed]
- Biagini, G.; Pich, E.M.; Carani, C.; Marrama, P.; Gustafsson, J.-Å.; Fuxe, K.; Agnati, L.F. Indole-Pyruvic Acid, a Tryptophan Ketoanalogue, Antagonizes the Endocrine but Not the Behavioral Effects of Repeated Stress in a Model of Depression. Biol. Psychiatry 1993, 33, 712–719. [Google Scholar] [CrossRef]
- Tigchelaar, E.F.; Zhernakova, A.; Dekens, J.A.M.; Hermes, G.; Baranska, A.; Mujagic, Z.; Swertz, M.A.; Muñoz, A.M.; Deelen, P.; Cénit, M.C.; et al. Cohort Profile: LifeLines DEEP, a Prospective, General Population Cohort Study in the Northern Netherlands: Study Design and Baseline Characteristics. BMJ Open 2015, 5, e006772. [Google Scholar] [CrossRef] [PubMed]
- Sudlow, C.; Gallacher, J.; Allen, N.; Beral, V.; Burton, P.; Danesh, J.; Downey, P.; Elliott, P.; Green, J.; Landray, M.; et al. UK Biobank: An Open Access Resource for Identifying the Causes of a Wide Range of Complex Diseases of Middle and Old Age. PLoS Med. 2015, 12, e1001779. [Google Scholar] [CrossRef] [PubMed]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-Level Analysis of Gut Microbiome Variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waclawiková, B.; El Aidy, S. Role of Microbiota and Tryptophan Metabolites in the Remote Effect of Intestinal Inflammation on Brain and Depression. Pharmaceuticals 2018, 11, 63. https://doi.org/10.3390/ph11030063
Waclawiková B, El Aidy S. Role of Microbiota and Tryptophan Metabolites in the Remote Effect of Intestinal Inflammation on Brain and Depression. Pharmaceuticals. 2018; 11(3):63. https://doi.org/10.3390/ph11030063
Chicago/Turabian StyleWaclawiková, Barbora, and Sahar El Aidy. 2018. "Role of Microbiota and Tryptophan Metabolites in the Remote Effect of Intestinal Inflammation on Brain and Depression" Pharmaceuticals 11, no. 3: 63. https://doi.org/10.3390/ph11030063
APA StyleWaclawiková, B., & El Aidy, S. (2018). Role of Microbiota and Tryptophan Metabolites in the Remote Effect of Intestinal Inflammation on Brain and Depression. Pharmaceuticals, 11(3), 63. https://doi.org/10.3390/ph11030063