Old Drugs as New Treatments for Neurodegenerative Diseases




Abstract
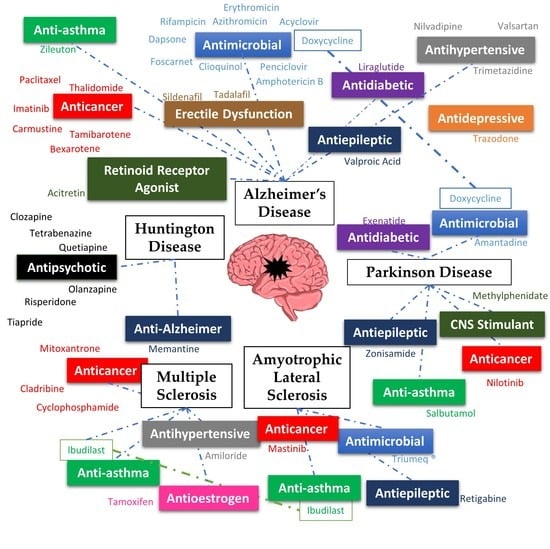
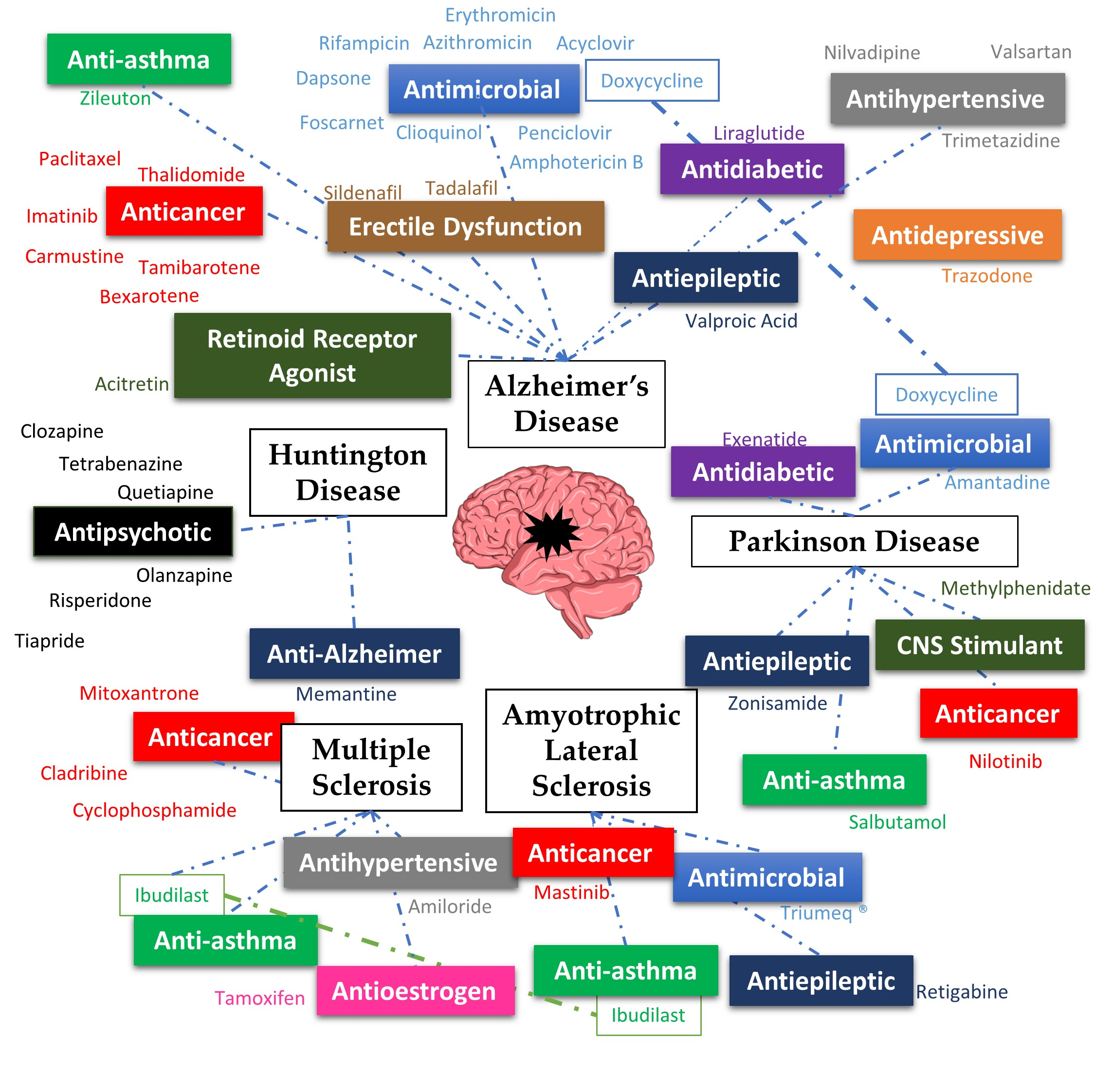
:
{kind=link}
{kind=link}
{kind=link}
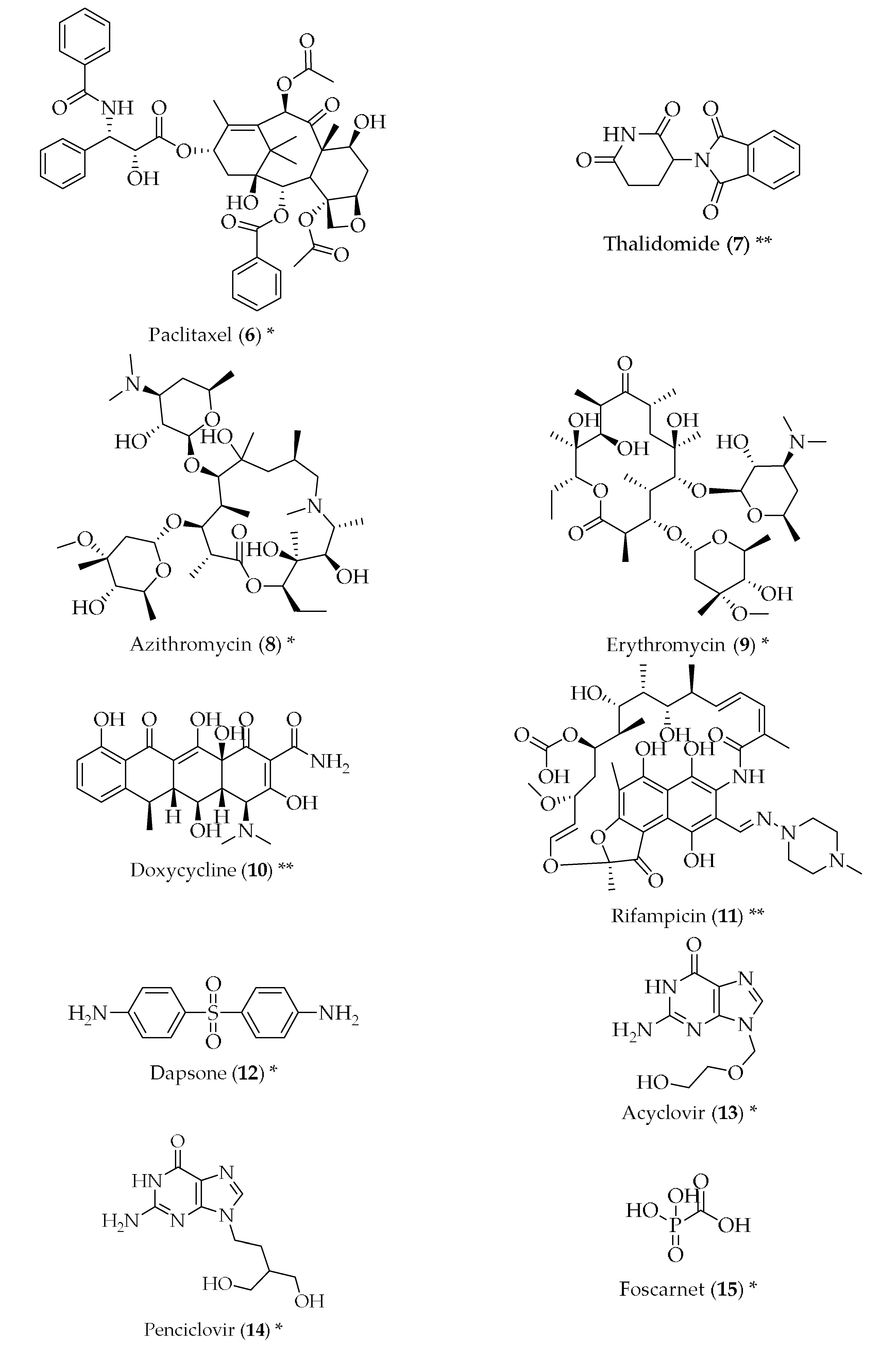{kind=link}
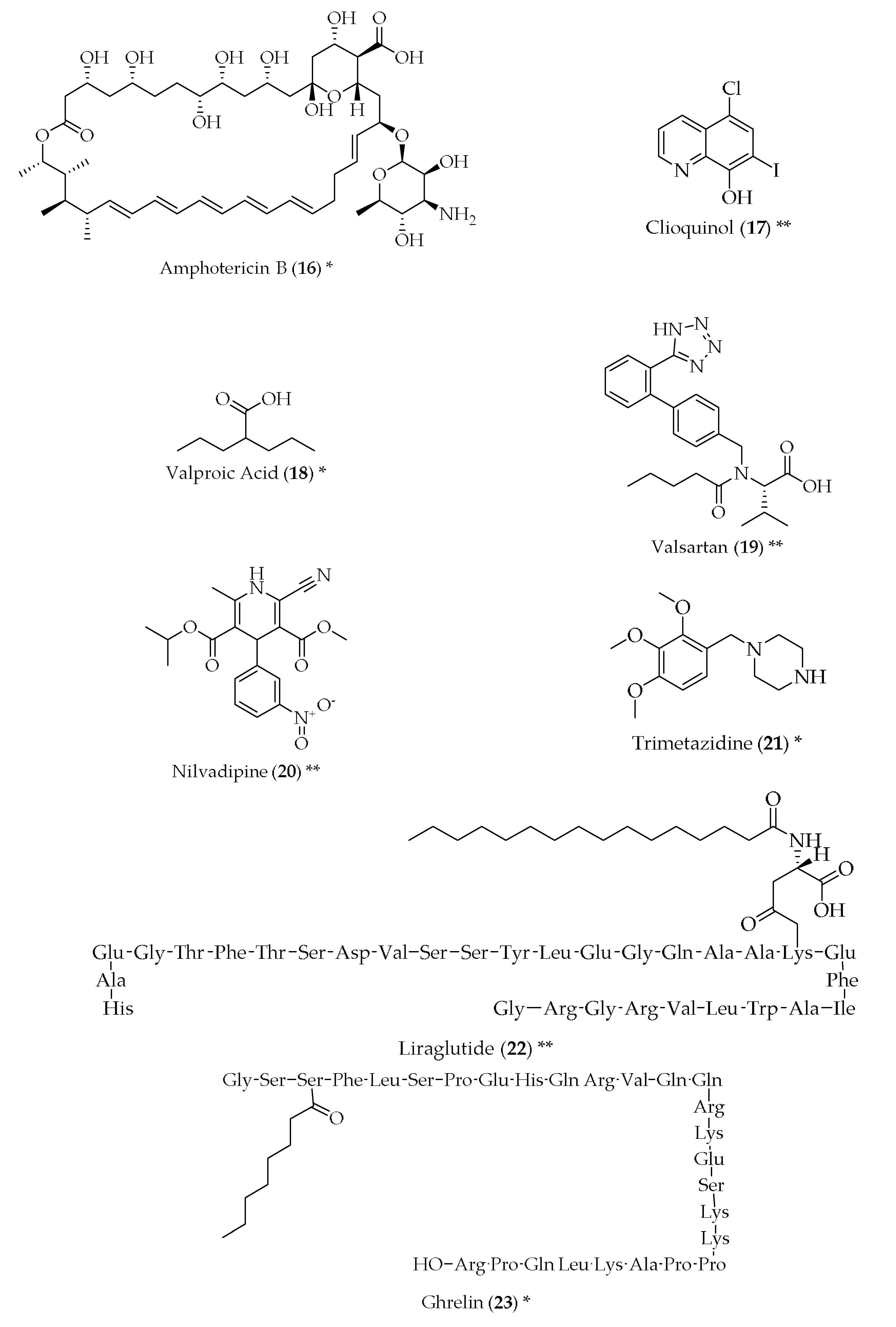{kind=link}
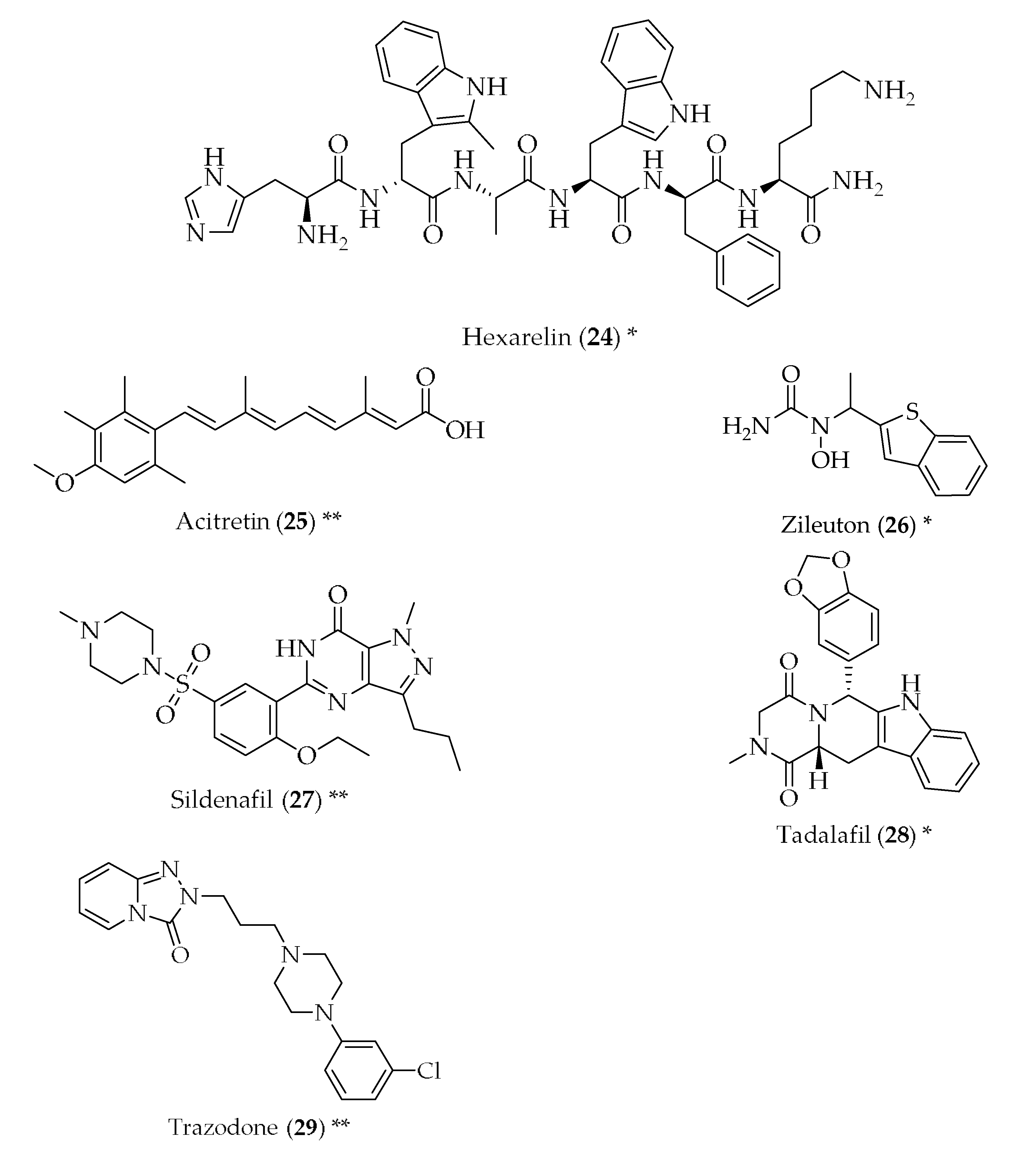{kind=link}
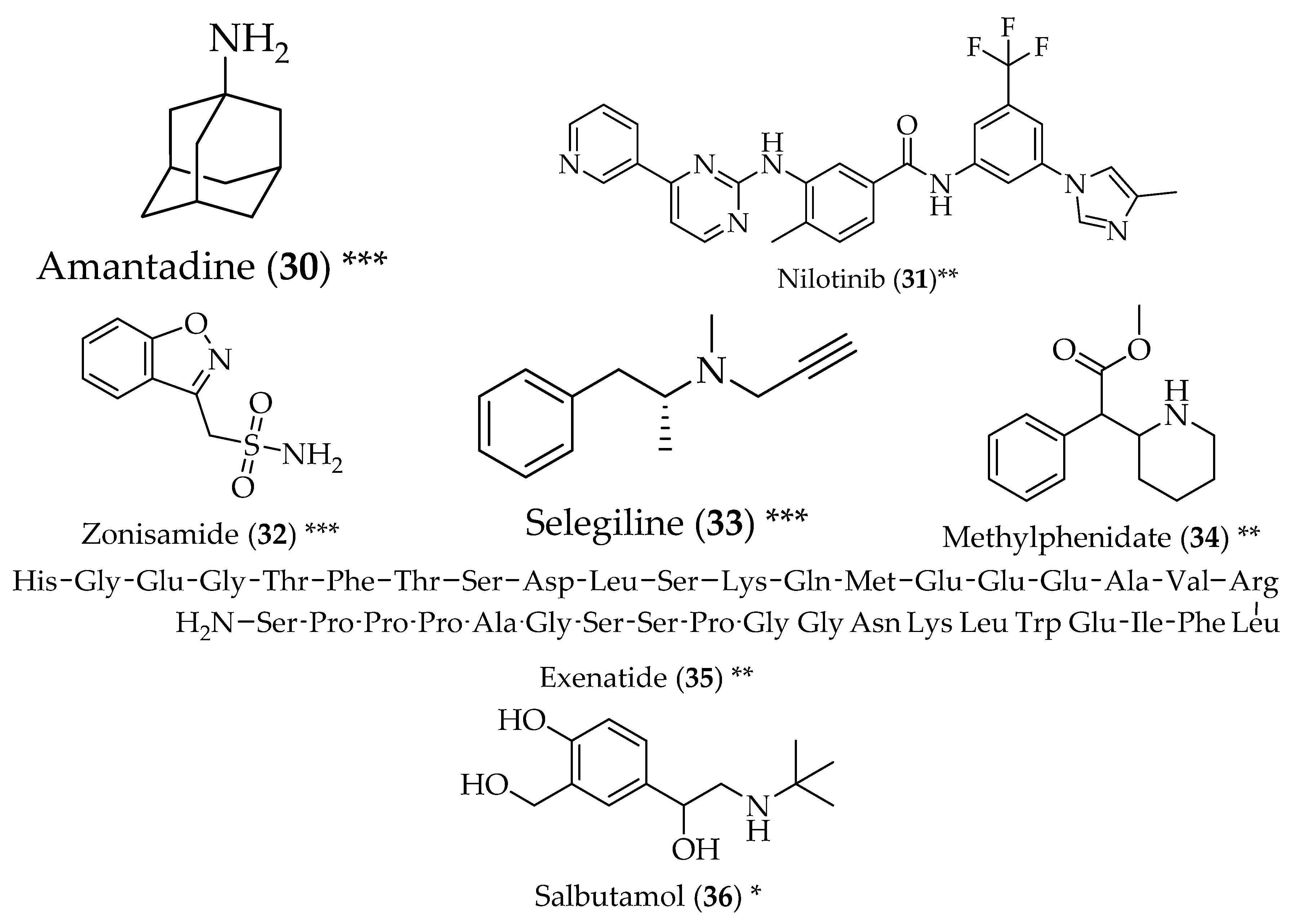{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
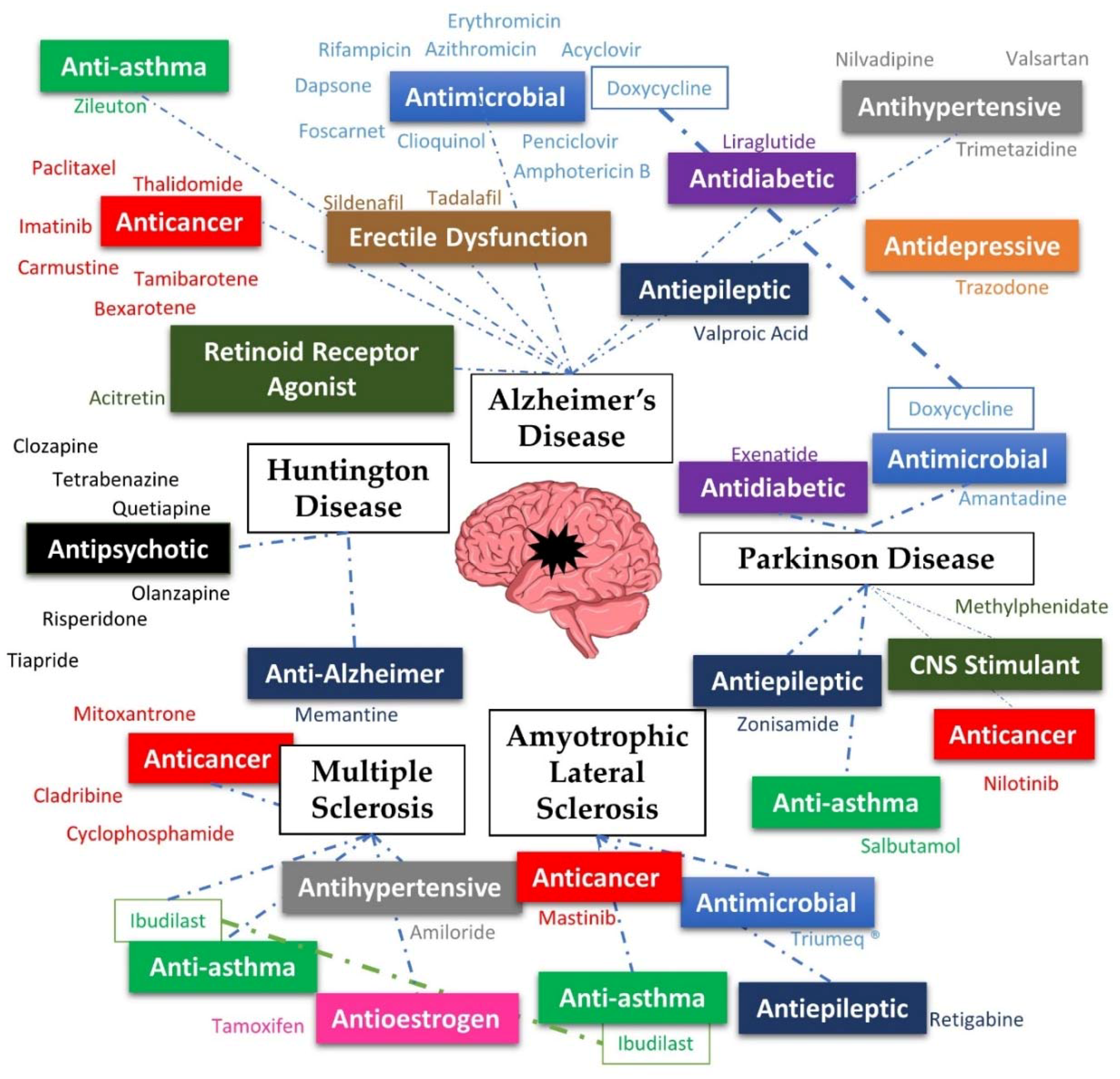
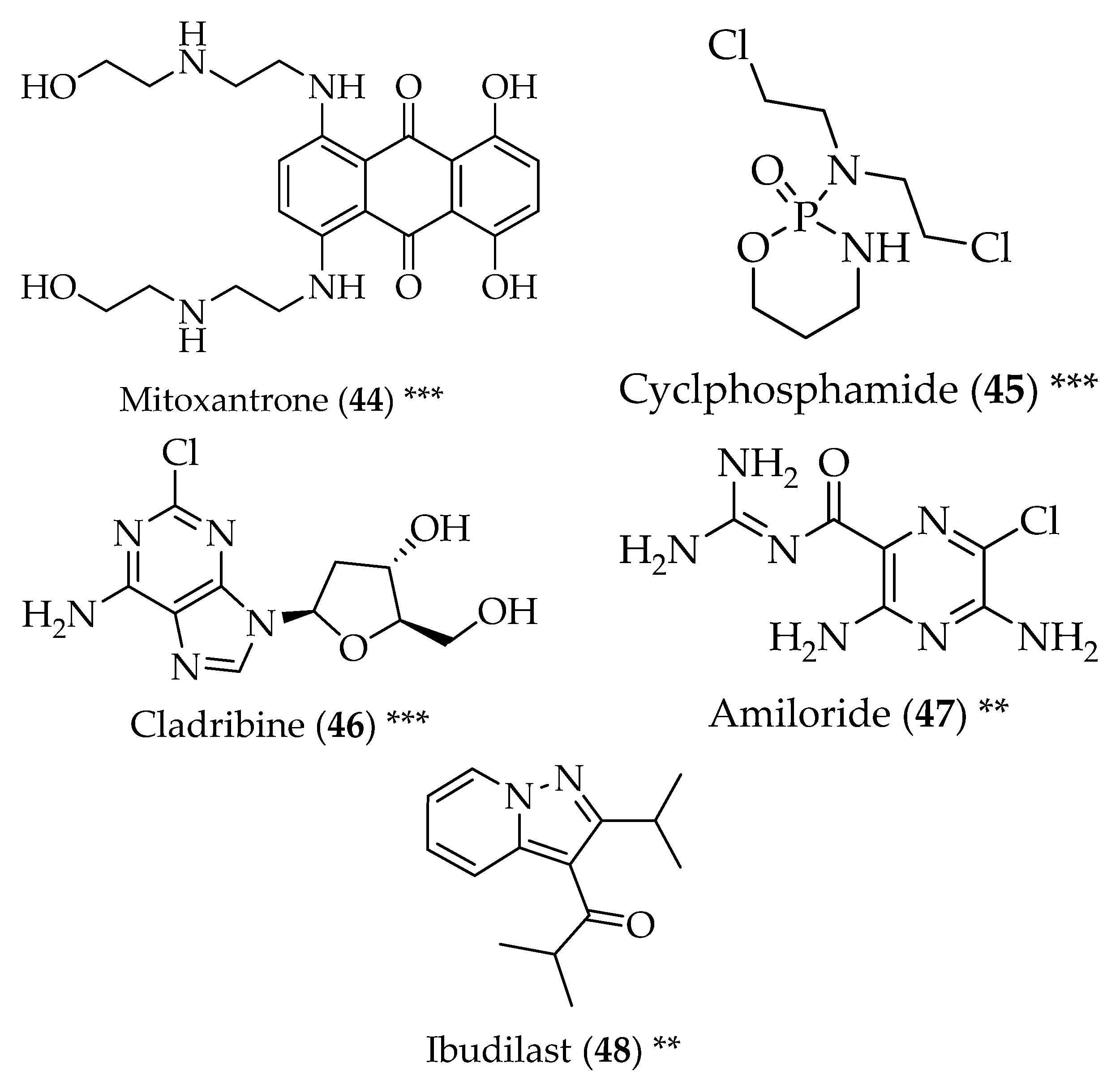
2. Alzheimer’s Disease
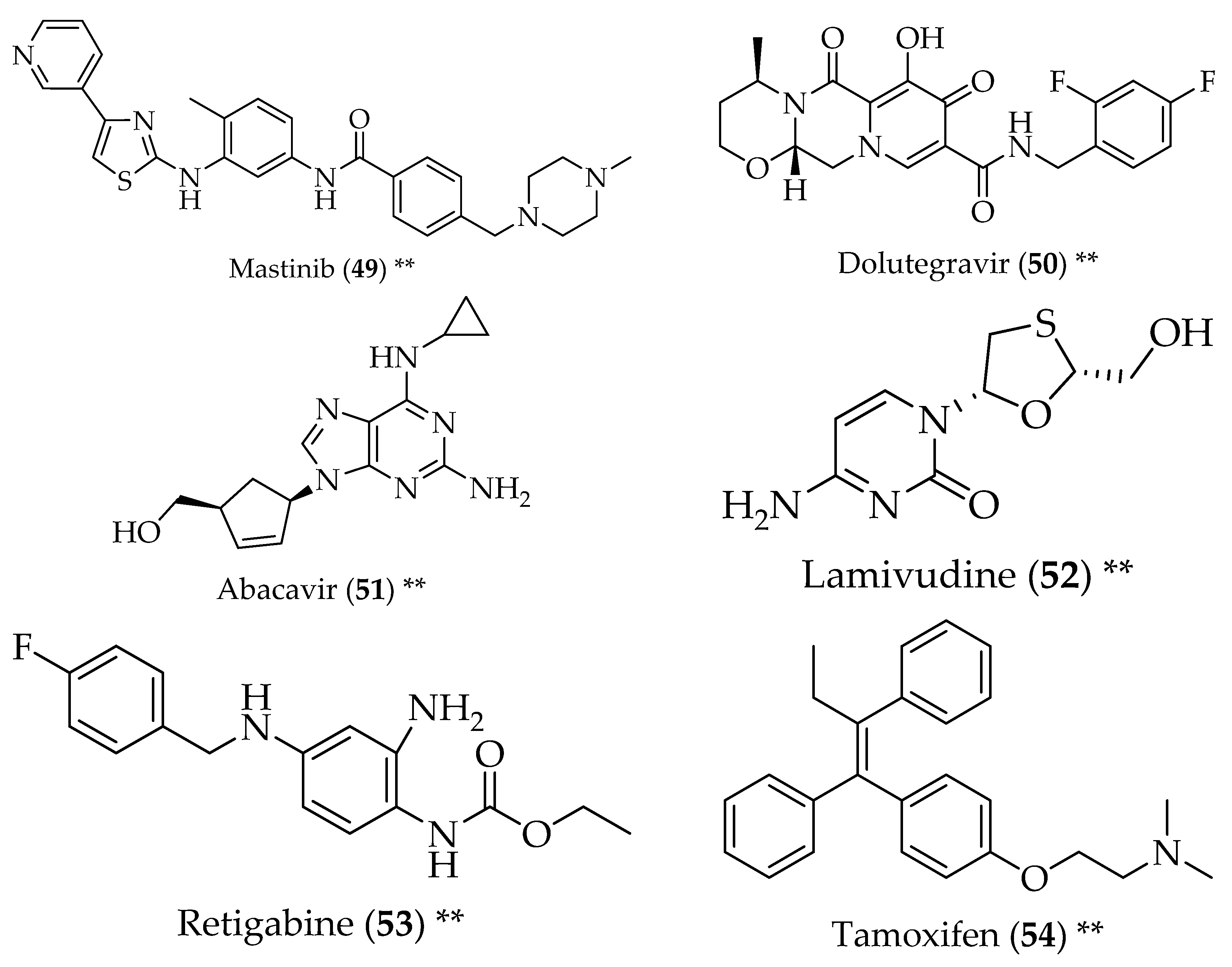
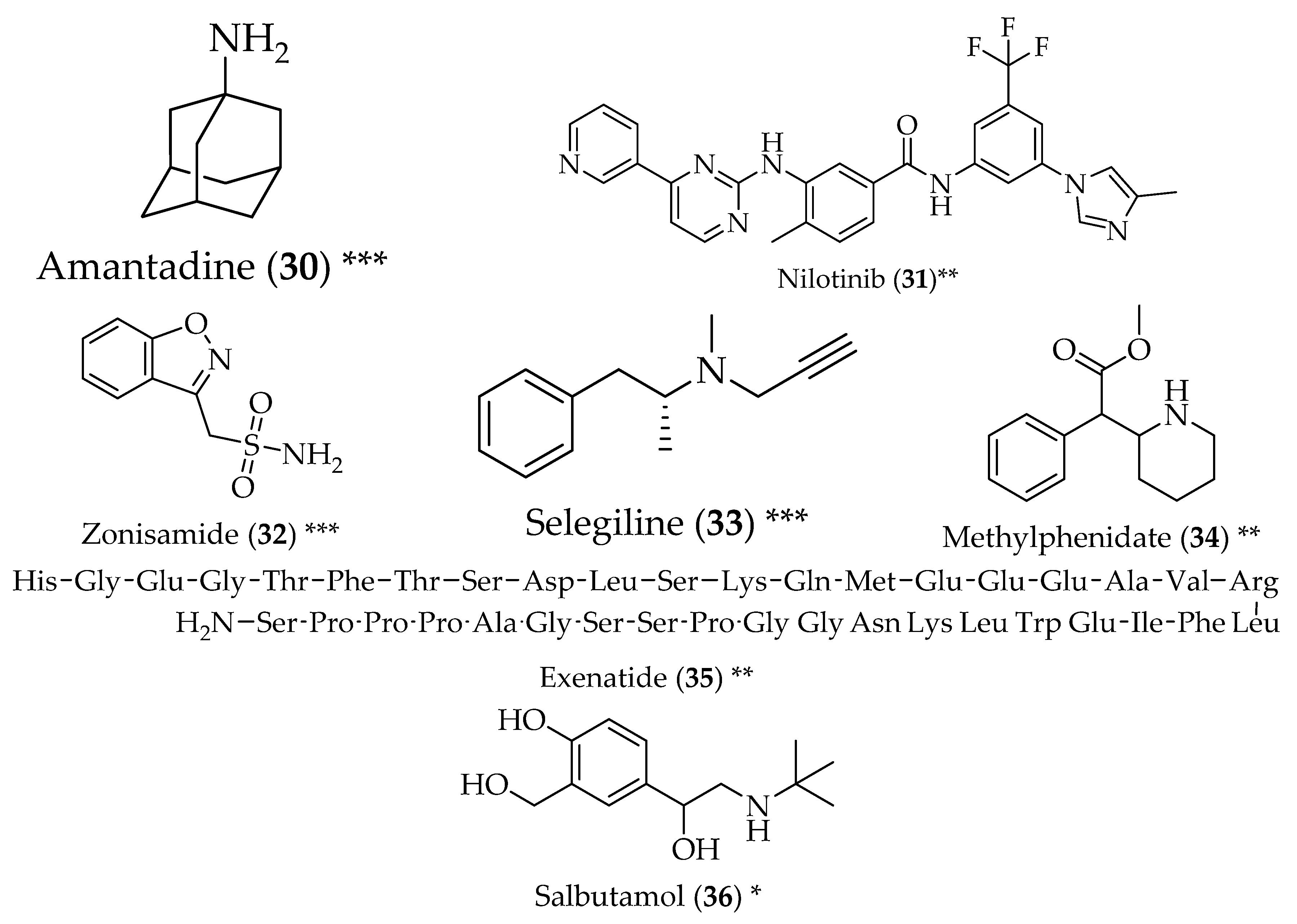
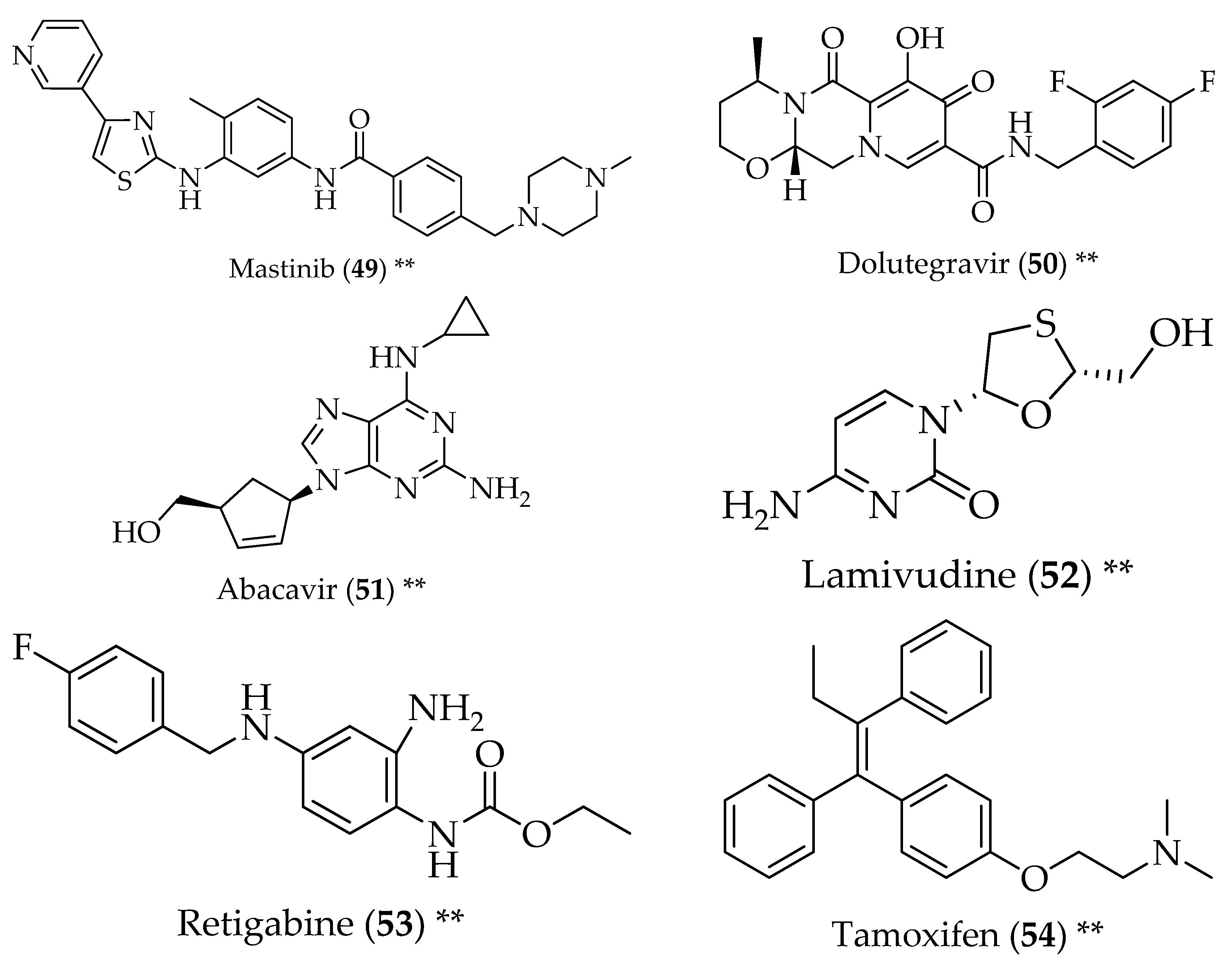
3. Parkinson’s Disease
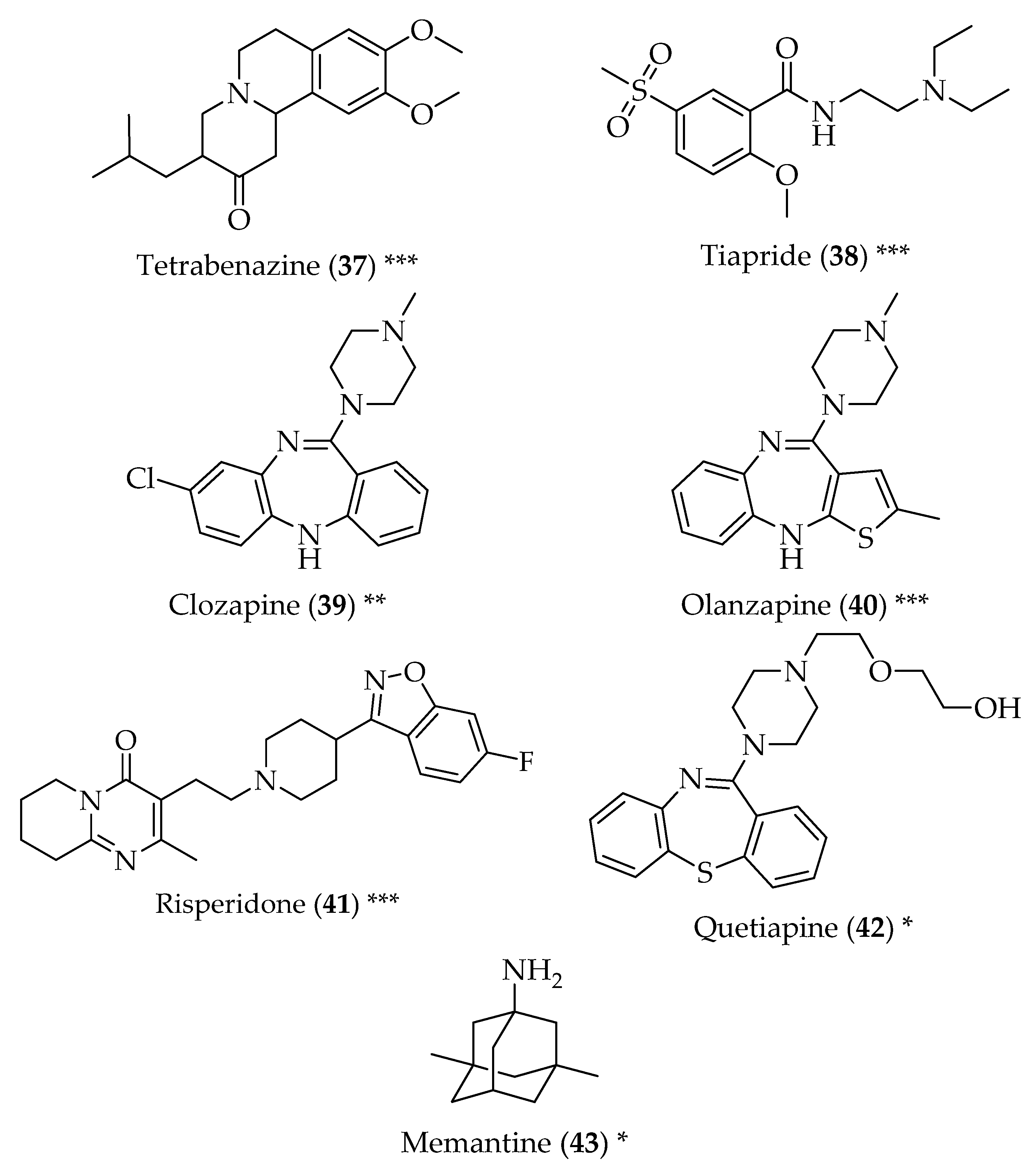
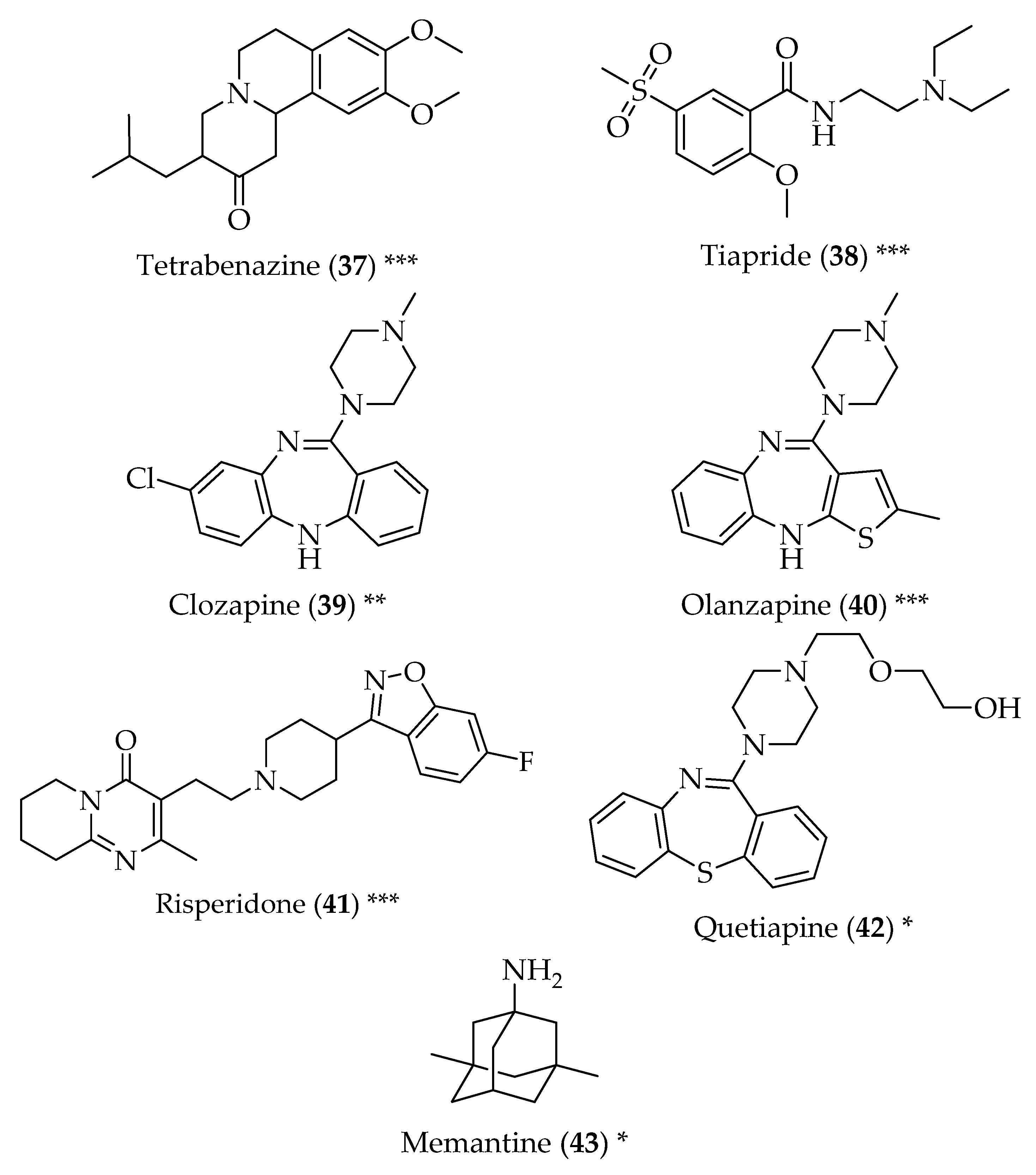
4. Huntington’s Disease
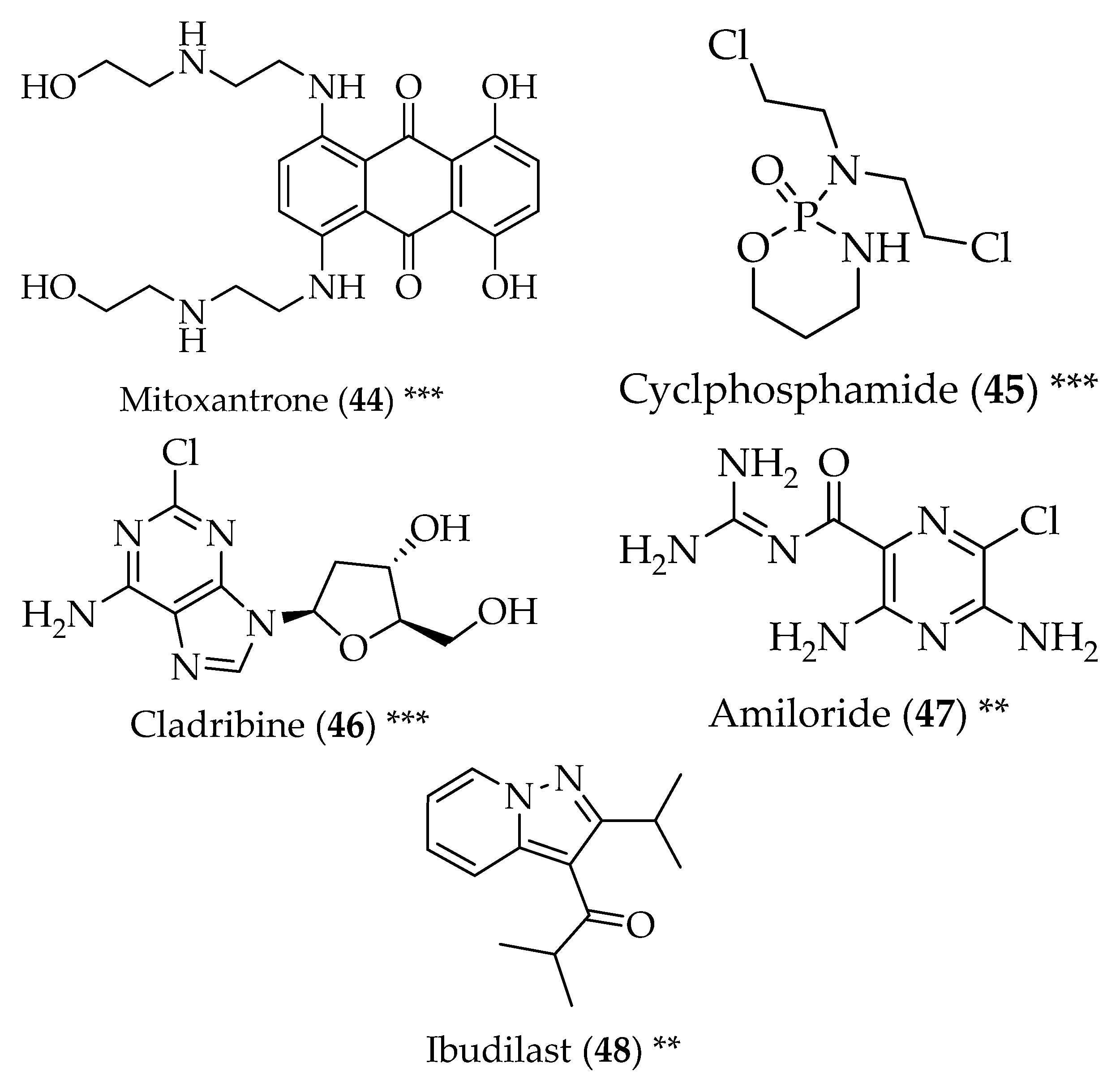
5. Multiple Sclerosis
6. Amyotrophic Lateral Sclerosis
7. Failed Repurposing
8. Conclusions
Funding
Conflicts of Interest
References
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Doan, T.L.; Pollastri, M.; Walters, M.A.; Georg, G.I. Chapter 23—The future of drug repositioning: Old drugs, new opportunities. In Annual Reports in Medicinal Chemistry; Macor, J.E., Ed.; Academic Press: Waltham, MA, USA, 2011; Volume 46, pp. 385–401. [Google Scholar]
- Fava, M. The promise and challenges of drug repurposing in psychiatry. World Psychiatry 2018, 17, 28–29. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.; Hideshima, T.; Anderson, K. Thalidomide: Emerging role in cancer medicine. Annu. Rev. Med. 2002, 53, 629–657. [Google Scholar] [CrossRef] [PubMed]
- Sleire, L.; Forde, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P.O. Drug repurposing in cancer. Pharmacol. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.C.; Ekins, S.; Williams, A.J.; Tropsha, A. A bibliometric review of drug repurposing. Drug Discov. Today 2018, 23, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Agnati, L.F.; Zoli, M.; Biagini, G.; Fuxe, K. Neuronal plasticity and ageing processes in the frame of the ‘red queen theory’. Acta Physiol. Scand. 1992, 145, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Agnati, L.F.; Benfenati, F.; Solfrini, V.; Biagini, G.; Fuxe, K.; Guidolin, D.; Carani, C.; Zini, I. Brain aging and neuronal plasticity. Ann. N. Y. Acad. Sci. 1992, 673, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Models Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Gammon, K. Neurodegenerative disease: Brain windfall. Nature 2014, 515, 299–300. [Google Scholar] [CrossRef] [PubMed]
- Appleby, B.S.; Nacopoulos, D.; Milano, N.; Zhong, K.; Cummings, J.L. A review: Treatment of alzheimer’s disease discovered in repurposed agents. Dement. Geriatr. Cogn. Disord. 2013, 35, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-M.; Kim, Y. Drug repurposing is a new opportunity for developing drugs against neuropsychiatric disorders. Schizophr. Res. Treat. 2016, 2016, 6378137. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Palomo Ruiz, M.D.; Perez, D.I.; Gil, C. Drugs in clinical development for the treatment of amyotrophic lateral sclerosis. Expert Opin. Investig. Drugs 2017, 26, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A. A review on alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.J. A systemic view of alzheimer disease—Insights from amyloid-beta metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 612–623. [Google Scholar] [CrossRef] [PubMed]
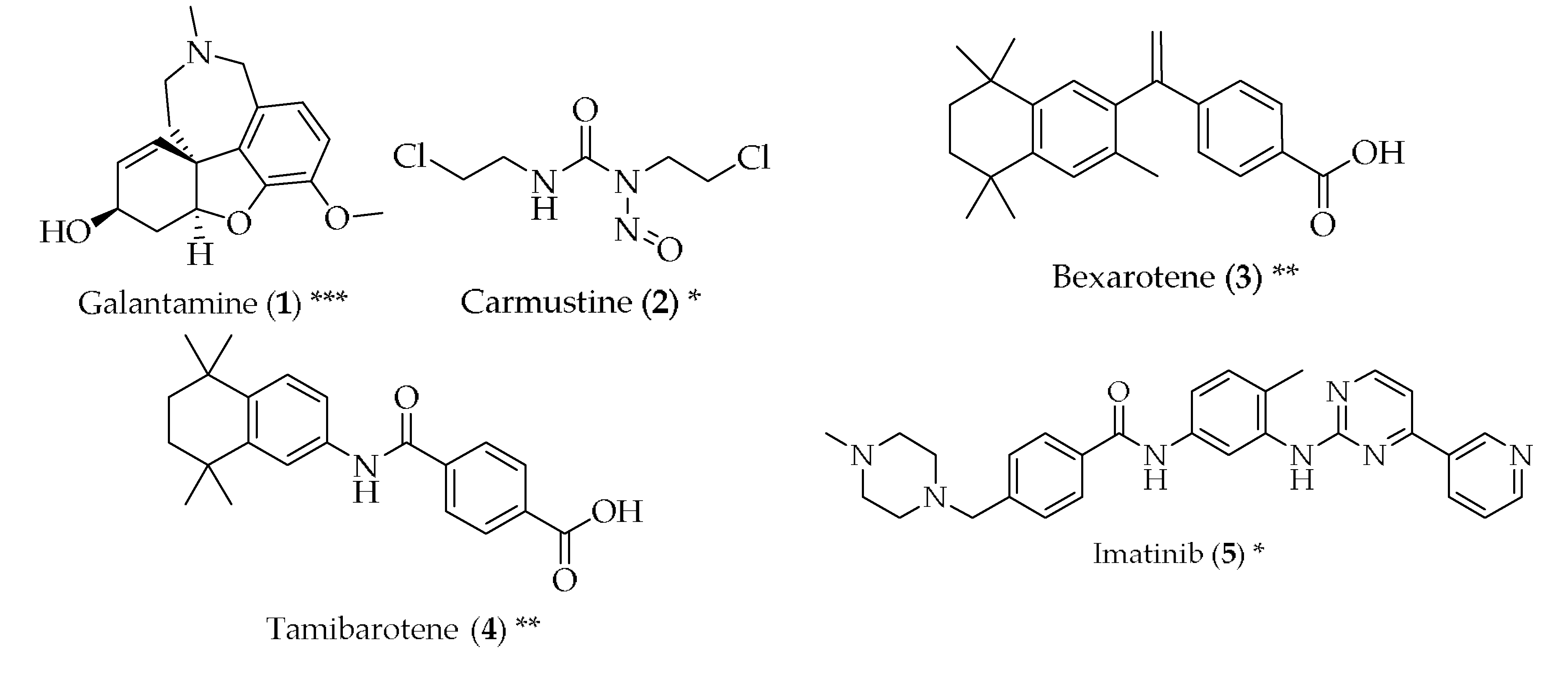
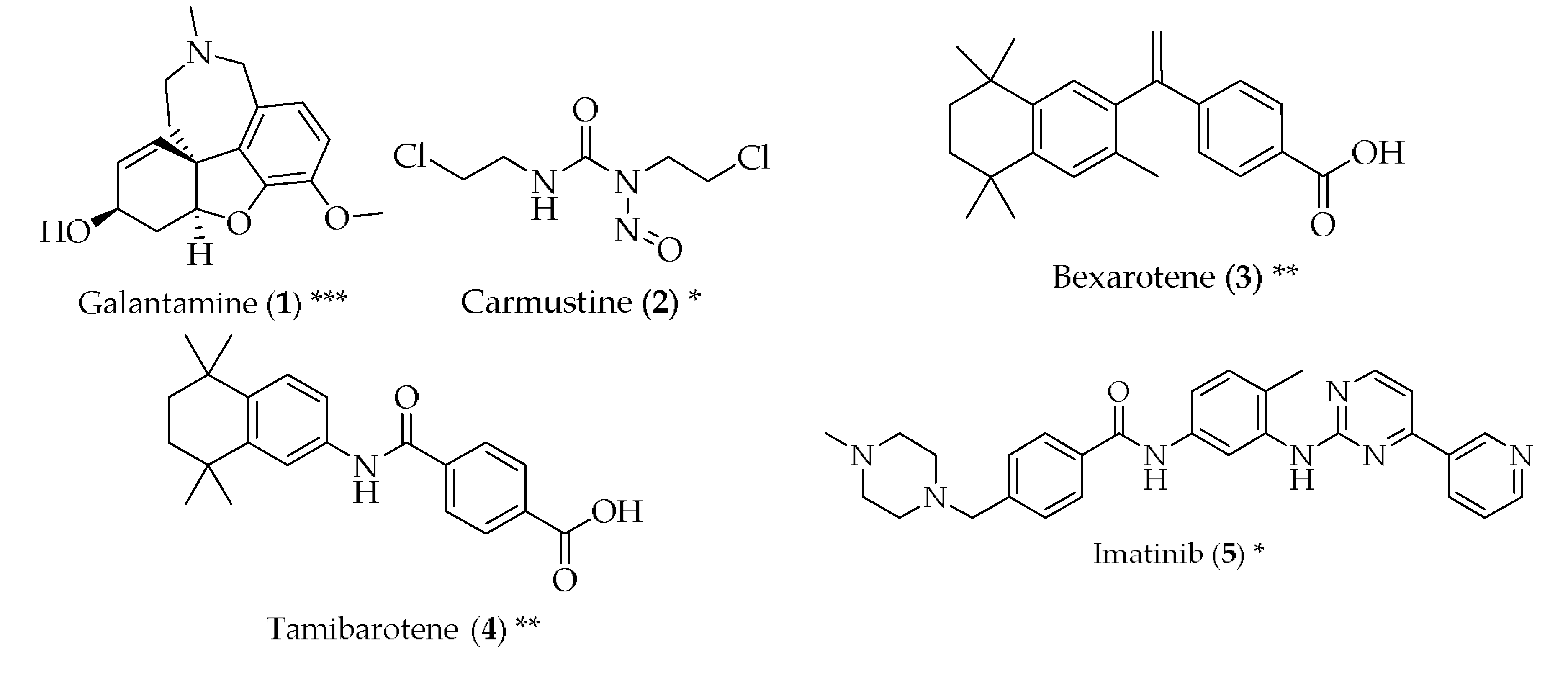
- Mucke, H.A.M. The case of galantamine: Repurposing and late blooming of a cholinergic drug. Future Sci. OA 2015, 1, FSO73. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M. Chapter 4—Galanthamine from galanthus and other amaryllidaceae—Chemistry and biology based on traditional use. In The Alkaloids: Chemistry and Biology; Cordell, G.A., Ed.; Academic Press: Waltham, MA, USA, 2010; Volume 68, pp. 157–165. [Google Scholar]
- Monacelli, F.; Cea, M.; Borghi, R.; Odetti, P.; Nencioni, A. Do cancer drugs counteract neurodegeneration? Repurposing for alzheimer’s disease. J. Alzheimers Dis. 2017, 55, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Blakeley, J.; Grossman, S.A. Chapter 17—Chemotherapy with cytotoxic and cytostatic agents in brain cancer. In Handbook of Clinical Neurology; Aminoff, M.J., Boller, F., Swaab, D.F., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 104, pp. 229–254. [Google Scholar]
- Hayes, C.D.; Dey, D.; Palavicini, J.P.; Wang, H.; Patkar, K.A.; Minond, D.; Nefzi, A.; Lakshmana, M.K. Striking reduction of amyloid plaque burden in an alzheimer’s mouse model after chronic administration of carmustine. BMC Med. 2013, 11, 81. [Google Scholar] [CrossRef] [PubMed]
- Tousi, B. The emerging role of bexarotene in the treatment of alzheimer’s disease: Current evidence. Neuropsychiatr. Dis. Treat. 2015, 11, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, H.; Nakagomi, M.; Yamagata, N.; Katsuki, H.; Kawahara, K.; Kitaoka, K.; Miki, T.; Shudo, K. Tamibarotene: A candidate retinoid drug for alzheimer’s disease. Biol. Pharm. Bull. 2012, 35, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Netzer, W.J.; Dou, F.; Cai, D.; Veach, D.; Jean, S.; Li, Y.; Bornmann, W.G.; Clarkson, B.; Xu, H.; Greengard, P. Gleevec inhibits β-amyloid production but not notch cleavage. Proc. Natl. Acad. Sci. USA 2003, 100, 12444–12449. [Google Scholar] [CrossRef] [PubMed]
- Brunden, K.R.; Yao, Y.; Potuzak, J.S.; Ferrer, N.I.; Ballatore, C.; James, M.J.; Hogan, A.M.; Trojanowski, J.Q.; Smith, A.B., III; Lee, V.M. The characterization of microtubule-stabilizing drugs as possible therapeutic agents for alzheimer’s disease and related tauopathies. Pharmacol. Res. 2011, 63, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Maiti, A.; Shively, S.; Lakhani, F.; McDonald-Jones, G.; Bruce, J.; Lee, E.B.; Xie, S.X.; Joyce, S.; Li, C.; et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc. Natl. Acad. Sci. USA 2005, 102, 227–231. [Google Scholar] [CrossRef] [PubMed]
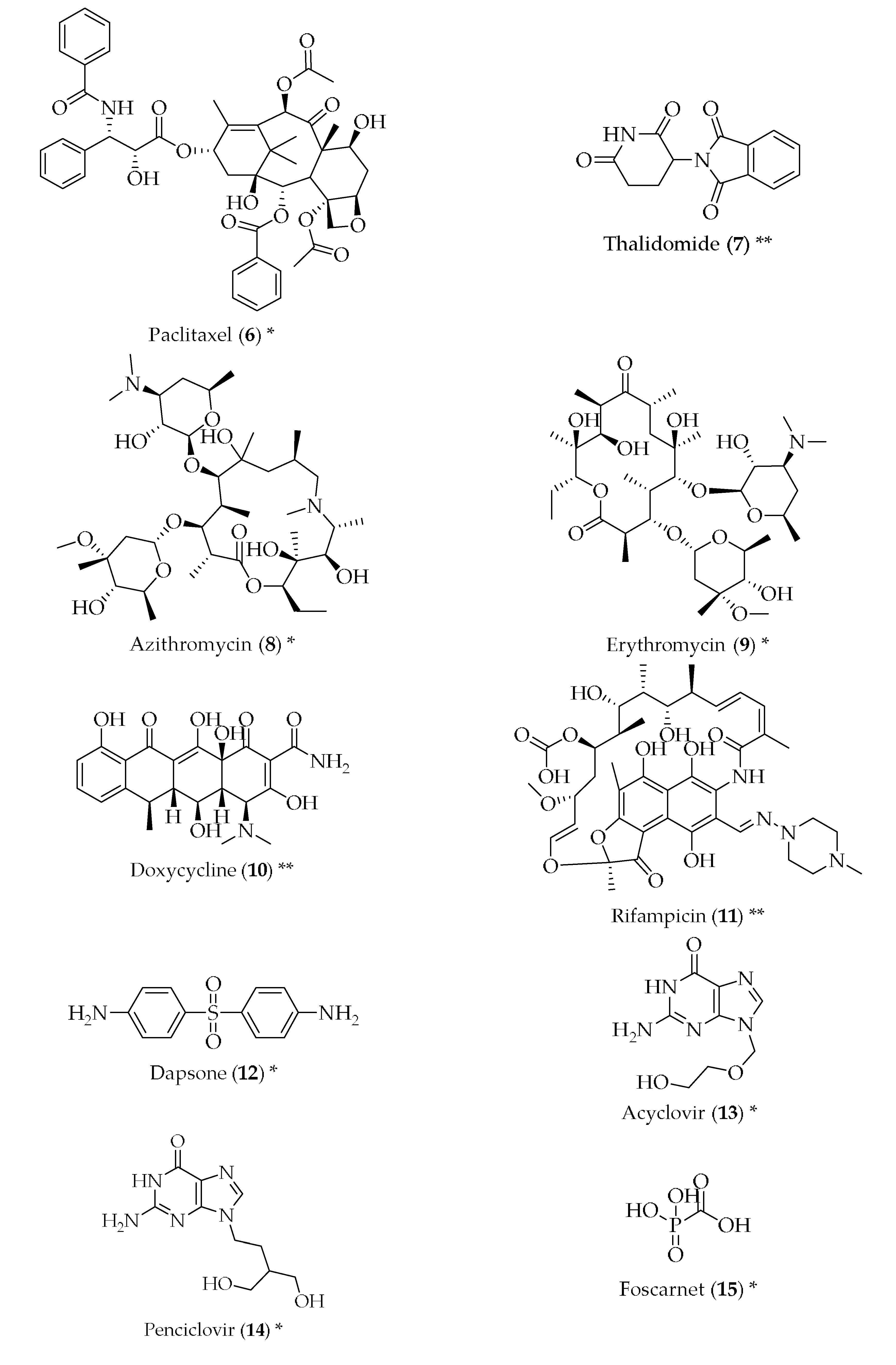
- Ryu, J.K.; McLarnon, J.G. Thalidomide inhibition of perturbed vasculature and glial-derived tumor necrosis factor-alpha in an animal model of inflamed alzheimer’s disease brain. Neurobiol. Dis. 2008, 29, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Diomede, L.; Cassata, G.; Fiordaliso, F.; Salio, M.; Ami, D.; Natalello, A.; Doglia, S.M.; De Luigi, A.; Salmona, M. Tetracycline and its analogues protect caenorhabditis elegans from beta amyloid-induced toxicity by targeting oligomers. Neurobiol. Dis. 2010, 40, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.; Speretta, E.; Crowther, D.C.; Cardoso, I. Testing the therapeutic potential of doxycycline in a drosophila melanogaster model of alzheimer disease. J. Biol. Chem. 2011, 286, 41647–41655. [Google Scholar] [CrossRef] [PubMed]
- Loeb, M.B.; Molloy, D.W.; Smieja, M.; Standish, T.; Goldsmith, C.H.; Mahony, J.; Smith, S.; Borrie, M.; Decoteau, E.; Davidson, W.; et al. A randomized, controlled trial of doxycycline and rifampin for patients with alzheimer’s disease. J. Am. Geriatr. Soc. 2004, 52, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Molloy, D.W.; Standish, T.I.; Zhou, Q.; Guyatt, G. A multicenter, blinded, randomized, factorial controlled trial of doxycycline and rifampin for treatment of alzheimer’s disease: The darad trial. Int. J. Geriatr. Psychiatry 2013, 28, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Shoji, A.; Kataoka, K.; Suwa, Y.; Asano, S.; Kaneko, H.; Endo, N. Inhibition of amyloid beta protein aggregation and neurotoxicity by rifampicin. Its possible function as a hydroxyl radical scavenger. J. Biol. Chem. 1996, 271, 6839–6844. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Goto, M. Existence of senile plaques in the brains of elderly leprosy patients. Lancet 1993, 342, 1364. [Google Scholar] [CrossRef]
- Chui, D.H.; Tabira, T.; Izumi, S.; Koya, G.; Ogata, J. Decreased beta-amyloid and increased abnormal tau deposition in the brain of aged patients with leprosy. Am. J. Pathol. 1994, 145, 771–775. [Google Scholar] [PubMed]
- Goto, M.; Kimura, T.; Hagio, S.; Ueda, K.; Kitajima, S.; Tokunaga, H.; Sato, E. Neuropathological analysis of dementia in a japanese leprosarium. Dementia 1995, 6, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Endoh, M.; Kunishita, T.; Tabira, T. No effect of anti-leprosy drugs in the prevention of alzheimer’s disease and beta-amyloid neurotoxicity. J. Neurol. Sci. 1999, 165, 28–30. [Google Scholar] [CrossRef]
- Wozniak, M.A.; Itzhaki, R.F. Antiviral agents in alzheimer’s disease: Hope for the future? Ther. Adv. Neurol. Disord. 2010, 3, 141–152. [Google Scholar] [CrossRef] [PubMed]
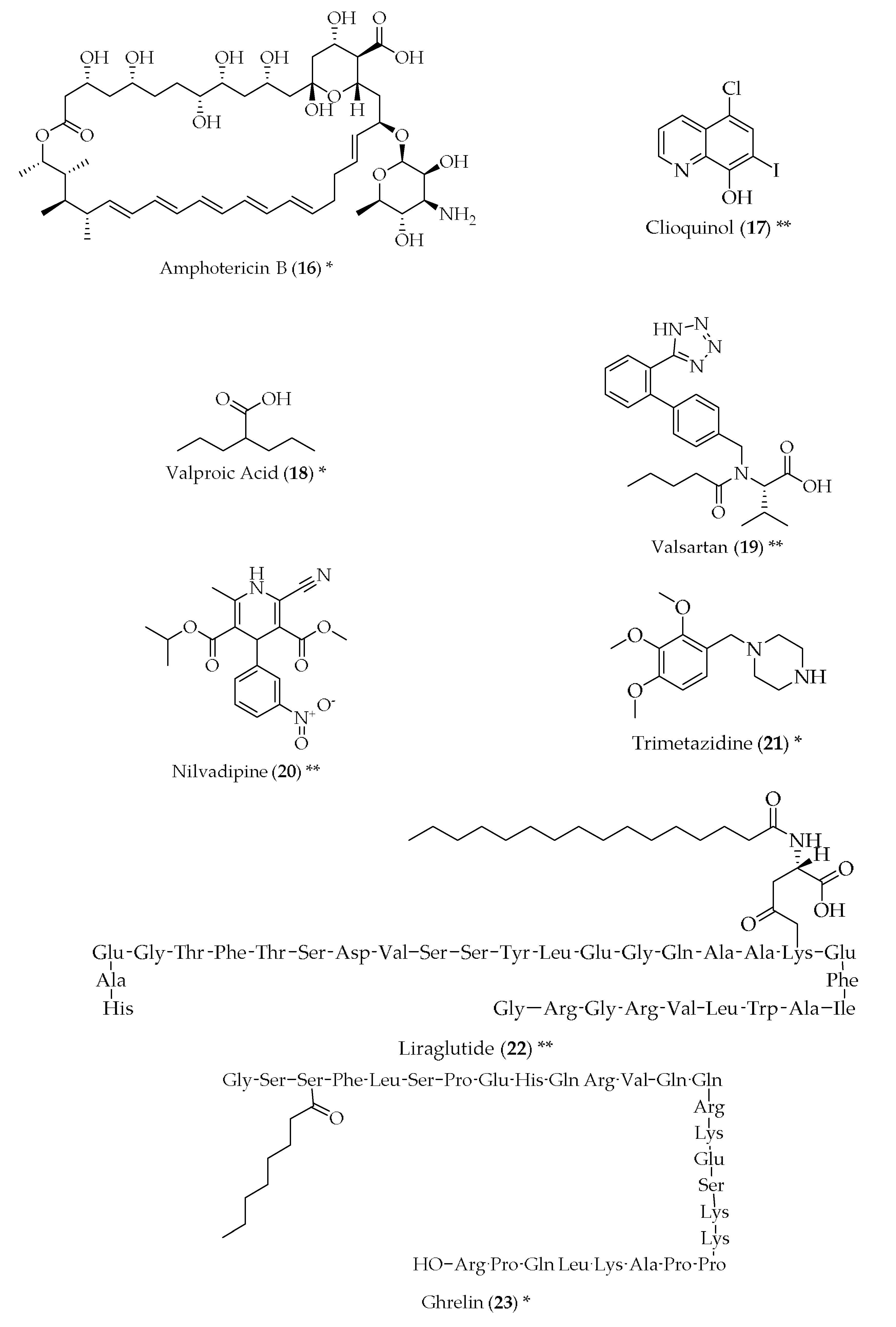
- Hartsel, S.C.; Weiland, T.R. Amphotericin b binds to amyloid fibrils and delays their formation: A therapeutic mechanism? Biochemistry 2003, 42, 6228–6233. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.W.; Annunziata, O.; Dzyuba, S.V. Amphotericin b interactions with soluble oligomers of amyloid abeta1-42 peptide. Bioorg. Med. Chem. 2009, 17, 2366–2370. [Google Scholar] [CrossRef] [PubMed]
- Grossi, C.; Francese, S.; Casini, A.; Rosi, M.C.; Luccarini, I.; Fiorentini, A.; Gabbiani, C.; Messori, L.; Moneti, G.; Casamenti, F. Clioquinol decreases amyloid-beta burden and reduces working memory impairment in a transgenic mouse model of alzheimer’s disease. J. Alzheimers Dis. 2009, 17, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Mark, R.J.; Ashford, J.W.; Goodman, Y.; Mattson, M.P. Anticonvulsants attenuate amyloid beta-peptide neurotoxicity, Ca2+ deregulation, and cytoskeletal pathology. Neurobiol. Aging 1995, 16, 187–198. [Google Scholar] [CrossRef]
- Smith, A.M.; Gibbons, H.M.; Dragunow, M. Valproic acid enhances microglial phagocytosis of amyloid-beta(1–42). Neuroscience 2010, 169, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Qing, H.; He, G.; Ly, P.T.; Fox, C.J.; Staufenbiel, M.; Cai, F.; Zhang, Z.; Wei, S.; Sun, X.; Chen, C.H.; et al. Valproic acid inhibits abeta production, neuritic plaque formation, and behavioral deficits in alzheimer’s disease mouse models. J. Exp. Med. 2008, 205, 2781–2789. [Google Scholar] [CrossRef] [PubMed]
- Tariot, P.N.; Schneider, L.S.; Cummings, J.; Thomas, R.G.; Raman, R.; Jakimovich, L.J.; Loy, R.; Bartocci, B.; Fleisher, A.; Ismail, M.S.; et al. Chronic divalproex sodium to attenuate agitation and clinical progression of alzheimer disease. Arch. Gen. Psychiatry 2011, 68, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Culman, J.; Blume, A.; Gohlke, P.; Unger, T. The renin-angiotensin system in the brain: Possible therapeutic implications for at(1)-receptor blockers. J. Hum. Hypertens. 2002, 16 (Suppl. 3), S64–S70. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.W.; Harding, J.W. Brain renin-angiotensin—A new look at an old system. Prog. Neurobiol. 2011, 95, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ho, L.; Chen, L.; Zhao, Z.; Zhao, W.; Qian, X.; Humala, N.; Seror, I.; Bartholomew, S.; Rosendorff, C.; et al. Valsartan lowers brain β-amyloid protein levels and improves spatial learning in a mouse model of alzheimer disease. J. Clin. Investig. 2007, 117, 3393–3402. [Google Scholar] [CrossRef] [PubMed]
- Li, N.C.; Lee, A.; Whitmer, R.A.; Kivipelto, M.; Lawler, E.; Kazis, L.E.; Wolozin, B. Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: Prospective cohort analysis. BMJ 2010, 340, b5465. [Google Scholar] [CrossRef] [PubMed]
- Danielyan, L.; Klein, R.; Hanson, L.R.; Buadze, M.; Schwab, M.; Gleiter, C.H.; Frey, W.H. Protective effects of intranasal losartan in the app/ps1 transgenic mouse model of alzheimer disease. Rejuvenation Res. 2010, 13, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Hanyu, H.; Hirao, K.; Shimizu, S.; Iwamoto, T.; Koizumi, K.; Abe, K. Favourable effects of nilvadipine on cognitive function and regional cerebral blood flow on spect in hypertensive patients with mild cognitive impairment. Nucl. Med. Commun. 2007, 28, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, J.; Ho, L.; Ono, K.; Teplow, D.B.; Pasinetti, G.M. Identification of antihypertensive drugs which inhibit amyloid-beta protein oligomerization. J. Alzheimers Dis. 2009, 16, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Bachmeier, C.; Beaulieu-Abdelahad, D.; Mullan, M.; Paris, D. Selective dihydropyiridine compounds facilitate the clearance of beta-amyloid across the blood-brain barrier. Eur. J. Pharmacol. 2011, 659, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Zhu, X.X.; Ding, Y.H.; Jin, Q.Y.; Qian, L.Y.; Huang, D.S.; Cen, X.J. Trimetazidine in conditions other than coronary disease, old drug, new tricks? Int. J. Cardiol. 2017, 234, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hassanzadeh, G.; Hosseini, A.; Pasbakhsh, P.; Akbari, M.; Ghaffarpour, M.; Takzare, N.; Zahmatkesh, M. Trimetazidine prevents oxidative changes induced in a rat model of sporadic type of alzheimer’s disease. Acta Med. Iran. 2015, 53, 17–24. [Google Scholar] [PubMed]
- Carro, E.; Torres-Aleman, I. The role of insulin and insulin-like growth factor i in the molecular and cellular mechanisms underlying the pathology of alzheimer’s disease. Eur. J. Pharmacol. 2004, 490, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.S.; Craft, S. Modulation of memory by insulin and glucose: Neuropsychological observations in alzheimer’s disease. Eur. J. Pharmacol. 2004, 490, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q.; Chen, H.; Quon, M.J.; Alkon, D.L. Insulin and the insulin receptor in experimental models of learning and memory. Eur. J. Pharmacol. 2004, 490, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.; Lahiri, D.K.; Sambamurti, K.; Chen, D.; Mattson, M.P.; Egan, J.M.; Greig, N.H. Glucagon-like peptide-1 decreases endogenous amyloid-beta peptide (abeta) levels and protects hippocampal neurons from death induced by abeta and iron. J. Neurosci. Res. 2003, 72, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.; Lahiri, D.K.; Chen, D.; Zhou, J.; Shaw, K.T.; Egan, J.M.; Greig, N.H. A novel neurotrophic property of glucagon-like peptide 1: A promoter of nerve growth factor-mediated differentiation in pc12 cells. J. Pharmacol. Exp. Ther. 2002, 300, 958–966. [Google Scholar] [CrossRef] [PubMed]
- McClean, P.L.; Parthsarathy, V.; Faivre, E.; Holscher, C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of alzheimer’s disease. J. Neurosci. 2011, 31, 6587–6594. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Zheng, C.; Wang, J.; Song, J.; Zhao, G.; Shen, H.; Deng, Y. The neuroprotection of liraglutide on alzheimer-like learning and memory impairment by modulating the hyperphosphorylation of tau and neurofilament proteins and insulin signaling pathways in mice. J. Alzheimers Dis. 2013, 37, 623–635. [Google Scholar] [PubMed]
- Wagner, J.; Vulinovic, F.; Grunewald, A.; Unger, M.M.; Moller, J.C.; Klein, C.; Michel, P.P.; Ries, V.; Oertel, W.H.; Alvarez-Fischer, D. Acylated and unacylated ghrelin confer neuroprotection to mesencephalic neurons. Neuroscience 2017, 365, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Lucchi, C.; Curia, G.; Vinet, J.; Gualtieri, F.; Bresciani, E.; Locatelli, V.; Torsello, A.; Biagini, G. Protective but not anticonvulsant effects of ghrelin and jmv-1843 in the pilocarpine model of status epilepticus. PLoS ONE 2013, 8, e72716. [Google Scholar] [CrossRef] [PubMed]
- Bulgarelli, I.; Tamiazzo, L.; Bresciani, E.; Rapetti, D.; Caporali, S.; Lattuada, D.; Locatelli, V.; Torsello, A. Desacyl-ghrelin and synthetic gh-secretagogues modulate the production of inflammatory cytokines in mouse microglia cells stimulated by beta-amyloid fibrils. J. Neurosci. Res. 2009, 87, 2718–2727. [Google Scholar] [CrossRef] [PubMed]
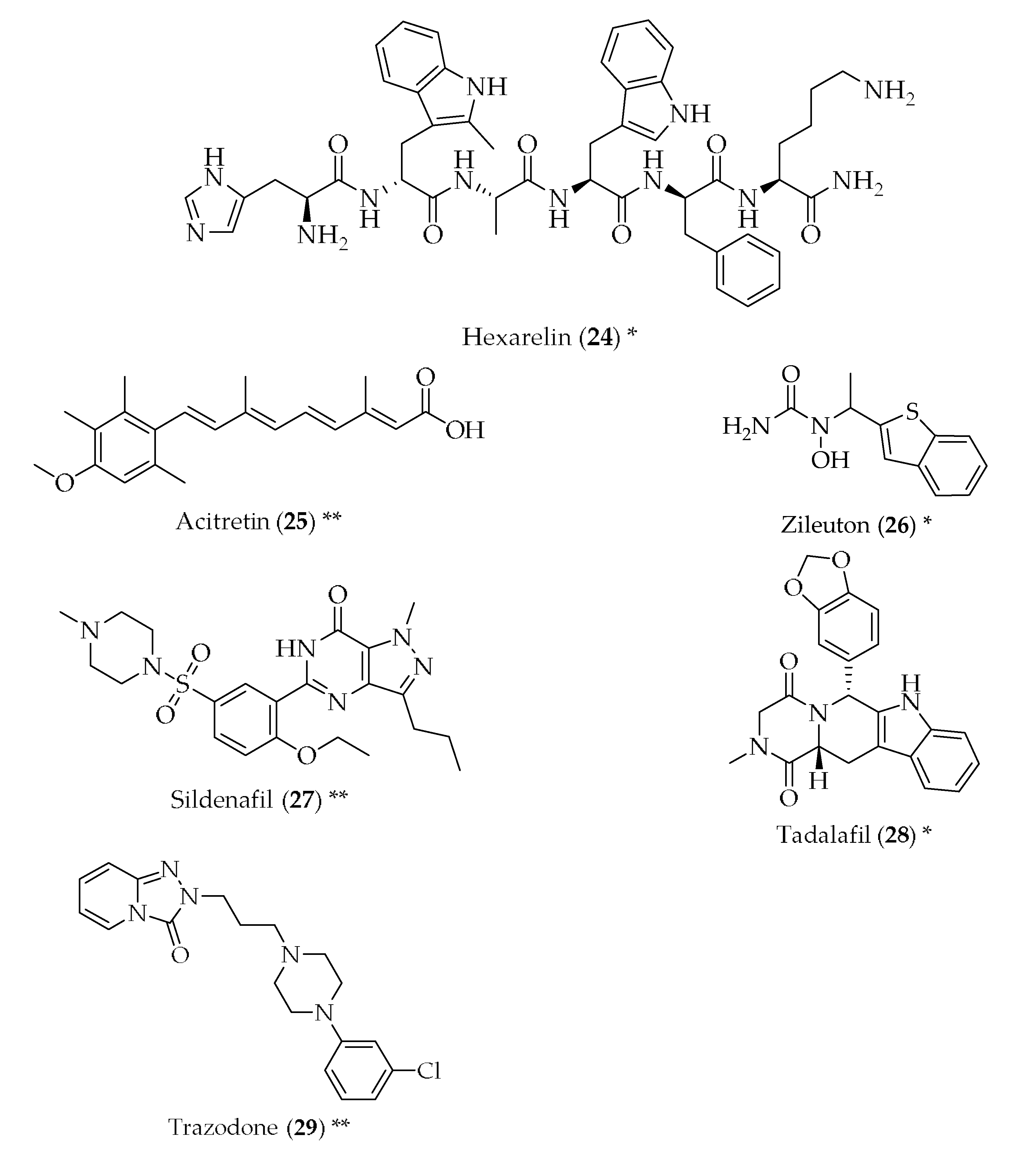
- Ding, Y.; Qiao, A.; Wang, Z.; Goodwin, J.S.; Lee, E.-S.; Block, M.L.; Allsbrook, M.; McDonald, M.P.; Fan, G.-H. Retinoic acid attenuates β-amyloid deposition and rescues memory deficits in an alzheimer’s disease transgenic mouse model. J. Neurosci. 2008, 28, 11622–11634. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, C.I.; Goncalves, M.B.; Clarke, E.; Dogruel, M.; Kalindjian, S.B.; Thomas, S.A.; Maden, M.; Corcoran, J.P. Retinoic acid receptor-alpha signalling antagonizes both intracellular and extracellular amyloid-beta production and prevents neuronal cell death caused by amyloid-beta. Eur. J. Neurosci. 2010, 32, 1246–1255. [Google Scholar] [CrossRef] [PubMed]
- Shudo, K.; Fukasawa, H.; Nakagomi, M.; Yamagata, N. Towards retinoid therapy for alzheimer’s disease. Curr. Alzheimer Res. 2009, 6, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Tippmann, F.; Hundt, J.; Schneider, A.; Endres, K.; Fahrenholz, F. Up-regulation of the alpha-secretase adam10 by retinoic acid receptors and acitretin. FASEB J. 2009, 23, 1643–1654. [Google Scholar] [CrossRef] [PubMed]
- Di Meco, A.; Lauretti, E.; Vagnozzi, A.N.; Praticò, D. Zileuton restores memory impairments and reverses amyloid and tau pathology in aged ad mice. Neurobiol. Aging 2014, 35, 2458–2464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guo, J.; Zhao, X.; Chen, Z.; Wang, G.; Liu, A.; Wang, Q.; Zhou, W.; Xu, Y.; Wang, C. Phosphodiesterase-5 inhibitor sildenafil prevents neuroinflammation, lowers beta-amyloid levels and improves cognitive performance in app/ps1 transgenic mice. Behav. Brain Res. 2013, 250, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Barroso, C.; Ricobaraza, A.; Pascual-Lucas, M.; Unceta, N.; Rico, A.J.; Goicolea, M.A.; Salles, J.; Lanciego, J.L.; Oyarzabal, J.; Franco, R.; et al. Tadalafil crosses the blood-brain barrier and reverses cognitive dysfunction in a mouse model of ad. Neuropharmacology 2013, 64, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Halliday, M.; Radford, H.; Zents, K.A.M.; Molloy, C.; Moreno, J.A.; Verity, N.C.; Smith, E.; Ortori, C.A.; Barrett, D.A.; Bushell, M.; et al. Repurposed drugs targeting eif2α-p-mediated translational repression prevent neurodegeneration in mice. Brain 2017, 140, 1768–1783. [Google Scholar] [CrossRef] [PubMed]
- Antony, P.M.; Diederich, N.J.; Kruger, R.; Balling, R. The hallmarks of parkinson’s disease. FEBS J. 2013, 280, 5981–5993. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Pagan, F.; Hebron, M.; Valadez, E.H.; Torres-Yaghi, Y.; Huang, X.; Mills, R.R.; Wilmarth, B.M.; Howard, H.; Dunn, C.; Carlson, A.; et al. Nilotinib effects in parkinson’s disease and dementia with lewy bodies. J. Parkinsons Dis. 2016, 6, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Hebron, M.L.; Lonskaya, I.; Moussa, C.E.H. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of α-synuclein in parkinson’s disease models. Hum. Mol. Genet. 2013, 22, 3315–3328. [Google Scholar] [CrossRef] [PubMed]
- González-Lizárraga, F.; Socías, S.B.; Ávila, C.L.; Torres-Bugeau, C.M.; Barbosa, L.R.S.; Binolfi, A.; Sepúlveda-Díaz, J.E.; Del-Bel, E.; Fernandez, C.O.; Papy-Garcia, D.; et al. Repurposing doxycycline for synucleinopathies: Remodelling of α-synuclein oligomers towards non-toxic parallel beta-sheet structured species. Sci. Rep. 2017, 7, 41755. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, P.E.; Anciones, B. A review of the use of zonisamide in parkinson’s disease. Ther. Adv. Neurol. Disord. 2009, 2, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.H.; Katzenschlager, R.; Lim, S.Y.; Barton, B.; de Bie, R.M.A.; Seppi, K.; Coelho, M.; Sampaio, C. International parkinson and movement disorder society evidence-based medicine review: Update on treatments for the motor symptoms of parkinson’s disease. Mov. Disord. 2018. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Muller, T. Monoamine oxidase-b inhibitors in the treatment of parkinson’s disease: Clinical-pharmacological aspects. J. Neural. Transm. 2018, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Biagini, G.; Zoli, M.; Fuxe, K.; Agnati, L.F. L-deprenyl increases gfap immunoreactivity selectively in activated astrocytes in rat brain. Neuroreport 1993, 4, 955–958. [Google Scholar] [CrossRef] [PubMed]
- Biagini, G.; Frasoldati, A.; Fuxe, K.; Agnati, L.F. The concept of astrocyte-kinetic drug in the treatment of neurodegenerative diseases: Evidence for l-deprenyl-induced activation of reactive astrocytes. Neurochem. Int. 1994, 25, 17–22. [Google Scholar] [CrossRef]
- Devos, D.; Moreau, C.; Delval, A.; Dujardin, K.; Defebvre, L.; Bordet, R. Methylphenidate: A treatment for parkinson’s disease? CNS Drugs 2013, 27, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Exenatide—A drug for diabetes and parkinson disease? Nat. Rev. Neurol. 2017, 13, 643–644. [Google Scholar] [CrossRef] [PubMed]
- Athauda, D.; Wyse, R.; Brundin, P.; Foltynie, T. Is exenatide a treatment for parkinson’s disease? J. Parkinsons Dis. 2017, 7, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Aviles-Olmos, I.; Dickson, J.; Kefalopoulou, Z.; Djamshidian, A.; Ell, P.; Soderlund, T.; Whitton, P.; Wyse, R.; Isaacs, T.; Lees, A.; et al. Exenatide and the treatment of patients with parkinson’s disease. J. Clin. Investig. 2013, 123, 2730–2736. [Google Scholar] [CrossRef] [PubMed]
- Bomba, M.; Ciavardelli, D.; Silvestri, E.; Canzoniero, L.M.; Lattanzio, R.; Chiappini, P.; Piantelli, M.; Di Ilio, C.; Consoli, A.; Sensi, S.L. Exenatide promotes cognitive enhancement and positive brain metabolic changes in ps1-ki mice but has no effects in 3xtg-ad animals. Cell Death Dis. 2013, 4, e612. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Bjørnevik, K.; Im, D.S.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. Β2-adrenoreceptor is a regulator of the α-synuclein gene driving risk of parkinson’s disease. Science 2017, 357, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Prim. 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Roos, R.A. Huntington’s disease: A clinical review. Orphanet J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Paleacu, D. Tetrabenazine in the treatment of huntington’s disease. Neuropsychiatr. Dis. Treat. 2007, 3, 545–551. [Google Scholar] [PubMed]
- Roos, R.A.; Buruma, O.J.; Bruyn, G.W.; Kemp, B.; van der Velde, E.A. Tiapride in the treatment of huntington’s chorea. Acta Neurol. Scand. 1982, 65, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Bonuccelli, U.; Ceravolo, R.; Maremmani, C.; Nuti, A.; Rossi, G.; Muratorio, A. Clozapine in huntington’s chorea. Neurology 1994, 44, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Paleacu, D.; Anca, M.; Giladi, N. Olanzapine in huntington’s disease. Acta Neurol. Scand. 2002, 105, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Coppen, E.M.; Roos, R.A.C. Current pharmacological approaches to reduce chorea in huntington’s disease. Drugs 2017, 77, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Duff, K.; Beglinger, L.J.; O’Rourke, M.E.; Nopoulos, P.; Paulson, H.L.; Paulson, J.S. Risperidone and the treatment of psychiatric, motor, and cognitive symptoms in huntington’s disease. Ann. Clin. Psychiatry 2008, 20, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Alpay, M.; Koroshetz, W.J. Quetiapine in the treatment of behavioral disturbances in patients with huntington’s disease. Psychosomatics 2006, 47, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Beister, A.; Kraus, P.; Kuhn, W.; Dose, M.; Weindl, A.; Gerlach, M. The n-methyl-d-aspartate antagonist memantine retards progression of huntington’s disease. J. Neural Transm. Suppl. 2004, 68, 117–122. [Google Scholar]
- Anitha, M.; Nandhu, M.S.; Anju, T.R.; Jes, P.; Paulose, C.S. Targeting glutamate mediated excitotoxicity in huntington’s disease: Neural progenitors and partial glutamate antagonist--memantine. Med. Hypotheses 2011, 76, 138–140. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, M.M. Multiple sclerosis review. P&T 2012, 37, 175–184. [Google Scholar]
- Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Hrynchak, I.; Sousa, E.; Pinto, M.; Costa, V.M. The importance of drug metabolites synthesis: The case-study of cardiotoxic anticancer drugs. Drug Metab. Rev. 2017, 49, 158–196. [Google Scholar] [CrossRef] [PubMed]
- Hartung, H.-P.; Gonsette, R.; Konig, N.; Kwiecinski, H.; Guseo, A.; Morrissey, S.P.; Krapf, H.; Zwingers, T. Mitoxantrone in progressive multiple sclerosis: A placebo-controlled, double-blind, randomised, multicentre trial. Lancet 2002, 360, 2018–2025. [Google Scholar] [CrossRef]
- Awad, A.; Stuve, O. Cyclophosphamide in multiple sclerosis: Scientific rationale, history and novel treatment paradigms. Ther. Adv. Neurol. Disord. 2009, 2, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Leist, T.P.; Weissert, R. Cladribine: Mode of action and implications for treatment of multiple sclerosis. Clin. Neuropharmacol. 2011, 34, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Holmoy, T.; Torkildsen, O.; Myhr, K.M. An update on cladribine for relapsing-remitting multiple sclerosis. Expert Opin. Pharmacother. 2017, 18, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Arun, T.; Tomassini, V.; Sbardella, E.; de Ruiter, M.B.; Matthews, L.; Leite, M.I.; Gelineau-Morel, R.; Cavey, A.; Vergo, S.; Craner, M.; et al. Targeting asic1 in primary progressive multiple sclerosis: Evidence of neuroprotection with amiloride. Brain 2013, 136, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Barkhof, F.; Hulst, H.E.; Drulovic, J.; Uitdehaag, B.M.; Matsuda, K.; Landin, R. Ibudilast in relapsing-remitting multiple sclerosis: A neuroprotectant? Neurology 2010, 74, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Zoccolella, S.; Beghi, E.; Palagano, G.; Fraddosio, A.; Guerra, V.; Samarelli, V.; Lepore, V.; Simone, I.L.; Lamberti, P.; Serlenga, L.; et al. Riluzole and amyotrophic lateral sclerosis survival: A population-based study in southern italy. Eur. J. Neurol. 2007, 14, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Sawada, H. Clinical efficacy of edaravone for the treatment of amyotrophic lateral sclerosis. Expert Opin. Pharmacother. 2017, 18, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Trias, E.; Ibarburu, S.; Barreto-Núñez, R.; Babdor, J.; Maciel, T.T.; Guillo, M.; Gros, L.; Dubreuil, P.; Díaz-Amarilla, P.; Cassina, P.; et al. Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J. Neuroinflamm. 2016, 13, 177. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A. Tamoxifen, a cancer therapy, explored for als. Neurol. Today 2005, 5, 22–26. [Google Scholar] [CrossRef]
- Hu, J.H.; Zhang, H.; Wagey, R.; Krieger, C.; Pelech, S.L. Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J. Neurochem. 2003, 85, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.F.; Guo, B.S.; Liu, Y.C.; Wu, C.C.; Yang, C.H.; Tsai, K.J.; Shen, C.K. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the tar DNA-binding protein 43. Proc. Natl. Acad. Sci. USA 2012, 109, 15024–15029. [Google Scholar] [CrossRef] [PubMed]
- Bezprozvanny, I. The rise and fall of dimebon. Drug News Perspect. 2010, 23, 518–523. [Google Scholar] [PubMed]
- Bharadwaj, P.R.; Bates, K.A.; Porter, T.; Teimouri, E.; Perry, G.; Steele, J.W.; Gandy, S.; Groth, D.; Martins, R.N.; Verdile, G. Latrepirdine: Molecular mechanisms underlying potential therapeutic roles in alzheimer’s and other neurodegenerative diseases. Transl. Psychiatry 2013, 3, e332. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Gavrilova, S.I.; Sano, M.; Thomas, R.G.; Aisen, P.S.; Bachurin, S.O.; Seely, L.; Hung, D. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate alzheimer’s disease: A randomised, double-blind, placebo-controlled study. Lancet 2008, 372, 207–215. [Google Scholar] [CrossRef]
- Cano-Cuenca, N.; Solis-Garcia del Pozo, J.E.; Jordan, J. Evidence for the efficacy of latrepirdine (dimebon) treatment for improvement of cognitive function: A meta-analysis. J. Alzheimers Dis. 2014, 38, 155–164. [Google Scholar] [PubMed]
- Sano, M.; Bell, K.L.; Galasko, D.; Galvin, J.E.; Thomas, R.G.; van Dyck, C.H.; Aisen, P.S. A randomized, double-blind, placebo-controlled trial of simvastatin to treat alzheimer disease. Neurology 2011, 77, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Sparks, D.L.; Sabbagh, M.N.; Connor, D.J.; Lopez, J.; Launer, L.J.; Petanceska, S.; Browne, P.; Wassar, D.; Johnson-Traver, S.; Lochhead, J.; et al. Atorvastatin therapy lowers circulating cholesterol but not free radical activity in advance of identifiable clinical benefit in the treatment of mild-to-moderate ad. Curr. Alzheimer Res. 2005, 2, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Nebes, R.D.; Pollock, B.G.; Houck, P.R.; Butters, M.A.; Mulsant, B.H.; Zmuda, M.D.; Reynolds, C.F., 3rd. Persistence of cognitive impairment in geriatric patients following antidepressant treatment: A randomized, double-blind clinical trial with nortriptyline and paroxetine. J. Psychiatr. Res. 2003, 37, 99–108. [Google Scholar] [CrossRef]
- Cudkowicz, M.E.; Titus, S.; Kearney, M.; Yu, H.; Sherman, A.; Schoenfeld, D.; Hayden, D.; Shui, A.; Brooks, B.; Conwit, R.; et al. Efficacy and safety of ceftriaxone for amyotrophic lateral sclerosis: Results of a multi-stage, randomised, double-blind, placebo-controlled, phase 3 study. Lancet 2014, 13, 1083–1091. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Durães, F.; Pinto, M.; Sousa, E. Old Drugs as New Treatments for Neurodegenerative Diseases. Pharmaceuticals 2018, 11, 44. https://doi.org/10.3390/ph11020044
Durães F, Pinto M, Sousa E. Old Drugs as New Treatments for Neurodegenerative Diseases. Pharmaceuticals. 2018; 11(2):44. https://doi.org/10.3390/ph11020044
Chicago/Turabian StyleDurães, Fernando, Madalena Pinto, and Emília Sousa. 2018. "Old Drugs as New Treatments for Neurodegenerative Diseases" Pharmaceuticals 11, no. 2: 44. https://doi.org/10.3390/ph11020044
APA StyleDurães, F., Pinto, M., & Sousa, E. (2018). Old Drugs as New Treatments for Neurodegenerative Diseases. Pharmaceuticals, 11(2), 44. https://doi.org/10.3390/ph11020044